

Abstract
There is an intensive need for the development of novel drugs for the treatment of epithelial ovarian cancer (EOC), the most lethal gynecologic malignancy due to the high recurrence rate. TP53 mutation is a common event in EOC, particularly in high-grade serous ovarian cancer, where it occurs in more than 90% of cases. Recently, PRIMA-1 and PRIMA-1MET (p53 reactivation and induction of massive apoptosis and its methylated form) were shown to have an antitumor effect on several types of cancer. Despite that PRIMA-1MET is the first compound evaluated in clinical trials, the antitumor effects of PRIMA-1MET on EOC remain unclear. In this study, we investigated the therapeutic potential of PRIMA-1MET for the treatment of EOC cells. PRIMA-1MET treatment of EOC cell lines (n=13) resulted in rapid apoptosis at various concentrations (24 h IC50 2.6–20.1 µM). The apoptotic response was independent of the p53 status and chemo-sensitivity. PRIMA-1MET treatment increased intracellular reactive oxygen species (ROS), and PRIMA-1MET-induced apoptosis was rescued by an ROS scavenger. Furthermore, RNA expression analysis revealed that the mechanism of action of PRIMA-1MET may be due to inhibition of antioxidant enzymes, such as Prx3 and GPx-1. In conclusion, our results suggest that PRIMA-1MET represents a novel therapeutic strategy for the treatment of ovarian cancer irrespective of p53 status and chemo-sensitivity.
Keywords: ovarian cancer, p53, PRIMA-1MET, ROS, antioxidant enzyme
Introduction
Each year, more than 100,000 women die of ovarian cancer worldwide (1). Epithelial ovarian cancer (EOC) accounts for the majority of all ovarian malignancies and is one of the most lethal among gynecologic malignancies among women. In many cases, the diagnosis is delayed due to its asymptomatic nature, and, as a consequence, approximately two-thirds of patients with EOC have already developed peritoneal carcinomatosis (2,3). The prognosis of EOC patients is closely related to the stage at diagnosis (4,5).
Most ovarian cancer patients are managed with surgical resection, followed by systemic chemotherapies. Despite recent advances in therapeutic agents, such as platinum-taxane combination chemotherapy, the 5-year survival rate is still less than 40% (6). EOC shows an unfavorable oncologic outcome, based on its asymptomatic features at an earlier clinical stage, and numerous intraperitoneal and/or distant metastases. Despite the relatively high susceptibility of EOC to paclitaxel plus platinum compounds, which are first-line chemotherapeutic agents against EOC, the intrinsic or acquired resistance of tumor cells to these chemotherapies makes the treatment of EOC difficult. In order to overcome chemo-resistance, various additional molecular-targeting therapies combined with conventional anti-neoplastic agents have been developed. High-grade serous ovarian cancer (HGSOC), which is observed much more frequently at an advanced stage, comprises approximately 60% of all histological subtypes of EOC. Recent studies have revealed that most cases of HGSOC carry TP53 mutations, in contrast to other types of EOC, which have a much lower incidence of TP53 mutations (7–9). A recent study using high-throughput sequencing technology revealed that TP53 mutations occurred in 96% of 316 HGSOC samples (10). This suggests that somatic mutation of TP53 is a nearly universal event in HGSOC.
TP53 is located on chromosome 17p and encodes the p53 protein. Wild-type p53 functions predominately as a transcriptional factor, with a potent tumor-suppressive function via its multiple activities, including induction of cell cycle arrest, apoptosis, differentiation and senescence (11). Recent studies have shown that missense TP53 mutations not only eliminate their own tumor-suppressive function, but also gain oncogenic properties that promote tumor growth, termed gain-of-function (GOF) (12-14). Furthermore, TP53 mutations may be associated with poor prognosis and malignant phenotypes in several types of cancers, including EOC (15–19). Considering the universality of the TP53 mutation in EOC, several novel drugs restoring the p53 pathway have been widely investigated to be utilized in cancer therapy.
PRIMA-1 (p53 reactivation and induction of massive apoptosis) and its methylated form PRIMA-1MET are small molecules that can convert mutant p53 to an active conformation, which restores wild-type functions of p53 in several types of cancers, such as breast, neck and thyroid cancer and melanoma (20–23). PRIMA-1MET is one of the most promising drugs for clinical use to restore wild-type functions to mutant p53 (24). Although PRIMA-1MET is the first compound of such drugs evaluated in clinical trials, the antitumor effects of PRIMA-1MET on EOC remain unclear.
In our present study, we investigated whether PRIMA-1MET induces growth suppression and apoptosis in EOC cells. Using EOC cells with either wild-type or mutant p53, and with either chemo-sensitivity or chemo-resistance, we demonstrated that PRIMA-1MET was able to effectively induce cell death. Therefore, PRIMA-1MET can be a promising therapeutic strategy to induce cytotoxic effects and reactivate the p53 pathway in EOC, particularly in HGSOC.
Materials and methods
Cell culture
Human EOC lines, A2780, OVCAR-3, ES-2, SKOV-3, CaOV-3, TOV21G and OV-90, were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). NOS2 and NOS3 cell lines derived from serous EOC were previously established in our institute (25). The NOS2CR and NOS2TR cells with chronic resistance to cisplatin and paclitaxel were previously established from the parental NOS2 cells in our institute (26). Furthermore, we recently established another two chronic cisplatin/paclitaxel-resistant cell lines from the parental NOS3 cells: NOS3CR (cisplatin) and NOS3TR (paclitaxel). All EOC cell lines were maintained at 37°C with 5% CO2 in RPMI-1640 medium (Sigma) supplemented with 10% fetal bovine serum (FBS), streptomycin (100 µg/ml), and penicillin (100 U/ml). PRIMA-1MET was purchased from Santa Cruz Biotechnology, Inc. PRIMA-1MET was diluted in dimethyl sulfoxide (DMSO) to create a 50-mmol/l stock solution and stored at −20°C. Antibodies to p53 (610184) were purchased from BD Pharmingen. Antibodies to cleaved-PARP, PARP and β-actin were purchased from Cell Signaling Technology.
Direct sequencing of TP53 mutations
Genomic DNA was extracted from the NOS2 and NOS3 cells using the Genomic DNA purification kit (Promega, Madison, WI, USA). The exons and flanking introns of TP53 were amplified by polymerase chain reaction (PCR). The primers used are shown in Table I. The resulting PCR products were sequenced and the mutation status was confirmed.
Table I.
The specific primers used for direct sequencing of the TP53 gene.
| Primer | Sequence | Length (bp) |
|---|---|---|
| p53 | ||
| Exon 2–4 | F: GTGTCTCATGCTGGATCCCCACT | 23 |
| R: GGATACGGCCAGGCATTGAAGT | 22 | |
| Exon 5–6 | F: TGCAGGAGGTGCTTACGCATGT | 22 |
| R: CCTTAACCCCTCCTCCCAGAGAC | 23 | |
| Exon 7–9 | F: ACAGGTCTCCCCAAGGCGCACT | 22 |
| R: TTGAGGCATCACTGCCCCCTGAT | 23 | |
| Exon 10 | F: GTCAGCTGTATAGGTACTTGAAGTGCAG | 28 |
| R: GCTCTGGGCTGGGAGTTGCG | 20 | |
| Exon 11 | F: CCTTAGGCCCTTCAAAGCATTGGTCA | 26 |
| R: GTGCTTCTGACGCACACCTATTGCAAG | 27 |
F, forward; R, reverse.
RNA extraction
RNA extraction from the cells was undertaken using the Qiagen RNeasy Mini kit according to the manufacturer's protocols. The cells were lysed in 250 µl of buffer RLT and filtered through the filtration spin column. The samples were applied to the RNeasy Mini spin column. Total RNA bound to the membrane and contaminants were removed by washing consecutively with buffers RW1 and RPE. RNA was eluted in RNase-free water. Extracted RNA was immediately stored at −80°C. The RNA concentration was determined with the NanoDrop 1000 spectrophotometer.
Reverse transcription
To obtain complementary DNA (cDNA), 1 µg of RNA and 0.2 µg of random primers (Promega) were used. After incubation at 72°C for 4 min, the mixture of RNA and random primers were placed on ice for 4 min. M-MLV RT 1X reaction buffer, M-MLV Reverse Transcriptase RNase Minus, and 10 mM dNTP (Promega) were added to the mixture and then incubated at 42°C for 90 min, followed by 70°C for 15 min. cDNA was stored at −20°C.
Quantitative real-time PCR (qRT-PCR)
Quantitative RT-PCR (qRT-PCR) was performed on the Takara PCR thermal cycler using the SYBR Green detection system (Takara, Tokyo, Japan). Cycling conditions consisted of a 3-min hot start at 95°C, followed by 40 cycles of denaturation at 95°C for 10 sec, annealing at 58–60°C for 10 sec, extension at 72°C for 10 sec, and then a final inactivation at 95°C for 10 sec. Dissociation curve analyses were carried out at the end of the cycling to confirm that one specific product was measured in each reaction. Relative quantification was performed using the ΔΔCT method (27). Expression normalization was conducted by the expression of GAPDH, a housekeeping gene shown to have stable expression in cancer cell lines (28). The specific primers for each gene are shown in Table II. All experiments were performed in triplicate.
Table II.
The specific primers used for quantitative real-time RT-PCR.
| Primer | Sequence | Length (bp) |
|---|---|---|
| PRX3 | F: GACGCTCAAATGCTTGATGA | 20 |
| R: GATTTCCCGAGACTACGGTG | 20 | |
| GPX1 | F: AAGAGCATGAAGTTGGGCTC | 20 |
| R: CAACCAGTTTGGGCATCAG | 19 | |
| GAPDH | F: TTGGTATCGTGGAAGGACTCA | 22 |
| R: TGTCATCATATTTGGCAGGTT | 21 |
F, forward; R, reverse.
Protein extraction and western blot analysis
Cultured ovarian cancer cells were washed with PBS and lysed in RIPA buffer (Millipore). The cells were scrapped into lysis buffer, centrifuged at 12,000 × g at 4°C for 15 min, and then diluted in 2X sample buffer [125 mM Tris-HCl (pH 6.8), 4% SDS, 10% glycerol, 0.01% bromophenol blue, and 10% 2-mercaptoethanol]. Equal amounts of protein (10 µg) were mixed with the 2X sample buffer and were boiled at 95°C for 5 min. The samples were loaded and separated by 7.5–15% SDS-polyacrylamide gel electrophoresis (PAGE) with running buffer. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 1% skim milk, incubated with each primary antibody overnight at 4°C, washed with TBS-T buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05% Tween-20) and incubated with the secondary antibodies. The proteins were visualized using enhanced chemiluminescence (GE Healthcare Bio-Sciences, Uppsala, Sweden).
Cell viability assay
The effect of PRIMA-1MET on the viability of human EOC cells was evaluated with the CellTiter-Glo Luminescent Cell Viability Assay (Promega), which quantifies living cells by ATP signal intensity. The luminescent signal was determined with a luminometer. Cells were seeded in triplicate in 96-well plates at a density of 2,000 cells/well. After a 24-h culture, the cells were treated with various concentrations of PRIMA-1MET, and then incubated for 24–72 h. Control cells were treated with the same concentration of DMSO as that of the PRIMA-1MET-treated cells.
Detection of apoptosis by staining with Annexin V-FITC and propidium iodide
Cells (2×105) were cultured in 6-well plates for 24 h before treatment with DMSO (control) or an appropriate concentration of PRIMA-1MET for 24 h. The cells were trypsinized, washed once with PBS, and then stained with Annexin V-FITC and propidium iodide (PI) to determine the early/late apoptotic cell population (MBL, Japan).
Results
Protein expression of p53 and the TP53 status in EOC cells
Firstly, we evaluated the levels of p53 protein expression in the EOC cells by western immunoblot analysis. The mutation status of TP53 in the EOC cells was acquired from previous studies. The mutation status of TP53 of NOS2 and NOS3 cells was evaluated by direct sequencing. The TP53 status of EOC cells is shown in Table III. The protein expression of p53 is shown in Fig. 1. EOC cells with wild-type p53, A2780 and NOS2, displayed a basal expression of p53 to some extent. On the contrary, we could not detect the protein expression of p53 in the EOC cells with mutant p53, except for the ES-2 cells. This result demonstrates that EOC cells bearing mutant p53 do not always express a higher level of p53 than those bearing wild-type p53.
Table III.
TP53 status of the EOC cell lines.
| Cancer cell lines | p53 status |
|---|---|
| ES-2 | S241F |
| OV-90 | S215R |
| OVCAR-3 | R248Q |
| TOV21G | Wild-type |
| A2780 | Wild-type |
| CaOV-3 | Q136Term |
| SKOV-3 | Null |
| NOS2 | Wild-type |
| NOS3 | L257P |
Figure 1.
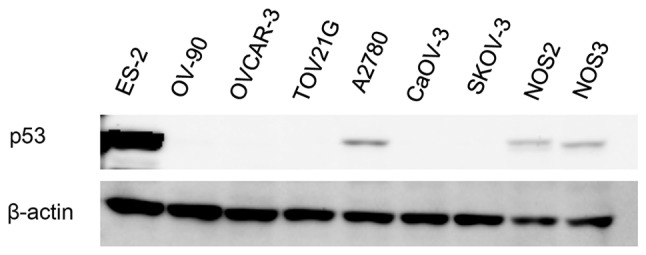
p53 protein expression in the EOC cells. EOC cells were culture in 6-well plates, and then lysed with lysis buffer. Immunoblot analysis revealed the p53 protein expression of EOC cells. The samples were loaded and separated by 10% SDS-PAGE with running buffer. The separated proteins were transferred to a PVDF membrane. After blocking with 1% skim milk, the membrane was incubated with anti-p53 or anti-β-actin at 4°C for 1 day. The proteins were visualized using enhanced chemiluminescence. β-actin was used as a loading control.
The effect of PRIMA-1MET on cell death and apoptotic morphological changes in EOC cells
To assess the effect of PRIMA-1MET on EOC cells, the anti-proliferative effects of various concentrations of PRIMA-1MET (approximately 0–100 µM) were determined in a total of 9 EOC cell lines: TOV21G, A2780, ES-2, OV-90, OVCAR-3, CaOV-3, SKOV-3, NOS2 and NOS3. Fig. 2a shows the cell viability of wild-type p53 cell lines (TOV21G, A2780, and NOS2) and mutant p53 cell lines (ES-2, OV-90, OVCAR-3, CaOV-3, NOS3 and SKOV-3) treated with PRIMA-1MET for 48 h. PRIMA-1MET reduced cell viability after 48 h in all EOC cell lines in a dose-dependent manner. The IC50 values of PRIMA-1MET ranged from 2.6 to 20.1 µM, which were independent of the mutation status of TP53 (Fig. 2b). Furthermore, PRIMA-1MET treatment induced a morphological change which was consistent with the apoptotic change within 6–24 h (Fig. 2c). We next investigated whether PRIMA-1MET had sufficient effects on cisplatin- and paclitaxel-resistant cell lines, which were previously developed from the parental NOS2 and NOS3 cells (NOS2CR, NOS2TR, NOS3CR, and NO3TR). Dose-responsive cell viability assays with PRIMA-1MET were performed to evaluate the sensitivities of the chemo-resistant cells. As shown in Fig. 2d, PRIMA-1MET displayed anti-proliferative effects on both the parental and chemo-resistant cells. The IC50 values of the NOS2, NOS2CR, and NOS2TR cells were 6.5, 7.4, and 8.8 µM, respectively. The IC50 value of the NOS3CR cells was slightly higher than the values of the NOS3 and NOS3TR cells (not significant). PRIMA-1MET had sufficient growth-suppressing activity regardless of the mutation status of the TP53 and the chemo-sensitivity in the EOC cells.
Figure 2.

The effects of PRIMA-1MET on tumor cell growth in ovarian cancer cells. (a) PRIMA-1MET-induced growth suppressive dose response curves of EOC cells following 48 h of PRIMA-1MET treatment. The IC50 values of PRIMA-1MET were determined in a total of 9 EOC cell lines by cell viability assay. Data represent the mean ± standard deviation (SD) from triplicate reactions. (b) Correlation between PRIMA-1MET 48 h IC50 values and TP53 status. EOC cell lines with wild-type p53 were slightly more sensitive than those with mutant p53 (not significant). (c) PRIMA-1MET-induced morphological changes in EOC cells. ES-2, OVCAR-3, and SKOV-3 cells were cultured in a 6-well plate, and then treated with 0, 50, 100 µM PRIMA-1MET for 24 h. PRIMA-1MET-induced morphological changes were observed using microscopy (scale bar, 200 µm). (d) PRIMA-1MET-induced growth suppressive dose response curves of NOS2, NOS3, and their chemo-resistant cells. The IC50 values of 48 h PRIMA-1MET were determined by cell viability assay. Data displays mean ± standard deviation (SD) from triplicate reactions.
PRIMA-1MET induces apoptosis in a dose-dependent manner in the EOC cells
We next performed an Annexin V-FITC/PI staining assay to investigate whether PRIMA-1MET actually induced apoptosis in the EOC cells. Treatment with PRIMA-1MET for 16 h against EOC cells, TOV21G and A2780, increased the fraction of early and late apoptotic cells (Fig. 3a). In the TOV21G cells, the fractions of early and late apoptotic cells were significantly increased from 1.1 and 4.3% following control vehicle treatment to 3.3 and 54.5% following 20 µM of PRIMA-1MET treatment, respectively (Fig. 3b). In the A2780 cells, the proportion of late apoptotic cells was significantly elevated from 5.3% following control vehicle treatment to 17.6% following 20 µM treatment (Fig. 3b). To determine whether PRIMA-1MET also induced apoptosis in chemo-resistant EOC cells, we evaluated cell apoptosis in another manner using fluorescence microscopy. The cells after a 24-h treatment with PRIMA-1MET were fixed with 4% paraformaldehyde, stained with Hoechst 33342, and then we identified apoptosis with fluorescence microscopy. The cells which had fragmented or condensed nuclei were defined as undergoing apoptosis and counted manually with fluorescence microscopy (29). The representative images of condensed nuclei are shown in Fig. 3c. PRIMA-1MET treatment significantly increased the fractions of apoptotic cells with fragmented or condensed nuclei in both parental cells and their chemo-resistant cells in a dose-dependent manner (Fig. 3d). These results indicate that PRIMA-1MET induces apoptotic cell death in both chemo-sensitive and chemo-resistant EOC cells.
Figure 3.

PRIMA-1MET induces apoptosis in a dose-dependent manner in EOC cell lines. (a) TOV21G and A2780 cells were treated with 0, 10, 20 µM of PRIMA-1MET for 16 h, and then stained with Annexin V-FITC and PI. The Annexin V-positive and PI-negative cells were defined as early apoptotic, and the Annexin V-positive and PI-positive cells were defined as late apoptotic (dead cells). (b) Viable, early apoptotic, and late apoptotic cells were quantified from triplicate samples. Asterisk indicates statistical significance (P<0.001). Error bars represent standard deviations. (c) Representative images of Hoechst 33342 staining of NOS3TR cells after 24 h of PRIMA-1MET treatment. Apoptotic cells were defined by their condensed and fragmented nuclei with fluorescence microscopy (scale bar, 200 µm). (d) Apoptosis levels of NOS2 and NOS3 cells, and their chemo-resistant cells after 0, 10, 25, 50 µM 20 h PRIMA-1MET treatment. Bars represent the percentages of apoptotic nuclei counted in each treatment group and are expressed as the mean ± SD.
PRIMA-1MET activates PARP cleavage
In order to confirm whether PRIMA-1MET-induced cell death is apoptotic, we evaluated the apoptosis-related protein levels after treatment with PRIMA-1MET in EOC cells by western blot analysis. Immunoblot analysis elucidated that PRIMA-1MET induced dose-dependent PARP cleavage in the NOS2 and NOS3 cells and in their chemo-resistant cells (Fig. 4). This result showed that PRIMA-1MET induces apoptosis in EOC cells through PARP cleavage.
Figure 4.

PRIMA-1MET induces PARP cleavage in EOC cells. NOS2 and NOS3 cells, and their chemo-resistant cells were treated with 0, 25 and 50 µM PRIMA-1MET for 24 h, and then lysed with lysis buffer. Cell lysates were subjected to western blot analysis. β-actin was used as a loading control.
PRIMA-1MET increases intracellular ROS in EOC cells
As it was reported that PRIMA-1MET induces intracellular ROS accumulation, we investigated intracellular ROS accumulation using 5–6-chloromethyl-2′7′-dichlorodihydroflorescein diacetate, acetyl ester (CM-H2DCFDA; Molecular Probes Invitrogen, Carlsbad, CA, USA) (20,30). PRIMA-1MET treatment for 24 h promoted intracellular ROS accumulation in the NOS2 and NOS3 cells, and their chemo-resistant cells (Fig. 5a). To quantify the proportion of fluorescence-positive cells in the TOV21G cells after a 24-h treatment with PRIMA-1MET, fluorescence activated cell sorting (FACS) was performed. The proportion of fluorescence-positive cells was increased in the cells treated with PRIMA-1MET in a dose-dependent manner, and the increase was significant (Fig. 5b and c). These results demonstrate that PRIMA-1MET promotes intracellular ROS accumulation in EOC cells.
Figure 5.

PRIMA-1MET induces intracellular ROS accumulation in EOC cells. (a) Intracellular ROS accumulation after 16 h of PRIMA-1MET treatment in the NOS2 and NOS3 cells, and their chemo-resistant cells. Intracellular ROS levels were detected by CM-H2DCFDA, resulting in fluorescence positivity under fluorescence microscopy. (b) Dose-dependent intracellular ROS accumulation in TOV21G cells. TOV21G cells were maintained in medium with the indicated concentrations of PRIMA-1MET, labeled with 5 µM CM-H2DCFDA, and then subjected to flow cytometry. The percentage of cells with fluorescence intensity above the level of 1,000 FL was measured. (c) Bars represent the mean percentage of fluorescence-positive cells. Error bars represent standard deviations.
ROS scavenger rescues apoptosis induced by PRIMA-1MET
To determine whether intracellular ROS accumulation by treatment with PRIMA-1MET induces apoptosis, we used a ROS scavenger N-acetyl cysteine (NAC). The compound NAC was added to cultured cells with 20 µM PRIMA-1MET medium at a final concentration of 10 mM. Sixteen hours after co-treatment with PRIMA-1MET and NAC, apoptotic cells were assessed by Annexin V-FITC and PI staining. The addition of NAC inhibited apoptosis and the growth-suppressing effect induced by PRIMA-1MET treatment (Fig. 6a and b). Furthermore, to examine the effect of PRIMA-1MET on the expression of antioxidant enzymes including Prx3 and GPx-1, which scavenge intracellular ROS to sustain homeostasis, we treated TOV21G and A2780 cells with PRIMA-1MET for 20 h and, thereafter, evaluated the mRNA levels of antioxidant enzymes by real-time RT-PCR. The mRNA levels of Prx3 and GPx-1 were significantly decreased after 20 h of treatment with PRIMA-1MET in a dose-dependent manner (Fig. 6c). Our results suggest that the antitumor effects of PRIMA-1MET may be mediated by intracellular ROS accumulation, and that the intracellular ROS accumulation and the cytotoxic effect induced by PRIMA-1MET may be due to downregulation of Prx3 and GPx-1.
Figure 6.

N-acetyl cysteine (NAC) inhibits the biological effect of PRIMA-1MET. (a) NAC completely blocked the cytotoxic effect of 16 h of PRIMA-1MET treatment in TOV21G cells, as shown by Annexin V-FITC and PI staining analysis. After 20 µM of PRIMA-1MET treatment with or without 10 mM NAC for 16 h, the fraction of viable, early apoptotic, and late apoptotic cells were examined. Bars represents the mean percentages. Error bars represent standard deviations. Asterisk indicates statistical significance (P<0.05). (b) Treatment with NAC prevented the PRIMA-1MET-induced growth suppressive effect in the TOV21G and A2780 cells according to the cell viability assay. After treatment with several concentrations of PRIMA-1MET with or without 10 mM NAC, viable cells were evaluated. Bars represents the mean percentages of viable cells. Error bars represent standard deviations. Asterisk indicates statistical significance (P<0.05). (c) Inhibition of antioxidant enzymes by PRIMA-1MET. TOV21G and A2780 cells were treated with PRIMA-1MET for 20 h and thereafter the mRNA levels of Prx3 and GPx-1 were evaluated by real-time RT-PCR. Data represent the mean ± SD from triplicate reactions.
Discussion
Most EOC patients experience recurrent disease, despite a high rate of complete clinical remission. Although recurrent EOC patients frequently receive chemotherapy, they are basically incurable due to the acquisition of chemo-resistance. Resistance to cytotoxic agents is a major obstacle to complete cure, and a number of attempts to overcome chemo-resistance have been made in EOC (31,32). While much effort has been made to restore chemo-sensitivity to resistant cells, no promising molecules have been identified. Thus, there is a need to develop novel therapeutics for EOC. Despite the fact that PRIMA-1MET has been confirmed to exhibit tumor-suppressing effects on various types of cancer cells, there have been few reports on the effect of PRIMA-1MET on chemo-resistant cells in EOC (33–35). In our present study, we attempted to verify whether PRIMA-1MET has antitumor effects on EOC cells.
PRIMA-1MET is a prodrug converted to MQ with potential to bind to cysteine residues and change the conformation of the core domain of mutant p53 (30). PRIMA-1/PRIMA-1MET has been reported to synergize with cytotoxic agents to induce apoptotic cell death (33,36,37). Recently, Mohell et al reported that combined treatment with APR-246 and platinum or other drugs could give rise to an improved strategy for recurrent high-grade serous ovarian cancer (37). In this study, chemo-resistant cells incubated with PRIMA-1MET exhibited apoptosis, which was characterized by morphological features, such as chromosomal DNA condensation and fragmentation. Furthermore, the effect of PRIMA-1MET on cell viability of chemo-resistant cells was similar to that of the parental cells with either wild-type p53 (NOS2) or mutant p53 (NOS3). These findings suggest that PRIMA-1MET may have the possibility to be used for patients with chemo-resistant EOC bearing not only mutant p53 but also wild-type p53.
EOC cells easily spread to the peritoneal cavity and form disseminated metastases with a large amount of ascites. During this metastatic process, tumors may gradually acquire stem-like properties and become chemo-resistant. To our knowledge, there has been no report examining the efficacy of PRIMA-1MET in cancer stem cells (or cancer stem-like cells). On the other hand, a recent study suggested the possibility that PRIMA-1MET may overcome the chemo-resistance of EOC cells (37). Indeed, PRIMA-1MET may be able to target cancer stem cells with chemo-resistant properties although additional studies are warranted.
In the present study, we investigated the efficacy of PRIMA-1MET in the growth suppression and apoptosis induction in ovarian cancer cell lines (n=9) in vitro. We demonstrated that PRIMA-1MET suppressed cell viability and induced massive apoptosis, regardless of the TP53 mutational status. Furthermore, ovarian cancer cell lines carrying wild-type p53 were slightly more sensitive than those carrying mutant p53 (not significant). To date, previous reports have shown that PRIMA-1/PRIMA-1MET were more effective on pancreatic and small cell lung cancer cells expressing mutant p53 than on those expressing wild-type p53 or null (38,39). Interestingly, despite the fact that there is evidence that PRIMA-1MET restores the wild-type p53 function to mutant p53, several recent studies have shown that PRIMA-1MET displayed cytotoxic effects on Ewing sarcoma cells, acute myeloid leukemia cells, and human myeloma cells irrespective of the TP53 mutational status (40–42). This controversy is because PRIMA-1MET not only restores wild-type p53 function to mutant p53, but also induces apoptosis in a p53-independent manner through intracellular ROS accumulation and endoplasmic reticulum (ER) stress (40,41). Indeed, in this study, we demonstrated that incubation with PRIMA-1MET resulted in an antitumor effect with intracellular ROS accumulation in ovarian cancer cells, and co-treatment with PRIMA-1MET and an ROS scavenger, NAC, blocked the cytotoxic effects, suggesting that the effects of PRIMA-1MET are due to an intracellular ROS increase in EOC cells. Our results were partly consistent with previous reports, and support the antitumor effects of PRIMA-1MET being universal irrespective of the TP53 mutational status in EOC cells. Unknown diverse mechanisms of PRIMA-1MET may provide a convincing strategy for overcoming chemo-resistance in not only EOC but also other cancers.
ROS can generate oxidative stress in cells inducing DNA damage, protein degradation, peroxidation of lipids, and finally cell death at a high concentration. It is well known that cancer cells are normally more tolerant to high levels of oxidative stress than normal cells (43). One of the underlying mechanisms of cancer cells to survive under high oxidative condition is overexpression of antioxidant enzymes to scavenge ROS (44). An inhibitor of glutathione synthesis, buthionine sulfoximine (BSO) was used in a clinical situation (45). In the present study, we demonstrated that PRIMA-1MET induced intracellular accumulation and suppressed the expression of antioxidant enzymes, Prx3 and GPx-1, in EOC cells. Prx3 is one of the 2-Cys peroxiredoxin family (PRX 1–4), and operates as a reductase to metabolize ROS (46). Cunniff et al reported that knockdown of Prx3 increased oxidative stress and mitochondrial dysfunction in malignant mesothelioma cells, suggesting that Prx3 plays a critical role in cell cycle progression and sustaining the mitochondrial structure (47). Furthermore, a recent report by Song et al showed that Prx3 was highly upregulated in colon cancer stem cells, and that knockdown of Prx3 led to decreased cellular viability (48). In addition, several studies have already shown that GPx-1 protects cancer cells upon exposure to severe oxidative stress (49,50). According to our findings, PRIMA-1MET suppressed the expression of both Prx3 and GPx-1, suggesting that it may induce an intracellular ROS increase mediated by downregulation of Prx3 and GPx-1.
To our knowledge, no previous studies have confirmed intracellular ROS accumulation in benign tumor cells or normal epithelial cells. In the present study, we did not evaluate whether PRIMA-1MET induces intracellular ROS accumulation in such non-malignant or normal cells. In the living body, the majority of ROS generated by various stimulation can be degenerated by a higher antioxidant capacity derived from intrinsic ROS scavengers, resulting in weakened efficacy. We believe that the actual effects may depend on the local balance between ROS and such intrinsic scavengers. Certainly, PRIMA-1MET may induce intracellular ROS accumulation in benign or normal cells as well as tumor cells. We speculate that the carcinogenetic effect of PRIMA-1MET in such cells may be minimal. However, we cannot deny that administration of PRIMA-1MET may have some risks. Therefore, further investigation concerning the effect of PRIMA-1MET against benign or normal cells is necessary when used clinically.
In conclusion, we demonstrated that PRIMA-1MET exhibited antitumor effects on chemo-resistant cells through intracellular ROS accumulation and repressed antioxidant enzymes. To utilize PRIMA-1MET for EOC patients including chemo-resistant cases, we need to investigate further how PRIMA-1MET suppresses Prx3 and GPx-1. PRIMA-1MET is a promising compound for further development as a potential cytotoxic agent against EOC.
Acknowledgments
The authors wish to thank Dr Kathleen Pishas for the study and technical support.
References
- 1.Sankaranarayanan R, Ferlay J. Worldwide burden of gynaecological cancer: The size of the problem. Best Pract Res Clin Obstet Gynaecol. 2006;20:207–225. doi: 10.1016/j.bpobgyn.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Masoumi Moghaddam S, Amini A, Morris DL, Pourgholami MH. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012;31:143–162. doi: 10.1007/s10555-011-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muñoz-Casares FC, Rufián S, Arjona-Sánchez Á, Rubio MJ, Díaz R, Casado Á, Naranjo Á, Díaz-Iglesias CJ, Ortega R, Muñoz-Villanueva MC, et al. Neoadjuvant intraperitoneal chemotherapy with paclitaxel for the radical surgical treatment of peritoneal carcinomatosis in ovarian cancer: A prospective pilot study. Cancer Chemother Pharmacol. 2011;68:267–274. doi: 10.1007/s00280-011-1646-4. [DOI] [PubMed] [Google Scholar]
- 4.Chan JK, Tian C, Monk BJ, Herzog T, Kapp DS, Bell J, Young RC, Gynecologic Oncology Group Prognostic factors for high-risk early-stage epithelial ovarian cancer: A Gynecologic Oncology Group study. Cancer. 2008;112:2202–2210. doi: 10.1002/cncr.23390. [DOI] [PubMed] [Google Scholar]
- 5.Chan JK, Teoh D, Hu JM, Shin JY, Osann K, Kapp DS. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol Oncol. 2008;109:370–376. doi: 10.1016/j.ygyno.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 7.Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al. Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Havrilesky L, Darcy M, Hamdan H, Priore RL, Leon J, Bell J, Berchuck A, Gynecologic Oncology Group Study Prognostic significance of p53 mutation and p53 overexpression in advanced epithelial ovarian cancer: A Gynecologic Oncology Group Study. J Clin Oncol. 2003;21:3814–3825. doi: 10.1200/JCO.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 9.Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Tang J, Li S, Zhang S, Shaw PA, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006;98:1694–1706. doi: 10.1093/jnci/djj465. [DOI] [PubMed] [Google Scholar]
- 10.Kang HJ, Chun SM, Kim KR, Sohn I, Sung CO. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS One. 2013;8:e72609. doi: 10.1371/journal.pone.0072609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 12.Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18:477–485. doi: 10.1038/sj.onc.1202314. [DOI] [PubMed] [Google Scholar]
- 14.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, Sudo S, Ju J, Sakuragi N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32:3286–3295. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Høgdall EV, Kjaer SK, Blaakaer J, Christensen L, Glud E, Vuust J, Høgdall CK. P53 mutations in tissue from Danish ovarian cancer patients: From the Danish 'MALOVA' ovarian cancer study. Gynecol Oncol. 2006;100:76–82. doi: 10.1016/j.ygyno.2005.07.131. [DOI] [PubMed] [Google Scholar]
- 16.Concin N, Hofstetter G, Berger A, Gehmacher A, Reimer D, Watrowski R, Tong D, Schuster E, Hefler L, Heim K, et al. Clinical relevance of dominant-negative p73 isoforms for responsiveness to chemotherapy and survival in ovarian cancer: Evidence for a crucial p53-p73 cross-talk in vivo. Clin Cancer Res. 2005;11:8372–8383. doi: 10.1158/1078-0432.CCR-05-0899. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Helland A, Holm R, Skomedal H, Abeler VM, Danielsen HE, Tropé CG, Børresen-Dale AL, Kristensen GB. TP53 mutations in early-stage ovarian carcinoma, relation to long-term survival. Br J Cancer. 2004;90:678–685. doi: 10.1038/sj.bjc.6601537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueno Y, Enomoto T, Otsuki Y, Sugita N, Nakashima R, Yoshino K, Kuragaki C, Ueda Y, Aki T, Ikegami H, et al. Prognostic significance of p53 mutation in suboptimally resected advanced ovarian carcinoma treated with the combination chemotherapy of paclitaxel and carboplatin. Cancer Lett. 2006;241:289–300. doi: 10.1016/j.canlet.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 19.Bartel F, Jung J, Böhnke A, Gradhand E, Zeng K, Thomssen C, Hauptmann S. Both germ line and somatic genetics of the p53 pathway affect ovarian cancer incidence and survival. Clin Cancer Res. 2008;14:89–96. doi: 10.1158/1078-0432.CCR-07-1192. [DOI] [PubMed] [Google Scholar]
- 20.Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, Arnér ES, Wiman KG. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013;4:e881. doi: 10.1038/cddis.2013.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 22.Russo D, Ottaggio L, Penna I, Foggetti G, Fronza G, Inga A, Menichini P. PRIMA-1 cytotoxicity correlates with nucleolar localization and degradation of mutant p53 in breast cancer cells. Biochem Biophys Res Commun. 2010;402:345–350. doi: 10.1016/j.bbrc.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 23.Roh JL, Kang SK, Minn I, Califano JA, Sidransky D, Koch WM. p53-Reactivating small molecules induce apoptosis and enhance chemotherapeutic cytotoxicity in head and neck squamous cell carcinoma. Oral Oncol. 2011;47:8–15. doi: 10.1016/j.oraloncology.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann S, Bykov VJ, Ali D, Andrén O, Cherif H, Tidefelt U, Uggla B, Yachnin J, Juliusson G, Moshfegh A, et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30:3633–3639. doi: 10.1200/JCO.2011.40.7783. [DOI] [PubMed] [Google Scholar]
- 25.Misawa T, Kikkawa F, Maeda O, Obata NH, Higashide K, Suganuma N, Tomoda Y. Establishment and characterization of acquired resistance to platinum anticancer drugs in human ovarian carcinoma cells. Jpn J Cancer Res. 1995;86:88–94. doi: 10.1111/j.1349-7006.1995.tb02992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–283. [PubMed] [Google Scholar]
- 27.Kapitanović S, Cacev T, Antica M, Kralj M, Cavrić G, Pavelić K, Spaventi R. Effect of indomethacin on E-cadherin and beta-catenin expression in HT-29 colon cancer cells. Exp Mol Pathol. 2006;80:91–96. doi: 10.1016/j.yexmp.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Sugiyama K, Kajiyama H, Shibata K, Yuan H, Kikkawa F, Senga T. Expression of the miR200 family of microRNAs in mesothelial cells suppresses the dissemination of ovarian cancer cells. Mol Cancer Ther. 2014;13:2081–2091. doi: 10.1158/1535-7163.MCT-14-0135. [DOI] [PubMed] [Google Scholar]
- 29.Nakahara T, Iwase A, Nakamura T, Kondo M, Bayasula, Kobayashi H, Takikawa S, Manabe S, Goto M, Kotani T, et al. Sphingosine-1-phosphate inhibits H2O2-induced granulosa cell apoptosis via the PI3K/Akt signaling pathway. Fertil Steril. 2012;98:1001.e1–1008.e1. doi: 10.1016/j.fertnstert.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Utsumi F, Kajiyama H, Nakamura K, Tanaka H, Mizuno M, Ishikawa K, Kondo H, Kano H, Hori M, Kikkawa F. Effect of indirect nonequilibrium atmospheric pressure plasma on anti-proliferative activity against chronic chemo-resistant ovarian cancer cells in vitro and in vivo. PLoS One. 2013;8:e81576. doi: 10.1371/journal.pone.0081576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan Z, Choy E, Hornicek FJ. NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One. 2009;4:e7415. doi: 10.1371/journal.pone.0007415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, Wiman KG. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24:3484–3491. doi: 10.1038/sj.onc.1208419. [DOI] [PubMed] [Google Scholar]
- 34.Supiot S, Zhao H, Wiman K, Hill RP, Bristow RG. PRIMA-1(met) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother Oncol. 2008;86:407–411. doi: 10.1016/j.radonc.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Ali D, Jönsson-Videsäter K, Deneberg S, Bengtzén S, Nahi H, Paul C, Lehmann S. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur J Haematol. 2011;86:206–215. doi: 10.1111/j.1600-0609.2010.01557.x. [DOI] [PubMed] [Google Scholar]
- 36.Nahi H, Lehmann S, Mollgard L, Bengtzen S, Selivanova G, Wiman KG, Paul C, Merup M. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br J Haematol. 2004;127:285–291. doi: 10.1111/j.1365-2141.2004.05210.x. [DOI] [PubMed] [Google Scholar]
- 37.Mohell N, Alfredsson J, Fransson Å, Uustalu M, Byström S, Gullbo J, Hallberg A, Bykov VJ, Björklund U, Wiman KG. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015;6:e1794. doi: 10.1038/cddis.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17:2830–2841. doi: 10.1158/1078-0432.CCR-10-3168. [DOI] [PubMed] [Google Scholar]
- 39.Izetti P, Hautefeuille A, Abujamra AL, de Farias CB, Giacomazzi J, Alemar B, Lenz G, Roesler R, Schwartsmann G, Osvaldt AB, et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs. 2014;32:783–794. doi: 10.1007/s10637-014-0090-9. [DOI] [PubMed] [Google Scholar]
- 40.Tessoulin B, Descamps G, Moreau P, Maïga S, Lodé L, Godon C, Marionneau-Lambot S, Oullier T, Le Gouill S, Amiot M, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014;124:1626–1636. doi: 10.1182/blood-2014-01-548800. [DOI] [PubMed] [Google Scholar]
- 41.Russo D, Ottaggio L, Foggetti G, Masini M, Masiello P, Fronza G, Menichini P. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim Biophys Acta. 2013;1833:1904–1913. doi: 10.1016/j.bbamcr.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Aryee DN, Niedan S, Ban J, Schwentner R, Muehlbacher K, Kauer M, Kofler R, Kovar H. Variability in functional p53 reactivation by PRIMA-1(Met)/APR-246 in Ewing sarcoma. Br J Cancer. 2013;109:2696–2704. doi: 10.1038/bjc.2013.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong L, Chuang CC, Wu S, Zuo L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367:18–25. doi: 10.1016/j.canlet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 44.Landry WD, Cotter TG. ROS signalling, NADPH oxidases and cancer. Biochem Soc Trans. 2014;42:934–938. doi: 10.1042/BST20140060. [DOI] [PubMed] [Google Scholar]
- 45.Bailey HH, Mulcahy RT, Tutsch KD, Arzoomanian RZ, Alberti D, Tombes MB, Wilding G, Pomplun M, Spriggs DR. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: An attempt at modulation of glutathione. J Clin Oncol. 1994;12:194–205. doi: 10.1200/JCO.1994.12.1.194. [DOI] [PubMed] [Google Scholar]
- 46.Cox AG, Peskin AV, Paton LN, Winterbourn CC, Hampton MB. Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry. 2009;48:6495–6501. doi: 10.1021/bi900558g. [DOI] [PubMed] [Google Scholar]
- 47.Cunniff B, Wozniak AN, Sweeney P, DeCosta K, Heintz NH. Peroxiredoxin 3 levels regulate a mitochondrial redox setpoint in malignant mesothelioma cells. Redox Biol. 2014;3:79–87. doi: 10.1016/j.redox.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song IS, Jeong YJ, Jeong SH, Heo HJ, Kim HK, Bae KB, Park YH, Kim SU, Kim JM, Kim N, et al. FOXM1-induced PRX3 regulates stemness and survival of colon cancer cells via maintenance of mitochondrial function. Gastroenterology. 2015;149:1006.e9–1016.e9. doi: 10.1053/j.gastro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Huang C, Ding G, Gu C, Zhou J, Kuang M, Ji Y, He Y, Kondo T, Fan J. Decreased selenium-binding protein 1 enhances glutathione peroxidase 1 activity and downregulates HIF-1α to promote hepatocellular carcinoma invasiveness. Clin Cancer Res. 2012;18:3042–3053. doi: 10.1158/1078-0432.CCR-12-0183. [DOI] [PubMed] [Google Scholar]
- 50.Gan X, Chen B, Shen Z, Liu Y, Li H, Xie X, Xu X, Li H, Huang Z, Chen J. High GPX1 expression promotes esophageal squamous cell carcinoma invasion, migration, proliferation and cisplatin-resistance but can be reduced by vitamin D. Int J Clin Exp Med. 2014;7:2530–2540. [PMC free article] [PubMed] [Google Scholar]