

Abstract
Lysosomal acid lipase (LAL) is a key enzyme controlling neutral lipid metabolic signaling in myeloid-derived suppressor cells (MDSCs). MDSCs from LAL-deficient (lal−/−) mice directly stimulate cancer cell proliferation. PPARγ ligand treatment inhibited lal−/− MDSCs stimulation of tumor cell growth and metastasis in vivo, and tumor cell proliferation and migration in vitro. In addition, PPARγ ligand treatment impaired lal−/− MDSCs transendothelial migration, and differentiation from lineage-negative cells. The corrective effects of PPARγ ligand on lal−/− MDSCs functions were mediated by regulating the mammalian target of rapamycin (mTOR) pathway, and subsequently blocking MDSCs ROS overproduction. Furthermore, in the myeloid-specific dominant-negative PPARγ (dnPPARγ) overexpression bitransgenic mouse model, tumor growth and metastasis were enhanced, and MDSCs from these mice stimulated tumor cell proliferation and migration. MDSCs with dnPPARγ overexpression showed increased transendothelial migration, overactivation of the mTOR pathway, and ROS overproduction. These results indicate that PPARγ plays a critical role in neutral lipid metabolic signaling controlled by LAL, which provides a mechanistic basis for clinically targeting MDSCs to reduce the risk of cancer proliferation, growth and metastasis.
Keywords: lysosomal acid lipase, lipid metabolic signaling, myeloid-derived suppressor cells, peroxisome proliferator-activated receptor-γ, tumor growth and metastasis
INTRODUCTION
Lysosomal acid lipase (LAL) hydrolyzes cholesteryl esters and triglycerides in the lysosome of cells to generate free fatty acids and cholesterol. Genetic ablation of the lal gene in mice results in a systemic increase of myeloid lineage cells, causing severe inflammation in multiple organs [1, 2]. Myeloid-derived suppressor cells (MDSCs), with co-expression of myeloid-cell lineage differentiation antigen Gr-1 and CD11b in mice, are a heterogeneous population of immature myeloid cells at different stages of differentiation [3]. We previously reported that the neutral lipid metabolic pathway controlled by LAL plays a critical role in the development and homeostasis of MDSCs, and demonstrated that LAL deficiency leads to the infiltration and accumulation of MDSCs in various organs of the mice, such as the lung, spleen, thymus, liver and small intestine [1, 4]. In addition to immune suppressive function, our recent studies showed that LAL-deficient (lal−/−) MDSCs possess direct tumor stimulatory function [5]. By Affymetrix GeneChip microarray, we have identified that many important gene pathways are involved in the dysfunctions of lal−/− MDSCs, notably among which, peroxisome proliferator-activated receptor-γ (PPARγ)-regulated gene expression is significantly altered [6].
PPARγ, a member of the nuclear receptor superfamily, serves as the receptor of free fatty acid derived compounds which arise downstream of LAL enzymatic action. After binding to these ligands, PPARγ plays an important role in limiting inflammation in various tissues by suppressing the expression of inflammatory cytokines [7, 8]. Overexpression of pro-inflammatory molecules (e.g., apoptosis inhibitor 6 and matrix metalloproteinase 12) that are negatively regulated by PPARγ has been reported to induce chronic inflammation and spontaneous tumor formation [9–13]. Our previous study has suggested that LAL deficiency causes inactivation of PPARγ by blocking ligand generation, which in turn promotes pulmonary inflammation and pathogenesis [14]. Furthermore, by using the dominant-negative PPARγ (dnPPARγ) myeloid-specific overexpression bitransgenic mouse model, we found that PPARγ plays a key role in controlling pro-inflammatory cytokine synthesis, MDSC expansion, immunosuppression, and the development of cancer [15].
Since LAL downstream metabolic derivatives serve as hormonal ligands for PPARγ, the current study examined if PPARγ plays an important role in LAL-mediated functions in MDSCs. Here, the corrective effects of the PPARγ ligand 9-hydroxyoctadecadienoic acid (9-HODE) on the neutral lipid metabolic signaling controlled by LAL are examined, including effects on the development and function of MDSCs, MDSCs transendothelial migration, tumor cell proliferation and metastasis. The results demonstrate that 9-HODE treatment corrected the defects in lal−/− MDSCs via effecting the mammalian target of rapamycin (mTOR) pathway and by inhibiting overproduction of reactive oxygen species (ROS). These findings provide novel mechanistic insights into the linkage between lipid metabolic signaling and PPARγ in MDSC dysfunction. Importantly, these studies reveal the critical role of LAL and PPARγ in checking MDSC functions and the potential as a therapeutic target to modulate tumor growth and spread associated with MDSCs.
RESULTS
PPARγ ligand impaired lal−/− Ly6G+ cell stimulation of tumor growth and metastasis in vivo
PPARγ inactivation has previously been reported to cause inflammation-triggered cell growth and emphysema in lal−/− mice, and treatment with the PPARγ ligand 9-HODE significantly rescued lal−/− pulmonary inflammation and aberrant gene expression [14]. lal−/− Ly6G+ MDSCs have recently been found to play a role in stimulating tumor growth and metastasis [5]. Based on the literature and our own experience, the wild type bone marrow is comprised of ∼50% CD11b+Ly6G+ myeloid precursor cells (with very low immunosuppressive function) and very few CD11b+ or Ly6G+ single cells. On the other hand, the lal−/− bone marrow is comprised of 70% CD11b+Ly6G+ cells (with very strong immunosuppression and cancer cell stimulation potential). These lal−/− bone marrow cells do not further differentiate into more mature myeloid cells as wild type cells do [1]. To see whether PPARγ inactivation within lal−/− Ly6G+ MDSCs contributes to their ability to stimulate tumor cells, freshly isolated bone marrow-derived lal+/+ or lal−/− Ly6G+ cells were pre-treated with 9-HODE or the vehicle, ethanol for 24 h. In lal−/− mice, since almost all Ly6G+ cells are positive for CD11b, a Ly6G-specific antibody was used for purification of Ly6G+CD11b+ cells. To examine tumor growth potential in vivo, pre-treated or untreated Ly6G+ cells were mixed with untreated B16 melanoma cells, and then co-injected subcutaneously into lal+/+ mice. One week after the injection, subcutaneous tumors detected in the lal−/− Ly6G+ cell-injected mice were significantly larger (tumor volume = 63.2 ± 11.7 mm3) than those tumors in lal+/+ Ly6G+ cell-injected mice (tumor volume = 34.6 ± 11.9 mm3, p < 0.01). However, the tumors from 9-HODE-treated lal−/− Ly6G+ cell-injected mice (tumor volume = 36.3 ± 12.4 mm3) were significantly smaller when compared with those developed in ethanol-treated lal−/− Ly6G+ cell-injected mice (tumor volume = 77.6 ± 16.4 mm3, p < 0.01) (Figure 1A). The similar effect of 9-HODE treatment on lal−/− ly6G+ cells to tumor size was also observed at 14 and 21 days post-injection (Figure 1A). Moreover, when B16 melanoma cells were co-injected with C57BL/6 Ly6G+ cells into C57BL/6 mice, similar results were observed that the tumors from 9-HODE-treated lal−/− Ly6G+ cell-injected mice were significantly smaller than those developed in ethanol-treated lal−/− Ly6G+ cell-injected mice at 7, 14 and 21 days post-injection (Figure 1B). As predicted, B16 melanoma tumor grew larger in C57BL/6 than that in FVB/N mice at 14 and 21 days post-injection.
Figure 1. PPARγ ligand reverses lal−/− MDSCs stimulation on tumor growth and metastasis in vivo.
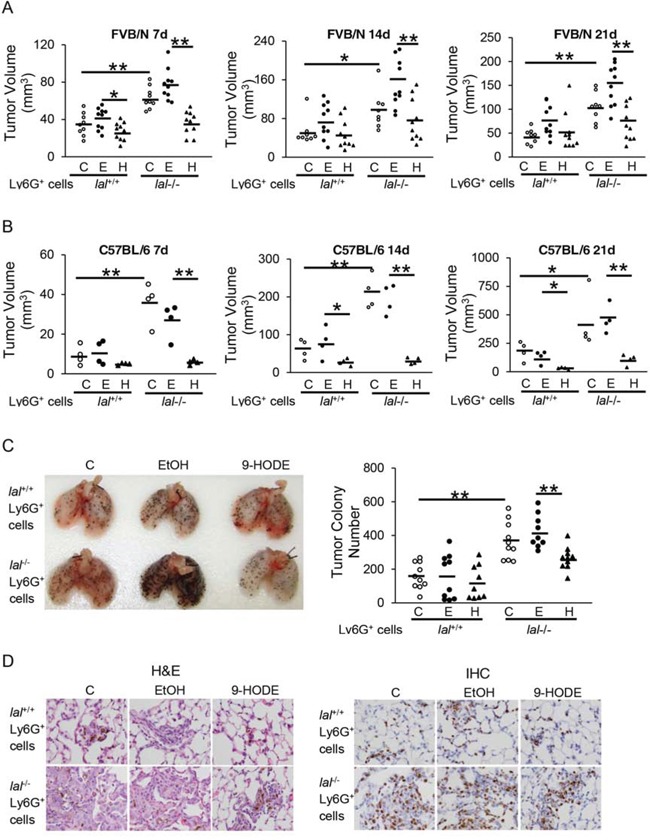
A. Ly6G+ cells from lal+/+ or lal−/− FVB/N mice were pre-treated with ethanol (E) or 20 μmol/L 9-HODE (H) or without treatment (C) for 24 h. Pre-treated Ly6G+ cells (6 × 105) and B16 melanoma cells (2 × 105, without any treatment) were mixed, and co-injected subcutaneously into the flank region of 3-month old lal+/+ FVB/N mice. n = 8∼10. B. Pre-treated C57BL/6 Ly6G+ cells (6 × 105) and B16 melanoma cells (2 × 105) were co-injected subcutaneously into the flank region of 3-month old lal+/+ C57BL/6 mice. n = 4. Tumor volume (in cubic millimeters) were measured and statistically analyzed at 7, 14, and 21 days post-injection. For statistical analyses, data were expressed as mean ± SD. **P < 0.01, *P < 0.05. C. Pre-treated Ly6G+ cells (2 × 106) and B16 melanoma cells (5 × 105, without any treatment) were intravenously co-injected into lal+/+ mice for 2 weeks. Representative lungs and quantitative analysis of the melanoma colony numbers in the lungs are shown. Data were expressed as mean ± SD; n = 9∼10. **P < 0.01. D. Representative H&E staining and IHC staining with Ki67 antibody of the lungs with metastasized melanoma are shown. Original magnification, × 400.
Next, the pre-treated Ly6G+ cells and B16 melanoma cells were injected into the tail veins of lal+/+ recipient mice to detect metastatic potential. Two weeks after injection, less B16 melanoma colonies were observed in the lungs of lal+/+ mice that received 9-HODE-treated lal−/− Ly6G+ and B16 cell co-injection than those received ethanol-treated lal−/− Ly6G+ and B16 cell co-injection (Figure 1C). 9-HODE treatment of lal+/+ ly6G+ cells did not affect B16 melanoma colonization in the lung (Figure 1C). Sections of the lungs showed less neoplastic cells by H&E staining and less Ki67 positive cells by IHC staining (Figure 1D). These observations suggest that ligand-induced activation of the PPARγ pathway in lal−/− Ly6G+ MDSCs impaired the ability of these myeloid cells to stimulate tumor growth and metastasis.
PPARγ ligand inhibited lal−/− Ly6G+ MDSCs stimulation of tumor proliferation and migration in vitro
The inhibitory effects of the PPARγ ligand on lal−/− Ly6G+ MDSCs stimulation of tumor growth were further examined by in vitro co-culture experiments. Ligand or vehicle pre-treated lal+/+ or lal−/− Ly6G+ cells were co-cultured with B16 melanoma cells for 72 h. As shown in Figure 2A, 9-HODE treatment of lal−/− Ly6G+ cells significantly decreased proliferation of B16 melanoma cells upon co-culture, compared with that of ethanol-treated lal−/− Ly6G+ cells. When 9-HODE-treated lal−/− Ly6G+ cells were co-cultured with Lewis lung cancer (LLC) cells, reduced proliferation of LLC cells was also observed (Figure 2B). Taken together, these results suggest that activation of the PPARγ pathway in lal−/− Ly6G+ cells impaired the capacity of these myeloid cells to stimulate tumor cell proliferation.
Figure 2. PPARγ ligand inhibits lal−/− MDSCs stimulation on tumor proliferation and migration in vitro.
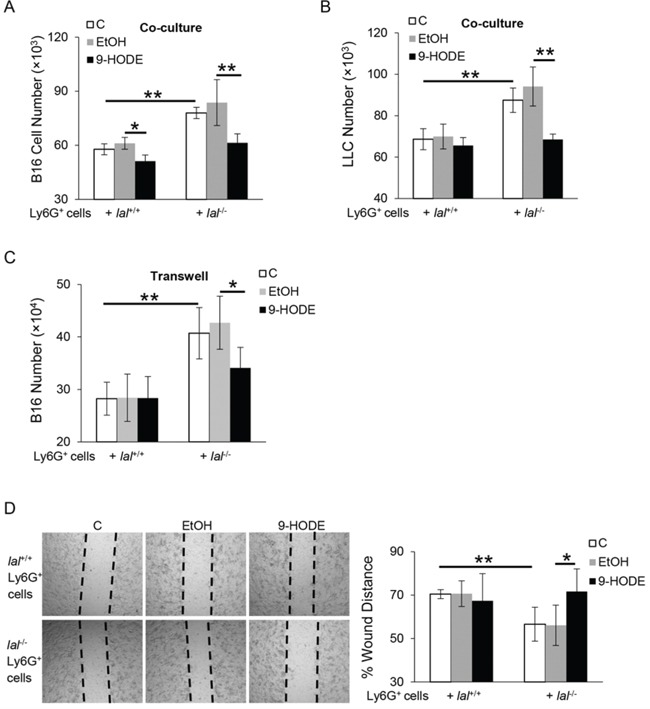
Ly6G+ cells from lal+/+ or lal−/− mice were pre-treated with ethanol (EtOH) or 20 μmol/L 9-HODE for 24 h. A. Pre-treated Ly6G+ cells (5 × 105) were co-cultured with B16 melanoma cells (5 × 103) in vitro for 72 h, and numbers of B16 melanoma cells were counted. n = 4∼5. B. Pre-treated Ly6G+ cells (5 × 105) were co-cultured with LLC cells (1 × 104) in vitro for 72 h, and numbers of LLC cells were counted. n = 4∼5. C. To see the effect of Ly6G+ cell-secreted cytokines on B16 melanoma cell proliferation, pre-treated Ly6G+ cells (1 × 106) were seeded into the upper chamber of transwells, in which B16 melanoma cells (2 × 104) were seeded in the lower chamber. After 72 h, the number of B16 melanoma cells was counted. n = 5. D. Left: in vitro migration of B16 melanoma cells with pre-treated Ly6G+ cells at 24 h after co-culture in the presence of mitomycin C. The dotted lines define the areas lacking cells. Right: Quantification of distance from one end of the wound area to the other end. Data were normalized to B16 melanoma cells co-cultured with control lal+/+ Ly6G+ cells at 0 h. Original magnification, × 40. n = 5. For statistical analyses, data were expressed as mean ± SD; **P < 0.01, *P < 0.05.
Cytokines secreted by lal−/− Ly6G+ MDSCs have been reported to be responsible for mediating their stimulatory effects on cancer cell proliferation [5]. To examine whether 9-HODE treatment has an effect on cytokine-mediated Ly6G+ MDSCs stimulation on cancer cell proliferation, transwell studies were performed with 9-HODE pre-treated Ly6G+ cells seeded in the upper chamber and melanoma cells seeded in the lower chamber. After 72 h co-culture, the number of B16 melanoma cells that were co-cultured with 9-HODE pre-treated lal−/− Ly6G+ cells was significantly less (Figure 2C), suggesting the ability of lal−/− Ly6G+ cells to promote melanoma cell proliferation was impaired by PPARγ ligand treatment.
Because cell migration contributes to metastasis, in vitro tumor cell migration assay was analyzed to determine whether PPARγ ligand treatment of lal−/− Ly6G+ cells influences B16 melanoma cell migration. Melanoma cells were treated with mitomycin C to eliminate the potential effects of cell proliferation in these assays. As shown in Figure 2D, 24 h after co-culture with lal−/− Ly6G+ cells, B16 melanoma cells migrated more efficiently into the area of an artificial wound area compared with those tumor cells co-cultured with lal+/+ Ly6G+ cells. However, delayed migration towards the scratch was observed in 9-HODE pre-treated lal−/− Ly6G+ cells, as revealed by a significant increase in the span of the wounded area. These results also suggest that activation of the PPARγ pathway in lal−/− Ly6G+ cells impaired the stimulatory effects of these MSDCs on B16 melanoma cell migration in vitro.
PPARγ ligand decreased lal−/− MDSC transendothelial migration capability and differentiation from lal−/− Lin− cells
Besides effects on tumor growth and metastasis, lal−/− Ly6G+ MDSCs displayed increased transendothelial migration capability [16], which likely results in the severe infiltration of MDSCs in multiple organs of lal−/− mice. To test whether PPARγ inactivation in lal−/− Ly6G+ cells plays a role in their increased transendothelial migration, transwell assays were performed with 9-HODE pre-treated CMFDA-labeled Ly6G+ cells seeded onto an endothelial monolayer in the upper chamber of the plates. Four hours later, the number of Ly6G+ cells that had migrated through to the lower chamber was determined. As shown in Figure 3A, there were less Ly6G+ cells in the lower chamber when lal−/− Ly6G+ cells were treated with 9-HODE compared with these cells treated with ethanol, suggesting that the PPARγ pathway is involved in Ly6G+ cell endothelial transmigration capability.
Figure 3. PPARγ ligand decreases lal−/− MDSCs transendothelial migration capacity and differentiation from lal−/− Lin- cells.
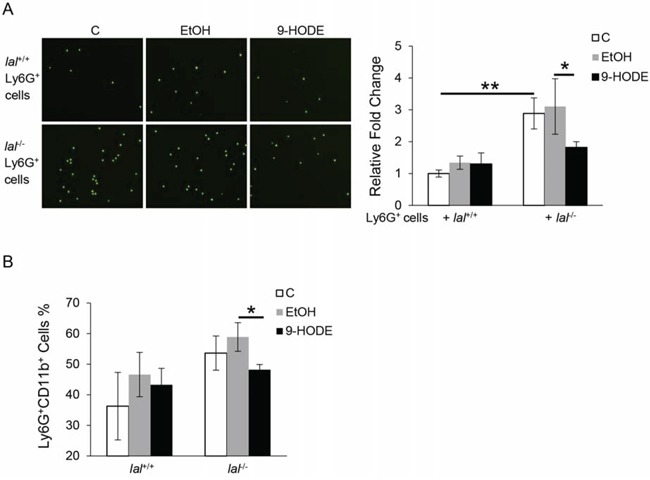
A. Transwell assay was performed to determine MDSCs transmigration across the endothelial monolayer. Ly6G+ cells from lal+/+ or lal−/− mice were pre-treated with ethanol (EtOH) or 20 μmol/L 9-HODE for 48 h, and then labeled with CMFDA and seeded onto the endothelial monolayer at a density of 2 × 104 cells/well. Four hours after seeding Ly6G+ cells on the EC monolayer, the number of Ly6G+ cells that have migrated to the lower chamber was counted. B. Statistical analysis of Ly6G+CD11b+ cells from Lin− cells that were treated with ethanol (EtOH) or 10 μmol/L 9-HODE for 5 days by flow cytometry. Data were expressed as mean ± SD; n = 3∼4. **P < 0.01, *P < 0.05.
Abnormal expansion of MDSCs was also observed in lal−/− mice, which was due to increased differentiation from Lin− cells [1]. PPARγ is known to be expressed in bone marrow progenitor cells and play a critical role in mesenchymal stem cell differentiation and adipogenesis [17–19]. To test the role of PPARγ in this process, bone marrow-derived Lin− cells from lal+/+ and lal−/− mice were isolated and treated with 9-HODE or ethanol. After 5 days of incubation, fewer Ly6G+CD11b+ cells were derived from 9-HODE-treated lal−/− Lin− cells compared with those with ethanol treatment (Figure 3B), suggesting that activation of the PPARγ pathway by 9-HODE prevented these Lin− cells from differentiating into MDSCs.
PPARγ ligand down-regulated mTOR pathway activation in lal−/− Ly6G+CD11b+ cells
We have previously reported that the tumor-promoting function of lal−/− MDSCs is mediated, at least in part, through enhanced activation of the mTOR pathway [5], and that the mTOR pathway is involved in the differentiation of Lin− cells into Ly6G+CD11b+ cells [20]. To test whether PPARγ has an effect on the mTOR pathway, bone marrow cells from lal+/+ and lal−/− mice were treated with 9-HODE or ethanol. After 2 h of incubation, the expression levels of phosphorylated mTOR (pmTOR) and phosphorylated S6 (pS6) in gated Ly6G+CD11b+ cells were measured by flow cytometry analysis. As shown in Figure 4, the increased levels of pmTOR and pS6 in lal−/− Ly6G+CD11b+ cells were not observed in cells following PPARγ ligand treatment. These results suggest that ligand-induced activation of the PPARγ pathway in lal−/− Ly6G+CD11b+ cells by 9-HODE downregulated mTOR pathway activation.
Figure 4. PPARγ ligand down-regulates the overactivation of the mTOR pathway in lal−/− MDSCs.

Bone marrow cells from lal+/+ or lal−/− mice were treated with ethanol (EtOH) or 20 μmol/L 9-HODE for 2 h. A. 9-HODE decreased phosphorylation of mTOR in gated lal−/− Ly6G+CD11b+ cells. B. 9-HODE decreased phosphorylation of S6 in gated lal−/− Ly6G+CD11b+ cells. Statistical analysis of mean fluorescent intensity (MFI) by flow cytometry is shown. Data were expressed as mean ± SD; n = 7. *P < 0.05.
PPARγ ligand reversed damaged mitochondrial membrane potential and suppressed ROS production in lal−/− Ly6G+CD11b+ cells
ROS is an important mediator for MDSCs functions, and its increase is often associated with mitochondrial damage. In lal−/− MDSCs, both damaged mitochondrial function and ROS overproduction have been observed, and inhibition of the mTOR pathway decreased the ROS levels and abnormal mitochondrial membrane potential in lal−/− MDSCs [6]. To see whether PPARγ ligand treatment corrects these defects, bone marrow cells were treated with 9-HODE or ethanol, and ROS levels and mitochondrial membrane potentials were measured by flow cytometry analysis. As demonstrated in Figure 5A, the impaired mitochondrial membrane potential in lal−/− Ly6G+ CD11b+ cells was partially recovered with 9-HODE treatment compared with that in ethanol-treated cells. In addition, 9-HODE treatment suppressed the increased ROS production in lal−/− Ly6G+CD11b+ cells (Figure 5B). These results suggest that ROS overproduction and damaged mitochondrial membrane potential associated with lal−/− MDSCs can be corrected by PPARγ ligand treatment.
Figure 5. PPARγ ligand reverses the damaged mitochondrial membrane potential and suppresses ROS production in lal−/− MDSCs.

Bone marrow cells from lal+/+ or lal−/− mice were treated with ethanol (EtOH) or 20 μmol/L 9-HODE for 48 h. A. 9-HODE increased the mitochondrial membrane potential in gated lal−/− Ly6G+CD11b+ cells. Left: Representative dot plot analysis of the JC-1 red and JC-1 green profiles by flow cytometry. Right: Statistical analysis of the mitochondrial membrane potential in Ly6G+CD11b+ cells. B. 9-HODE decreased ROS production in lal−/− Ly6G+CD11b+ cells. Left: Representative analysis of MFI by flow cytometry. Right: Statistical analysis of MFI in Ly6G+CD11b+ cells. Data were expressed as mean ± SD; n = 5∼6. **P < 0.01, *P < 0.05.
Overexpression of dnPPARγ in myeloid cells facilitated tumor growth and metastasis in vivo, and tumor proliferation and migration in vitro
To further confirm the critical role of PPARγ in myeloid-lineage cells, a doxycycline-inducible c-fms-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mouse model was used, in which a dominant negative PPARγ (dnPPARγ) was overexpressed in myeloid cells under the control of the c-fms promoter [15]. As we published before when the endogenous PPARγ signaling pathway was inhibited by overexpression of dnPPARγ in myeloid cells, the MDSCs level increased in bone marrow, spleen, blood and lung [15]. Here we assess whether the disruption of PPARγ function by expression of dnPPARγ in myeloid cells has similar effect on tumor cell growth and metastasis in vivo and tumor cell proliferation and migration in vitro. In tumor growth assessment, B16 melanoma cells were subcutaneously injected into the flank region of the bitransgenic mice. Figure 6A showed that the tumor volume from doxycycline-treated bitransgenic mice was significantly increased compared with those in untreated mice at 4 weeks post-injection. For the tumor metastasis potential, statistical analysis revealed that two weeks after intravenous injection of B16 melanoma cells, the doxycycline-treated bitransgenic mice showed increased number of melanoma colonies in the lungs compared with untreated mice (Figure 6B). These results suggest that PPARγ inactivation in myeloid cells contributed to the increased tumor growth and metastasis.
Figure 6. Overexpression of dnPPARγ in myeloid cells facilitates tumor growth and metastasis in vivo, and tumor proliferation and migration in vitro.
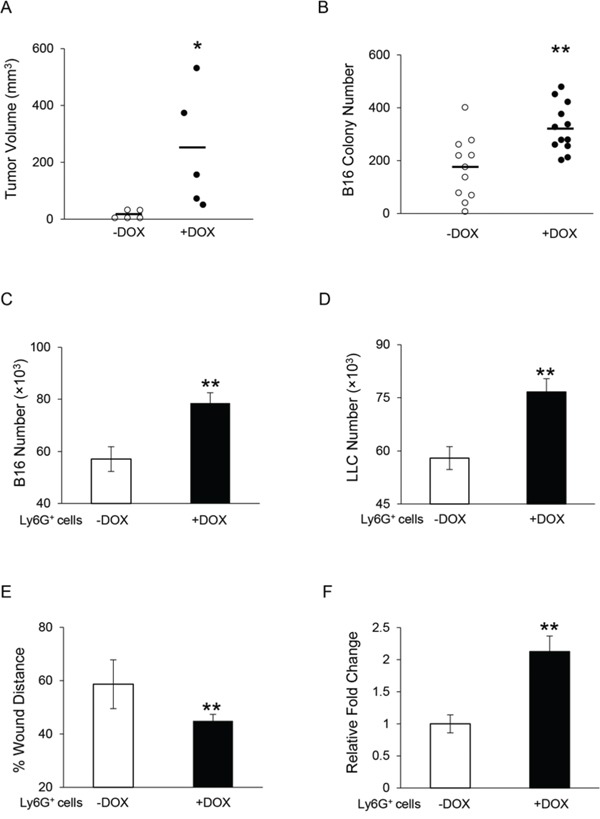
A. Statistical analysis of tumor volume (in cubic millimeters) at 4 weeks after B16 melanoma cells (2 × 105) were subcutaneously injected into doxycycline-treated or untreated c-fmsrtTA/(tetO)7-dnPPARγ bitransgenic mice. n = 5. *P < 0.05. B. Quantitative analysis of metastasized B16 melanoma colonies in the lungs of doxycycline-treated or untreated bitransgenic mice with intravenous injection of 5 × 105 B16 melanoma cells for 2 weeks. n = 11∼12. **P < 0.01. C. B16 melanoma cells (5 × 103) were co-cultured with Ly6G+ cells (5 × 105) from doxycycline-treated or untreated bitransgenic mice in vitro for 72 h, and numbers of B16 melanoma cells were counted. D. LLC cells (1 × 104) were co-cultured with doxycycline-treated or untreated Ly6G+ cells (5 × 105) in vitro for 72 h, and the numbers of LLC cells were counted. E. In vitro migration of B16 melanoma cells with doxycycline-treated or untreated Ly6G+ cells at 24 h after co-culture in the presence of mitomycin C. Data were normalized to B16 melanoma cells co-cultured with untreated Ly6G+ cells at 0 h. F. Ly6G+ cell transendothelial migration was determined. Data are normalized to untreated Ly6G+ cells. In the above experiments (C-F), data were expressed as mean ± SD; n = 4. **P < 0.01.
When bone marrow Ly6G+ cells from doxycycline-treated bitransgenic mice were co-cultured with B16 melanoma cells in vitro, increased proliferation of B16 melanoma cells was observed in comparison with those cells from untreated bitransgenic mice (Figure 6C). Similarly, proliferation of LLC was significantly increased after co-cultured with bone marrow Ly6G+ cells from doxycycline-treated bitransgenic mice (Figure 6D). Furthermore, the in vitro wound healing assay showed accelerated migration towards the scratch in B16 melanoma cells co-cultured with bone marrow Ly6G+ cells from doxycycline-treated bitransgenic mice 24 h after creating the scratch, with a significant decrease of distance in the wounding area (Figure 6E). In addition, the transendothelial migration capability of Ly6G+ cells from doxycycline-treated bitransgenic mice was obviously increased as shown in Figure 6F. Taken together, these results indicate that PPARγ inactivation in Ly6G+ cells facilitated their transendothelial migration, and stimulation of tumor cell proliferation and migration.
Overexpression of dnPPARγ in myeloid cells overactivated the mTOR pathway, increased ROS production and impaired maintenance of mitochondrial membrane potential
To explore the potential mechanisms underlying the dysfunctions of MDSCs from doxycycline-treated dnPPARγ bitransgenic mice, changes in the mTOR pathway were explored. As determined above using PPARγ ligands, the pathogenic function of MDSCs could be linked to mTOR activation in lal−/− MDSCs. Results showed that the phosphorylation levels of mTOR and S6 in gated doxycycline-treated Ly6G+CD11b+ cells were increased significantly compared with those of untreated Ly6G+CD11b+ cells, with no statistically significant change of mTOR and S6 protein levels (Figure 7A). As a consequence, the mitochondrial membrane potential in doxycycline-treated Ly6G+CD11b+ cells was impaired (Figure 7B). In addition, the ROS production in doxycycline-treated Ly6G+CD11b+ cells was significantly increased compared with untreated Ly6G+CD11b+ cells (Figure 7C). These results support that the PPARγ pathway regulates MDSCs functions by modulating mTOR, ROS production and mitochondrial membrane potential.
Figure 7. Overexpression of dnPPARγ in myeloid cells overactivates the mTOR pathway, increases ROS production and impairs mitochondrial membrane potential.
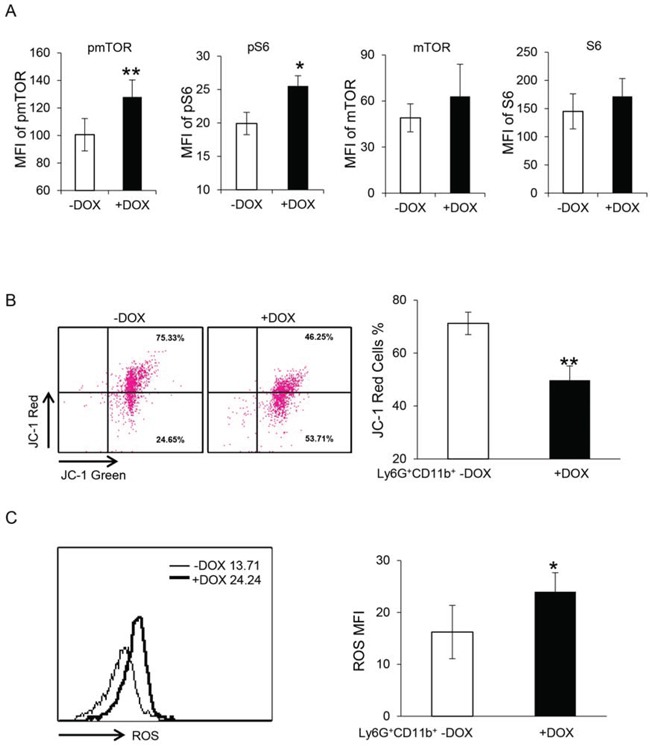
A. The mTOR pathway was overactivated in doxycycline-treated bone marrow Ly6G+CD11b+ cells. Statistical analysis of MFI by flow cytometry is shown. Data were expressed as mean ± SD; n = 4∼5. **P < 0.01, *P < 0.05. B. The mitochondrial membrane potential was impaired in doxycycline-treated bone marrow Ly6G+CD11b+ cells. Left: Representative dot plot analysis of the JC-1 red and JC-1 green profiles by flow cytometry. Right: Statistical analysis of the mitochondrial membrane potential in Ly6G+CD11b+ cells. C. ROS production was increased in doxycycline-treated Ly6G+CD11b+ cells. Left: Representative analysis of MFI by flow cytometry. Right: Statistical analysis of MFI in Ly6G+CD11b+ cells. For statistical analyses, data were expressed as mean ± SD; n = 5. **P < 0.01, *P < 0.05.
DISCUSSION
Activating PPARγ can prevent cancer development and spread in tissues such as colon, breast, prostate and lung, while ligand-induced activation of PPARγ in cancer cell lines is associated with the induction of cell cycle arrest, the increased expression of mRNAs and proteins required for terminal differentiation, as well as changes in cell morphology that are consistent with a differentiated phenotype [21]. Yet understanding pathways by which PPARγ contributes to the spread and progression of cancer in vivo, including its effects on immune cells is less well understood. LAL is a key enzyme which functions in the metabolism of neutral lipids, and its role in inflammation has been widely studied [1, 4, 20, 22]. Genetic ablation of the lal gene in mice results in a systemic increases in MDSCs and decreases in T cell populations, causing severe inflammation and pathogenesis in multiple organs [1, 23]. LAL deficiency causes inactivation of PPARγ by blocking PPARγ ligand synthesis [14]. The PPARγ signaling pathway has recently been reported to play a key role in controlling MDSC expansion and T cell proliferation [15]. Here, 9-HODE, a PPARγ ligand [24], reversed the increased MDSC expansion (Figure 3B) and decreased T cell numbers in lal−/− mice (data not shown), suggesting that PPARγ signaling is critical in regulating LAL-mediated metabolic pathways central to immune suppression [15]. Interestingly and importantly, our previous [5] and present studies have shown that lal−/− MDSCs were able to also overcome potent immune rejection and destruction of tumors in allogeneic mouse models.
In addition to immunosuppression, we have shown that lal−/− MDSCs have a second potent effect on tumors, by directly stimulating tumor cell proliferation, growth and metastasis [5]. B16 melanoma cells grew quickly and metastasized massively in allogeneic lal−/− mice, yet this effect was suppressed in allogeneic lal+/+ mice where immune destruction of the tumor took place. However, lal−/− MDSCs facilitated melanoma cells' growth and metastasis in allogeneic lal+/+ mice [5]. In our preliminary study, B16 melanoma cells' metastasis was delayed in lal−/− mice pre-treated with PPARγ ligand 9-HODE. In the present study, activation of the PPARγ pathway in lal−/− MDSCs with its ligand 9-HODE not only impaired their stimulatory effects on in vivo tumor growth and metastasis (Figure 1), but also significantly retarded the ability of these MSDCs to block in vitro tumor cell proliferation and migration (Figure 2). We have reported that cytokines, (especially TNFα) secreted by lal−/− Ly6G+ cells are, at least in part, responsible for mediating stimulatory effects on cancer cells [5]. In the present study, 9-HODE inhibited the cytokine-mediated stimulatory effects of lal−/− MDSCs on melanoma cell proliferation (Figure 2C). Therefore, PPARγ inactivation in lal−/− MDSCs represents a major mechanism underlying the stimulatory effects of MSDCs on cancer cell proliferation and metastasis.
One major manifestation during LAL deficiency is systemic MDSCs expansion and dysfunction in multiple organs of the mice [1, 2, 14, 22, 25], which arises from dysregulated production of hematopoietic myeloid progenitor cells in the bone marrow [1]. When lal−/− Lin− cells were treated with 9-HODE to activate the PPARγ pathway, reduced differentiation of Lin− cells into Ly6G+CD11b+ MDSCs was observed (Figure 3B). Kaipainen et al showed that PPARγ ligands inhibit primary tumor growth and metastasis by targeting endothelial cells to inhibit angiogenesis [26]. Abnormal accumulation of MDSCs in the lungs of lal−/− mice has been linked to both endothelial cells (ECs) and MDSCs [16]. Activation of the PPARγ pathway in lal−/− MDSCs with 9-HODE decreased the transendothelial migration of MDSCs through the EC monolayer (Figure 3A). These results collectively indicate that activation of the PPARγ pathway corrects lal−/− MDSCs dysfunction and abnormal expansion during LAL deficiency.
In addition to inhibition of the PPARγ pathway, enhanced activation of the mTOR pathway was associated with lal−/− MDSC dysfunction as detected by Affymetrix GeneChip microarray and Ingenuity analyses [6]. Thus, multiple pathways may contribute to regulate MSDCs functions. Studies have shown that the mTOR pathway regulates PPARγ activation during adipogenesis by targeting the transactivation activity of PPARγ [27, 28]. Interaction between mTOR and PPARγ has been reported before in hepatocytes [29]. Deficiency of PPARγ in chondrocytes resulted in aberrant activation of mTOR signaling pathway [30]. The present study shows that the mTOR pathway in lal−/− MDSCs is regulated by PPARγ. Incubation with 9-HODE not only significantly decreased the phosphorylation levels of mTOR and S6, but also reduced the overall levels of mTOR and S6 in lal−/− MDSCs (Figure 4). ROS production has been reported to be one mechanism underlying MDSCs functions [3]. In our previous studies, ROS production was increased in lal−/− MDSCs with impaired mitochondrial function, which mediated the mTOR-regulated lal−/− MDSCs dysfunctions [6, 20]. Activation of the PPARγ pathway in lal−/− bone marrow cells with its ligand 9-HODE effectively improved the mitochondrial function and blocked ROS overproduction in lal−/− Ly6G+CD11b+ MDSCs (Figure 5), suggesting that ROS overproduction by lal−/− MDSCs is controlled by the PPARγ pathway. Therefore, the mTOR-ROS pathway serves as a potential mechanism to mediate the LAL-PPARγ axis in MDSC dysfunctions. Kittler et al recently found that PPARγ inhibits cancer cell proliferation by a metabolic switch, including suppressing pyruvate oxidation and reducing glutathione levels, which results in a marked increase of ROS levels, leading to rapid hypophosphorylation of retinoblastoma protein and cell-cycle rest [31]. Similarly, in a “lal−/− MDSCs-like cell line”, we have observed an mTOR-controlled metabolic switch towards increased glycolysis and ROS levels 30. In the current study, with PPARγ ligand treatment of lal−/− MDSCs we found these cells displayed reduced ROS and were unable to effectively stimulate tumor cell proliferation.
The role of the PPARγ pathway in MDSC functions was further investigated using an established bitransgenic mouse model, in which dnPPARγ was overexpressed in myeloid-lineage cells, resulting in blockade of endogenous PPARγ function [15]. In this mouse model, the function of the receptor of PPARγ pathway is impaired rather than the ligand expression which is perturbed in lal−/− mouse model. When melanoma cells were injected subcutaneously into these mice, larger tumor developed in the mice with myeloid-specific dnPPARγ overexpression induced by doxycycline than non-induced bitransgenic mice (Figure 6A). In addition, after intravenous injection of melanoma cells, more melanoma developed in the lungs of mice with myeloid-specific dnPPARγ overexpression (Figure 6B). When MDSCs from the mice with myeloid-specific dnPPARγ overexpression were co-cultured with B16 melanoma cells or LLC cells in vitro, cancer cell proliferation was enhanced (Figure 6C and 6D). Moreover, these MDSCs facilitated melanoma cell migration (Figure 6E), possessed increased transmigration through the EC monolayer (Figure 6F), over-activated the mTOR pathway, and impaired mitochondrial function and ROS overproduction (Figure 7), similar to the characteristics observed in lal−/− MDSCs, which showed inactivation of the PPARγ pathway.
In conclusion, the PPARγ pathway plays a critical role in metabolic signaling controlled by LAL through regulating the function of MDSCs. The PPARγ pathway served as a novel target to modulate the emergence of MDSCs to reduce the risk of cancer progression and metastasis. It has been extensively reported that PPARγ ligands have a direct inhibitory effect on tumor cells [32–40]. But their effect in MDSCs of the tumor microenviroment is poorly understood, and should be given a special attention. Therefore, PPARγ may impact cancer cell proliferation through both direct and indirect mechanisms including effects on MSDCs. The study outlined here indicates that enhancing PPARγ function in MDSCs should prove to be a highly effective strategy in blocking tumor cell growth and spread even in cases where tumors may not respond directly to PPARγ ligands. Among collection of ligands to PPARγ, those more likely to achieve this therapeutic outcome in MDSCs remain to be tested.
MATERIALS AND METHODS
Animals and cell lines
Wild-type (lal+/+) and lal−/− mice of the FVB/N background were bred in house. c-fms-rtTA/(TetO)7-CMV-dnPPARγ bitransgenic mice of the FVB/N background is a previously generated bitransgenic mouse model [15]. All scientific protocols involving the use of animals have been approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine and followed guidelines established by the Panel on Euthanasia of the American Veterinary Medical Association. Animals were housed under Institutional Animal Care and Use Committee-approved conditions in a secured animal facility at Indiana University School of Medicine.
The murine B16 melanoma cell line, Lewis lung carcinoma (LLC) cell line, and murine endothelial cell (SVEC) line (purchased from ATCC, Manassas, VA, USA) were cultured in DMEM supplemented with 10% FBS (Gibco, Grand Island, NY, USA).
PPARγ ligand treatment
For in vitro PPARγ ligand treatment, 9-HODE (Cayman Chemical Co., Ann Arbor, MI, USA) was added into the culture medium of MDSCs to a final concentration of 20 μmol/L for 24 or 48 h. For the study of the effect of PPARγ ligand on the mTOR signaling pathway, bone marrow cells were treated with 9-HODE (20 μmol/L) for 2 h.
Isolation of bone marrow-derived MDSCs
MDSCs were isolated as we previously described [5, 6]. Unlike those being classified into monocytic and granulocytic MDSCs, almost all lal−/− MDSCs are Ly6G+Ly6C+, and almost all lal−/− MDSCs are CD11b+Ly6G+ cells. Therefore, to simplify the lal−/− MDSCs isolation procedure, Ly6G antibody-coupled magnetic beads were used and sufficient to isolate lal−/− MDSCs from the lal−/− bone marrow, and equivalent control from the wild type bone marrow [1, 2]. Briefly, bone marrow cells were isolated from the femurs and tibias of mice. Cells were first incubated with biotin-conjugated anti-Ly6G antibody at 4°C for 15 min. After washed with PBS, cells were incubated with anti-biotin microbeads at 4°C for another 15 min. Subsequently, cells were subjected to magnetic bead sorting according to the manufacturer's instructions (Miltenyi Biotec., Auburn, CA, USA).
Mouse tumor growth and metastasis model
The tumor growth and metastasis model have been described recently [5]. MDSCs and B16 melanoma cells were collected separately. A pilot study has been performed to determine the best ratio between MDSCs and B16 melanoma cells. To test the tumor growth potential, 6 × 105 pre-treated MDSCs and 2 × 105 B16 melanoma cells were mixed, centrifuged and re-suspended in 100 μL PBS, and then injected subcutaneously into the flank region of 3-month old recipient lal+/+ mice. Tumor volume (length × width2 × π/6) was monitored every week for 4 weeks. To test the metastasis potential, 2 × 106 pre-treated MDSCs and 5 × 105 B16 melanoma cells were mixed and incubated at 37°C, 5% CO2 for 30 min. After the incubation, cells were centrifuged, re-suspended, and injected intravenously into 3-month old lal+/+ mice. Two weeks after the injection, the mice were sacrificed and the lungs were inflated with 4% paraformaldehyde for examination of metastasis.
Histology and immunohistochemical staining
The harvested lungs were fixed with 4% paraformaldehyde in PBS at 4°C for overnight. After fixation and embedding in paraffin, tissue sections were cut to 5 μm thick. Hematoxylin and eosin (H&E) staining and immunohistochemical (IHC) staining with anti-Ki67 antibody were performed by the Histological Core Facility, Department of Pathology and Laboratory Medicine, Indiana University. Images were taken by Olympus microscopy image system (Olympus, Tokyo, Japan).
In vitro co-culture of MDSCs and B16 melanoma cells
Previous study has determined the best ratio between MDSCs and B16 melanoma cells [5]. Ethanol or 20 μmol/L 9-HODE pre-treated (for 24 h) MDSCs (5 × 105) and B16 melanoma cells (5 × 103) were mixed, and seeded into a well of 96-well plates in DMEM supplemented with 10% FBS. Seventy-two hours later, unattached MDSCs were removed by washing with PBS, and the number of attached B16 melanoma cells was counted. Morphologically, MDSCs are much smaller than B16 melanoma cells for exclusion.
In vitro migration assay
In vitro wound healing assay was performed to analyze B16 melanoma cell migration as previously described [16, 41]. Briefly, B16 melanoma cells were seeded at a density of 1.5 × 105 cells/well into a 24-well plate and incubated overnight to form a confluent monolayer. Scratch was created by scraping the cell monolayer in a straight line with a p200 pipet tip. After washing 3 times with DMEM, the medium was changed with DMEM containing 10% FBS and 5 μg/mL mitomycin C (Sigma-Aldrich, St. Louis, MO, USA), and MDSCs pre-treated with 9-HODE or ethanol for 24 h were added onto B16 melanoma cell monolayer at a density of 1 × 106 cells/well. Images were taken at 0 and 24 h after creating the scratch. Migration was estimated by measuring the distances from one side of scratch to the other side using Image Pro-Plus software (Media Cybernetics, Rockville, MD, USA).
Transwell assay
Transwell assay was used to determine MDSC transendothelial migration [16]. SVECs were added to the upper chamber of 24-well 8.0-μm-pore Transwell plates (Corning, Corning, NY, USA), and incubated at 37°C, 5% CO2 for 48 h to form an EC monolayer. The supernatant was then removed, and CellTrackerTM Green 5-Chloromethylfluorescein Diacetate (CMFDA) (Invitrogen, Grand Island, NY, USA)-labeled MDSCs (2 × 104 cells in 200 μL media) were added to the upper well. After 4 h, transendothelial migration of MDSCs was determined by counting their numbers in the lower chamber under 5 random microscopic fields.
To observe the effect of MDSCs-secreted cytokines on melanoma cell proliferation, transwell assay was performed with 0.4-μm-pore 6.5-mm diameter Transwell plates (Corning) to separate MDSCs and B16 melanoma cells. One million pre-treated MDSCs in 200 μL media were seeded into the upper chamber of the plates, while 2 × 104 melanoma cells in 500 μL media were placed in the lower chamber. After 72 hours' culture, the transwells were removed, and the number of B16 melanoma cells in the lower chamber was counted.
Isolation of bone marrow lineage-negative cells
Lineage-negative (Lin−) cells were isolated from the bone marrow by removing blood lineage marker-positive cells with an immunomagnetic microbead technique as we previously described [20]. Briefly, bone marrow cells were first incubated with a cocktail of biotin-conjugated antibodies against lineage specific antigens: CD11b, GR-1, B220, TER-119, and CD3ε (Mouse Lineage Panel Kit, BD Pharmingen, San Diego, CA, USA) at 4°C for 15 min. After washed with PBS, cells were then incubated with anti-biotin microbeads at 4°C for another 15 min. Subsequently, cells were subjected to magnetic bead sorting according to the manufacturer's instructions (Miltenyi Biotec.). The resulting Lin− cells were cultured in RPMI1640 with 10% FBS. Five days later, Ly6G+CD11b+ cells derived from Lin− cells were analyzed by flow cytometry analysis.
ROS and mitochondrial membrane potential measurement
The reactive oxygen species (ROS) level and mitochondrial membrane potential in MDSCs was measured by flow cytometry as we previously described [20]. Briefly, bone marrow cells were first treated with or without 20 μmol/L 9-HODE or ethanol for 2 days. For ROS level detection, cells were harvested, washed, and stained with 2′, 7′-dichlorofluorescein diacetate (2 μmol/L, Invitrogen), allophycocyanin cy7-conjugated anti-Ly6G Ab, and phycoerythrin cy7-conjugated anti-CD11b Ab (eBioscience) at 37°C for 15 min. After PBS wash, the ROS level in Ly6G+CD11b+ cells was analyzed using a LSRII machine (BD Biosciences).
For mitochondrial membrane potential measurement, cells were stained with the fluorescent dye JC-1 (2 μmol/L, Molecular Probes, Eugene, OR, USA), allophycocyanin cy7-conjugated anti-Ly6G antibody, and phycoerythrin cy7-conjugated anti-CD11b antibody (eBioscience) at 37°C for 15 min, and then analyzed for phycoerythrin (JC-1 red) and fluorescein isothiocyanate (JC-1 green) fluorescent cells in Ly6G+CD11b+ cells by flow cytometry. Cells treated with 50 μmol/L carbonyl cyanide 3-chlorophenylhydrazone for 5 min were served as a fluorescein isothiocyanate-positive control.
Flow cytometry analysis
Single cells from the bone marrow of 5-month-old lal+/+ and lal−/− mice were prepared as previously described [9]. After 20 μmol/L 9-HODE treatment for 2 h, cells were harvested, and labeled with anti-Ly6G and CD11b cell surface antibody (eBioscience) at 4°C for 15 min. Cells were then fixed and permeabilized using BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer's instructions, and incubated with Alexa Fluor 647-conjugated anti-mTOR antibody, Alexa Fluor 488-conjugated anti-S6 antibody, Alexa Fluor 488-conjugated anti-pS6 (Ser235/236) antibody, and rabbit anti-pmTOR (Ser2448) antibody (Cell Signaling Technology, Beverly, MA, USA) at 4°C overnight. For anti-pmTOR antibody staining, cells were incubated with Alexa Fluor 647-conjugated anti-rabbit IgG in the following day. Cells were washed and ready for flow cytometry analysis. Mean fluorescence intensities of the proteins in the gated Ly6G+CD11b+ area were analyzed. For flow cytometry analysis, ≥ 10,000 cells were acquired and scored using a LSRII machine (BD Biosciences). Data were processed using the CellQuest software program (BD Biosciences).
Statistics
Data were expressed as mean ± SD. Differences between two treatment groups were compared by Student's t-test. When more than two groups were compared, one-way ANOVA with post-hoc Newman-Keul's multiple comparison test was used. Results were considered statistically significant when P < 0.05. All analyses were performed with GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA).
Acknowledgments
This work was supported by the National Institutes of Health Grants CA138759, CA152099 (to C. Y.) and HL087001 (to H. D.). We thank Michele Klunk and Katlin L. Walls for animal maintenance.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by National Institutes of Health Grants HL087001 (to H. D.), and CA138759, CA152099 (to C. Y.).
REFERENCES
- 1.Qu P, Shelley WC, Yoder MC, Wu L, Du H, Yan C. Critical roles of lysosomal acid lipase in myelopoiesis. Am J Pathol. 2010;176:2394–2404. doi: 10.2353/ajpath.2010.091063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qu P, Yan C, Blum JS, Kapur R, Du H. Myeloid-Specific Expression of Human Lysosomal Acid Lipase Corrects Malformation and Malfunction of Myeloid-Derived Suppressor Cells in lal−/− Mice. The Journal of Immunology. 2011;187:3854–3866. doi: 10.4049/jimmunol.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan C, Lian X, Li Y, Dai Y, White A, Qin Y, Li H, Hume DA, Du H. Macrophage-specific expression of human lysosomal acid lipase corrects inflammation and pathogenic phenotypes in lal−/− mice. Am J Pathol. 2006;169:916–926. doi: 10.2353/ajpath.2006.051327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao T, Du H, Ding X, Walls K, Yan C. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal−/− mice. Oncogene. 2015;34:1938–1948. doi: 10.1038/onc.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan C, Ding X, Dasgupta N, Wu L, Du H. Gene Profile of Myeloid-Derived Suppressive Cells from the Bone Marrow of Lysosomal Acid Lipase Knock-Out Mice. PLoS ONE. 2012;7:e30701. doi: 10.1371/journal.pone.0030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang C, Ting AT, Seed B. PPAR-[gamma] agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 8.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-[gamma] is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 9.Qu P, Du H, Li Y, Yan C. Myeloid-specific expression of Api6/AIM/Sp alpha induces systemic inflammation and adenocarcinoma in the lung. J Immunol. 2009;182:1648–1659. doi: 10.4049/jimmunol.182.3.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu P, Du H, Wang X, Yan C. Matrix metalloproteinase 12 overexpression in lung epithelial cells plays a key role in emphysema to lung bronchioalveolar adenocarcinoma transition. Cancer Res. 2009;69:7252–7261. doi: 10.1158/0008-5472.CAN-09-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Du H, Qin Y, Roberts J, Cummings OW, Yan C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007;67:8494–8503. doi: 10.1158/0008-5472.CAN-07-0647. [DOI] [PubMed] [Google Scholar]
- 12.Qu P, Yan C, Du H. Matrix metalloproteinase 12 overexpression in myeloid lineage cells plays a key role in modulating myelopoiesis, immune suppression, and lung tumorigenesis. Blood. 2011;117:4476–4489. doi: 10.1182/blood-2010-07-298380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Qu P, Wu L, Li B, Du H, Yan C. Api6/AIM/Spalpha/CD5L overexpression in alveolar type II epithelial cells induces spontaneous lung adenocarcinoma. Cancer Res. 2011;71:5488–5499. doi: 10.1158/0008-5472.CAN-10-4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian X, Yan C, Qin Y, Knox L, Li T, Du H. Neutral lipids and peroxisome proliferator-activated receptor-{gamma} control pulmonary gene expression and inflammation-triggered pathogenesis in lysosomal acid lipase knockout mice. Am J Pathol. 2005;167:813–821. doi: 10.1016/s0002-9440(10)62053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L, Yan C, Czader M, Foreman O, Blum JS, Kapur R, Du H. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119:115–126. doi: 10.1182/blood-2011-06-363093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao T, Ding X, Du H, Yan C. Myeloid-derived suppressor cells are involved in lysosomal acid lipase deficiency-induced endothelial cell dysfunctions. J Immunol. 2014;193:1942–1953. doi: 10.4049/jimmunol.1301941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahman S, Czernik PJ, Lu Y, Lecka-Czernik B. β-Catenin Directly Sequesters Adipocytic and Insulin Sensitizing Activities but Not Osteoblastic Activity of PPARγ2 in Marrow Mesenchymal Stem Cells. PLoS ONE. 2012;7:e51746. doi: 10.1371/journal.pone.0051746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rippo MR, Babini L, Prattichizzo F, Graciotti L, Fulgenzi G, Tomassoni Ardori F, Olivieri F, Borghetti G, Cinti S, Poloni A, Fazioli F, Procopio AD. Low FasL levels promote proliferation of human bone marrow-derived mesenchymal stem cells, higher levels inhibit their differentiation into adipocytes. Cell Death Dis. 2013;4:e594. doi: 10.1038/cddis.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARγ2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. Journal of Cellular Biochemistry. 2009;106:232–246. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding X, Du H, Yoder MC, Yan C. Critical role of the mTOR pathway in development and function of myeloid-derived suppressor cells in lal−/− mice. Am J Pathol. 2014;184:397–408. doi: 10.1016/j.ajpath.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lian X, Yan C, Yang L, Xu Y, Du H. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L801–807. doi: 10.1152/ajplung.00335.2003. [DOI] [PubMed] [Google Scholar]
- 23.Qu P, Du H, Wilkes DS, Yan C. Critical roles of lysosomal acid lipase in T cell development and function. Am J Pathol. 2009;174:944–956. doi: 10.2353/ajpath.2009.080562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schupp M, Lazar MA. Endogenous ligands for nuclear receptors: digging deeper. J Biol Chem. 2010;285:40409–40415. doi: 10.1074/jbc.R110.182451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. Journal of Lipid Research. 2001;42:489–500. [PubMed] [Google Scholar]
- 26.Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, Fletcher JA, Hlatky L, Hahnfeldt P, Folkman J, Kaipainen A. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JE, Chen J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–2756. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Xu G, Qin Y, Zhang C, Tang H, Yin Y, Xiang X, Li Y, Zhao J, Mulholland M, Zhang W. Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARγ signaling pathway. Proceedings of the National Academy of Sciences. 2014;111:13163–13168. doi: 10.1073/pnas.1411571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasheghani F, Zhang Y, Li Y-H, Blati M, Fahmi H, Lussier B, Roughley P, Lagares D, Endisha H, Saffar B, Lajeunesse D, Marshall WK, Rampersaud YR, Mahomed NN, Gandhi R, Pelletier J-P, et al. PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Annals of the Rheumatic Diseases. 2015;74:569–578. doi: 10.1136/annrheumdis-2014-205743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava N, Kollipara RK, Singh DK, Sudderth J, Hu Z, Nguyen H, Wang S, Humphries CG, Carstens R, Huffman KE, DeBerardinis RJ, Kittler R. Inhibition of Cancer Cell Proliferation by PPARgamma Is Mediated by a Metabolic Switch that Increases Reactive Oxygen Species Levels. Cell Metab. 2014;20:650–661. doi: 10.1016/j.cmet.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paulitschke V, Gruber S, Hofstatter E, Haudek-Prinz V, Klepeisz P, Schicher N, Jonak C, Petzelbauer P, Pehamberger H, Gerner C, Kunstfeld R. Proteome analysis identified the PPARgamma ligand 15d-PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor-stroma interaction. PLoS One. 2012;7:e46103. doi: 10.1371/journal.pone.0046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han SW, Roman J. Anticancer actions of PPARgamma ligands: Current state and future perspectives in human lung cancer. World journal of biological chemistry. 2010;1:31–40. doi: 10.4331/wjbc.v1.i3.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol Ther. 2005;4:687–693. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- 35.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8:6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 36.Rashid-Kolvear F, Taboski MA, Nguyen J, Wang DY, Harrington LA, Done SJ. Troglitazone suppresses telomerase activity independently of PPARgamma in estrogen-receptor negative breast cancer cells. BMC cancer. 2010;10:390. doi: 10.1186/1471-2407-10-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang M, Fernandes S., Jerome W.G., He Y., Yu X., Cai H., Boone B., Yi Y., Magnuson M.A., Roy-Burman P., Matusik R.J., Shappell S.B., Hayward S.W. Disruption of PPARgamma signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. Cell Death Differ. 2010;17:469–481. doi: 10.1038/cdd.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehta RG, Williamson E., Patel M.K., Koeffler H.P. A ligand of peroxisome proliferator-activated receptor gamma, retinoid, and prevention of preneoplastic mammary lesions. J Natl Cancer Inst. 2000;92:418–423. doi: 10.1093/jnci/92.5.418. [DOI] [PubMed] [Google Scholar]
- 39.Bren-Mattison Y, Meyer AM, Van Putten V, Li H, Kuhn K, Stearman R, Weiser-Evans M, Winn RA, Heasley LE, Nemenoff RA. Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Molecular pharmacology. 2008;73:709–717. doi: 10.1124/mol.107.042002. [DOI] [PubMed] [Google Scholar]
- 40.Sawayama H, Ishimoto T, Watanabe M, Yoshida N, Sugihara H, Kurashige J, Hirashima K, Iwatsuki M, Baba Y, Oki E, Morita M, Shiose Y, Baba H. Small molecule agonists of PPAR-gamma exert therapeutic effects in esophageal cancer. Cancer Res. 2014;74:575–585. doi: 10.1158/0008-5472.CAN-13-1836. [DOI] [PubMed] [Google Scholar]
- 41.Liang C-C, Park AY, Guan J-L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protocols. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]