

Abstract
In patients with malignancy, the major barrier to achieving complete response is emergence of resistance to current chemotherapeutic agents. One of the major mechanisms by which tumour cells become resistant to therapies is by altering cellular drug targets through mutations and/or deletions. Resistance by this mechanism is achieved more easily if the drug has limited cellular targets and/or processes. We hypothesized that as Pseudomonas aeruginosa exotoxin T (ExoT) targets six proteins that are required for cancer cell survival and proliferation, it is highly unlikely for cancer cells to develop resistance to this toxin. We assessed ExoT’s cytotoxicity against multiple invasive and highly resistant tumour cell lines in order to evaluate its potential as a chemotherapeutic agent. Our data demonstrated that ExoT induced potent cytotoxicity in all tumour cell lines that we examined. Collectively, our data highlighted the potential of ExoT as a possible chemotherapeutic candidate for the treatment of cancer.
Introduction
Resistance to cancer chemotherapeutic drugs is a frequent cause of cancer treatment failure (Gonzalez-Angulo et al., 2007) and highlights the need for novel therapies. Tumour cells can utilize a number of mechanisms to develop resistance to cancer drugs (Gottesman, 2002; Calcagno & Ambudkar, 2010). One of the main mechanisms by which resistance to therapy develops in cancer is by modifications in the cellular drug target through mutations, deletions and/or gene amplification (Gottesman, 2002; Calcagno & Ambudkar, 2010). Development of drug resistance by this mechanism is achieved more easily if the therapy has limited cellular targets and/or processes. For instance, mutations in the epidermal growth factor receptor (EGFR) gene have been identified in specimens from patients with non-small-cell lung cancer treated with anilinoquinazoline EGFR inhibitors (Kobayashi et al., 2005). Similarly, resistance to the Abl tyrosine kinase inhibitor STI-571 in chronic myeloid leukaemia patients was shown to occur through either bcr–abl gene amplification or a single C→T nucleotide change within the abl locus at nt 944 (Gorre et al., 2001).
Bacterial toxins have been, and continue to be, evaluated in various clinical trials as potential cancer therapeutics (Kawakami et al., 2006; Kreitman, 2006, 2009). For example, Pseudomonas exotoxin A and diphtheria toxin are the most common bacterial toxins that have been or are currently under clinical evaluation against a variety of haematologic malignancies and solid tumours with promising results (reviewed by Becker & Benhar, 2012). Although these new recombinant immunotoxins have shown high potency in killing tumour cells with high specificity, they too share the limitation of targeting a single cellular substrate, eEF-2 (Jørgensen et al., 2008). In fact, mutations in eEF-2 have been shown to emerge easily, conferring resistance to these toxins in cancer cells (Foley et al., 1995; Jørgensen et al., 2008; Wei et al., 2012).
Pseudomonas aeruginosa exotoxin T (ExoT) is different from exotoxin A and diphtheria toxin in that instead of a single putative target (e.g. eEF-2), it has at least six cellular proteins (Krall et al., 2000; Kazmierczak & Engel, 2002; Sun & Barbieri, 2003; Garrity-Ryan et al., 2004), which play important roles in survival, proliferation, metastasis and angiogenesis in cancer. ExoT’s substrates include the small GTPase proteins, Ras homologue gene family, member A (RhoA), Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division control protein 42 homologue (Cdc42); which serve many important functions in cancer, such as regulation of actin cytoskeletal dynamics, activation of protein kinases, cell cycle progression, cytokinesis, metastasis and cellular survival (del Peso et al., 1997; Gómez et al., 1997; Bagrodia et al., 1998; Murga et al., 2002; Feng et al., 2010); the C10 regulator of kinases (CrkI, CrkII adaptor proteins), which are important for the formation and maintenance of cellular focal adhesions and cytokinesis (Cho & Klemke, 2000; Lamorte et al., 2002; Rodrigues et al., 2005; Heasman & Ridley, 2008); and the glycolytic enzyme phosphoglycerate kinase 1 (PGK1), which is important for angiogenesis in cancer (Lay et al., 2000; Hwang et al., 2006). Therefore, development of resistance in cancer cells to ExoT-induced cytotoxicity is highly unlikely.
We have shown previously that ExoT intoxication results in potent cell death in human cervical adenocarcinoma, HeLa cells (Shafikhani et al., 2008). In this report, we investigated ExoT’s cytotoxicity against a number of invasive and highly resistant skin, breast, lung and ovarian tumour cell lines in order to evaluate ExoT’s potential as a possible candidate for chemotherapy. We found that in vitro, ExoT was capable of causing potent cytotoxicity in all cell lines studied. We also found that ExoT was sufficient to induce cytotoxicity and to reduce tumour establishment and growth of B16 melanoma in vivo. Collectively, our data suggested that P. aeruginosa ExoT may be an attractive novel candidate as a cancer drug.
Methods
Transformed and non-transformed cell lines.
Tumour cell lines MCF-7 [human metastatic breast adenocarcinoma (Soule et al., 1973)], MDA-MB-231 [triple-negative human metastatic breast adenocarcinoma (Cailleau et al., 1974)], EMT6 [murine breast carcinoma (Rockwell et al., 1972)], 4T1 [murine metastatic breast cancer (Aslakson & Miller, 1992)], MCA-205 [murine-derived fibrosarcoma cell line (Korrer & Routes, 2014)], B16 murine melanoma [WT BRAF (Wellbrock et al., 2008)], A375 human melanoma [with BRAF(V600E) mutation (Alcazar et al., 2011)], Calu-3 [human lung adenocarcinoma (Fogh et al., 1977)], LLC1 [murine lung carcinoma (Bertram & Janik, 1980)], SK-OV-3 [human ovarian adenocarcinoma (Fogh et al., 1977)] and HeLa [human cervical adenocarcinoma (Scherer et al., 1953)] were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with phenol red supplemented with 10 % FBS, 1 % l-glutamine (Gibco) and 1 % penicillin/streptomycin (Gibco). The non-transformed cell types used in these studies were mouse embryonic fibroblasts, isolated as described previously (Park et al., 2006), and peripheral blood mononuclear cells, cultured as described above. Cells were treated with bacteria when they were ~80–90 % confluent and transfected when they were ~60–70 % confluent (see below).
Plasmids.
Expression vectors pIRES2-ExoT-EGFP (pExoT-GFP) and the empty expression vector (pGFP) were used in experiments involving transient transfection, and were described previously (Shafikhani & Engel, 2006).
Cytotoxicity assessment by time-lapse immunofluorescent video microscopy.
ExoT was delivered into cancer cells either by P. aeruginosa bacteria or by transient transfection using pIRES2 mammalian expression vector and cytotoxicity was assessed as described previously (Shafikhani et al., 2008; Wood et al., 2013). Briefly, bacteria were grown overnight in LB media, adjusted to OD600 0.05, corresponding to m.o.i.~10. The bacterial strains used in these studies were an ExoT-expressing ExoU-deleted PA103 strain (ΔU) or its isogenic ExoT-defective type III secretion system (T3SS) mutant strain [PA103 pscJ-gentR (pscJ)]. These strains were described previously (Shafikhani & Engel, 2006; Shafikhani et al., 2008; Mahmood et al., 2013). Cancer cells, infected with ExoT-expressing and ExoT-defective P. aeruginosa, were placed under 5 % CO2 at 37 °C in an incubation chamber (Pecon) fitted to an AxioVert Z1 (Zeiss) microscope using AxioVision version 4.2 software. Time-lapse video microscopy was performed at ×100 magnification (unless otherwise specified) and images were acquired at 15 min intervals, as described previously (Shafikhani & Engel, 2006; Shafikhani et al., 2008; Wood et al., 2013). Cytotoxicity associated with ExoT was assessed by the uptake of propidium iodide (PI) impermeant dye using time-lapse video microscopy as previously described (Wood et al., 2013). Briefly, the number of red pixels (PI) for each video frame was measured by thresholding the red fluorescence channel using ImageJ version 1.48 (http://imagej.nih.gov/ij/). For transfection experiments, cells were grown as described above and transfected with Effectene (Qiagen) as per the manufacturer’s protocol 1 h prior to the addition of PI and the start of time-lapse video microscopy. Time-lapse videos were assessed for cytotoxicity by determining the percentage of transfected GFP that became PI-positive, as described previously (Shafikhani & Engel, 2006; Shafikhani et al., 2008; Wood et al., 2013).
Cytotoxicity assessment by flow cytometry.
Transformed cells were transfected with Effectene, whilst non-transformed cells were transfected using an electroporation kit (Lonza). At 24 h after transient transfection, cytotoxicity was also assessed using Fixable Viability Dye (eBiosciences) stain to determine the per cent cytotoxicity in transfected cells and the data were analysed by flow cytometry (FACSCanto II; BD Biosciences) as per the manufacturer’s protocol. After 24 h, peripheral blood mononuclear cells were additionally stained with anti-CD3 antibody (eBiosciences) in order to specifically gate on the expression of ExoT in T-lymphocytes alone.
Cytotoxicity assessment by lactate dehydrogenase (LDH) release assay.
LDH release was measured using an LDH Cytotoxicity Detection kit (Clontech). In a 24-well plate, cancer cells were seeded at 8×104 cells per well and cultured in 1 ml DMEM (Gibco) media without phenol red, sodium pyruvate and penicillin/streptomycin antibiotics, and supplemented with 1 % FBS. To further assess the cytotoxic effect ExoT had on cancer, tumour cell lines were infected with either ExoT-expressing ExoU-deleted PA103 strain (ΔU) or its isogenic ExoT-defective T3SS mutant strain (pscJ) at m.o.i.~10 (Shafikhani & Engel, 2006; Shafikhani et al., 2008; Mahmood et al., 2013). To measure the amount of LDH release, the suspensions of uninfected and infected cells were collected and centrifuged at 2.5 g for 10 min to remove bacteria. Uninfected and infected samples, along with LDH-high control samples treated with 1 % Triton X-100 and LDH-low control samples with media alone, were added into a 96-well plate and treated with LDH detection reagents as outlined in the LDH Cytotoxicity Detection kit user manual. A490 was measured using a Thermo Scientific Multiskan Spectrum spectrophotometer and SkanIt software. Per cent LDH release was calculated by substituting the appropriate absorbance values into the following formula:
Animal experiments.
All mouse experiments were approved by the Rush University Medical Center Institutional Animal Care and Use Committee. Female C57BL/6 mice (aged 6–8 weeks) were obtained from Harlan Laboratories. B16 tumours were established by injecting mice subcutaneously with 1×106 cells in 200 µl sterile PBS. Tumour size was determined as the product of the two longest dimensions as measured by callipers. For in vivo assessment of ExoT cytotoxicity, tumours were generated as above and allowed to grow until they reached 50 mm2. At this time, the tumours were injected with 250 ng plasmid DNA (Shafikhani et al., 2008) packaged in Lipofectamine transfection reagent (Invitrogen) to facilitate uptake. At 24 h after transfection, the mice were injected (intravenously) with 200 µl SR-FLIVO (ImmunoChemistry Technologies) cellular death dye and then euthanized after 1 h. Tumours were assessed by flow cytometry or fixed and mounted with VectaShield (Vector Laboratories) containing DAPI. For tumour growth studies, B16 cells were transfected with plasmid DNA, as described previously (Shafikhani et al., 2008), for 15 h prior to injection. Cell viability was assessed using Trypan blue dye staining and transfection efficiency was determined by identifying GFP expression by flow cytometry. ExoT-GFP- or GFP-transfected B16 cancer cells (1×105) were injected into the right flank of C57BL/6 mice. Tumour surface area was measured daily with the use of callipers. Animals were euthanized using CO2 asphyxiation at the indicated time, or when tumour size reached >150 mm2 or animals were moribund.
Statistical analysis.
All studies were performed in triplicate or as indicated. Statistical significance was determined by Student’s two-tailed t-tests using Prism 6 (GraphPad). P≤0.05 was considered significant.
Results
P. aeruginosa ExoT induces potent cytotoxicity in a variety of murine and human tumour cell lines
ExoT has previously been shown to induce potent cytotoxicity in HeLa cells (Shafikhani et al., 2008). As ExoT targets six cellular proteins (Shafikhani & Engel, 2006; Shafikhani et al., 2008) that are essential for cancer cell survival and proliferation (del Peso et al., 1997; Gómez et al., 1997; Cho & Klemke, 2000; Lay et al., 2000; Lamorte et al., 2002; Murga et al., 2002; García et al., 2006; Hwang et al., 2006; Heasman & Ridley, 2008; Zhu et al., 2008; He et al., 2010), we hypothesized that resistance to ExoT-induced cytotoxicity would be highly unlikely and therefore it may be able to induce cytotoxicity in a wide range of tumour cell lines.
To gain insight into the efficacy of the ExoT-induced cytotoxicity, we first used P. aeruginosa to deliver ExoT into a number of highly resistant cancer cell lines, including B16, HeLa, EMT6, 4T1, MDA-MB-231, SK-OV-3, MCA-205 and Calu-3 (see Methods).
Tumour cells were treated with either an ExoT-expressing P. aeruginosa PA103 strain ΔU or ExoT-deficient P. aeruginosa PA103 strain pscJ, or left untreated (see Methods). We assessed ExoT-mediated cytotoxicity by measuring the extracellular release of the cytoplasmic protein LDH into the culture media. Treatment with the ExoT-expressing P. aeruginosa strain (ΔU) resulted in significantly higher LDH release in all the tumour cells, as early as 10 h post-infection (Fig. 1).
Fig. 1.

Infection with ExoT-expressing P. aeruginosa can cause cytotoxicity in a number of cancer cell lines. The indicated cancer cells were either infected with ExoT-expressing (ΔU) or ExoT-defective T3SS mutant (pscJ) P. aeruginosa isogenic strains at m.o.i.~10, or cultured in media alone. Media were collected at 10 h intervals over a 30 h period and the amount of LDH released into media was analysed using the LDH release assay. The per cent LDH release was determined by the ratio of experimental LDH release to the total LDH release in the presence of Triton X-100. Analysis of LDH release of infected cancer cells, especially at later time points, indicated that infection with ExoT-expressing ΔU resulted in significantly more cytotoxicity compared with cells treated with the pscJ strain or left uninfected. *P<0.05, n = 6, Student’s t-test.
In order to home in on the kinetics of ExoT-induced cytotoxicity in these tumour cell lines, we assessed cytotoxicity at 15 min intervals using PI impermeant nuclear dye uptake as a marker for cell death. PI uptake and subsequent PI fluorescence is an established and irreversible marker for cell death (Shafikhani et al., 2008; Kroemer et al., 2009; Wood et al., 2013). ExoT-expressing P. aeruginosa was able to induce cytotoxicity in all the tumour cell lines we tested within 15–25 h post-infection (Fig. 2, Movies S1, S2 and S3, available in the online Supplementary Material; only the representative movie frames of B16 are shown in Fig. 2b).
Fig. 2.

Kinetics of ExoT-induced cytotoxicity in cancer cell lines. (a) The indicated cancer cell lines were treated with ExoT-expressing (ΔU) or ExoT-defective (pscJ) P. aeruginosa strains at m.o.i.~10, or left untreated. Cells were observed by time-lapse fluorescence video microscopy in the presence of PI (×100 magnification, 15 min intervals). Cytotoxicity was assessed by total PI uptake at 15 min intervals (cytotoxicity is shown as the mean of three fields of view). (b) Cytotoxicity analysis of B16 melanoma cells as assessed by PI staining (red). (c) Representative frames from B16 time-lapse video microscopy.
ExoT is sufficient to induce cytotoxicity in tumour cells
Although LDH release is routinely used as a measure of cytotoxicity (Korzeniewski & Callewaert, 1983; Decker & Lohmann-Matthes, 1988; Arechabala et al., 1999), injured but viable cells can also release LDH as a result of damage to their membrane (Reddy et al., 2001). Moreover, LDH release could potentially underestimate cytotoxicity in apoptotic cells, because activated caspases, which are proteases that mediate apoptosis (Fiers et al., 1999; Lavrik et al., 2005), could degrade LDH peptides, thus reducing the amount of LDH released into the medium. ExoT is known to induce apoptosis (Shafikhani et al., 2008). In addition, the use of bacteria to deliver ExoT into tumour cells could introduce additional bacterial factors, which may contribute and/or synergize the ExoT-mediated killing of tumour cells. In fact, treatment with the ExoT-defective pscJ strain resulted in increased LDH release and cytotoxicity in some tumour cell lines, albeit at significantly lower levels than the LDH release or the cytotoxicity observed in the presence of the ExoT-expressing ΔU strain (Figs 1 and 2).
To determine if ExoT was sufficient to cause cytotoxicity in the aforementioned tumour cell lines and to account for the ExoT-independent cytotoxic effects of P. aeruginosa on tumour cells, we transfected the tumour cell lines with an expression vector harbouring either the exoT gene, C-terminally fused to gfp (pExoT-GFP), or the gfp empty vector control (pGFP) and assessed cytotoxicity by time-lapse fluorescence video microscopy, using PI uptake as a marker for cell death. This technique allowed us to quantify ExoT’s cytotoxic effect in tumour cells on a per cell basis. Some cancer cell lines were poorly transfectable and thus were not included in these analyses. However, in those tumour cell lines that we were able to transfect, ExoT expression resulted in significantly more killing than the GFP control vector, indicating that ExoT was sufficient to cause cytotoxicity in these cancer cells (Fig. 3, Movies S4 and S5; only selected frames from B16 melanoma cells are shown in Fig. 3b).
Fig. 3.

ExoT is sufficient to cause cytotoxicity in cancer cells in vitro. Indicated cancer cells were transfected with pGFP vector control or pExoT-GFP and cytotoxicity was assessed by time-lapse fluorescence video microscopy in the presence of PI. (a) Tabulated data for A375, MCF-7, EMT6 and Calu-3. (b) Representative frames of B16 transfected with pExoT-GFP or pGFP. (c) Tabulated data for B16. *P<0.0001; n = 3 independent experiments each with ~100 events counted; Student’s t-test.
We further corroborated the cytotoxic effects of ExoT in transfected tumour cells by flow cytometry using a Viability Dye (eBiosciences), which, instead of using PI staining as a marker for cell death, works by binding to free amino acids in the cytosol (Perfetto et al., 2010). As expected, cells intoxicated with ExoT exhibited significantly higher cytotoxicities as than the GFP control at 24 h post-transfection (Fig. 4). Depending on the techniques, there were some variations in the ExoT-induced cytotoxicities in these cell lines (compare Figs 1, 2 and 3). This is likely due to sensitivity of the techniques and/or the limitation of the use of peptides (i.e. LDH or Viability Dye) as markers for cell death in apoptotic cells as discussed above. Nevertheless, these data indicate that ExoT is sufficient to induce potent cytotoxicity in all tumour cell lines we tested.
Fig. 4.

Assessment of ExoT-induced cytotoxicity in cancer by Viability Dye. Cancer cells were transfected with pGFP vector control or pExoT-GFP. At 17 h after transfection, cells were stained with Viability Dye and analysed by flow cytometry. (a) Representative FACS plots for B16 show ExoT was sufficient to cause cytotoxicity in transfected cells. SSC, side scatter; FSC, forward scatter. (b) FACS analysis of transfected B16 cells indicated that pExoT-GFP resulted in significantly more cytotoxicity compared with pGFP-transfected cells. *P<0.0001; n = 7; Student’s two-tailed t-test.
Most cancer chemotherapeutics exert cytotoxicity in normal non-transformed cells, leading to well-chronicled side-effects associated with cancer therapy. To gain insight into potential side-effects of ExoT-based therapy, we transfected mouse embryonic fibroblasts and human T-lymphocytes with an ExoT expression vector as described above. Not surprisingly, ExoT also induced comparable cytotoxicity in these cell lines (Fig. S1). We were unable to evaluate ExoT’s cytotoxicity in human or mouse primary epithelial cell lines due to low transfection efficiencies as well as their sensitivity to bacterial infection as ExoT-deficient bacteria also caused massive cytotoxicity in these cell lines. Nevertheless, these results highlighted the need to target this toxin directly into tumour cells (see Discussion for possible approaches).
ExoT is sufficient to induce cytotoxicity in vivo
We next assessed the ability of ExoT to induce cytotoxicity in vivo, using the B16 melanoma tumour model (Overwijk & Restifo, 2001). We transplanted B16 melanoma cells subcutaneously in the flanks of C57BL/6 mice. Once the tumours reached 50 mm2, the expression vector expressing ExoT-GFP was packaged within a lipid-based transfection reagent to facilitate its uptake and injected directly into the lesion. After 24 h, the animals were injected systemically (intravenously) with SR-FLIVO, a marker for apoptotic cell death in vivo (Riol-Blanco et al., 2009), and the tumours were analysed by fluorescence microscopy. The data indicated that transfection with ExoT resulted in cell death in B16 cells in vivo as indicated by co-localization of ExoT-GFP (green) and SR-FLIVO (red) (Fig. S2).
As in situ transfection efficiencies into the tumours by this method were extremely low (~3 %), ExoT failed to control overall tumour growth (data not shown). We modified our experimental approach to enhance transfection efficiencies in order to better evaluate the ability of ExoT to affect tumour establishment and growth in vivo. B16 cells were transfected with either pExoT-GFP or pGFP vector control ex vivo prior to subcutaneous injection in the mice. This approach increased the transfection efficiencies to ~20 %. Next, pExoT-GFP- or pGFP-transfected B16 tumour cells were transplanted into mice. Despite these low transfection efficiencies, the mean size of the ExoT-transfected B16 tumours was significantly less than pGFP-transfected B16 tumours (Fig. 5a, n = 5 per group, P<0.05; mean tumour size was analysed up to the first tumour end point). Tumour measurements for individual animals showed that tumours transfected with pExoT-GFP reached their end point significantly slower than pGFP tumours. Collectively, these data indicated that ExoT was sufficient to induce cytotoxicity in B16 tumour cells and delay tumour growth in vivo.
Fig. 5.
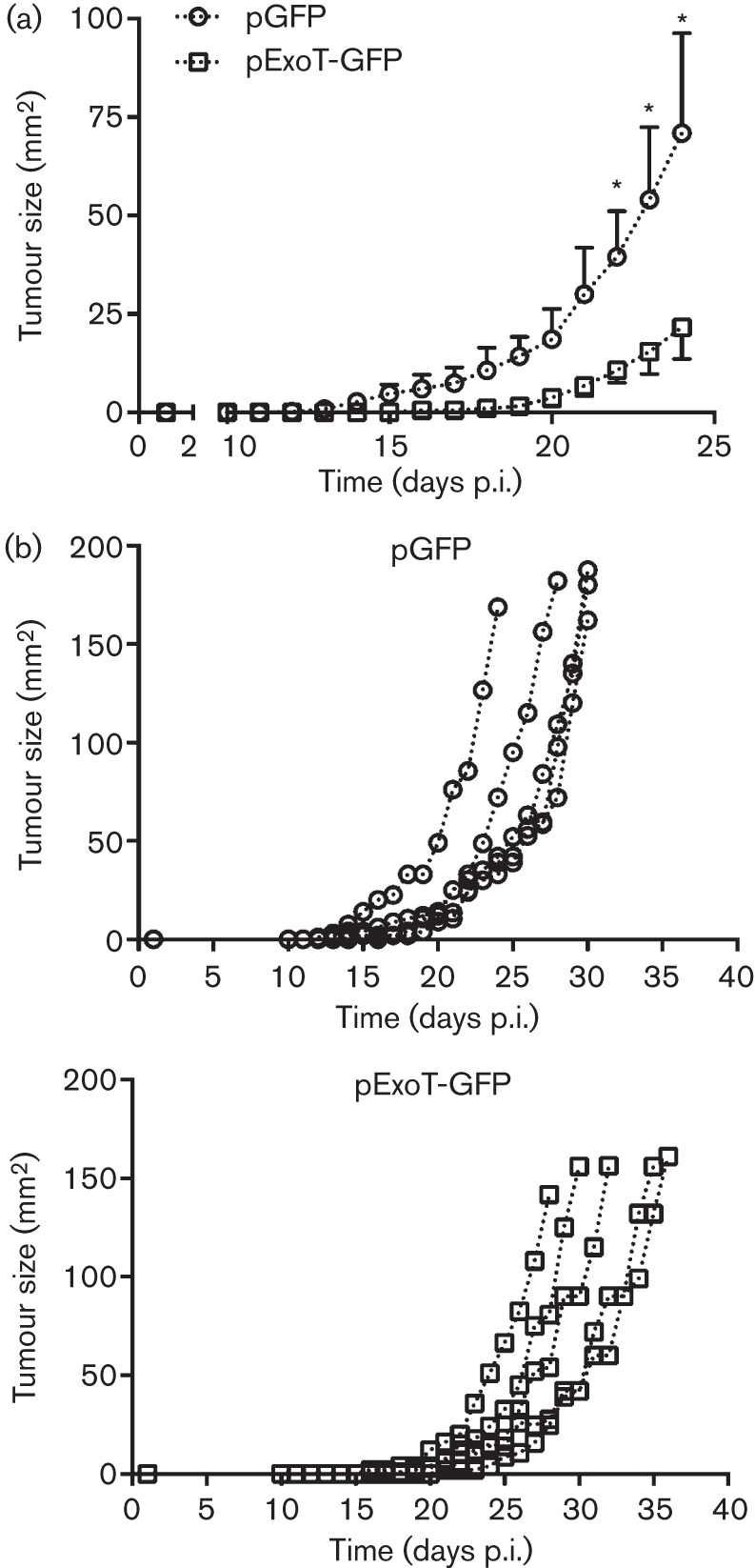
ExoT slows B16 tumour burden in vivo. (a) B16 tumour cells were transfected with pGFP or pExoT ex vivo. At 15 h after transfection, 1×105 pGFP- or pExoT-GFP-transfected B16 tumour cells were transferred subcutaneously by injection in the flank of C57BL/6 mice. Tumour size was measured daily by use of callipers until tumours reached 150 mm2. Tumour sizes are shown as mean±sem from the time of injection to the first animal death. n = 5 mice per group, *P<0.05. (b) Individual tumour growth curves for the experiment performed in (a).
Discussion
Bacteria have been and continue to be used to deliver toxins into tumours (Dang et al., 2001; Van Mellaert et al., 2006; Patyar et al., 2010; Karbach et al., 2012). This study is the first evaluation of ExoT as a potential candidate for cancer therapy. Our results demonstrate that ExoT is capable of causing potent cytotoxicity in all murine and human tumour cell lines that we have examined thus far (Figs 1, 2, 3 and 4, Movies S2 and S5, and data not shown). ExoT-induced cytotoxicity occurred across a wide range of tumour cell lines, including breast, lung, cervical and even melanoma that is largely refractory to current cytotoxic drugs in the clinical setting (Atkins et al., 2008; Flaherty et al., 2012; Homet & Ribas, 2013).
Our in vivo results are encouraging in that they demonstrate that ExoT can significantly slow B16 melanoma tumour growth in vivo (Fig. 5). Low transfection efficiency is likely a major reason why these tumours eventually grew out and reached their end point. Nevertheless, the ExoT-mediated reduction in tumour growth in vivo highlights the potential of ExoT in cancer therapy.
The primary challenge concerning the formulation of ExoT as a potential cancer therapy is the development of means to safely deliver this toxin and specifically target it to tumours in vivo. There are multiple approaches that have been or are currently being investigated in several clinical trials that may accomplish this task. One approach is conjugation with tumour-specific antibodies (reviewed by Becker & Benhar, 2012). Conjugating ExoT to tumour-specific antibodies or the immunoglobulin variable fragment regions can enhance the delivery of the toxin (e.g. intravenously) as well as improve the specificity of ExoT to target the tumour. Indeed, engineered toxins combined to a receptor ligand or the variable domain of a mAb can differentiate between normal and malignant cells by binding to tumour-associated cell surface receptors (e.g. CD22, CD25, claudin-4, glycoprotein-NMB and mesothelin) (Chaudhary et al., 1987, 1989; Pastan et al., 2004).
Another approach is the use of recombinant viral delivery systems, such as vaccinia virus, with inherent tropism toward tumour cells (Baguley, 2010). A viral-based delivery platform has a number of unique biological properties that make it ideally suited for delivery and amplification of transgenes within tumours. These include intravenous stability and the ability to spread to distant tissues (Vanderplasschen et al., 1998), preferential accumulation in solid tumours where neovasculature shows increased permeability (Kirn & Thorne, 2009), tropism toward cancerous cells (Park et al., 2008; Breitbach et al., 2011), and only minor side-effects in cancer patients even at extremely high infection titres (107–109 p.f.u.) (Park et al., 2008; Breitbach et al., 2011).
Another potential translational approach is the use of image-guided ultrasound-based microbubble technology to deliver and activate ExoT selectively in the tumour microenvironment. Gene therapy using this technology has gained significant momentum in recent years (Smith et al., 2011; Chen et al., 2012; Sirsi & Borden, 2012; Smith & Land, 2012).
In summary, we propose that P. aeruginosa ExoT possesses attractive anti-cancer properties that make it an attractive cancer drug candidate. ExoT’s attractive anti-cancer properties include: (1) its ability to induce potent cytotoxicity in all cancer cell lines, as we demonstrated in this report; (2) its anti-proliferative effect in cancer (Shafikhani & Engel, 2006); (3) its ability to inhibit cell migration (Garrity-Ryan et al., 2004), thus potentially being able to interfere with metastasis; and (4) the low probability of tumour resistance to ExoT due to its multiple cellular targets.
Acknowledgements
This work was supported by a grant from the Brian Piccolo Cancer Research Fund to S. H. S. and The Rush University Committee on Research, Young Investigator Award to S. H. S. A provisional patent has been filed on the findings of this manuscript by Rush University Medical Center (Attorney Docket Number 61/792,606).
Abbreviations:
- EGFR
epidermal growth factor receptor
- ExoT
exotoxin T
- LDH
lactate dehydrogenase
- PI
propidium iodide
- T3SS
type III secretion system
References
- Alcazar O., Achberger S., Aldrich W., Hu Z., Negrotto S., Saunthararajah Y., Triozzi P. (2011). Epigenetic regulation by decitabine of melanoma differentiation in vitro and in vivo. Int J Cancer 131, 18–29. 10.1002/ijc.26320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arechabala B., Coiffard C., Rivalland P., Coiffard L. J. M., de Roeck-Holtzhauer Y. (1999). Comparison of cytotoxicity of various surfactants tested on normal human fibroblast cultures using the neutral red test, MTT assay and LDH release. J Appl Toxicol 19, 163–165. [DOI] [PubMed] [Google Scholar]
- Aslakson C. J., Miller F. R. (1992). Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52, 1399–1405. [PubMed] [Google Scholar]
- Atkins M. B., Hsu J., Lee S., Cohen G. I., Flaherty L. E., Sosman J. A., Sondak V. K., Kirkwood J. M., Eastern Cooperative Oncology Group (2008). Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 26, 5748–5754. 10.1200/JCO.2008.17.5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagrodia S., Taylor S. J., Jordon K. A., Van Aelst L., Cerione R. A. (1998). A novel regulator of p21-activated kinases. J Biol Chem 273, 23633–23636. 10.1074/jbc.273.37.23633 [DOI] [PubMed] [Google Scholar]
- Baguley B. C. (2010). Multiple drug resistance mechanisms in cancer. Mol Biotechnol 46, 308–316. 10.1007/s12033-010-9321-2 [DOI] [PubMed] [Google Scholar]
- Becker N., Benhar I. (2012). Antibody-based immunotoxins for the treatment of cancer. Antibodies 1, 39–69. 10.3390/antib1010039 [DOI] [Google Scholar]
- Bertram J. S., Janik P. (1980). Establishment of a cloned line of Lewis lung carcinoma cells adapted to cell culture. Cancer Lett 11, 63–73. 10.1016/0304-3835(80)90130-5 [DOI] [PubMed] [Google Scholar]
- Breitbach C. J., Burke J., Jonker D., Stephenson J., Haas A. R., Chow L. Q., Nieva J., Hwang T. H., Moon A., et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477, 99–102. 10.1038/nature10358 [DOI] [PubMed] [Google Scholar]
- Cailleau R., Young R., Olivé M., Reeves W. J., Jr (1974). Breast tumor cell lines from pleural effusions. J Natl Cancer Inst 53, 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno A. M., Ambudkar S. V. (2010). Molecular mechanisms of drug resistance in single-step and multi-step drug-selected cancer cells. Methods Mol Biol 596, 77–93. 10.1007/978-1-60761-416-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary V. K., FitzGerald D. J., Adhya S., Pastan I. (1987). Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc Natl Acad Sci U S A 84, 4538–4542. 10.1073/pnas.84.13.4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary V. K., Queen C., Junghans R. P., Waldmann T. A., FitzGerald D. J., Pastan I. (1989). A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 339, 394–397. 10.1038/339394a0 [DOI] [PubMed] [Google Scholar]
- Chen W. J., Xiong Z. A., Tang Y., Dong P. T., Li P., Wang Z. G. (2012). Feasibility and effect of ultrasound microbubble-mediated wild-type p53 gene transfection of HeLa cells. Exp Ther Med 3, 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. Y., Klemke R. L. (2000). Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol 149, 223–236. 10.1083/jcb.149.1.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L. H., Bettegowda C., Huso D. L., Kinzler K. W., Vogelstein B. (2001). Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A 98, 15155–15160. 10.1073/pnas.251543698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T., Lohmann-Matthes M. L. (1988). A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 115, 61–69. 10.1016/0022-1759(88)90310-9 [DOI] [PubMed] [Google Scholar]
- del Peso L., Hernández-Alcoceba R., Embade N., Carnero A., Esteve P., Paje C., Lacal J. C. (1997). Rho proteins induce metastatic properties in vivo. Oncogene 15, 3047–3057. 10.1038/sj.onc.1201499 [DOI] [PubMed] [Google Scholar]
- Feng Y. X., Zhao J. S., Li J. J., Wang T., Cheng S. Q., Yuan Y., Wang F., Wang X. F., Xie D. (2010). Liver cancer: ephrinA2 promotes tumorigenicity through Rac1/Akt/NF-kappaB signaling pathway. Hepatology 51, 535–544. 10.1002/hep.23313 [DOI] [PubMed] [Google Scholar]
- Fiers W., Beyaert R., Declercq W., Vandenabeele P. (1999). More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18, 7719–7730. 10.1038/sj.onc.1203249 [DOI] [PubMed] [Google Scholar]
- Flaherty K. T., Infante J. R., Daud A., Gonzalez R., Kefford R. F., Sosman J., Hamid O., Schuchter L., Cebon J., et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367, 1694–1703. 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogh J., Fogh J. M., Orfeo T. (1977). One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59, 221–226. [DOI] [PubMed] [Google Scholar]
- Foley B. T., Moehring J. M., Moehring T. J. (1995). Mutations in the elongation factor 2 gene which confer resistance to diphtheria toxin and Pseudomonas exotoxin A. Genetic and biochemical analyses. J Biol Chem 270, 23218–23225. 10.1074/jbc.270.39.23218 [DOI] [PubMed] [Google Scholar]
- García Z., Silio V., Marqués M., Cortés I., Kumar A., Hernandez C., Checa A. I., Serrano A., Carrera A. C. (2006). A PI3K activity-independent function of p85 regulatory subunit in control of mammalian cytokinesis. EMBO J 25, 4740–4751. 10.1038/sj.emboj.7601324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity-Ryan L., Shafikhani S., Balachandran P., Nguyen L., Oza J., Jakobsen T., Sargent J., Fang X., Cordwell S., et al. (2004). The ADP ribosyltransferase domain of Pseudomonas aeruginosa ExoT contributes to its biological activities. Infect Immun 72, 546–558. 10.1128/IAI.72.1.546-558.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez J., Martínez C., Giry M., García A., Rebollo A. (1997). Rho prevents apoptosis through Bcl-2 expression: implications for interleukin-2 receptor signal transduction. Eur J Immunol 27, 2793–2799. 10.1002/eji.1830271108 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Angulo A. M., Morales-Vasquez F., Hortobagyi G. N. (2007). Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol 608, 1–22. 10.1007/978-0-387-74039-3_1 [DOI] [PubMed] [Google Scholar]
- Gorre M. E., Mohammed M., Ellwood K., Hsu N., Paquette R., Rao P. N., Sawyers C. L. (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. 10.1126/science.1062538 [DOI] [PubMed] [Google Scholar]
- Gottesman M. M. (2002). Mechanisms of cancer drug resistance. Annu Rev Med 53, 615–627. 10.1146/annurev.med.53.082901.103929 [DOI] [PubMed] [Google Scholar]
- He X., Liu J., Qi Y., Brakebusch C., Chrostek-Grashoff A., Edgar D., Yurchenco P. D., Corbett S. A., Lowry S. F., et al. (2010). Rac1 is essential for basement membrane-dependent epiblast survival. Mol Cell Biol 30, 3569–3581. 10.1128/MCB.01366-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman S. J., Ridley A. J. (2008). Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9, 690–701. 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- Homet B., Ribas A. (2013). New drug targets in metastatic melanoma. J Pathol 232, 134–141. 10.1002/path.4259 [DOI] [PubMed] [Google Scholar]
- Hwang T. L., Liang Y., Chien K. Y., Yu J. S. (2006). Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics 6, 2259–2272. 10.1002/pmic.200500345 [DOI] [PubMed] [Google Scholar]
- Jørgensen R., Wang Y., Visschedyk D., Merrill A. R. (2008). The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep 9, 802–809. 10.1038/embor.2008.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbach J., Neumann A., Brand K., Wahle C., Siegel E., Maeurer M., Ritter E., Tsuji T., Gnjatic S., et al. (2012). Phase I clinical trial of mixed bacterial vaccine (Coley’s toxins) in patients with NY-ESO-1 expressing cancers: immunological effects and clinical activity. Clin Cancer Res 18, 5449–5459. 10.1158/1078-0432.CCR-12-1116 [DOI] [PubMed] [Google Scholar]
- Kawakami K., Nakajima O., Morishita R., Nagai R. (2006). Targeted anticancer immunotoxins and cytotoxic agents with direct killing moieties. ScientificWorldJournal 6, 781–790. 10.1100/tsw.2006.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak B. I., Engel J. N. (2002). Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun 70, 2198–2205. 10.1128/IAI.70.4.2198-2205.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn D. H., Thorne S. H. (2009). Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer 9, 64–71. 10.1038/nrc2545 [DOI] [PubMed] [Google Scholar]
- Kobayashi S., Boggon T. J., Dayaram T., Jänne P. A., Kocher O., Meyerson M., Johnson B. E., Eck M. J., Tenen D. G., Halmos B. (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352, 786–792. 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- Korrer M. J., Routes J. M. (2014). Possible role of arginase-1 in concomitant tumor immunity. PLoS One 9, e91370. 10.1371/journal.pone.0091370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski C., Callewaert D. M. (1983). An enzyme-release assay for natural cytotoxicity. J Immunol Methods 64, 313–320. 10.1016/0022-1759(83)90438-6 [DOI] [PubMed] [Google Scholar]
- Krall R., Schmidt G., Aktories K., Barbieri J. T. (2000). Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect Immun 68, 6066–6068. 10.1128/IAI.68.10.6066-6068.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman R. J. (2006). Immunotoxins for targeted cancer therapy. AAPS J 8, E532–E551. 10.1208/aapsj080363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman R. J. (2009). Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr Pharm Des 15, 2652–2664. 10.2174/138161209788923949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., El-Deiry W. S., Golstein P., et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16, 3–11. 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L., Royal I., Naujokas M., Park M. (2002). Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell 13, 1449–1461. 10.1091/mbc.01-10-0477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrik I. N., Golks A., Krammer P. H. (2005). Caspases: pharmacological manipulation of cell death. J Clin Invest 115, 2665–2672. 10.1172/JCI26252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay A. J., Jiang X. M., Kisker O., Flynn E., Underwood A., Condron R., Hogg P. J. (2000). Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 408, 869–873. 10.1038/35048596 [DOI] [PubMed] [Google Scholar]
- Mahmood F., Hakimiyan A., Jayaraman V., Wood S., Sivaramakrishnan G., Rehman T., Reuhs B. L., Chubinskaya S., Shafikhani S. H. (2013). A novel human antimicrobial factor targets Pseudomonas aeruginosa through its type III secretion system. J Med Microbiol 62, 531–539. 10.1099/jmm.0.051227-0 [DOI] [PubMed] [Google Scholar]
- Murga C., Zohar M., Teramoto H., Gutkind J. S. (2002). Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-kappaB. Oncogene 21, 207–216. 10.1038/sj.onc.1205036 [DOI] [PubMed] [Google Scholar]
- Overwijk W. W., Restifo N. P. (2001). B16 as a mouse model for human melanoma. Curr Protoc Immunol 39, 20.1.1–20.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T. J., Boyd K., Curran T. (2006). Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol 26, 6272–6282. 10.1128/MCB.00472-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. H., Hwang T., Liu T. C., Sze D. Y., Kim J. S., Kwon H. C., Oh S. Y., Han S. Y., Yoon J. H., et al. (2008). Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 9, 533–542. 10.1016/S1470-2045(08)70107-4 [DOI] [PubMed] [Google Scholar]
- Pastan I., Beers R., Bera T. K. (2004). Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol 248, 503–518. [DOI] [PubMed] [Google Scholar]
- Patyar S., Joshi R., Byrav D. S., Prakash A., Medhi B., Das B. K. (2010). Bacteria in cancer therapy: a novel experimental strategy. J Biomed Sci 17, 21. 10.1186/1423-0127-17-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto S. P., Chattopadhyay P. K., Lamoreaux L., Nguyen R., Ambrozak D., Koup R. A., Roederer M. (2010). Amine-reactive dyes for dead cell discrimination in fixed samples. Curr Protoc Cytom 53, 9.34.1–9.34.14. 10.1186/1423-0127-17-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A., Caler E. V., Andrews N. W. (2001). Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106, 157–169. 10.1016/S0092-8674(01)00421-4 [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L., Delgado-Martín C., Sánchez-Sánchez N., Alonso-C L. M., Gutiérrez-López M. D., Del Hoyo G. M., Navarro J., Sánchez-Madrid F., Cabañas C., et al. (2009). Immunological synapse formation inhibits, via NF-kappaB and FOXO1, the apoptosis of dendritic cells. Nat Immunol 10, 753–760. 10.1038/ni.1750 [DOI] [PubMed] [Google Scholar]
- Rockwell S. C., Kallman R. F., Fajardo L. F. (1972). Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J Natl Cancer Inst 49, 735–749. [PubMed] [Google Scholar]
- Rodrigues S. P., Fathers K. E., Chan G., Zuo D., Halwani F., Meterissian S., Park M. (2005). CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Mol Cancer Res 3, 183–194. [DOI] [PubMed] [Google Scholar]
- Scherer W. F., Syverton J. T., Gey G. O. (1953). Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med 97, 695–710. 10.1084/jem.97.5.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani S. H., Engel J. (2006). Pseudomonas aeruginosa type III-secreted toxin ExoT inhibits host-cell division by targeting cytokinesis at multiple steps. Proc Natl Acad Sci U S A 103, 15605–15610. 10.1073/pnas.0605949103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani S. H., Morales C., Engel J. (2008). The Pseudomonas aeruginosa type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cell Microbiol 10, 994–1007. 10.1111/j.1462-5822.2007.01102.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirsi S. R., Borden M. A. (2012). Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics 2, 1208–1222. 10.7150/thno.4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B., Land H. (2012). Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep 2, 580–590. 10.1016/j.celrep.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. H., Fujii H., Kuliszewski M. A., Leong-Poi H. (2011). Contrast ultrasound and targeted microbubbles: diagnostic and therapeutic applications for angiogenesis. J Cardiovasc Transl Res 4, 404–415. 10.1007/s12265-011-9282-2 [DOI] [PubMed] [Google Scholar]
- Soule H. D., Vazguez J., Long A., Albert S., Brennan M. (1973). A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst 51, 1409–1416. [DOI] [PubMed] [Google Scholar]
- Sun J., Barbieri J. T. (2003). Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. J Biol Chem 278, 32794–32800. 10.1074/jbc.M304290200 [DOI] [PubMed] [Google Scholar]
- Van Mellaert L., Barbé S., Anné J. (2006). Clostridium spores as anti-tumour agents. Trends Microbiol 14, 190–196. 10.1016/j.tim.2006.02.002 [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A., Mathew E., Hollinshead M., Sim R. B., Smith G. L. (1998). Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci U S A 95, 7544–7549. 10.1073/pnas.95.13.7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H., Xiang L., Wayne A. S., Chertov O., FitzGerald D. J., Bera T. K., Pastan I. (2012). Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proc Natl Acad Sci U S A 109, 6898–6903. 10.1073/pnas.1204523109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C., Rana S., Paterson H., Pickersgill H., Brummelkamp T., Marais R. (2008). Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One 3, e2734. 10.1371/journal.pone.0002734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S., Pithadia R., Rehman T., Zhang L., Plichta J., Radek K. A., Forsyth C., Keshavarzian A., Shafikhani S. H. (2013). Chronic alcohol exposure renders epithelial cells vulnerable to bacterial infection. PLoS One 8, e54646. 10.1371/journal.pone.0054646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Korzh V., Gong Z., Low B. C. (2008). RhoA prevents apoptosis during zebrafish embryogenesis through activation of Mek/Erk pathway. Oncogene 27, 1580–1589. 10.1038/sj.onc.1210790 [DOI] [PubMed] [Google Scholar]