

Abstract
Background
Mitochondrial autophagy is an important mediator of mitochondrial quality control in cardiomyocytes. The occurrence of mitochondrial autophagy and its significance during cardiac hypertrophy are not well understood.
Methods and Results
Mice were subjected to transverse aortic constriction (TAC) and observed at multiple time points up to 30 days. Cardiac hypertrophy developed after 5 days, the ejection fraction was reduced after 14 days, and heart failure (HF) was observed 30 days after TAC. General autophagy was upregulated between 1 and 12 hours after TAC but was downregulated below physiological levels 5 days after TAC. Mitochondrial autophagy, evaluated by electron microscopy, mitochondrial content, and Mito-Keima, was transiently activated around 3–7 days post-TAC, coinciding with mitochondrial translocation of Drp1. However, it was downregulated thereafter, followed by mitochondrial dysfunction. Haploinsufficiency of Drp1 abolished mitochondrial autophagy and exacerbated the development of both mitochondrial dysfunction and HF after TAC. Injection of Tat-Beclin 1, a potent inducer of autophagy, but not control peptide, on Day 7 after TAC partially rescued mitochondrial autophagy, and attenuated mitochondrial dysfunction and HF induced by pressure overload (PO). Haploinsufficiency of either drp1 or beclin 1 prevented the rescue by Tat-Beclin 1, suggesting that its effect is mediated in part through autophagy, including mitochondrial autophagy.
Conclusions
Mitochondrial autophagy is transiently activated and then downregulated in the mouse heart in response to PO. Downregulation of mitochondrial autophagy plays an important role in mediating the development of mitochondrial dysfunction and HF, whereas restoration of mitochondrial autophagy attenuates dysfunction in the heart during PO.
Keywords: Pressure overload, hypertrophy, autophagy, mitochondria, Drp1
Introduction
Autophagy is an important mechanism for degradation of intracellular components, in which cytosolic proteins and organelles are sequestered in double-membrane vesicles called autophagosomes and delivered to lysosomes for degradation1. Autophagy helps maintain the quality of the intracellular environment by eliminating misfolded proteins and malfunctioning organelles, and preserves the ATP level by recycling extracted amino acids and fatty acids for ATP synthesis. Increasing evidence suggests that autophagy plays an important role in protecting the heart during heart failure (HF)2, 3.
Although autophagy can degrade bulk intracellular materials in a relatively non-specific manner, it can also specifically target malfunctioning organelles. For example, activation of a series of events, including stabilization of Pink1 on depolarized mitochondria, phosphorylation of Mfn2, recruitment of Parkin to mitochondria, and recognition of depolarized mitochondria by autophagosomes, specifically eliminates damaged mitochondria; this process is termed mitochondrial autophagy or mitophagy4, 5. Mitochondria are the primary source of cellular ATP, but when they malfunction, they become a major source of oxidative stress and trigger both apoptosis and necrosis. Mitochondria are therefore normally regulated by quality control mechanisms, including fission and fusion, biogenesis, and autophagy6. Impairment of any of these mitochondrial quality control processes leads to mitochondrial dysfunction and cell death.
The heart undergoes hypertrophy in response to hemodynamic overload, such as pressure overload (PO), initially with the purpose of reducing wall stress. The heart also initiates various adaptive mechanisms, including autophagy, to cope with energetic stress, increased oxidative stress, and cell death. Previous studies have shown that general autophagy is activated during cardiac hypertrophy and HF, and plays both protective and detrimental roles in the heart3, 7. However, although cardiac hypertrophy and HF are commonly accompanied by general and non-specific autophagy3, 7, the presence of organelle-specific mitochondrial autophagy and its role during pathological hypertrophy have not been well characterized.
Drp1 is a small GTPase that mediates mitochondrial fission. Drp1 also plays an important role in mitochondrial autophagy in cardiomyocytes (CMs) at baseline and in response to energy stress6. Although Drp1 is localized primarily in the cytosol, it is recruited to mitochondria in response to stress, thereby inducing mitochondrial fission and/or mitochondrial autophagy in the heart6. However, whether Drp1 is involved in mitochondrial autophagy or the maintenance of mitochondrial function during PO-induced cardiac hypertrophy is currently unknown.
Using mice with transverse aortic constriction (TAC) as a model, we investigated the role of mitochondrial autophagy during PO-induced hypertrophy and HF. The goals in this study were to 1) demonstrate activation of mitochondrial autophagy, 2) clarify the role of Drp1 in mediating mitochondrial autophagy, and 3) elucidate the functional significance of mitochondrial autophagy in the development of mitochondrial dysfunction and HF in response to PO.
Methods
An expanded Methods section is available in the Online Data Supplement.
Mouse models
Cardiac-specific heterozygous Drp1 knockout (Drp1-hetCKO) mice were generated by crossing Drp1 flox heterozygous (fl/+) mice with αMHC-Cre mice6. Transgenic mice expressing GFP-LC3 (Tg-GFP-LC3) or mRFP-GFP-LC3 (Tg-tf-LC3)8, beclin 1 heterozygous knockout mice (beclin 1-hetKO)9 and cardiac-specific mammalian sterile 20-like kinase 1 (Mst1) homozygous knockout (Mst1-CKO) mice2 have been described. All experiments involving animals were approved by the Rutgers–New Jersey Medical School’s Institutional Animal Care and Use Committee.
Keima with mitochondrial localization signal (Mito-Keima)
Mito-Keima is a mitochondrially localized pH-indicator protein10. The method used to detect lysosomal delivery of Mito-Keima has been described6.
Tat-Beclin 1 (TB1)
The retro-inverso TB1 D-amino acid sequence was RRRQRRKKRGYGGTGFEGDHWIEFTANFVNT11. The control peptide, Tat-scrambled (TS), consisted of the Tat protein transduction domain, a GG linker, and a scrambled version of the C-terminal 18 amino acids of TB1 (RRRQRRKKRGYGGWETAFGTTEHIFFDNGV). For in vivo experiments, D-amino acid peptides were dissolved in H2O and stored at −80°C until use. For induction of autophagy in vitro, CMs were washed with PBS(-) and treated for 3 hours with peptides (25 μM) dissolved in OPTI-MEM acidified with (0.15% v/v) 6 N HCl11. For in vivo induction, mice were injected intraperitoneally with TS or TB1 at 20 mg/kg daily for 2 weeks, beginning 1 week after TAC, or daily for 1 week, beginning immediately after TAC. To evaluate autophagic flux in vivo, chloroquine was injected (10 mg/kg) intraperitoneally12 30 days after TAC. Four hours later, animals were euthanized for immunoblot detection of autophagy markers.
Electron microscopy (EM)
Conventional EM was performed as described previously9. Mitochondrial mass was analyzed using ImageJ6. The average mitochondrial mass was calculated from 250 mitochondria per slide on four different slides.
Real-time qPCR for mitochondrial DNA
Total DNA was extracted from mouse hearts using the DNeasy Blood & Tissue kit (QIAGEN). The mtDNA content was quantified by real-time qPCR of cytochrome b and β-actin as described6.
AAV transduction
AAV vectors were generated from AAV9 vectors. Doses of 1 × 1011 particles of each vector were administered intravenously through the jugular vein.13
Mitochondrial assays
The mitochondrial fraction was prepared from mouse hearts (40–50 mg) as described6. Mitochondrial electron transport chain complex activity was evaluated with the MitoCheck Complex Activity Assay Kit (Cayman Chemical Company). ATP production was measured with an ATP Bioluminescent Assay (Sigma) in which 25 μg of mitochondrial protein was incubated with ATP assay mix and MSH buffer containing 625 μM ADP and substrate (10 mM pyruvate and 10 mM malate). A Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience) was used to measure the rate of oxidative phosphorylation in intact CMs14. The data are expressed as relative experimental values per heart.
Immunoblot analyses
The methods used for preparation of cell lysates from in vitro and in vivo samples and for immunoblot analyses have been described previously15. The antibodies used include Drp1 (BD Transduction, 611112), phospho-Drp1 (Ser616) (Cell Signaling, 4494S), phospho-Drp1 (Ser637) (Cell Signaling, 4867S), LC3 (BML, M186-3), p62 (ORIGENE, TA307334), Cox I (abcam, ab110413), Beclin 1 (Cell signaling, 3738S), PGC-1α (Santa Cruz, sc-13067), Cox IV (Cell Signaling, 4844S), Mst1 (BD Transduction, 611052), phospho-Mst1 (Cell Signaling, 3681S) and GAPDH (Cell Signaling, 2118S).
Human heart samples
Heart lysates from explanted hearts were obtained from patients who received heart transplants and from age-matched donors. The study was approved by the institutional Ethics Committee (VGHIRB No.:2012-06-028D). All patients or their families expressed their willingness to participate through an informed consent form. Detailed information regarding the human samples was provided previously16.
Statistical Analysis
Statistical analyses were conducted with the SPSS 22.0 J software program (SPSS Japan Institute, Tokyo, Japan) or the JMP software program (SAS Japan Institute, Tokyo, Japan). Data are expressed as mean ± SEM for the indicated number of experiments or mice. The difference in means between 2 groups and multiple groups was evaluated using the Welch’s t-test and the Welch’s ANOVA, respectively. Post-test comparisons for multiple analysis were performed using the Tukey test. P values were two-sided and p values of <0.05 were considered statistically significant.
Results
Basal characterization of C57BL/6J mice after PO
In order to evaluate the effects of PO, C57BL/6J mice were subjected to either TAC or sham operation and followed up for 1 hour to 30 days. There was no significant difference in cardiac hypertrophy or function between mice subjected to sham operation and mice that had not undergone any operation at any time point (not shown). Therefore, “sham” refers to the combined data from the sham operated mice at all time points. The heart is enlarged in response to TAC in a time-dependent manner (Figure 1A). Postmortem assessment showed that left ventricular (LV) weight/tibia length (TL) was significantly greater 5 days after TAC and thereafter than in sham (Figure 1B). Wheat germ agglutinin (WGA) staining showed that CM cross-sectional area was significantly greater 7, 14, and 30 days after TAC than in sham (Supplemental Figure 1A). Myocardial fibrosis, evaluated with Picric Acid Sirius Red (PASR) staining, was also significantly greater 7, 14 and 30 days after TAC than in sham (Supplemental Figure 1B). Echocardiographic measurements showed that although LV ejection fraction (LVEF) was maintained until 7 days after TAC, it was decreased significantly 14 and 30 days after TAC compared to sham (Figure 1CD). Lung weight/TL was significantly elevated 1, 3, 6, and 12 hours after TAC compared to sham. This index was then normalized at 24 hours of TAC but was again elevated at 14 and 30 days (Figure 1E). LV end-diastolic pressure (LVEDP) was significantly elevated 6 hours after TAC compared to sham. Although the LVEDP declined at 24 hours of TAC to a level slightly higher than that in sham, it rose again and remained elevated compared to sham until 30 days (Figure 1FG). The pressure gradient was between 70 and 80 mmHg except at 6 hours, and 14 and 30 days (Supplemental Figure 1C). We speculate that constriction is still evolving at 6 hours, whereas LV dysfunction decreases the pressure gradient at 14 and 30 days. CM apoptosis, evaluated with TUNEL staining, was increased significantly 14 and 30 days after TAC compared to sham (Supplemental Figure 1D). These results suggest that TAC induces cardiac hypertrophy within 5 days, LV dysfunction within 14 days, and HF at 14–30 days.
Figure 1.
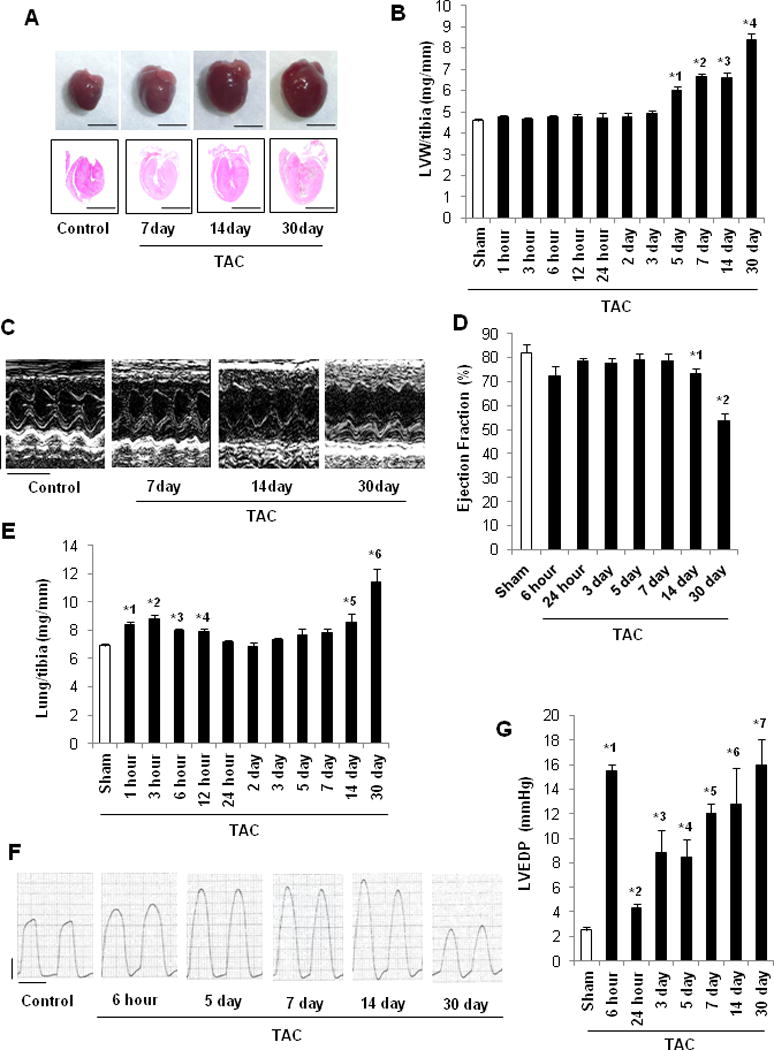
Cardiac function of C57BL/6J mice after PO. In A–C and E, C57BL/6J mice were subjected to either sham operation (n=43) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n=8, 7, 12, 8, 7, 8, 18, 11, 13, 12, and 27, respectively). In D, mice were subjected to either sham operation (n=6) or TAC for 6 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n=7, 6, 6, 6, 6, 8, 8, and 8, respectively). A. Gross morphology and longitudinal heart sections of C57BL/6J mice stained with HE. Scale bars, 5.0 mm. B. LV weight to TL (LV/tibia) ratio. * p<0.05 vs. sham at each time point. *1 p<0.001, *2 p<0.001, *3 p<0.001, *4 p<0.001. C. Representative echocardiographs. Scale bars, vertical 2.5 mm and horizontal 250 ms. D. LVEF, evaluated by echocardiography. * p<0.05 vs. sham at each time point. *1 p=0.047, *2 p<0.001. E. Lung weight to TL (lung/tibia) ratio. * p<0.05 vs. sham at each time point. *1 p<0.001, *2 p=0.004, *3 p<0.001, *4 p<0.001, *5 p=0.045, *6 p=0.025. In F–G, mice were subjected to either sham operation (n=16) or TAC for 6 or 24 hours, or 3, 5, 7, 14 or 30 days (n=6, 6, 5, 8, 6, 5, and 6, respectively). F. Representative LV pressure wave forms. Scale bars, vertical 40 mmHg and horizontal 100 ms. G. LVEDP, evaluated with hemodynamic measurements. * p<0.05 vs. sham at each time point. *1 p<0.001, *2 p=0.001, *3 p=0.027, *4 p=0.004, *5 p<0.001, *6 p=0.025, *7 p=0.001.
Autophagy is transiently upregulated in the early phase but downregulated in the chronic phase of PO
We next investigated how TAC affects autophagy in the heart. LC3-II was significantly increased 3, 6, and 12 hours after TAC, returned to sham level by 24 hours, and then was significantly decreased from 5 days on (Figure 2A). p62/SQSTM1, a protein known to be degraded by autophagy, was significantly decreased 1 and 3 hours after TAC and increased at 6 hours and 3, 5, and 7 days after TAC compared to sham (Figure 2B). These results suggest that autophagy is transiently activated by TAC during the initial 12 hours but is suppressed after 5 days. To further validate our results, autophagic flux was evaluated with tandem fluorescent LC3 (tf-LC3). At 6 hours, both GFP/RFP double-positive autophagosomes (yellow) and RFP-positive autolysosomes (red) were significantly increased in the TAC group compared to in the sham group. Furthermore, chloroquine induced a significantly greater increase in the number of yellow dots at 6 hours after TAC than after sham operation. However, after 7 days of TAC, both yellow and red dots were significantly decreased in the absence of chloroquine compared to sham, and the chloroquine-induced increase in the number of yellow dots was significantly smaller, consistent with the notion that autophagic flux was decreased 7 days after TAC (Figure 2C, Supplemental Figure 2). Together, these data suggest that autophagic flux is transiently activated after TAC, peaking within 3–12 hours, but inactivated after 7 days.
Figure 2.
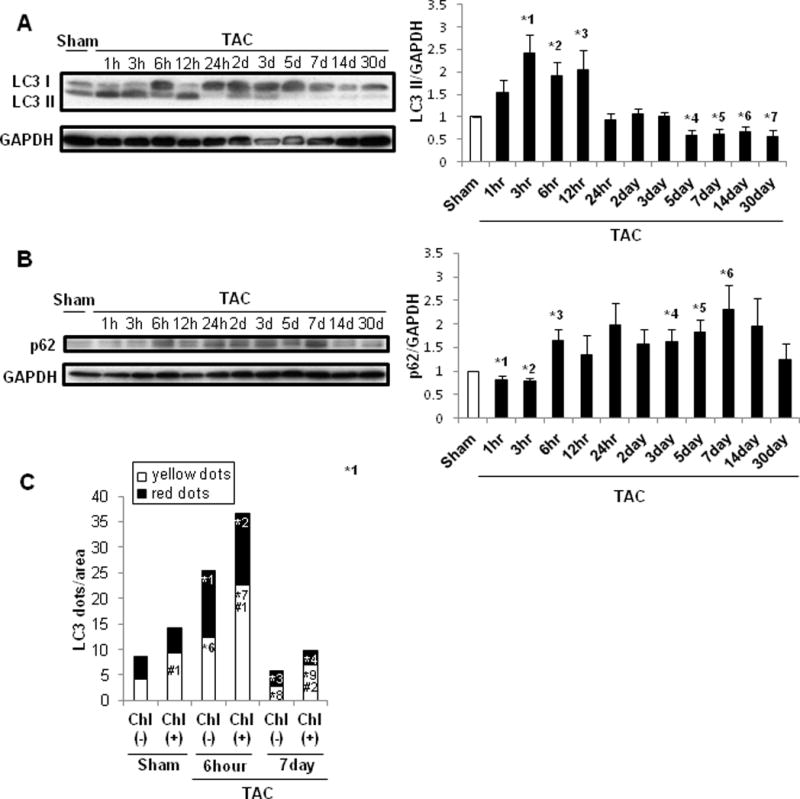
Autophagy was upregulated in the early phase but downregulated in the chronic phase of PO. C57BL/6J mice were subjected to either sham operation (n=43) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n= 4, 4, 4, 4 4, 4, 4, 4, 3, 4, and 4, respectively). A. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for LC3 and GAPDH. * p<0.05 vs. sham-operated mice. *1 p=0.018, *2 p=0.018, *3 p=0.049, *4 p=0.007, *5 p=0.012, *6 p=0.032, *7 p=0.035. B. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for p62 and GAPDH. * p<0.05 vs. sham-operated mice. *1 p=0.043, *2 p=0.015, *3 p=0.032, *4 p=0.044, *5 p=0.016, *6 p=0.031. C. Bar graph indicates mean number of autophagosomes (yellow dots) and autolysosomes (red dots) per cell. Chl: Chloroquine. * p<0.05 vs. sham operation, # p<0.05 vs. Chl (-) at each time point. Data were obtained from 3 experiments. *1 p=0.008, *2 p<0.001, *3 p=0.031, *4 p=0.049, *5 p=0.001, *6 p=0.010, *7 p=0.001, *8 p=0.022, *9 p=0.004,#1 p=0.041,#2 p<0.001.
Activation of Mst1 suppresses autophagy in CMs2. Mst1-CKO mice were used to test the involvement of Mst1 in the suppression of autophagy after TAC. In control hearts, Mst1 protein and Thr183-phosphorylated Mst1 were increased 7 days after TAC (Supplemental Figure 3AB). Although LC3-II was decreased and p62 was increased 7 days after TAC in control hearts, LC3-II was significantly increased and p62 was decreased in Mst1-CKO mice (Supplemental Figure 3CD). These results suggest that activation of endogenous Mst1 partly mediates the suppression of autophagy in response to PO.
Mitochondrial autophagy is stimulated after transient activation of general autophagy ceases
EM analyses showed that autophagosomes containing only mitochondria were observed after 3, 5 and 7 days of TAC (Figure 3A), but not after 6 or 24 hours or 14 or 30 days of TAC or after sham operation (data not shown). Quantitative analysis indicated that mitochondrial autophagy was increased significantly after 3, 5 and 7 days of TAC compared to sham (Figure 3B). Mitochondrial DNA content, evaluated through real-time PCR of cytochrome b, was significantly greater 1, 3, 6, and 24 hours and 2 days after TAC than in sham (Figure 3C). However, it transiently decreased below control levels 3 and 5 days after TAC, and returned to control levels after 7 days (Figure 3C). The level of Cox I, a mitochondrial matrix protein, normalized by the level of GAPDH, a cytosolic protein, decreased transiently 5–7 days after TAC compared to sham (Figure 3D). Cardiac PGC-1α protein expression did not change significantly at any time point, suggesting that PO may not affect mitochondrial biogenesis (Figure 3E). In order to further evaluate mitochondrial autophagy, we expressed Mito-Keima6 in the heart, using AAV-mediated gene delivery. To confirm the lysosomal localization of Mito-Keima, we co-injected AAV-YFP-Lamp1. Puncta with a high 560/440 nm excitation ratio, reflecting increased lysosomal localization of Mito-Keima, were increased 3–7 days after TAC, compared to sham operation. The high ratio Mito-Keima dots were co-localized with YFP-Lamp1 dots, suggesting lysosomal localization (Supplemental Figure 4A and 7B). These results further suggest that lysosomal degradation of mitochondria is stimulated 3–7 days after TAC. Although mitochondrial localization of Parkin and polyubiquitination of mitochondrial proteins, indicators of Parkin-mediated mitophagy17, were both increased 6 hours after TAC, they were downregulated at 7 days (Supplemental Figure 4BC). Beclin1 was increased significantly 1 and 6 hours after TAC, preceding the initial peak in autophagy, briefly significantly decreased below baseline at 12 hours, and then increased compared to sham again after 5–7 days, coincident with the occurrence of mitochondrial autophagy (Supplemental Figure 5). These results suggest that mitochondrial autophagy is stimulated transiently between 3 and 7 days after TAC, accompanied by transient decreases in mitochondrial content.
Figure 3.
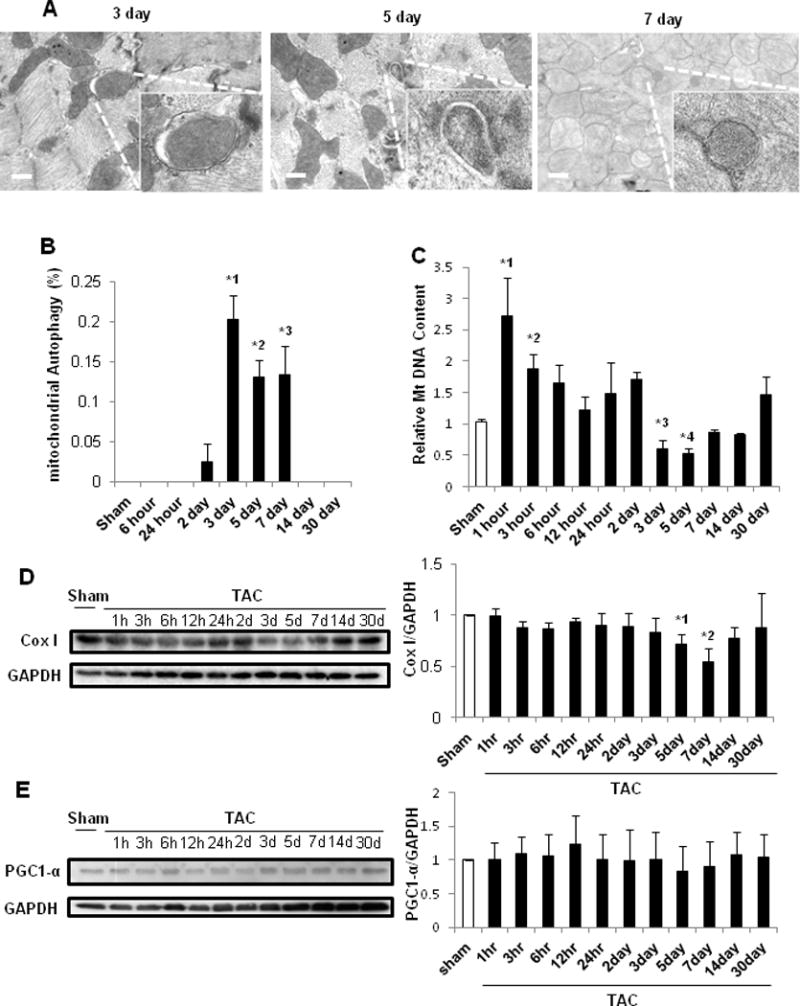
Mitophagy was upregulated 3 days after PO. In A–B, C57BL/6J mouse hearts were examined by EM after sham operation (n=16) or TAC (n=4, 4, 3, and 4 for 6 hours, and 5, 7, and 30 days, respectively). A. Representative EM images of C57BL/6J mouse hearts after TAC. Insets show mitophagy, seen only 3, 5 and 7 days after TAC. Scale bar, 500 nm. B. Bar graphs indicate the number of autophagosomes containing mitochondria per total number of mitochondria. * p<0.05 vs. sham operation. *1 p=0.003, *2 p=0.004, *3 p=0.018. C. Relative mitochondrial DNA content after TAC. C57BL/6J mice were subjected to either sham operation (n=43) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n= 4, 4, 4, 4, 3, 4, 4, 8, 7, 4, and 4, respectively). * p<0.05 vs. sham operation. *1 p=0.025, *2 p=0.044, *3 p=0.040, *4 p=0.043. In D–E, C57BL/6J mice were subjected to either sham operation (n=4) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n= 4, 4, 4, 4, 4, 4, 4, 4, 3, 4, and 4, respectively). D. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for Cox I and GAPDH. * p<0.05 vs. sham operation. *1 p=0.030, *2 p=0.015. E. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for PCG1α and GAPDH.
Mitochondrial dysfunction develops after mitochondrial autophagy is suppressed
Mitochondrial ATP production and ATP content were both attenuated significantly 5 days after TAC and thereafter (Figure 4AB). The activities of mitochondrial complexes I, II + III, and IV were also significantly attenuated 5–7 days after TAC compared to sham (Figure 4C–E). We also evaluated the rate of oxidative phosphorylation in isolated mitochondria, using a Seahorse analyzer. The basal oxygen consumption rate (OCR) and the maximum OCR were significantly lower 14 days after TAC than in sham (Figure 4FG), as was the level of proton leak, determined by subtracting OCR-linked ATP synthesis from basal OCR (Figure 4H). These data indicate that TAC induces mitochondrial dysfunction in the mouse heart after 5–7 days of TAC.
Figure 4.
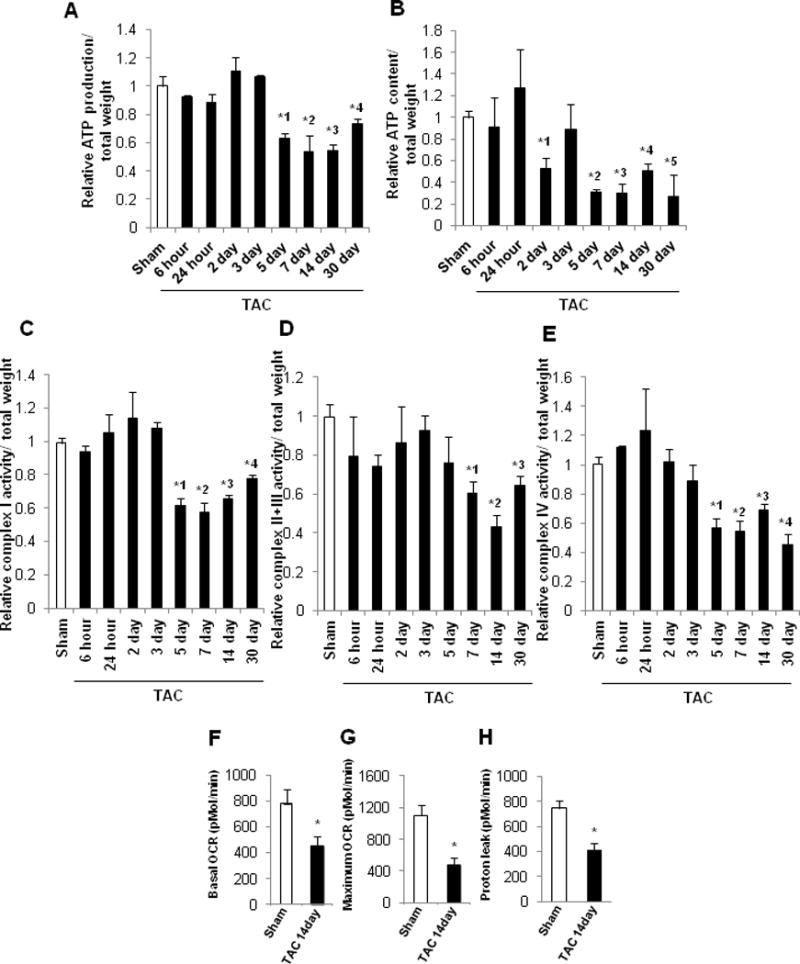
Time course of mitochondrial function in the mouse heart after PO. In A–E, the values from sham-operated mouse hearts at each time point were averaged and expressed as 1. * p<0.05 vs. sham at each time point. In A–B, C57BL/6J mice were subjected to either sham operation (n=34) or TAC for 6 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n=4, 4, 4, 4, 4, 6, 4, and 4, respectively). A. Relative mitochondrial ATP production in the mouse heart. *1 p=0.021, *2 p=0.027, *3 p=0.009, *4 p=0.025. B. Relative mitochondrial ATP content in the mouse heart. *1 p=0.036, *2 p=0.029, *3 p=0.001, *4 p=0.049, *5 p=0.043. In C–E, C57BL/6J mice were subjected to either sham operation (n=29) or TAC for 6 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n=4, 4, 3, 4, 3, 3, 4, and 4, respectively). C. Relative respiratory chain complex I activity in mitochondria. *1 p=0.043, *2 p=0.032, *3 p=0.007, *4 p=0.022. D. Relative respiratory chain complex II+III activity in mitochondria. *1 p=0.022, *2 p=0.003, *3 p=0.004. E. Relative respiratory chain complex IV activity in mitochondria. *1 p=0.014, *2 p=0.012, *3 p=0.014, *4 p=0.002. In F–H, mice were subjected to either sham operation (n=5) or 14 days of TAC (n=4). The mitochondrial fraction prepared from heart homogenates was subjected to Seahorse analyses. * p<0.05 vs. sham at 14 days. F. Basal respiration.* p=0.013 vs. sham at 14 days. G. Maximal respiration.* p=0.006 vs. sham at 14 days. H. Proton leak.* p=0.004 vs. sham at 14 days.
Foreshortening of mitochondria and mitochondrial translocation of Drp1 coincide with activation of mitochondrial autophagy
EM analyses showed that mitochondria in control hearts were primarily rectangular or spherical at baseline, with tubular mitochondria observed less frequently. Mitochondria were enlarged transiently around 24 hours after TAC, shrunk to a spherical shape again 3–5 days after TAC, and then elongated/enlarged again 30 days after TAC (Figure 5A, Supplemental Figure 6A). Quantitative analyses revealed that the mitochondrial mass was significantly greater 24 hours and 30 days after TAC, but was significantly smaller 3 and 5 days after TAC, than in sham (Figure 5AB). The longitudinal length of individual mitochondria compared to that of sarcomeres was evaluated as described previously6. Although the proportion of elongated mitochondria was significantly greater 24 hours and 30 days after TAC than in sham, that of foreshortened mitochondria was significantly greater 7 days after TAC than in sham (Supplemental Figure 6B). Drp1 plays an important role in mediating mitochondrial fission and autophagy in the heart6. Drp1 is primarily cytosolic at baseline but translocates to mitochondria in response to stress6. Total Drp1 in the heart did not change significantly after TAC (Supplemental Figure 6C). However, mitochondrial accumulation of Drp1 was significantly increased 2 and 3 days after TAC, whereas it was significantly suppressed 14 and 30 days after TAC compared to sham (Figure 5C). Conversely, cytosolic Drp1 was significantly decreased 2–5 days after TAC, whereas it was significantly increased 14 and 30 days after TAC compared to in sham (Figure 5D). Phosphorylation of Drp1 at S616 and at S637 positively and negatively correlates with mitochondrial translocation of Drp1, respectively18, 19. Drp1 S616 phosphorylation was increased significantly 1, 2, 3, and 5 days after TAC compared to sham (Figure 5E). In contrast, Drp1 S637 phosphorylation was decreased significantly 12 hours and 2 days after TAC compared to sham (Figure 5F). Together, these results suggest that mitochondrial translocation of Drp1 is induced between 2 and 5 days, peaking 3 days after TAC, but that Drp1 is located primarily in the cytosol again thereafter. Furthermore, the mitochondrial translocation of Drp1 after TAC at least partially coincided with S616 phosphorylation/S637 dephosphorylation of Drp1. In myocardial sections obtained from human HF patients, phosphorylation of Drp1 at S616 was significantly lower than in normal controls (Supplemental Figure 6D), whereas phosphorylation of Drp1 at S637 did not differ significantly. Since significant mitochondrial dysfunction and HF only developed in mice after mitochondrial foreshortening and mitochondrial translocation of Drp1 returned to normal levels, we next investigated the functional significance of Drp1 in the heart during PO-induced cardiac hypertrophy and HF6.
Figure 5.
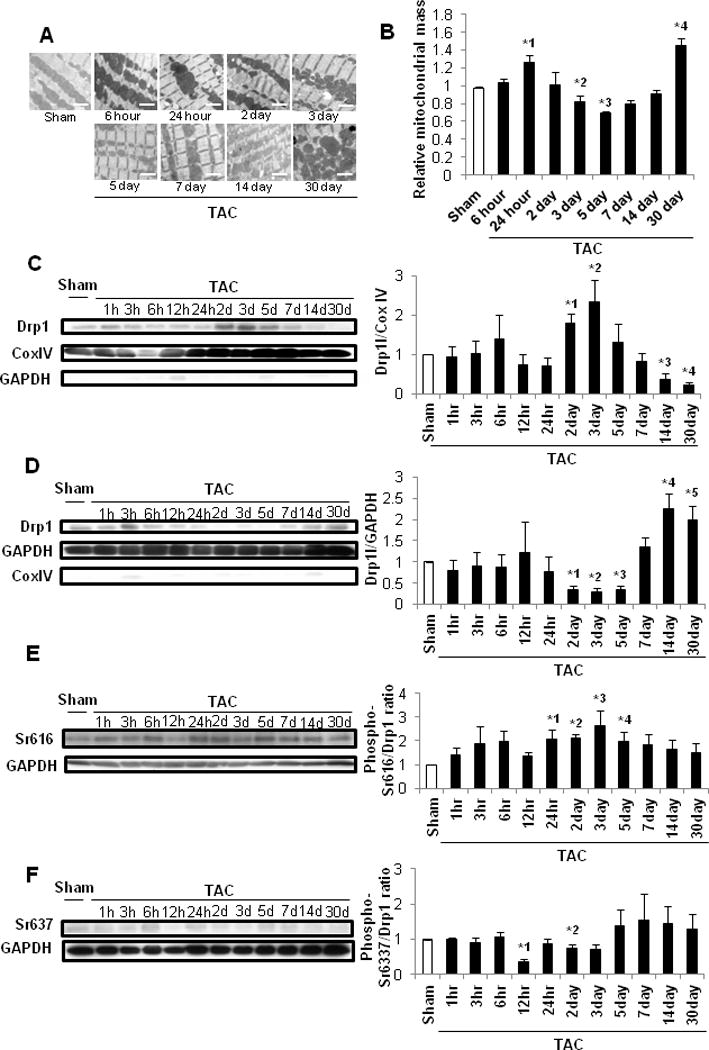
Mitochondrial morphology and mitochondrial translocation of Drp1 after PO. In A–B, C57BL/6J mice were subjected to either sham operation (n =32) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n= 4, 4, 4, 4, 4, 4, 4, 4, 3, 4, and 4, respectively). A. EM images of C57BL/6J mouse hearts after TAC. Scale bar, 2 μm. B. Mitochondrial mass in control mouse hearts is expressed as 1. * p<0.05 vs. sham at each time point. *1 p=0.048, *2 p=0.047, *3 p=0.027, *4 p=0.010. In C–F, C57BL/6J mice were subjected to either sham operation (n =4) or TAC for 1, 3, 6, 12 or 24 hours, or 2, 3, 5, 7, 14 or 30 days (n= 4, 4, 4, 4, 4, 4, 4, 4, 3, 4, and 4, respectively). C. Representative immunoblots and quantitative analysis of Drp1, GAPDH and Cox IV in the mitochondrial fraction prepared from heart homogenates. * p<0.05 vs. sham. *1 p=0.013, *2 p=0.040, *3 p=0.007, *4 p<0.001. D. Representative immunoblots and quantitative analysis of Drp1, GAPDH and Cox IV in the cytosolic fraction prepared from heart homogenates. *1 p<0.001, *2 p<0.001, *3 p<0.001, *4 p=0.013, *5 p=0.023. In E–F, whole-cell heart homogenates were subjected to immunoblot analyses. Representative immunoblots and quantitative analyses are shown. E. Drp1 Ser616 phosphorylation. *1 p=0.026, *2 p=0.001, *3 p=0.048, *4 p=0.049. F. Drp1 Ser637 phosphorylation. *1 p<0.001, *2 p=0.049.
Downregulation of Drp1 exacerbates PO-induced cardiac dysfunction
We used 16-week-old cardiac-specific conditional Drp1 heterozygous knockout (Drp1-hetCKO) mice6 to evaluate the role of endogenous Drp1 during PO-induced cardiac hypertrophy. Immunoblot analyses confirmed that there was significantly less Drp1 protein in Drp1-hetCKO mice than in control mice (Supplemental Figure 7A). Drp1 fl/fl mice or Drp1 flox heterozygous (fl/+) mice (without Cre) were used as controls. These mice were subjected to either TAC or sham operation for 1, 3, or 24 hours, or 3 or 7 days, but not longer due to the high mortality rate observed in Drp1-hetCKO mice after TAC at later time points. Neither control nor Drp1-hetCKO mice showed any significant difference in cardiac parameters after sham operation. Drp1-hetCKO mouse hearts were enlarged compared to control mouse hearts after TAC (Figure 6A). LVEF was significantly lower in Drp1-hetCKO mice than in control mice 3 and 7 days after TAC (Figure 6BC). LV weight/TL, lung weight/TL, and LVEDP were significantly greater in Drp1-hetCKO mice than in control mice 3 and 7 days after TAC (Figure 6D–F). Mitochondrial ATP production was significantly attenuated 3 and 24 hours and 7 days after TAC in Drp1-hetCKO mice compared to in control mice (Figure 6G). These data indicate that TAC-induced cardiac hypertrophy, dysfunction, HF, and mitochondrial dysfunction were rapidly developed in Drp1-hetCKO mice compared to in control mice.
Figure 6.
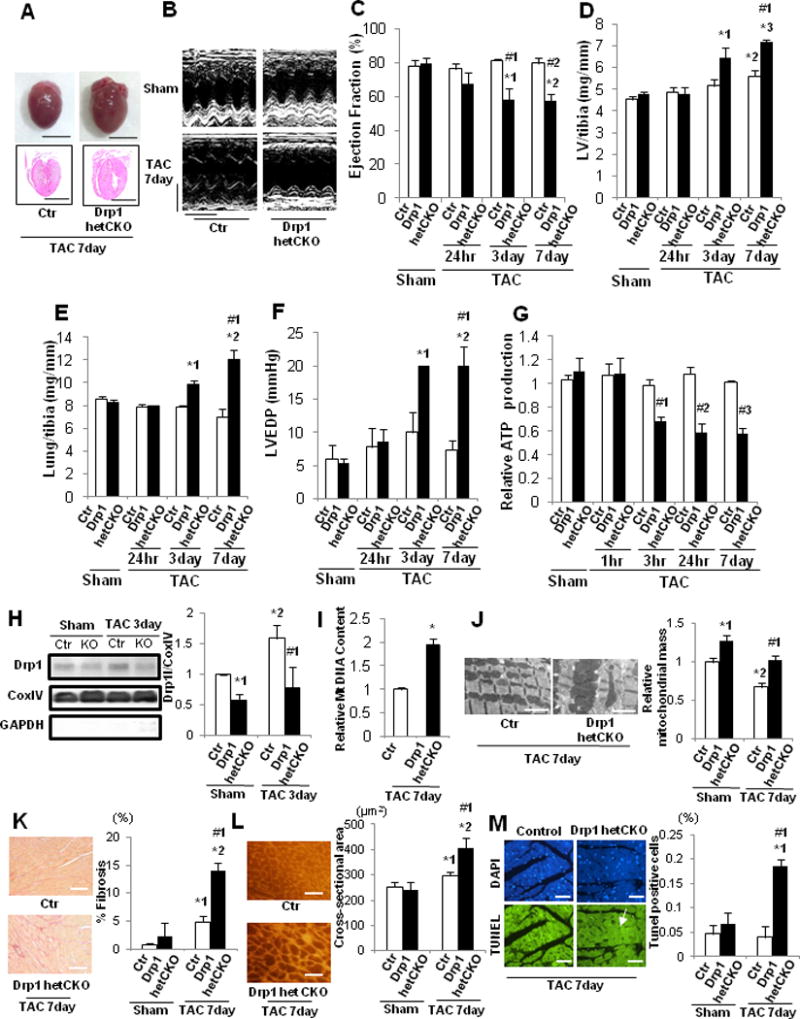
HF and mitochondrial dysfunction after PO were exacerbated in cardiac-specific Drp1 heterozygous knockout mice. Drp1-hetCKO mice were subjected to either sham operation (n=20) or TAC for 1, 3, 6, or 24 hours, or 3 or 7 days (n=3, 3, 3, 3, 3 and 9, respectively). A. Representative gross morphology and longitudinal heart sections of Drp1-hetCKO and control mice stained with HE 7 days after TAC. Scale bars, 5.0 mm. B. Representative echocardiographs. Scale bars, horizontal 250 ms and vertical 2.5 mm. In C–G, * p<0.05 vs. sham operation at each time point, # p<0.05 vs. control in each group. C. LVEF, evaluated by echocardiography. *1 p=0.045, *2 p=0.014,#1 p=0.007,#2 p=0.001. D. LV weight to TL (LV/tibia) ratio. *1 p=0.001, *2 p=0.027, *3 p<0.003,#1 p=0.003. E. Lung weight to TL (lung/tibia) ratio. *1 p=0.036, *2 p=0.015,#1 p<0.001. F. LVEDP, evaluated by hemodynamic measurement. *1 p=0.008, *2 p=0.004,#1 p=0.003. G. Relative mitochondrial ATP content in the mouse heart.#1 p=0.014,#2 p=0.004,#3 p=0.006. H. Representative immunoblots and quantitative analysis of Drp1, GAPDH, and Cox IV. Mice were subjected to either TAC or sham operation for 3 days. Mitochondrial fractions prepared from the heart homogenates were subjected to immunoblot analyses. * p<0.05 vs. control in sham group, # p<0.05 vs. control in sham and TAC. *1 p=0.036, *2 p=0.019,#1 p=0.007. I. Relative mitochondrial DNA content in Drp1-hetCKO mice after 7 days of TAC. * p=0.048 vs. control group. J. Representative EM images of Drp1-hetCKO and control mouse hearts after TAC and quantitative analysis of mitochondrial mass. Scale bar, 2 μm. Mitochondrial mass in control mouse hearts is expressed as 1. * p<0.05 vs. control in sham group, # p<0.05 vs. control in sham and TAC. *1 p=0.012, *2 p=0.025,#1 p=0.043. K. PASR staining to assess cardiac fibrosis. * p<0.05 vs. control in sham group, # p<0.05 vs. control in sham and TAC. Scale bar, 200 μm. *1 p=0.025, *2 p=0.001,#1 p=0.009. L. WGA staining to assess CM cross-sectional area. * p<0.05 vs. control in sham group, # p<0.05 vs. control in sham and TAC. Scale bar, 500 μm. *1 p=0.041, *2 p=0.003,#1 p=0.010. M. TUNEL staining to assess apoptosis in CMs. # p<0.05 vs. control in sham and TAC. Scale bar, 50 μm. *1 p=0.005,#1 p=0.012.
There was significantly less Drp1 in mitochondria in Drp1-hetCKO hearts than in control hearts 3 days after TAC and sham (Figure 6H). Mitochondrial DNA content and mitochondrial mass were significantly greater in Drp1-hetCKO hearts 7 days after TAC than in control hearts or sham (Figure 6IJ). The number of high ratio Mito-Keima dots and their co-localization with YFP-Lamp1 dots were significantly greater in control hearts than in Drp1-hetCKO mice after 7 days of TAC, suggesting that TAC-induced lysosomal translocation of Mito-Keima is Drp1-dependent (Supplemental Figure 7B). Myocardial fibrosis, CM cross-sectional area and the number of TUNEL-positive nuclei in CMs were also significantly greater in Drp1-hetCKO hearts than in control hearts 7 days after TAC (Figure 6K–M, Supplemental Figure 7C). The TAC-induced increases in the number of cells with foreshortened mitochondria observed in control mice at 7 days were significantly attenuated in Drp1-hetCKO mice, suggesting that endogenous Drp1 is involved in mitochondrial fission during PO (Supplemental Figure 7D). The activity of mitochondrial complex I was significantly attenuated and that of complexes II + III and IV also tended to be attenuated in Drp1-hetCKO mouse hearts compared to in control mouse hearts 7 days after TAC (Supplemental Figure 7E–G). These results are consistent with the notion that downregulation of endogenous Drp1 induces accumulation of damaged mitochondria and mitochondrial dysfunction and exacerbates the development of HF after TAC.
Tat-Beclin 1 peptide (TB1) treatment restores mitochondrial autophagy and attenuates cardiac dysfunction after PO
To elucidate whether downregulation of autophagy and mitochondrial autophagy contributes to the development of HF, we conducted rescue experiments, using TB111. TB1 contains 18 amino acids derived from Beclin 1 (267–284) and potently stimulates autophagy in HeLa and other cells by mobilizing endogenous Beclin 1 from the Golgi apparatus where it is tethered through Golgi-associated plant pathogenesis-related protein 111. In order to evaluate the effect of TB1 upon autophagic flux, CMs were transduced with Ad-tf-LC3. Treatment of CMs with 25 μM TB1 for 3 hours significantly increased the numbers of both GFP and RFP puncta compared to treatment with Tat-scrambled peptide (TS) (Figure 7A, Supplemental Figure 8A). Similarly, daily treatment of GFP-LC3 mice with TB1 for 2 weeks significantly increased the number of GFP-LC3 dots in the heart compared to treatment with TS, both in the absence and presence of chloroquine (Figure 7B, Supplemental Figure 8B). The chloroquine-induced increase in the number of GFP-LC3 dots in the heart was greater after TB1 treatment than after TS treatment, suggesting that TB1 increases autophagic flux in the mouse heart. We evaluated whether lysosomal clearance of mitochondria is stimulated by TB1 using Mito-Keima10. High ratio puncta were significantly increased after 3-hour treatment with 25 μM TB1 compared to after treatment with TS, suggesting that TB1 stimulates lysosomal translocation of Mito-Keima in CMs (Figure 7C, Supplemental Figure 8C). Treatment of cardiac fibroblasts with TB1 also significantly increased the number of GFP-LC3 puncta compared to treatment with TS in the presence or absence of chloroquine treatment (Supplemental Figure 8D). In order to normalize the cardiac autophagy level, which is downregulated after 5 days of TAC (Figure 2), we treated mice with TB1 or TS once daily on Days 7–21 (Supplemental Figure 9A). After 30 days of TAC, LVEF was significantly higher, lung weight/TL and LVEDP were significantly lower, and the pressure gradient was significantly higher in TB1-treated mice than in TS-treated mice (Figure7E–I). Myocardial fibrosis, CM cross-sectional area, and CM apoptosis were all significantly reduced in TB1-treated mice compared to in TS-treated mice (Figure 7J–L, Supplemental Figure 9BC). TB1 significantly increased LC3-II and decreased p62 in sham operated mice, suggesting that TB1 stimulates general autophagy at baseline (Figure 7MN). In contrast, TB1 significantly increased LC3-II but did not decrease p62 in the presence of TAC compared to TS, suggesting that TB1 failed to increase the level of general autophagy above control in the presence of TAC. Importantly, however, DNA content and Cox I in mitochondria were significantly lower in TB1-treated mice than in TS-treated mice (Figure 7OP). Furthermore, EM analyses of the mouse heart showed that autophagosomes containing mitochondria were observed in TB1-treated but not in TS-treated hearts (Figure 7Q). Quantitative analysis indicated that mitochondrial autophagy was increased significantly in TB1-treated hearts compared to in TS-treated ones (Figure 7R). Mitochondrial ATP production after TAC was greater in TB1-treated than in TS-treated mice (Figure 7S). These results suggest that TB1 treatment stimulates mitochondrial autophagy, thereby preserving overall mitochondrial health in the presence of PO.
Figure 7.
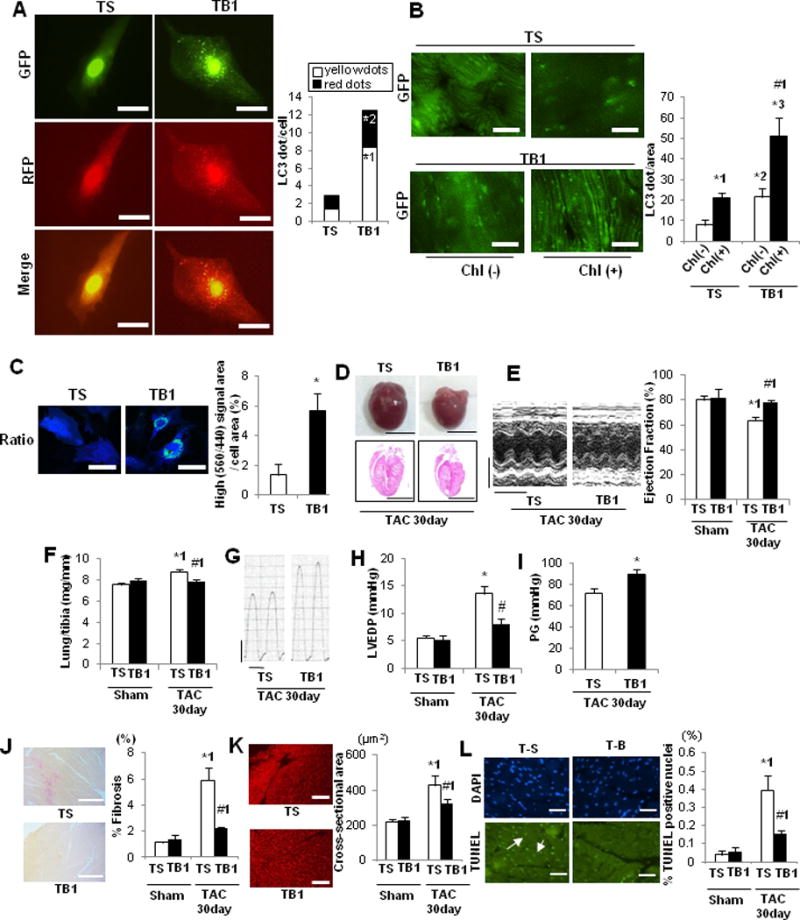
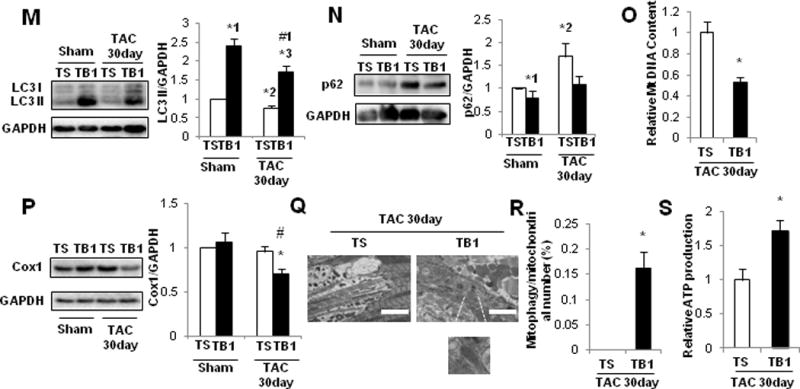
TB1 attenuates mitochondrial dysfunction and development of HF after PO. A. Cultured CMs were transduced with Ad-tf-LC3 and then treated with TB1 or TS. Representative images of mRFP-GFP-LC3 puncta. Red puncta indicate autolysosomes, whereas yellow puncta indicate autophagosomes. Scale bar, 50 μm. The bar graph indicates mean numbers of autophagosomes and autolysosomes per cell obtained from 3 experiments. * p<0.05 vs. TS. *1 p=0.007, *2 p=0.048. B. Tg-GFP-LC3 mice were treated with TB1 or TS for 14 days. Mice were treated with chloroquine (10 mg/kg, ip) for 4 hours and then euthanized. Representative images of GFP-LC3 puncta. Chl: chloroquine. Bar graph indicates mean number of LC3 puncta per area. n= 3 (TS with Chl), 3 (TS without Chl), 3 (TB1 with Chl), and 3 (TB1 without Chl). * p<0.05 vs. TS, # p<0.05 Chl (+) vs. Chl (−) in each group. *1 p=0.009, *2 p=0.031, *3 p=0.002,#1 p=0.025. C. Cultured CMs were transduced with Ad-Keima-MLS and then treated with TB1 or TS. Representative images of Keima-MLS. Puncta with high 560/440 indicate mitochondrial autophagy. The ratio of the area of puncta with high 560/440 vs. the total cell area obtained from 3 experiments is shown. Scale bar, 50 μm. * p=0.037 vs. TS. In D–N, mice were subjected to either sham operation or TAC for 30 days. These mice were then treated daily with either TB1 (n=12) or TS (n=13) on Days 7–21 and euthanized on Day 30. D. Gross morphology and HE staining of longitudinal sections of the mouse hearts. Scale bars, 5.0 mm. E. Representative M-mode tracings of echocardiographs and LVEF. * p<0.05 vs. TS with sham operation, # p<0.05 vs. TS with 30 days TAC. Scale bars, horizontal 250 ms and vertical 2.5 mm. *1 p=0.003,#1 p<0.001. F. Lung weight to TL (lung/tibia) ratio. # p<0.05 vs. TS. *1 p=0.007,#1 p=0.001. G. Representative LV pressure wave forms. Scale bars, vertical 40 mmHg and horizontal 60 ms. H. LVEDP, evaluated by hemodynamic measurements. # p<0.05 vs. TS with 30 days TAC. * p=0.035, #p=0.034. I. Pressure gradient at TAC. *p=0.010 vs. TS. J. PASR staining to assess cardiac fibrosis. * p<0.05 vs. TS with sham operation, # p<0.05 vs. TS with 30 days TAC. Scale bar, 500 μm. *1 p=0.001,#1 p=0.005. K. WGA staining to assess CM cross-sectional area. * p<0.05 vs. TS with sham operation, # p<0.05 vs. TS with 30 days TAC. Scale bar, 200 μm. *1 p=0.009,#1 p=0.011. L. TUNEL staining to assess CM apoptosis. * p<0.05 vs. TS with sham operation, # p<0.05 vs. TS with 30 days TAC. Scale bar, 50 μm. *1 p=0.003,#1 p=0.028. In M–N, whole-cell heart homogenates were subjected to immunoblots. M. Representative immunoblots and quantitative analysis of LC3 and GAPDH. * p<0.05 vs. TS with sham operation. # p<0.05 vs. TS with 30 days TAC. *1 p=0.001, *2 p=0.030, *3 p=0.016,#1 p=0.002. N. Representative immunoblots and quantitative analysis of p62 and GAPDH. * p<0.05 vs. TS with sham operation. *1 p=0.012, *2 p=0.036. In O–S, mice were subjected to either sham operation (TS: n=3, TB: n=3) or TAC (TS: n=3, TB: n=3). O. Relative mitochondrial DNA content. * p=0.032 vs. TS. P. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for Cox I and GAPDH. * p<0.05 vs. TS with sham operation, # p<0.05 vs. TS with 30 days TAC. * p=0.042, #p=0.013. Q. Representative EM images of the mouse heart. The inset shows autophagosomes containing mitochondria, observed only in TB1-treated mice. Scale bar, 2 μm. R. Bar graphs indicate the number of autophagosomes containing mitochondria per total number of mitochondria. * p=0.039 vs. sham operation. S. Relative mitochondrial ATP production. * p=0.034 vs. TS.
The protective effect of TB1 against PO-induced cardiac hypertrophy and HF is abolished in the presence of Drp1 or Beclin 1 downregulation
In order to test whether the aforementioned effect of TB1 is mediated through general autophagy or mitochondrial autophagy, we treated Drp1-hetCKO, beclin 1-hetKO, and respective control mice with TB1 or TS. Due to the high susceptibility of these mice to TAC-induced cardiac dysfunction, these mice were subjected to only 7 days of TAC. The mice were treated daily with either TB1 or TS on Days 1–7 and euthanized on Day 7 (Supplemental Figure 10A). Neither TB1 nor TS affected cardiac function, mitochondrial content or mitochondrial function in sham operated Drp1-hetCKO or beclin 1-hetKO mice. TAC induced cardiac dysfunction and signs of HF in both Drp1-hetCKO and beclin 1-hetKO mice treated with either TB1 or TS. Importantly, LVEF, lung weight/TL, pressure gradient, and LVEDP did not significantly differ between TB1-treated and TS-treated mice after TAC when these peptides were injected into Drp1-hetCKO (Figure 8A–E) or beclin 1-hetKO mice (Figure 8I–M). Furthermore, TB1 failed to restore mitochondrial ATP production or reduce mitochondrial content or Cox I in Drp1-hetCKO mice (Figure 8F–H). Thus, although TB1 prevented exacerbation of PO-induced cardiac dysfunction in control mice, it failed to do so in Drp1-hetCKO or beclin 1-hetKO mice. Taken together, these data suggest that TB1 attenuates the development of HF by stimulating Drp1- or Beclin 1-dependent activation of autophagy and/or mitochondrial autophagy in response to PO.
Figure 8.
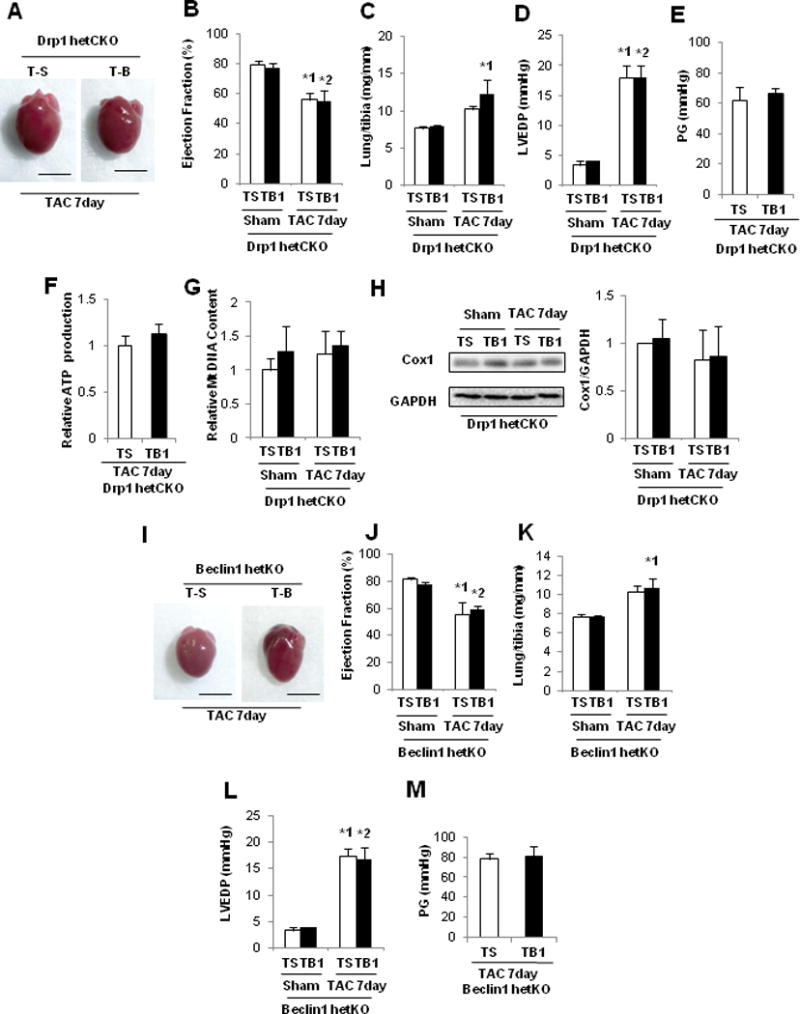
The protective effect of TB1 in the PO heart was abrogated in Drp1-hetCKO and beclin 1-hetKO mice. In A–H, Drp1-hetCKO and control mice were subjected to either sham operation or TAC for 7 days. These mice were then treated daily with either TB1 (n=3) or TS (n=4) on Days 1–7 and euthanized on Day 7. A. Gross morphology of mouse hearts. Scale bars, 5.0 mm. B. LVEF, evaluated by echocardiography. *1 p=0.037, *2 p=0.029. C. Lung weight to TL (lung/tibia) ratio. *1 p=0.040. D. LVEDP, evaluated by hemodynamic measurement. E. Pressure gradient at TAC. F. Relative mitochondrial ATP production. G. Relative mitochondrial DNA content. H. Representative immunoblots and quantitative analysis of whole-cell heart homogenates for Cox I and GAPDH. In I–M, beclin 1-hetKO and control mice were subjected to either sham operation or TAC for 7 days. These mice were then treated daily with either TB1 (n=6) or TS (n=7) on Days 1–7 and euthanized on Day 7. I. Gross morphology of the mouse hearts. Scale bars, 5.0 mm. J. LVEF, evaluated by echocardiography. *1 p=0.040, *2 p=0.044. K. Lung weight to TL (lung/tibia) ratio. *1 p=0.040. L. LVEDP, evaluated by hemodynamic measurement. *1 p=0.011, *2 p=0.021. M. Pressure gradient at TAC.
Discussion
The major findings of the present study are: 1) PO initially activates general autophagy but suppresses it thereafter, 2) the time course of mitochondrial autophagy is distinct from that of general autophagy, 3) PO-induced suppression of general and mitochondrial autophagy is followed by the development of mitochondrial dysfunction and HF, 4) endogenous Drp1 plays an essential role in mediating mitochondrial autophagy and maintaining both mitochondrial and cardiac function during PO, and 5) restoration of mitochondrial autophagy attenuates progression of HF during PO (Supplemental Figure 10B).
We here show that PO activates autophagy 3–12 hours after TAC but suppresses autophagy below baseline after 5 days. One report showed that autophagy is upregulated7, whereas others have demonstrated that autophagy is downregulated 7 days after PO20. However, the time-dependent changes in cardiac autophagic activity during PO that were observed in this study have not been shown previously. In the acute phase, PO elicited transient LV dysfunction, as indicated by an increase in lung weight/TL and LVEDP, but the mice recovered within 24 hours. During this period, the heart appeared to retain the ability to activate autophagy. However, autophagy was suppressed below physiological levels after 3–5 days and the flux was significantly suppressed at 7 days. Acute mechanical stress activates Nox2-dependent reactive oxygen species production at the junctional sarcoplasmic reticulum21, while energy stress stimulates Nox4 at the endoplasmic reticulum22, and we have shown previously that oxidative stress stimulates autophagy8, 22. In contrast, chronic activation of either Mst1 or mTOR suppresses autophagy2, 23. PO chronically activates mTOR and Mst1, both of which are mechanistically involved in the pathogenesis of HF2, 24. We here showed that activation of Mst1 partly mediates the suppression of general autophagy 7 days after PO, but further investigation is required to elucidate the signaling mechanisms mediating the phase-dependent changes in autophagic activity in response to PO.
Mitochondrial autophagy was upregulated 3–7 days after TAC and downregulated thereafter. Autophagosomes containing only mitochondria were observed 3–7 days after TAC but not at other time points. This was accompanied by decreases in mitochondrial content. This time course of mitochondrial autophagy is distinct from that of general autophagy. Dissociation between mitophagy and conventional autophagy was also observed in mouse embryonic fibroblasts subjected to starvation or hypoxia25. Since mitochondrial autophagy acts only to eliminate damaged mitochondria, mitochondrial autophagy likely occurs on a much smaller scale than general autophagy. Thus, mitochondrial autophagic activity may not be faithfully reflected by changes in markers of general autophagy. Alternatively, mitochondrial autophagy may be regulated by mechanisms distinct from those regulating general autophagy, such as an Atg5- or Atg7-independent form of autophagy (alternative autophagy), that cannot be evaluated with conventional markers such as LC3-II25.
Activation of mitochondrial autophagy in response to PO coincides with mitochondrial translocation of Drp1, which was also accompanied by phosphorylation of Ser616 and dephosphorylation of Ser637 of Drp1. Furthermore, mitochondrial autophagy is abrogated in Drp1-hetCKO mice. These results suggest that Drp1 plays an important role in mediating mitochondrial autophagy in response to PO. We also found that Beclin 1, a key mediator of autophagy, is upregulated at Days 5–7 of TAC, as well as during the initial 24 hours, despite downregulation of general autophagy at Days 3–7. We have shown previously that both Drp1 and Beclin 1 are important mediators of mitochondrial autophagy in the heart6. Thus, Beclin 1 may also be critical in mediating mitochondrial autophagy during PO. In contrast, Parkin, a well-known mediator of autophagy may not be involved in mitochondrial autophagy in response to PO, since neither mitochondrial localization of Parkin nor increased ubiquitination of mitochondrial proteins was observed when mitochondrial autophagy was induced after PO.
Previous reports suggested that mitochondrial autophagy is stimulated rather than inhibited when Drp1 or Miff, a major receptor for Drp1, is downregulated26, 27. Our study used Mito-Keima, EM, and evaluation of mitochondrial content, while the other study used mitochondrial accumulation of p62 and LC3-II, aggregation of Parkin, and co-localization of mitochondrial proteins and lysosomes as indicators of mitophagy26. Our methods broadly detect mitochondrial degradation by both conventional and non-conventional28 autophagy, whereas the methods used by the other study may be more specific to conventional (LC3- and Parkin-dependent) autophagy. We found that mitochondrial autophagy is upregulated only after general autophagy is downregulated in response to TAC. Thus, we speculate that our methods may detect clearance of mitochondria by non-conventional autophagy.
Currently, the molecular mechanisms by which PO leads to Ser616 phosphorylation and Ser637 dephosphorylation of Drp1 are unknown. Cdks, ERKs, and PKCdelta have been shown to be involved in Ser616 phosphorylation19, 29, whereas PKA, ROCK1, calcineurin, and PGAM5 are involved in Ser637 phosphorylation/dephosphorylation30, 31. We predict that the PO-regulated endogenous kinases/phosphatases responsible for Drp1 phosphorylation/dephosphorylation play an important role in mediating mitochondrial autophagy in the heart. Thus, future studies should test whether manipulating the responsible kinase and/or phosphatase is sufficient to control mitochondrial autophagy in the heart during PO.
Understanding whether stress-induced autophagy is protective or detrimental in the heart is important but complex, since it depends upon the type of stress. Here we show that both general and mitochondrial autophagy are downregulated in the chronic phase of PO, when mice subjected to TAC develop LV dysfunction. In particular, both mitochondrial and LV dysfunction developed only after mitochondrial autophagy was downregulated. Drp1 haploinsufficiency exacerbated PO-induced mitochondrial dysfunction and HF in the mouse heart, whereas stimulation of mitochondrial autophagy with TB1 partially normalized cardiac function in the presence of PO. Importantly, the beneficial effect of TB1 was abrogated when either Drp1 or Beclin 1, molecules shown to be involved in glucose-deprivation-induced mitochondrial autophagy in CMs6, was downregulated. These results suggest that downregulation of mitochondrial autophagy plays an essential role in mediating cardiac dysfunction during PO. The decrease in mitochondrial autophagy is accompanied by increases in mitochondrial mass and mitochondrial dysfunction, suggesting that it causes accumulation of damaged mitochondria and consequent increases in oxidative stress and cell death.
Since the acute cardiac dysfunction during the initial 24 hours of TAC was alleviated after transient activation of general autophagy, peaking at 3–6 hours, early activation of general autophagy may also be protective. However, given that the functional significance of general autophagy during PO remains controversial7, more studies are needed to clarify this issue. In any case, it would be important to conduct loss-of-function studies by selectively inhibiting general autophagy without affecting mitochondrial autophagy. A better understanding of the molecular mechanisms mediating general and mitochondrial autophagy is essential for addressing this issue.
Treatment with TB1, a potent inducer of autophagy11, attenuated the progression of HF in the presence of PO. Although excessive autophagy can be detrimental in the presence of PO7, we here show that an intervention that stimulates autophagy can be used to rescue PO-induced cardiac dysfunction if given when mitochondrial autophagy is suppressed below physiological levels. Since TB1 was not effective in Drp1-hetCKO and beclin 1-hetKO mice, we believe that TB1 is particularly effective in stimulating a Drp1- and Beclin 1-dependent form of autophagy such as mitochondrial autophagy6. In fact, TB1 given on Days 7–21 of TAC promoted mitochondrial degradation by stimulating mitochondrial autophagy. TB1 also increased general autophagy, but its effect upon general autophagy was not as prominent as upon mitochondrial autophagy. Although it is unclear why TB1’s effect upon general autophagy in the presence of TAC was less prominent, it is possible that other mediators of general autophagy are actively suppressed in the presence of TAC. Alternatively, endogenous Beclin 1 released from the Golgi apparatus may be utilized more effectively for mitochondrial autophagy. Because downregulation of autophagy and consequent mitochondrial dysfunction underlie the pathogenesis of HF, autophagy-inducing agents with proven effects upon mitochondrial quality control mechanisms, such as TB1, may have potential for the prevention of HF. It will be interesting to test whether other interventions known to stimulate autophagy equally stimulate mitochondrial autophagy.
It should be noted that TB1 also stimulates autophagy in cardiac fibroblasts. However, the functional significance of fibroblasts in mediating the salutary effect of TB1 remains to be elucidated.
Downregulation of endogenous Drp1 inhibited increases in the number of CMs with fragmented mitochondria 7 days after TAC, suggesting that Drp1 regulates mitochondrial fission during PO. It has been shown recently that cardiac and mitochondrial dysfunction caused by downregulation of Miff can be rescued by concomitant downregulation of Mfn127, suggesting that balancing mitochondrial fusion and fission is critical for the maintenance of mitochondrial function in the heart. Aside from the effect on mitochondrial autophagy, whether or not the lack of appropriate fission in Drp1-hetCKO mice also contributes to the enhancement of HF in these mice during PO remains to be elucidated.
In summary, our results suggest that mitochondrial autophagy is chronically suppressed following transient activation 3–5 days after TAC, and that suppression of mitochondrial autophagy plays an important role in mediating mitochondrial dysfunction and the development of HF in response to PO.
Supplementary Material
Clinical Perspectives.
Mitochondria are intracellular organelles involved in metabolism and ATP production that are vitally important in cardiomyocytes. Heart failure is commonly accompanied by mitochondrial dysfunction. Dysfunctional mitochondria are the major source of reactive oxygen species, and trigger both apoptosis and necrosis of cardiomyocytes. Mitochondrial damage spreads rapidly through depolarization of the mitochondrial membrane potential and causes deterioration of the function of healthy mitochondria. Thus, cardiomyocytes possess robust quality control mechanisms, and autophagy, which degrades protein and organelles through lysosomes, is a major mechanism for elimination of dysfunctional mitochondria. In this study, we show that both general autophagy and a mitochondria-specific form of autophagy (mitochondrial autophagy) are downregulated after pressure overload and that mitochondrial dysfunction develops only after mitochondrial autophagy is downregulated. Mitochondrial autophagy was induced in response to pressure overload through a mechanism dependent upon Drp1, a protein known to mediate mitochondrial fission. Treatment of animals with TAT-Beclin 1 peptide, known to stimulate autophagy through mobilization of endogenous Beclin 1, a protein essential for autophagy, increased the level of mitochondrial autophagy in the heart and partially rescued the animals from the development of pressure overload-induced heart failure. These results suggest that interventions to restore the level of mitochondrial autophagy should be considered for potential treatment for heart failure patients.
Acknowledgments
The authors thank Daniela Zablocki and Christopher D. Brady for critical reading of the manuscript. We also thank Dr. Toshiaki Otsuka (Department of Hygiene and Public Health, Nippon Medical School, Tokyo) for statistical advices.
Funding Sources: This work was supported in part by U.S. Public Health Service Grants HL67724, HL91469, HL102738, HL112330 and AG23039 and by the Leducq Foundation Transatlantic Network of Excellence (J.S.). A.S. has been supported by a Postdoctoral Fellowship from the Uehara Memorial Foundation.
Footnotes
Disclosures: None.
References
- 1.Gatica D, Chiong M, Lavandero S, Klionsky DJ. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015;116:456–467. doi: 10.1161/CIRCRESAHA.114.303788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting Beclin1-Bcl-2 interaction. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 4.Saito T, Sadoshima J. The molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477–1490. doi: 10.1161/CIRCRESAHA.116.303790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 7.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hariharan N, Zhai P, Sadoshima J. Oxidative Stress Stimulates Autophagic Flux during Ischemia/Reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Roles of AMP-Activated Protein Kinase and Beclin 1 in Mediating Autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 10.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doroudgar S, Volkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, Wang W, Gude N, Muller OJ, Wehrens XH, Sussman MA, Glembotski CC. Hrd1 and ER-Associated Protein Degradation, ERAD, Are Critical Elements of the Adaptive ER Stress Response in Cardiac Myocytes. Circ Res. 2015;117:536–546. doi: 10.1161/CIRCRESAHA.115.306993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 20.Hariharan N, Ikeda Y, Hong C, Alcendor RR, Usui S, Gao S, Maejima Y, Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One. 2013;8:e51632. doi: 10.1371/journal.pone.0051632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 22.Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 2013;113:1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a Critical Regulator of Autophagy during Myocardial Ischemia: Pathophysiological Implications in Obesity and Metabolic Syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T, Kang D, Kanki T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11:332–343. doi: 10.1080/15548627.2015.1023047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, Chan DC. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol. 2015;211:795–805. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 29.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.