

Abstract
Structure-based drug design was utilized to develop novel, 1-hydroxy-2-naphthoate-based small-molecule inhibitors of Mcl-1. Ligand design was driven by exploiting a salt bridge with R263 and interactions with the p2 and p3 pockets of the protein. Significantly, target molecules were accessed in just two synthetic steps, suggesting further optimization will require minimal synthetic effort. Molecular modeling using the Site-Identification by Ligand Competitive Saturation (SILCS) approach was used to qualitatively direct ligand design as well as develop quantitative models for inhibitor binding affinity to Mcl-1 and the Bcl-2 relative Bcl-xL as well as for the specificity of binding to the two proteins. Results indicated hydrophobic interactions with the p2 pockets dominate the affinity of the most favourable binding ligand (3bl: Ki = 31 nM). Compounds were up to 20-fold selective for Mcl-1 over Bcl-xL. Selectivity of the inhibitors was driven by interactions with the deeper p2 pocket in Mcl-1 versus Bcl-xL. The SILCS-based SAR of the present compounds represents the foundation for the development of Mcl-1 specific inhibitors with the potential to treat a wide range of solid tumours and hematological cancers, including acute myeloid leukaemia.
Keywords: Mcl-1, Bcl-xL, protein–protein interaction, apoptosis, cancer
Introduction
The B-cell lymphoma-2 (Bcl-2) family of proteins regulates the intrinsic apoptosis pathway that is responsible for programmed cell death, or apoptosis. The pathway involves protein–protein interactions (PPIs) between pro-apoptotic members of the Bcl-2 family, such as Bim, Bak and Bad, and anti-apoptotic members, such as Bcl-xL and myeloid cell leukemia-1 (Mcl-1).1,2 More specifically, through conserved hydrophobic crevices, the anti-apoptotic Bcl-2 proteins capture the BH3 α-helical domains of their pro-apoptotic counterparts, effectively “neutralizing” their cell killing functions. Evasion of apoptosis is a hallmark of cancer, and is also one culprit for the development of resistance to current chemo- and radiotherapies.3 More than a decade of research has seen the emergence of potent inhibitors of Bcl-xL, including the prototypical BH3 mimetic ABT-737.4-6 In particular ABT263, or Navitoclax, which is a dual inhibitor of Bcl-xL and Bcl-2, but not of Mcl-1,7 has performed well in phase I/II clinical trials for several malignancies, validating the inhibition of the anti-apoptotic Bcl-2 proteins as a strategy to treat cancer.8-11
Mcl-1 overexpression and/or amplification of the Mcl-1 gene immortalizes cells, and has been observed in many human solid tumours, including pancreatic, prostate, cervical, lung and breast cancers,12-19 as well as B-cell lymphomas and hematological cancers, including acute myeloid leukemia (AML).20,21 While Navitoclax continues to perform well in clinical trials, its low affinity for Mcl-1 is a contributing factor to the observed resistance of several tumour cell lines.22-26 Moreover, the upregulation of Mcl-1 has been directly linked to the reduced efficacy of several FDA-approved anti-cancer chemotherapies. Meanwhile, Zhang and colleagues demonstrated that RNAi-mediated downregulation of Mcl-1 decreased tumorigenicity of a mouse xenograft model.27 Taken together, these findings indicate that the pharmacologic inhibition of Mcl-1 is an attractive, complementary and/or adjuvant strategy towards the execution of cancer cells by re-activating apoptosis.
In a similar vein to the inhibition of Bcl-xL, it is envisaged that the development of synthetic agents capable of disrupting the interaction between Mcl-1 and the BH3 α-helical “death” domains of pro-apoptotic Bcl-2 proteins will “neutralize” Mcl-1’s cell survival role. Indeed, several groups have implemented this stratagem and successfully developed effective inhibitors of Mcl-1.26,28-40 We herein report a structure-based design approach that has led to the discovery of potent inhibitors of Mcl-1 based on a novel 1-hydroxy-2-naphthoate scaffold.
Design
Fesik and co-workers recently reported the identification of potent and selective inhibitors of Mcl-1 through fragment-based drug design (FBDD).28 Their inhibitor design, for example 1 (Scheme 1), encompasses a hydrophobic bi-aryl scaffold, projected from which is a carboxylic acid that recognizes R263 (bound by D67 of Bim-BH3), and also a hydrophobic linker and “tail” that probes into the p2 pocket (bound by L62 of Bim-BH3). Meanwhile, researchers at AbbVie, discovered 5-substituted salicylates, such as 2, as potent inhibitors of Mcl-1 wherein the carboxylic acid also binds R263 and the 5-substituent delves into the p2 and/or p1 pockets.37 Inspired by these reports, we considered that merging the two scaffolds to afford a 1-hydroxy-2-naphthoic acid core would provide a novel and alternative platform from which to inhibit Mcl-1, wherein the carboxylic acid was predicted to bind R263, and the distal phenyl ring towards and/or in the p3 pocket. Further engineering to gain access to the p2 pocket was driven by the hydroxyl group and the carboxylic acid whose ortho- and meta-directing effects, respectively, synergize to promote the regioselective 4-chlorosulfonylation of the scaffold;41 subsequent amination of this readily introduced functional group is expected to facilitate occupancy of the p2 pocket. In addition, although not discussed by Abbvie, we reasoned that the hydroxyl group of their salicylate moiety might promote cell permeability through an intramolecular hydrogen bond that serves to mask the negative charge of the carboxylic acid. Whilst this work was in progress, similar small-molecules derived from 2-((1-hydroxynaphthalen-2-yl)thio)acetic acid were reported to inhibit Mcl-1,38,42 although the present work differs in that the bicycle core is unique and the synthetic route to access target molecules is simpler and shorter (Scheme 2).
Scheme 1.

Structure-based design of novel 1-hydroxy-4-sulfamoyl-2-naphthoates as Mcl-1 inhibitors.
Scheme 2.
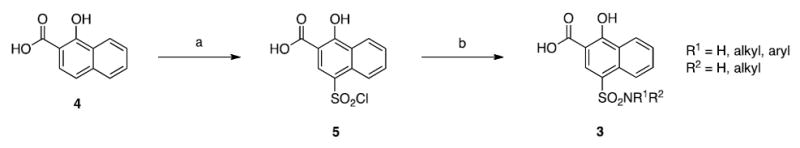
(a) ClSO3H, 0 °C, 1 h; (b) ArNH2, pyr, acetone, 50 °C, 3 h or R1NH2/R1R2NH, DIPEA, acetone, RT, 5 h.
To facilitate ligand design, the Site Identification by Ligand Competitive Saturation (SILCS) method43-46 was used. SILCS FragMaps represent the 3D free energy functional group requirements for different types of functional groups in and around the protein. The FragMaps include contributions from solute-protein interactions and both solute and protein desolvation in the context of protein flexibility.44,45 In addition, to assure that all regions accessible to solute atoms can be occupied by the studied ligands, the protein surface is defined based on SILCS exclusion maps.47 Notably, the information content of SILCS may be used to qualitatively direct ligand design as well as to rapidly make quantitative estimates of relative ligand affinities.48
As motivation for the present ligand design was in part based on the compound reported by Fesik and coworkers (1), initial analysis of the SILCS maps on Mcl-1 was performed with respect to this compound. Figure 1 shows the generic FragMaps overlaid on Mcl-1 along with the crystallographic and docked orientations of 1. The docked orientation closely mimics the crystallographic orientation of the ligand. The ligand occupies nonpolar FragMaps, including a region in the upper left of panel A that is occupied by the chlorine of 1. In addition, a negative (NEG) FragMap (orange) is occupied by the acid moiety of the ligand. While these features largely directed the docked orientation of 1, the inclusion of protein flexibility in the SILCS method is essential. Shown in Figure 1B is a surface representation of the crystal structure of Mcl-1 used to initiate the SILCS simulations onto which the crystallographic orientation of 1 is overlaid. As can be seen, the inhibitor structure overlaps with the protein surface, such that if the protein surface from the crystal structure was used for docking, it would not be possible to attain the experimental binding orientation. However, as the SILCS method includes protein flexibility allowing for solutes in the SILCS simulations to penetrate the initial protein surface the surface may alternatively be defined based on the region not sampled by the solutes or water during the simulations. This allows the protein surface to be defined in terms of an exclusion map that, as shown in Figure 1C, accounts for the structural changes in the protein required to properly dock 1. A notable feature of the bound orientation of 1 and the SILCS FragMaps is the pocket into which the 4-chloro-3,5-dimethylphenyl moiety binds. This is occupied by a nonpolar FragMap, which extends beyond the 4-chloro-3,5-dimethylphenyl ring, suggesting that larger nonpolar groups could facilitate binding in that region consistent with the ligand design strategy.
Figure 1.

(A) Predicted binding orientation (cyan carbons) of 1 overlaid on its crystal orientation (pink carbons). FragMaps are shown as wire mesh in the following colours and GFE cutoff for generic nonpolar (APOLAR, green, −1.2 kcal/mol), neutral donor (HBDON, blue, −1.0 kcal/mol), neutral acceptor (HBACC, red, −1.0 kcal/mol), and negative acceptor (NEG, orange, -1.3 kcal/mol). (B & C) The crystallographic orientation of the inhibitor 1 (from PDB ID: 4HW3) overlaid (B) on the crystal protein surface of the Mcl-1-BH3 peptide complex structure (PDB ID: 4HW4) used to initiate the SILCS simulations and (C) on the SILCS exclusion map.
Results and Discussion
Molecular modeling and SILCS functional group affinity mapping (FragMaps)43-46 of the Mcl-1 binding site indicated that the carboxylic acid of designed molecule 3a (Figure 2) would occupy an energetically favourable region indicated by a negative FragMap, associated with a salt bridge interaction with R263, while the distal ring of the naphthyl core is close to the p3 pocket demarcated by a favourable non-polar FragMap, consistent with the published studies.27,37,38 As anticipated, the aniline was directed into the hydrophobic p2 pocket, which is also demarcated by a nonpolar FragMap. With the molecular modeling data in hand, compound 3a was then synthesized according to Scheme 2. Briefly, commercially available 1-hydroxy-2-naphthoic acid (4) was regioselectively 4-chlorosulfonylated to yield 5, which was isolated by pouring over ice and used without further purification. Sulfonyl chloride 5 was next reacted with 4-bromoaniline to furnish the target molecule 3a in excellent overall yield (83%). This convenient and simple 2-step synthesis allowed us to access the range of ligands presented below, resulting in the discovery of a low-nanomolar binder, and is in sharp contrast to the more hazardous chemistry27 or lengthier synthesis of other families of Mcl-1 inhibitors.38
Figure 2.
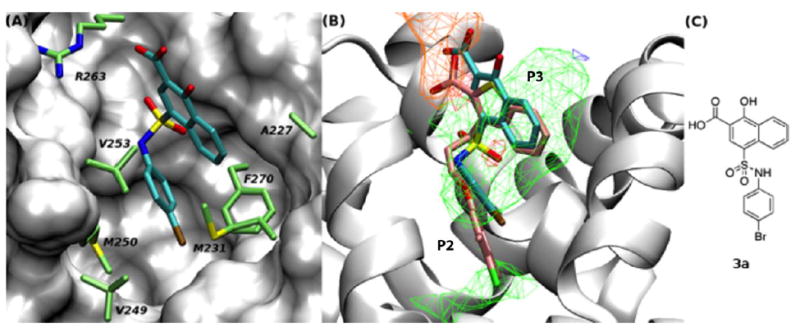
(A) The binding mode of 3a, as predicted by SILCS. Important residues within the p2 binding pocket are shown in stick representation with the carbon atoms coloured in green. (B) Compound 3a and 2 are shown in stick representation with the carbon atoms coloured in cyan and pink, respectively. Colours and cutoffs used to show the FragMaps are the same as in Figure 1. (C) 2D chemical structure of 3a.
Evaluation of 3a in a fluorescence polarization competition assay (FPCA)49 indicated that it disrupted the Mcl-1–Bak-BH3 PPI with an IC50 of 10.9 μM, corresponding to a Ki of 2.76 μM. Given the ability of 3a to inhibit Mcl-1, we embarked on a structure-activity relationship (SAR) campaign, the results of which are presented in Tables 1-3. The target molecules in Table 1 were all prepared according to the concise synthetic route depicted in Scheme 2, those in Table 2 were synthesized using anilines prepared (or purchased) as described in Scheme 3, whilst those in Table 3 were generated by following the synthetic route in Scheme 4. SAR model development was based on visual inspection of predicted binding orientations based on the SILCS modeling, with quantitative analysis based on the SILCS ligand grid free energy (LGFE) scores, as described in more detail below.
Table 1.
Experimental and computational Mcl-1 and Bcl-xL inhibitory profiles of first generation inhibitors. Experimental data determined by a fluorescence polarization competition assay.
| Compound | R1 | R2 | Mcl-1 Ki (μM)a |
Bcl-xL Ki (μM)a |
Selectivity | Mcl-1 LGFE (kcal mol-1) |
Bcl-xL LGFE (kcal mol-1) |
|---|---|---|---|---|---|---|---|
| 3a | 4-Br-C6H4 | H | 2.76 ± 1.26 | 51.4 ± 31.3 | 19 | -39 | -36 |
| 3b | H | H | 99.0 ± 11.2 | NA | -31 | ||
| 3c | Bn | H | 56.3 ± 2.1 | NA | -39 | ||
| 3d | CH2-(2-Cl-C6H4) | H | 50.3 ± 11.8 | - | - | -37 | |
| 3e | CH2-(3-Cl-C6H4) | H | 5.03 ± 2.78 | - | - | -39 | |
| 3f | CH2-(4-Cl-C6H4) | H | 21.0 ± 3.9 | 338 ± 203 | 16 | -41 | -39 |
| 3g | CH2-C6H11 | H | 7.31 ± 1.42 | 77.6 ± 14.3 | 11 | -42 | -41 |
| 3h | Me | Me | 25.7 ± 4.86 | NA | -36 | ||
| 3i | -CH2CH2CH2CH2CH2- | 2.95 ± 0.43 | - | -43 | |||
| 3j | -CH2CH2N(Ph)CH2CH2- | 4.54 ± 3.86 | - | -45 | |||
| 3k | -CH2CH2N(Bn)CH2CH2- | 3.41 ± 0.37 | - | -34 | |||
| 3l | Ph | H | 106 ± 26 | NA | -37 | ||
| 3m | 2-Br-C6H5 | H | 6.84 ± 2.23 | - | -37 | ||
| 3n | 3-Br-C6H5 | H | 5.64 ± 1.76 | - | -39 | ||
| 3o | 2,4-di-Br-C6H4 | H | 0.420 ± 0.163 | 2.31 ± 1.35 | 5.5 | -39 | -38 |
| 3p | 1-Naphthyl | H | 11.2 ± 2.0 | 110 ± 45 | 10 | -40 | -38 |
| 3q | 2-Naphthyl | H | 4.58 ± 1.56 | 68.5 ± 36.3 | 15 | -39 | -37 |
| 3r | 2-Ph-C6H4 | H | 8.50 ± 1.34 | - | -43 | ||
| 3s | 3-Ph-C6H4 | H | 1.88 ± 0.62 | -41 | |||
| 3t | 4-Ph-C6H4 | H | 1.54 ± 0.46 | - | -44 | ||
| 3u | 2-CF3-C6H4 | H | 4.05 ± 2.11 | 19.3 ± 9.0 | 4.8 | -42 | -41 |
| 3v | 4-Cl-C6H4 | H | 1.91 ± 0.26 | - | -40 | ||
| 3w | 4-CF3-C6H4 | H | 2.50 ± 0.65 | - | -43 | ||
| 3x | 4-Me-C6H4 | H | 34.3 ± 1.0 | - | -39 | ||
| 3y | 4-(iPr)-C6H4 | H | 3.86 ± 1.53 | - | -42 | ||
| 3z | 4-OMe-C6H4 | H | 11.9 ± 2.5 | 71.9 ± 21.0 | 6 | -38 | -36 |
| 3aa | 4-(OiPr)-C6H4 | H | 54.0 ± 4.6 | NA | -41 | ||
| 3ab | 4-CN-C6H4 | H | 22.2 ± 1.6 | - | -37 | ||
| 3ac | 4-NO2-C6H4 | H | 4.49 ± 1.20 | 23.3 ± 2.5 | 5.2 | -39 | -38 |
| 3ad | 3-CN-C6H4 | H | 26.9 ± 0.6 | -36 | |||
Ki values determined by Nikolovska-Coleska equation from IC50 values.50 Data represent the average of at least two independent assays; errors are standard deviations.
Selectivity is defined as the Ki (Bcl-xL) divided by the Ki (Mcl-1). NA, no activity.
Table 3.
Investigation into the significance of the inhibitor’s carboxylic acid and hydroxyl group towards the inhibition of Mcl-1. Experimental data determined by a fluorescence polarization competition assay.

| Compound | X | Y | Mcl-1 Ki (μM)a |
Mcl-1 LGFE (kcal mol-1) |
|---|---|---|---|---|
| 3ca | CO2H | OH | 0.566 ± 0.031 | -42 |
| 10 | CO2Me | OH | >500 | -34 |
| 11 | CO2Me | OMe | >500 | -35 |
| 12 | CO2H | OMe | 23.4 ± 2.6 | -42 |
Ki values determined by Nikolovska-Coleska equation from IC50 values.50 Data represent the average of at least two independent assays; errors are standard deviations.
Table 2.
Experimental and computational Mcl-1 and Bcl-xL inhibitory profiles of second-generation inhibitors. Experimental data determined by a fluorescence polarization competition assay.

| Compound | R2 | R3 | Mcl-1 Ki (μM)a |
Bcl-xL Ki (μM)a |
Selectivityb | Mcl-1 LGFE (kcal mol-1) |
Bcl-xL LGFE (kcal mol-1) |
|---|---|---|---|---|---|---|---|
| 3ba | iBu | iPr | 0.487 ± 0.035 | 6.08 ± 0.81 | 12 | -39 | -36 |
| 3bb | H | Ph | 1.15 ± 0.28 | ND | - | -44 | |
| 3bc | H | 4-Me-C6H4 | 0.335 ± 0.027 | 0.84 ± 0.05 | 2.5 | -44 | -43 |
| 3bd | H | 1-Naphthyl | 0.082 ± 0.012 | 0.22 ± 0.06 | 3.7 | -47 | -48 |
| 3be | H | 3-Br-C6H4 | 0.114 ± 0.017 | 0.30 ± 0.04 | 3.8 | -45 | -44 |
| 3bf | H | 3,5-di-Me-C6H3 | 0.284 ± 0.125 | 0.39 ± 0.10 | 1.4 | -46 | -46 |
| 3bg | H | 2,4-di-Cl-C6H3 | 0.079 ± 0.010 | 0.19 ± 0.02 | 2.4 | -47 | -46 |
| 3bh | H | 4-Cl-C6H4 | 0.173 ± 0.070 | ND | - | -46 | |
| 3bi | H | 4-Cl-3,5-di-Me-C6H2 | 0.117 ± 0.060 | 0.48 ± 0.05 | 4.1 | -47 | -47 |
| 3bj | iBu | 4-Cl-3,5-di-Me-C6H2 | 0.080 ± 0.019 | 0.17 ± 0.06 | 2.1 | -48 | -47 |
| 3bk | Cp | 4-Cl-3,5-di-Me-C6H2 | 0.033 ± 0.025 | 0.29 ± 0.05 | 8.8 | -40 | -39 |
| 3bl | Bn | 4-Cl-3,5-di-Me-C6H2 | 0.031 ± 0.017 | 0.34 ± 0.13 | 11 | -52 | -51 |
Ki values determined by Nikolovska-Coleska equation from IC50 values.50 Data represent the average of at least two independent assays; errors are standard deviations.
Selectivity is defined as the Ki (Bcl-xL) divided by the Ki (Mcl-1). ND, not determined.
Scheme 3.
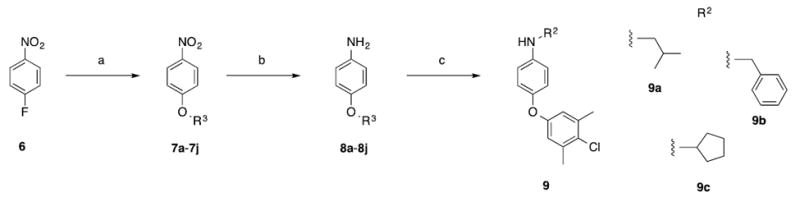
(a) ArOH, K2CO3, 100 °C, 16 h; (b) SnCl2•2H2O, EtOAc, 50 °C, 16 h; (c) isobutyraldehyde, benzaldehyde or cyclopentanone, NaBH(OAc)3, 1,2-dichloroethane, RT, 16 h.
Scheme 4.
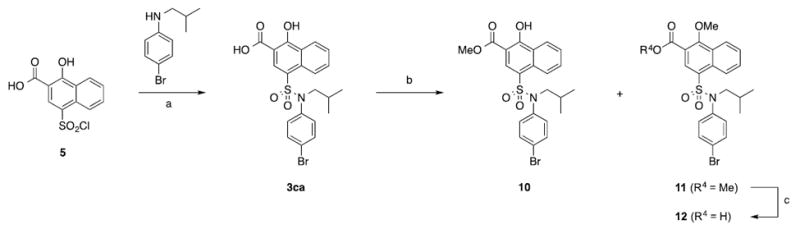
(a) ArNH2, pyr, acetone, 50 °C, 3 h; (b) MeI, K2CO3, DMF, rt, 16 h; (c) NaOH, MeOH/THF/H2O, 3:1:1, rt, 16 h.
Unsubstituted sulfonamide 3b was the weakest Mcl-1 inhibitor with a Ki of 99.0 μM, consistent with the less favourable LGFE score as compared to 1 (Table 1, see supporting information). Replacement of the NH2 group with benzylic amines and anilines improved Mcl-1 inhibitory activity in every case, which is attributed to the occupancy of the apolar FragMaps with the apolar FragMap in the p2 pocket yielding more favourable LGFE scores. In addition, the FragMaps extend further towards the interior of the protein, a region partially occupied by the 4-chloro-3,5-dimethylphenyl of 2, suggesting that further hydrophobic extension of benzylic amines would improve activity. Indeed, chlorobenzylic amine derivatives 3d–3f were more potent than unsubstituted benzylic amine derivative 3c by up to 8-fold. Replacement of the benzyl group in 3c with a cyclohexylmethyl group (3g) also afforded an 8-fold improvement in inhibitory activity with a Ki of 7.31 μM. Piperidine derivative 3i achieved good inhibition (Ki = 2.95 μM), as did the neutral and basic piperazines 3j and 3k, respectively, indicating a positively-charged group here is not detrimental to binding. Substitution of the aniline ring in 3l with bromine atoms afforded between 10 to 54-fold improvement in activity, with the most profound effect observed with para-substituted derivative 3a (Ki = 2.76 μM cf 83.8 μM for 3l). Introduction of an additional bromine atom into the ortho position of 3a to afford 2,4-dibromo derivative 3o led to an even more potent inhibitor with a Ki of 420 nM. Concomitantly, however, this change also led to an erosion in selectivity of more than ten-fold for Mcl-1 over Bcl-xL. Naphthalene derivatives 3p and 3q were also more active than unsubstituted 3l. Functionalization of the aniline ring in 3l with additional phenyl rings to afford biphenyls 3r–3t resulted in improved inhibition in each case due to occupancy of the apolar map adjacent to residues in the p2 pocket, which include V249 and M250, as well as residues in a non-peptide-binding sub-pocket (A227 and M231) in the vicinity of the p3 pocket. Once again, the para-substituted derivative 3u proved the most potent of the series (Ki = 1.54 μM). It is noteworthy that this sub-pocket was recently shown to be important in the recognition of Fesik’s inhibitor 1.27 For the remainder of the compounds in Table 1, substitution of the aniline ring in 3l with hydrophobic and polar groups resulted in enhanced activities in all instances with the greatest improvements observed with hydrophobic groups in the para position.
Motivated by the improved activity of extended hydrophobic substituents on the sulfonamide phenyl ring (e.g. 3o and 3s) along with the extended apolar FragMap in the p2 pocket (Figure 2b), we chose to expand our library of para-substituted anilines by introduction of various aryloxy groups into the para position. Requisite anilines were synthesized according to Scheme 3, and then coupled to sulfonyl chloride 5 according to Scheme 2. As shown in Table 2, compounds 3ba–3bi afforded potent inhibition of Mcl-1 with the tightest binder delivering a Ki of 79 nM. This ~1000-fold enhancement in inhibitory activity relative to unsubstituted aniline 3l is associated with the increased overlap of the nonpolar groups with the second apolar FragMaps in the p2 pocket, as quantified by the systematically more favourable LGFE scores (Table 2). Simultaneously, these modifications also greatly increased affinity of the compounds to Bcl-xL, which is associated with a change in the predicted binding orientation with Bcl-xL as discussed below. The sulfonamide NH group in 3bi offers an additional position from which further inhibitory activity might be acquired associated with occupancy of the apolar FragMap in the vicinity of residue V253, as shown in Figure 2B for 3a. Indeed, alkylation of this nitrogen atom with cyclopentyl (3bk) and benzyl (3bl) resulted in our most potent inhibitors yet with Ki’s of 33 nM and 31 nM, respectively. Pleasingly, these alkylations also led to a recovery of selectivity for Mcl-1 over Bcl-xL of up to eleven-fold.
Finally, we wanted to verify the importance of the carboxylic acid and hydroxyl groups of the inhibitor scaffold. Analysis of Figure 2B showing overlap of the negative FragMaps with the acid moiety indicate the importance of that group to binding while analysis of the acceptor FragMaps at a lower contour level of -0.3 kcal/mol (not shown) indicated small, but favourable contributions from the alcohol moiety. Accordingly, we synthesized the compounds shown in Scheme 4. As seen in Table 3, the carboxylic acid is critical to activity, which is consistent with its likelihood of forming a salt bridge with R263. Methylation of the hydroxyl group to deliver compound 12 resulted in around a 40-fold reduction in activity indicating its importance, too. Although SILCS predicted the methoxy derivative 12 (LGFE = -42 kcal/mol) to bind with similar affinity than its hydroxy counterpart 3ca (LGFE = -42 kcal/mol), the observed disparity may be explained by an anticipated greater acidity for the latter through a six-membered intramolecular hydrogen bond between the hydroxyl and the carboxylate that affords a stronger salt bridge with R263.
NMR HSQC experiments using 15N-labeled Mcl-1172-327 (recombinant human Mcl-1 residues 172 to 327) confirmed the direct interaction of this class of compounds with the protein. Specifically, the HSQC spectra of 15N-labeled Mcl-1172-327 (Figure 3: black = Mcl-1; red = Mcl-1 and inhibitor) shows the addition of compound 3ba causes perturbations in the chemical shifts or loss of Mcl-1 backbone 1H-15N correlations due to chemical exchange broadening in amino acids consistent with the predicted binding mode of 3a shown in Figure 2. When the residues that are perturbed or lost due to chemical exchange broadening are mapped onto the 3D structure of Mcl-1 (Figure 4) their positions support the model shown in Figure 2. The majority of the changes occur in residues near the predicted binding mode of the compound including R263 and N260, and residues in the p2 pocket (V249, M250, and F270). There are also chemical shift perturbations near the p3 pocket (A227 and M231), although these may be associated with indirect effects. The residues undergoing chemical shift changes are also consistent with the X-ray crystal structure of 1 bound to Mcl-1 (PDB ID: 4HW3)28 with most changes located on the end of helix 2 (residues 204 to 224) and in most of helices 3 (residues 226 to 236), 4 (residues 239 to 255), and 5 (residues 260 to 280) that form the BH3-binding groove.
Figure 3.
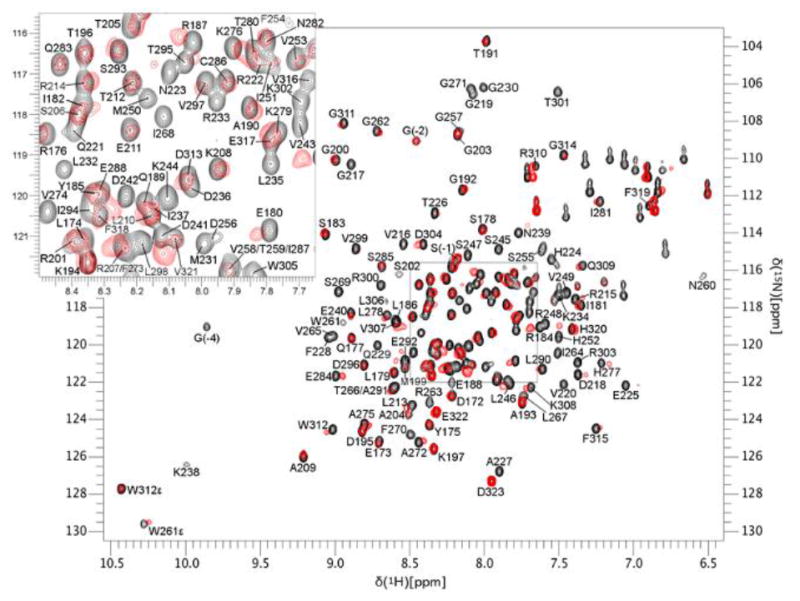
The NMR HSQC spectra shows chemical shift perturbations caused by the direct interaction of compound 3ba with Mcl-1. The spectrum of the control sample (black) is overlaid with the spectrum of Mcl-1 bound to 3ba (red). Inset is enlargement of boxed region.
Figure 4.
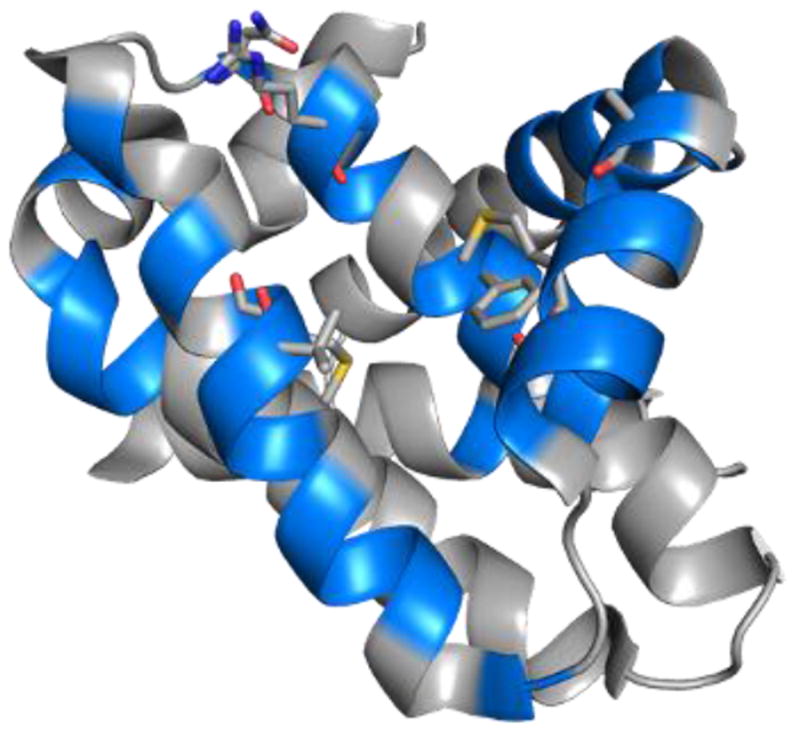
Mcl-1 residues (blue) whose chemical shifts are perturbed or lost in the presence of compound 3ba.
Comprehensive SILCS SAR analysis of Mcl-1 binding
Additional SILCS based modeling was undertaken to predict the bound orientation of the active ligands, to quantitatively predict the relative affinities of the ligands as well as the contributions of the different regions of the ligands to binding to Mcl-1 and Bcl-xL and to develop a quantitative model of differential binding of the compounds to Mcl-1 versus Bcl-xL. This information would be of utility in designing ligands with improved potency and selectivity for Mcl-1. Thus, subsequent modeling involved docking of each studied compound using the SILCS-Pharm approach as described in the supporting information (Figure S1).46,47 The docked orientation of each compound was then subjected to MC-SILCS sampling to allow the molecules to conformationally sample the local binding region as defined by the FragMaps and the exclusion maps, with the LGFE scores calculated from the MC SILCS conformational sampling. Comparison of the LGFE values for all the tested compounds with the experimental affinities converted to free energies (ΔG = -RTlnKi, where R is the Boltzmann constant and T is the temperature) for Mcl-1 yields a correlation of R2 = 0.53 and a high predictive index51 of 0.70, (Figure S2, supporting information). Thus, the LGFE scores correlate with the Mcl-1 experimental data, indicating the quality of the bound orientations from the SILCS-Pharm-MC-SILCS protocol.45-47
Given the predictive capabilities of the LGFE scores, further analysis was undertaken to use the SILCS modeling to further interpret the experimental SAR data. This analysis focused on four compounds: 3b, 3a, 3bi and 3bl. These compounds differ in the number of phenyl rings attached to the sulfonamide moiety going from 0 to 3 rings, in that order, with the binding affinities varying from 99 μM down to 31 nM. For these compounds a correlation of R2 = 0.99 between the LGFE and experimental G values is obtained.
The predicted conformation of the four compounds is presented in Figure 5 along with the crystallographic orientation of 1.28 For all four compounds, the carboxylic acid overlaps with the negative FragMap (orange arrows), associated with its interaction with R263 of Mcl-1 and the aromatic rings suitably overlap with hydrophobic FragMaps (green arrows), binding within the hydrophobic groove. For 3b, the sulfonamide group is placed outside of the hydrophobic binding pocket corresponding to an H-bond donor FragMap (blue arrow in (A)) indicating its possible H-bonding interaction with residue H224. The hydroxyl group on the naphthalene ring overlaps with an H-bond acceptor FragMap (red arrow in (A)), possibly making an H-bond to residue T266. For the other three compounds, the naphthalene group flipped over relative to 3b, maintaining the position of the carboxylic acid while allowing the additional aromatic groups to interact with the hydrophobic binding pocket, consistent with that observed with 1. Notably the sulfonamide oxygen atoms in this orientation overlap with an acceptor FragMap associated with interactions with residue T266 (red arrows).
Figure 5.

The orientations of the four compounds binding to Mcl-1 predicted by SILCS. (A) 3b; (B) 3a; (C) 3bi; (D) 3bl. Compounds shown in stick format with atom-based colouring. The compound, 2, from the previously reported complex structure 4HW3 is also shown, with the carbon atoms coloured in pink. Mcl-1 residue R263 is shown. Same colour and GFE cutoff is used as in Figure 1.
Additional analysis of the contribution of individual atoms and functional groups on the compounds to binding was performed by analyzing the atom-based GFE contributions of the individual atoms to the overall LGFEs. Presented in Figure 6 are the atom-based GFE contributions for the most favourable binding conformations of the four compounds. For all compounds, the naphthyl and acid moieties make favourable contributions to binding. With 3b, significant favourable contributions occur with both the sulfonamide and hydroxyl moieties. In the remaining three compounds with the naphthyl moiety flipped; the sulfonamide and hydroxyl moieties still make favourable contributions to binding though the magnitude is generally less than that of 3b. For example, while some of the sulfonamide oxygens make more favourable contributions with the larger compounds, the amide NH makes a very unfavourable contribution. However, the less favourable interactions of the hydroxyl and sulfonamide moieties are overcome by favourable contributions from the additional aromatic groups, which lead to the improved binding of the larger inhibitors. Interestingly, the quite favourable contributions from the second phenyl ring coming off the sulfonamide at the R2 position was counter-balanced by decreased contributions from the aromatics group at the R1 position upon going from 3bi to 3bl. This is consistent with the experimental free energy of binding changing by only -0.8 kcal/mol versus 3bi while differences going from 3b to 3a and 3a to 3bi are -2.1 and -1.9 kcal/mol, respectively. This atomic detailed interpretation of the experimental SAR data is anticipated to facilitate further improvements in the compounds.
Figure 6.

Atom GFE contributions for the most LGFE favourable binding conformation of the four compounds. (A) 3b; (B) 3a; (C) 3bi; (D) 3bl.
SILCS quantitatively captures the binding specificity of compounds for Mcl-1 over Bcl-xL
A long-standing challenge in targeting the anti-apoptotic Bcl-2 proteins is achieving family member specificity, particularly Mcl-1 specificity, although selective ligands are beginning to emerge.5,6,39,52 To investigate the binding specificity of compounds for Mcl-1 versus Bcl-xL, SILCS simulations and GFE FragMap generation followed by LGFE scoring were also done for Bcl-xL (Table S1 of the supporting information). A correlation of R2 = 0.58 and a high predictive index of 0.76 was found between the LGFE and experimental binding data for Bcl-xL (Figure S3 of the supporting information). Difference FragMaps, ΔFragMaps, were then calculated as GFEBcl-xl – GFEMcl-1 such that positive ΔFragMaps favour Mcl-1 over Bcl-xL. The ΔFragMaps may then be used to qualitatively understand the contributions driving specificity as well as quantitatively obtain difference LGFE scores (ΔLGFE). Correlation analysis between the ΔLGFE scores and the experimental differences in the binding affinities for Mcl-1 and Bcl-xL yielded an R2 = 0.57 and predictive index of 0.83 (Figure S4 in supporting information), indicating the quality of the SILCS ΔFragMaps in modeling the relative affinities for Mcl-1 versus Bcl-xL. Additional analysis was therefore undertaken on 3a as this compound showed the largest specificity for Mcl-1 over Bcl-xL. Figure 7A shows the ΔFragMaps between Mcl-1 and Bcl-xL, and includes the docked orientation of 3a with the atom contributions to the ΔLGFE between Mcl-1 and Bcl-xL mapped onto this orientation. The p-bromophenyl group (green arrow) that binds deeply in the p2 pocket between helices α4 and α5 is in a Mcl-1 positive ΔFragMap indicating that region to favour binding to Mcl-1. Similarly, the sulfonamide oxygen (red arrow) contributes to the binding of 3a to Mcl-1 over Bcl-xL. Alternatively, the NH moiety of the sulfonamide contributes to more favourable binding to Bcl-xL over Mcl-1. The acid moiety in the lower right of the figure is also predicted to favour binding to Bcl-xL over Mcl-1. Summing over the different ΔFragMap classes showed the overall ΔLGFE score of 3.0 kcal/mol favouring binding to Mcl-1 to have favourable contributions from the hydrophobic and neutral acceptor terms, 4.4 and 1.6 kcal/mol, respectively, while the neutral donor, negatively charged acceptor and hydroxyl groups favour Bcl-xL binding, with values of -1.8, -0.7 and -0.5 kcal/mol, respectively. Similar trends are observed for the other compounds for which a selectivity of Mcl-1 over Bcl-xL was found, as shown in Table S2 in the supporting information. Indeed, given that all the tested compounds favoured Mcl-1 binding over Bcl-xL, these results indicate that the exploitation of the binding pocket inherently favours Mcl-1 binding, as previously discussed by Fesik.28 The docked orientation of 3a to the two proteins in Figure 7B shows that this pocket is not being exploited with Bcl-xL. This is consistent with the fact that Mcl-1 shows more opening at the p2 pocket as compared to Bcl-xL as illustrated in Figure 7B,28 disallowing access of 3a deep into the hydrophobic pocket in the p2 site of Bcl-xL.
Figure 7.

(A) Difference FragMaps (ΔFragMaps) between Mcl-1 and Bcl-xL showing the favourable binding patterns for Mcl-1. Hydrophobic and neutral donor ΔFragMaps are shown in green and blue, respectively. The atom GFE contributions to the binding affinity difference are also shown. (B) Experimental structures of Mcl-1 (blue, 4HW4) and Bcl-xL (red, 1BXL) shows that the binding pocket between helix α4 (upper right) and α5 (bottom left) is larger for Mcl-1 than Bcl-xL. The predicted binding modes of 3a for Mcl-1 (blue) and Bcl-xL (red) differs due to this p2 pocket difference. The 4-bromophenyl ring of 3a binds deeply to the opened pocket for Mcl-1 while it binds on the protein surface for Bcl-xL. (C) Binding orientations of 3bl to Mcl-1 (blue, 4HW4) and Bcl-xL (red, 1BXL) along with the ΔFragMaps. (D) Binding orientations of 3bl to Bcl-xL (1BXL) along with the Bcl-xL APOLAR and NEG FragMaps at the contour levels specified in Figure 1.
However, further extension of the ligands (e.g. 3bl) leads to improved binding affinity, but not improved specificity. This may be explained by different predicted binding orientations of the compound to the two proteins. As seen in Figure 7C, 3bl binds with its additional 4-chloro-3,5-dimethylphenyl R3 group deep in the p2 pocket of Mcl-1, and with the R2 benzyl group occupying the upper region of the p2 pocket. In contrast, the orientation of 3bl bound to Bcl-xL shows the 4-chloro-3,5-dimethylphenyl still interacting with the p2 site, though not as deep as that occurring with Mcl-1. This leads to a shift in the location of the naphthyl ring as well as the R2 phenyl group into the p3 site where they can exploit the hydrophobic character of that sub-pocket as well as interactions with R263, as indicated by the apolar and negative Bcl-xL FragMaps in Figure 7D, respectively. Thus, the significant increase in the size of 3bl leads to increased affinity with respect to both Mcl-1 and Bcl-xL, with that larger size predicted to change the binding orientation with Bcl-xL such that further increased specificity for Mcl-1 is not gained over 3a.
Cellular activity of Mcl-1 inhibitors
To investigate the cellular activity of our Mcl-1 inhibitors, the most potent compound 3bl was evaluated for its ability to inhibit the proliferation of A375 and SK-MEL-5 melanoma cells, both of which express high levels of Mcl-1.53 Unfortunately, the IC50 for 3bl was 50 μM and 90 μM in A375 and SK-MEL-5 cells, respectively (Table 4). We attributed the poor efficacy in cells to the charged carboxylic acid of 3bl. To test this hypothesis, we prepared two ester prodrugs of the potent, but more hydrophilic, inhibitor 3ba, methyl ester 13 and acetoxymethyl ester 14, according to Scheme 3. As expected, both prodrugs 13 and 14 demonstrated no binding affinity to Mcl-1 in vitro. (data not shown). Naphthoate derivative 3ba exhibited worse activity than 3bl in cells, consistent with its weaker affinity to Mcl-1 (Table 3). However, the acetoxymethyl ester 14 displayed an IC50 of 15 μM in both A375 and SK-MEL-5 cells, presumably due to improved cell penetration and then intracellular hydrolysis to the active metabolite 3ba (Table 4). We postulate that the lack of cellular activity for methyl ester 13 might be due to limited hydrolysis to 3ba, as we have observed elsewhere.54 These results suggest a path forward with respect to rationally improving the cell penetration capabilities of this class of compounds based on a prodrug strategy.
Table 4.
Inhibition of cell proliferation by select compounds, as determined by a cell titer blue assay.
| Compound | IC50 (μM) | |
|---|---|---|
| A375 | SK-MEL-5 | |
| 3a | ~300 | ~300 |
| 3bl | 50 | 50 |
| 3ba | 150 | ~300 |
| 13 | ~300 | ~300 |
| 14 | 15 | 15 |
Conclusions
Presented is a novel class of Mcl-1 inhibitors that target R263 and the p2 and p3 pockets within the hydrophobic BH3-binding crevice on the surface of the protein. Inhibitor design was motivated by compounds reported by Fesik and coworkers and by AbbVie that are known to exploit the p2 pocket and interactions with R263. SILCS analysis of this region of the BH3 binding site supported this strategy, with the use of the SILCS exclusion maps indicating significant additional opening of the p2 pocket that could be exploited in ligand design. Accordingly, a series of compounds were designed, synthesized and subjected to experimental evaluation using a FP assay with binding of 3ba to targeted pocket of Mcl-1 verified using NMR. From this strategy, 3bl was identified with a Ki = 31 nM for Mcl-1 with a specificity of 11-fold over Bcl-xL. Although only moderate cell activity was observed with the 2-hydroxynaphthoate-based Mcl-1 inhibitors, this was enhanced by the preparation of an acetoxymethyl ester prodrug of the critical carboxylic acid.
The two-step synthesis developed in this study allowed us to rapidly synthesize and test over 40 analogs, with the selected compounds based, in part, on commercially available starting materials. These synthetic capabilities allowed us to readily synthesize compunds that would be able to reach into and complement the hydrophobic p2 pocket. This yielded compounds with affinities spanning over a 10,000-fold range. Detailed SILCS analysis of all of these compounds quantified the contributions of the different moieties leading to improved affinity. These results suggest that fine tuning of interactions with the p2 pocket via appropriate modifications of the R3 ring (Table 2) as well as of the R1 ring will likely lead to improved affinity. Additional SILCS analysis on the differential binding of selected compounds to Mcl-1 versus Bcl-xL indicates that fine tuning of the R3 ring occupying the p2 pocket may also improve specificity. Further modifications that may contribute to improved specificity include alkylation of the NH moiety of the sulfonamide moiety and additions of both hydrogen bond acceptor (e.g. OMe) or hydrophobic (e.g. Cl) functional groups to the naphthyl ring as indicated by the differential Fragmaps in the top, central region of Figure 7A. Future studies will involve the design of new compounds using the validated SILCS affinity and specificity models for quantitative analysis of synthetically accessible compounds.
Experimental
Computational
Molecular modeling studies were initiated with the crystal structure of the Mcl-1-BH3 peptide complex (PDB ID: 4HW4),28 following removal of the 16mer BH3 peptide, and with the Bcl-xL-Bak complex structure (PDB ID: 1BXL), following removal of the Bak peptide. For both proteins, the Reduce software55 was used to choose optimal Asn, Gln, and His side-chain orientations and determine the optimal protonation states of His residues. The protein was immersed in a box of an aqueous solution containing eight small probe molecules at approximately 0.25 M each with water at ~55 M. The size of the simulation box was chosen so as to have the protein extrema separated from the edges by a minimum of 8 Å based on non-hydrogen atoms. The small molecules, or solutes, include benzene, propane, methanol, formamide, acetaldehyde, methylammonium and acetate, as previously used,45 along with imidazole. Ten such protein-small molecule aqueous systems were generated for each protein with each simulation system differing in the initial positions and orientations of the small molecules and water. The SILCS43-46 molecular dynamics (MD) simulations were performed using the GROMACS56 simulation program with the CHARMM36 force field,57 CHARMM general force field (CGenFF)58,59 and TIP3P water model60 to describe the protein, small molecules and water, respectively. The simulations were each extended for 40 ns, yielding a total of 400 ns of simulation time for each protein. Additional MD simulation details can be found in Reference 45.45
From the simulation, 3D probability distributions of selected atoms from the small molecules were constructed to form the FragMaps, yielding a total of ten different FragMaps. The final FragMaps are converted to free energies, termed grid free energies (GFE), by normalizing the distributions with respect to the distributions of the solutes in an aqueous solution in the absence of the proteins followed by Boltzmann transformation to yield the GFE values.45 As the GFE FragMaps are normalized with respect to the fragment probabilities in solution, they contain both favourable regions as well as unfavourable regions. The unfavourable regions typically range from 0 to 3 kcal/mol, with the upper limit based on sampling issues. In addition to the FragMaps, exclusion maps were constructed by calculating the 3D probability distributions of all non-hydrogen atoms of the water and solutes together and identifying those voxels with zero occupancies, which defines the exclusion maps. This exclusion map represents regions forbidden to the small molecules and water and may be considered an alternate to more traditional representations of the protein surface. For quantitative analysis, these voxels were assigned a very high energetic penalty (1000 kcal/mol) while the remaining voxels were assigned energies associated with the specific FragMaps. The availability of the GFE FragMaps and the exclusion maps allows for quantitative estimates of binding affinities to be made, referred to as ligand grid free energies (LGFE). These are a simple summation of the GFE energy contribution of all the atoms in each ligand that are classified with respect to the FragMap types followed by normalization of the summed energies by the number of classified ligands atoms and subsequent multiplication by the total number of non-hydrogen atoms, yielding the final LGFE values. Note that the LGFE scores are not directly equivalent to experimental free energies due to the additive approximation of the LGFE scores (i.e. the individual atom-based GFEs are summed to yield the LGFE), the lack of accounting for the energy cost of connecting the fragments that comprise the full compounds and issues associated with the standard state in the experimental and computational conditions.
In the present study the specific FragMaps were used for calculation of GFE and LGFE scores, while generic FragMaps are used for visualization. The specific and generic FragMap types that were used include: (1) aromatic, AROM (benzene carbons); (2) aliphatic, ALIP (propane carbons); (3) dual role hydrogen bonding atom, MEOO (methanol oxygens); (4) FORN, (formamide nitrogen); (5) FORO, (formamide oxygen); (6) IMIN, (imidazole acceptor nitrogen); (7) IMIH, (imidazole donor nitrogen); (8) AALO, (acetaldehyde oxygen); (9) positive donor, POS (methylammonium nitrogen); (10) negative acceptor, NEG (acetate oxygens); (11) generic nonpolar, APOLAR (benzene and propane carbons); (12) generic neutral hydrogen-bond donor, HBDON (formamide and imidazole donor nitrogen); and (13) generic neutral hydrogen-hydrogen acceptor, HBACC (formamide, acetaldehyde oxygens and imidazole acceptor nitrogen). The convergence of the FragMaps was examined by calculating the overlap coefficient between two sections of the simulations (trajectories 1–5 and trajectories 6–10), as previously described.45 All the generic FragMaps show an overlap coefficient of greater than 0.6, indicating satisfactory convergence.
To identify the binding modes of the tested compounds with Mcl-1, the SILCS-Pharm protocol46,47 was used in which the FragMaps are used to define the pharmacophore features. This involved identifying all possible pharmacophore features in the peptide binding pocket based on the region close to inhibitor 2 (PDB ID: 4HW3).28 The selected pharmacophore was used to direct docking of the studied compounds in the Mcl-1 binding pocket. Generation of 250 conformations of each compound and pharmacophore docking were conducted using MOE.61 For each compound, the best conformation, based on the smallest RMSD with the pharmacophore was retained for SILCS ligand grid free energy (LGFE) scoring. The compounds were locally relaxed and minimized using FragMap-based Monte Calro (MC) sampling in the field of the GFE FragMaps (MC-SILCS) from which the LGFE scores were obtained.45 The MC-SILCS was conducted for 50000 steps under slow cooling mode and was repeated 20 times for each conformation with different random seeds to get a Boltzmann averaged LGFE value, along with the corresponding standard deviation. The lowest energy LGFE conformation for each compound was chosen for visual presentation and analysis. The SILCS-MC was also performed in the same way using FragMaps of Bcl-xL to calculate Bcl-xL related LGFEs for the compounds that have available Bcl-xL experimental data. Bcl-xL SILCS-MC calculations were initiated from the Mcl-1 SILCS-PHARM orientations in the context of the Bcl-xL FragMaps and exclusion maps.
Chemistry
General
Unless otherwise stated, all reactions were performed under an inert atmosphere (N2). Reagents and solvents were ACS grade, and purchased from Sigma-Aldrich, Alfa Aesar, Oakwood and TCI America. Anhydrous solvents were used as provided without further purification. Reactions were monitored by thin-layer chromatography (TLC), visualizing with a UV lamp and/or KMnO4 stain. Flash column chromatography was performed with silica gel 60 Å (70-230 mesh, Merck). 1H and 13C NMR spectra were recorded on a Varian INOVA 400 MHz NMR spectrometer at 25 °C. Chemical shifts are reported in parts per million (ppm). Data for 1H NMR are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration), where multiplicities are: s = singlet, d = doublet, t = triplet, sep = septet, m = multiplet. The residual solvent peak was used as an internal reference: CDCl3 (δH 7.26; δC 77.21) and d6-DMSO (δH 2.50; δC 39.51). Mass spectra were obtained on an Electrospray TOF (ESI-TOF) mass spectrometer (Bruker AmaZon X). All final molecules were deemed to be >95% pure by reversed-phased HPLC using a Waters 1525 analytical/preparative HPLC fitted with a C18 reversed-phase column (Atlantis T3: 4.6 mm × 150 mm) according to the following conditions with solvents (A) H2O/0.1% TFA, (B) CH3CN–H2O, 9:1 with 0.1% TFA at 1 ml min-1: (I) a gradient of 100% A to 100% B over 22 min; (II) a gradient of 50% A to 100% B over 22 min; (III) a gradient of 25% A to 100% B over 22 min; (IV) an isocratic gradient of 100% B over 22 min; (V) a gradient of 100% A to 100% B over 22 min, then 100% B for 13 min. Data are presented as retention time (tR (min)), purity (%), condition (I or II).
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5)
1-Hydroxy-2-naphthoic acid (4; 5 g, 26.6 mmol) was added portionwise with stirring over the course of 1 h to chlorosulfonic acid (25 mL) at -10 °C. Once the addition was complete, TLC of the reaction mixture confirmed all starting material had been consumed (Acetone/EtOAc, 1:1). The reaction mixture was carefully poured over ice. The resulting pinkish-grey solid was collected by vacuum filtration, washing several times with ice-cold water. After the product was allowed to dry on the filter for 1 h, it was transferred to a vacuum oven where it was dried further at 50 °C for 16 h.
General Procedure A: Amination with Anilines
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5; 1 eq) was suspended in anhydrous acetone (0.2 M), and then the requisite aniline (1.2 eq) and pyridine (3 eq) were added under an inert (N2) atmosphere. The reaction mixture was stirred at 50 °C for 3 h, by which time TLC (acetone/EtOAc, 1:1) indicated that the reaction was complete. The reaction was concentrated to dryness and then suspended in a 1:1 mixture of EtOAc and 1 M HCl and was vigorously stirred for 5 min. The mixture was transferred to a separatory funnel, then partitioned between EtOAc and 1 M HCl. The organic layer was washed three times with 1 M HCl, dried over Na2SO4, filtered, concentrated to deliver a residue that was purified by column chromatography over silica gel using an eluent of CH2Cl2/MeOH/AcOH 92:7:1 to provide the title compound.
General Procedure B: Amination with Primary Aliphatic Amines
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5; 1 eq) was suspended in anhydrous acetone (0.2 M). The requisite benzylic amine (1.2 eq) and DIPEA (2.5 eq) were added under an inert (N2) atmosphere at room temperature (RT). The reaction was stirred at RT for 16 h. TLC (acetone/EtOAc, 1:1) indicated that reaction was complete. The reaction was concentrated to dryness and then suspended in a 1:1 mixture of EtOAc and 1 M HCl and was vigorously stirred for 5 min. The mixture was transferred to a separatory funnel, then partitioned between EtOAc and 1 M HCl. The organic layer was washed three times with 1 M HCl, dried over Na2SO4, filtered, concentrated to deliver a residue that was purified by column chromatography over silica gel using an eluent of CH2Cl2/MeOH/AcOH 92:7:1 to provide the title compound.
General Procedure C: Amination with Secondary Aliphatic Amines
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5; 1 eq) was suspended in anhydrous acetone (0.2 M), and then the requisite secondary amine (3 eq) was added under an inert (N2) atmosphere at room temperature. After 1 h, TLC (acetone/EtOAc, 1:1) indicated that the reaction was complete. The reaction was concentrated to dryness and then suspended in a 1:1 mixture of EtOAc and 1 M HCl and was vigorously stirred for 5 min. The mixture was transferred to a separatory funnel, then partitioned between EtOAc and 1 M HCl. The organic layer was washed three times with 1 M HCl, dried over Na2SO4, filtered, concentrated to deliver a residue that was purified by column chromatography over silica gel using an eluent of CH2Cl2/MeOH/AcOH 92:7:1 to provide the title compound.
General Procedure D: Nucleophilic aromatic substitution (SNAr)
4-Fluoronitrobenzene (1 eq) and the requisite phenol (1 eq) were dissolved in anhydrous DMSO (0.3 M). K2CO3 (2 eq) was added, and then the reaction was stirred at 120 °C for 16 h. The next day, TLC (Hex/EtOAc, 9:1) indicated the reaction was complete. The reaction was quenched with water and ice, which resulted in precipitation. The precipitate was collected by vacuum filtration and then dried overnight in a vacuum oven at 50 °C to furnish compounds 7 that were sufficiently pure to be advanced to the next step.
General Procedure E: Reduction of Nitro Group with SnCl2.2H2O
The appropriate nitroarene 7 was dissolved in EtOAc (0.1 M), and then SnCl2.2H2O (5 eq) was added. The reaction mixture was stirred at 50 °C for 16 h, by which time TLC confirmed reaction completion. The reaction mixture was partitioned between EtOAc and sat. NaHCO3. The organic layer was collected and the aqueous layer was extracted two times with EtOAc. The organics were combined, washed with sat. NaHCO3, brine, dried with Na2SO4, filtered and concentrated. No further purification was required.
General Procedure F: Reductive Amination
The aniline (1 eq) was dissolved in 1,2-dichloroethane (0.1 M) and the required aldehyde (1 eq) was added, followed by NaBH(OAc)3 (2 eq). The reaction mixture stirred for 16 h at room temperature. TLC (Hex/EtOAc, 1:1) indicated that the reaction was complete. The reaction mixture was diluted with CH2Cl2 and then partitioned with sat. NaHCO3. The aqueous layer was extracted with further CH2Cl2 (x2), and then the organic layers were combined, washed with brine, dried (Na2SO4) filtered and concentrated. The crude material was purified by flash column chromatography over silica gel using an eluent of Hex/EtOAc, 1:1 to provide the title compound.
4-(N-(4-bromophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3a)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-bromoaniline according to General Procedure A on a 1 mmol scale to yield the title compound as an off-white solid (300 mg, 71%): δH (400 MHz, d6-DMSO) 10.47 (s, 1H, SO2NH), 8.48 – 8.45 (m, 2H, Ar), 8.32 (d, 1H, Ar, J = 8.0 Hz), 7.67 (t, 1H, Ar, J = 7.4 Hz), 7.49 (t, 1H, Ar, J = 7.4 Hz), 7.31 (d, 2H, Ph, J = 8.4 Hz), 6.93 (d, 2H, Ph, J = 8.8 Hz); δC (100 MHz, d6-DMSO) 171.0, 138.0, 133.8, 132.2, 131.5, 130.0, 128.1, 125.4, 125.2, 124.5, 120.7, 117.5, 115.3, 107.3, 95.9; Calcd (M+): 421.0, Found: 420.0 ([M-H]-); tR = 8.1 min (100%, III).
1-Hydroxy-4-sulfamoyl-2-naphthoic acid (3b)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5; 287 mg, 1 eq) was suspended in dioxane (5 mL) at 0 °C, then NH4OH (1 mL) was added dropwise. After 1 h, TLC (Acetone/EtOAc 1:1) confirmed the reaction was complete. The reaction mixture was concentrated to dryness, then partitioned between EtOAc (50 mL) and 1 M HCl (25 mL). The aqueous layer was extracted with further EtOAc (50 mL), then the organic layers were washed with brine, dried (Na2SO4), filtered and concentration to afford the title compound light orange solid (160 mg, 60%): δH (400 MHz, d6-DMSO) 8.43 – 8.40 (m, 2H, Ar), 8.34 (d, 1H, Ar, J = 8.4 Hz), 7.60 (t, 1H, Ar, J = 7.4 Hz), 7.46 (t, 1H, Ar, J = 8.0 Hz), 7.13 (s, 2H, SO2NH2); δC (100 MHz, d6-DMSO) 171.3, 170.6, 131.5, 130.6, 128.9, 128.4, 125.3, 125.0, 124.6, 122.6, 107.9; Calcd (M+): 267.0, Found: 266.0 ([M-H]-).
4-(N-Benzylsulfamoyl)-1-hydroxy-2-naphthoic acid (3c)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to benzylamine according to General Procedure B on a 1 mmol scale to yield the title compound as a dark yellow solid (232 mg, 65%): δH (400 MHz, d6-DMSO) 8.45 (d, J = 8.0, 1 H, Ar), 8.43 (s, 1 H, Ar), 8.36 (d, J = 8.0, 1 H, Ar), 7.95 (t, J = 6.4, 1 H, NH), 7.62 (t, J = 8.0, 1 H, Ar), 7.48 (t, J = 8.0, 1 H, Ar), 7.22-7.10 (m, 5 H, Ar), 3.89 (d, J = 6.4, 2 H, CH2); δC (100 MHz, d6-DMSO) 171.2, 138.6, 132.9, 131.7, 129.3, 128.4, 127.8, 127.3, 125.1, 125.0, 124.8, 46.2; Calcd (M+): 357.1, Found: 356.0 ([M-H]-); tR = 11.5 min (98.9%, II).
4-(N-(2-Chlorobenzyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3d)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2-chlorobenzylamine according to General Procedure B on a 1 mmol scale to yield the title compound as a light purple solid (274 mg, 70%): δH (400 MHz, d6-DMSO) 8.45-8.33 (m, 2 H, Ar), 8.35 (d, J = 7.6, 1 H, Ar), 7.99 (t, J = 6.0, 1 H, NH), 7.61 (t, J = 7.6, 1 H, Ar), 7.46 (t, J = 7.6, 1 H, Ar), 7.40-7.37 (m, 1 H, Ar), 7.32-7.29 (m, 1 H, Ar), 7.22-7.17 (m, 2 H, Ar), 3.98 (d, J = 6.0, 2 H, CH2); δC (100 MHz, d6-DMSO) 171.0, 135.7, 133.1, 132.2, 131.7, 129.9, 129.3, 129.1, 128.6, 127.2, 125.1, 125.0, 124.7, 107.8, 43.6; Calcd (M+): 391.0, Found: 414.1 ([M+Na]+); tR = 10.2 min (97.2%, II).
4-(N-(3-Chlorobenzyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3e)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 3-chlorobenzylamine according to General Procedure B on a 1 mmol scale to yield the title compound as a dark purple solid (286 mg, 73%): δH (400 MHz, d6-DMSO) 8.43-8.39 (m, 2 H, Ar), 8.35 (d, J = 7.6, 1 H, Ar), 8.04 (t, J = 6.4, 1 H, NH), 7.63 (t, J = 7.6, 1 H, Ar), 7.47 (t, J = 7.6, 1 H, Ar), 7.21-7.15 (m, 3 H, Ar), 7.13-7.09 (m, 1 H, Ar), 3.91 (d, J = 6.4, 2 H, CH2; δC (100 MHz, d6-DMSO) 171.2, 141.2, 133.1, 132.8, 131.6, 130.1, 129.4, 128.3, 127.6, 127.1, 126.4, 125.1, 125.0, 124.9, 107.6, 45.5; Calcd (M+): 391.0, Found: 414.1 ([M+Na]+); tR = 21.5 min (98.8%, I).
4-(N-(4-Chlorobenzyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3f)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-chlorobenzylamine according to General Procedure B on a 1 mmol scale to yield the title compound as a light brown solid (294 mg, 75%): δH (400 MHz, d6-DMSO) 8.56 (d, 1H, Ar, J = 8.4 Hz), 8.46 (t, 1H, SO2NH, J = 5.8Hz), 8.39 (d, 1H, Ar, J = 8.8 Hz), 8.32 (s, 1H, Ar), 7.84 (t, 1H, Ar, J = 7.6 Hz), 7.71 (t, 1H, Ar, J = 7.4 Hz), 7.11, 7.06 (ABq, 4H, Ar, JAB = 8.8 Hz), 3.97 (d, 2H, Ar, J = 6.4 Hz); δC (100 MHz, CDCl3) 177.1, 169.4, 141.6, 136.7, 136.1, 136.0, 134.6, 134.4, 134.2, 132.3, 131.9, 130.6, 130.1, 129.1, 109.8, 50.3; Calcd (M+): 391.0, Found: 392.0 ([M+H]+); tR = 12.8 min (100%, II).
4-(N-(Cyclohexylmethyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3g)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to cyclohexylmethylamine according to General Procedure B on a 1 mmol scale to yield the title compound as a brown solid (218 mg, 60%): δH (400 MHz, d6-DMSO) 8.61 (d, 1H, Ar, J = 8.8 Hz), 8.42 – 8.39 (m, 3H, Ar), 7.86 – 7.81 (m, 2H, Ar, SO2NH), 7.71 (t, 1H, Ar, J = 7.4 Hz), 2.56 (t, 1H, CHCH2, J = 5.8 Hz), 1.55 – 1.45 (m, 5H, cyclohexyl), 1.30 – 1.18 (m, 1H, CHCH2), 1.05 – 0.90 (m, 3H, cyclohexyl), 0.62 – 0.74 (m, 2H, cyclohexyl); δC (100 MHz, d6-DMSO) 172.1, 164.1, 131.2, 130.8, 129.1, 126.8, 125.8, 125.2, 125.1, 124.0, 104.5, 48.6, 37.2, 30.1, 25.9, 25.3; Calcd (M+): 363.1, Found: 386.3 ([M+Na]+); tR = 13.7 min (98.2%, II).
4-(N,N-Dimethylsulfamoyl)-1-hydroxy-2-naphthoic acid (3h)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to dimethylamine hydrochloride according to General Procedure B on a 1 mmol scale to yield the title compound as a dark purple solid (170 mg, 58%): δH (400 MHz, d6-DMSO) 8.58 (d, 1H, Ar, J = 8.4 Hz), 8.43 (d, 1H, Ar, J = 8.0 Hz), 8.40 (s, 1H, Ar), 7.86 (t, 1H, Ar, J = 7.8 Hz), 7.71 (t, 1H, Ar, J = 7.4 Hz), 2.70 (s, 6H, N(CH3)2); δC (100 MHz, d6-DMSO) 172.2, 165.3, 132.3, 131.5, 127.3, 127.2, 125.7, 125.5, 124.5, 121.7, 105.6, 37.4; Calcd (M+): 295.1, Found: 296.2 ([M+H]+); tR = 20.5 min (98.3%, I).
1-Hydroxy-4-(piperidin-1-ylsulfonyl)-2-naphthoic acid (3i)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to piperidine according to General Procedure C on a 0.8 mmol scale to yield the title compound as a light brown solid (265 mg, 79%): δH (400 MHz, d6-DMSO) 8.58 (d, 1H, Ar, J = 8.4 Hz), 8.43 (d, 1H, Ar, J = 8.8 Hz), 8.40 (s, 1H, Ar), 7.88 (t, 1H, Ar, J = 7.8 Hz), 7.73 (t, 1H, Ar, J = 7.6 Hz), 3.05 – 3.03 (m, 4H, CH2NCH2), 1.50 – 1.32 (m, 6H, piperidinyl); δC (100 MHz, d6-DMSO) 172.3, 164.7, 132.1, 131.5, 131.2, 127.4, 125.5, 125.4, 124.5, 122.9, 105.4, 46.2, 25.4, 23.3; Calcd (M+): 335.1, Found: 336.2 ([M+H]+); tR = 16.5 min (95.1%, II).
1-Hydroxy-4-((4-phenylpiperazin-1-yl)sulfonyl)-2-naphthoic acid (3j)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 1-phenylpiperizine according to General Procedure C on a 0.8 mmol scale to yield the title compound as a brown-orange solid (264 mg, 64%): δH (400 MHz, d6-DMSO) 8.70 (d, 1H, Ar, J = 8.8 Hz), 8.46 – 8.43 (m, 2H, Ar), 7.88 (t, 1H, Ar, J = 8.0 Hz), 7.73 (t, 1H, Ar, J = 7.4 Hz), 7.17 (t, 2H, Ph, J = 7.8 Hz), 6.86 (d, 2H, Ph, J = 8.0 Hz), 6.78 (d, 1H, Ph, J = 7.2 Hz) 3.19 – 3.12 (m, 8H, piperazinyl); δC (100 MHz, d6-DMSO) 171.9, 165.0, 150.4, 131.8, 131.5, 131.3, 129.0, 127.0, 125.3, 125.1, 124.2, 121.3, 119.8, 116.2, 105.3, 48.3, 45.2; Calcd (M+): 412.1, Found: 413.3 ([M+H]+); tR = 19.4 min (100%, II).
4-((4-Benzylpiperazin-1-yl)sulfonyl)-1-hydroxy-2-naphthoic acid (3k)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to N-benzylpiperazine according to General Procedure C on a 1 mmol scale to yield the title compound as a light yellow solid (303 mg, 71%): δH (400 MHz, d6-DMSO) 8.41 – 8.37 (m, 3H, Ar), 7.62 (t, 1H, Ar, J = 7.6 Hz), 7.47 (t, 1H, Ar, J = 7.6 Hz), 7.43 – 7.36 (m, 5H, Ph), 4.22 (br s, 2H, CH2Ph), 3.70 – 2.80 (m, 8H, piperazinyl); δC (100 MHz, d6-DMSO) 173.4, 170.4, 158.7, 135.3, 132.6, 131.3, 131.0, 129.9, 129.2, 129.1, 125.6, 125.5, 124.9, 124.8, 107.9; Calcd (M+): 426.1, Found: 427.2 ([M+H]+); tR = 6.5 min (95.3%, II).
1-Hydroxy-4-(N-phenylsulfamoyl)-2-naphthoic acid (3l)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to aniline according to General Procedure A on a 1 mmol scale to yield the title compound as a cream solid (275 mg, 80%): δH (400 MHz, d6-DMSO) 10.58 (s, 1H, SO2NH), 8.66 (d, 1H, Ar, J = 8.4 Hz), 8.47 (s, 1H, Ar), 8.38 (d, 1H, Ar, J = 8.8 Hz), 7.87 (t, 1H, Ar, J = 8.0 Hz), 7.71 (t, 1H, Ar, J = 7.8 Hz), 7.15 (t, 2H, Ph, J = 7.8 Hz), 6.99 (d, 2H, Ph, J = 8.0 Hz), 6.92 (t, 1H, Ph, J = 7.0 Hz);δC (100 MHz, d6-DMSO) 172.2, 164.9, 137.9, 131.6, 131.3, 130.9, 129.5, 127.4, 125.3, 125.0, 124.7, 124.6, 124.1, 119.7, 105.2; Calcd (M+): 343.1, Found: 344.1 ([M+H]+); tR = 3.9 min (100%, II).
4-(N-(2-bromophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3m)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2-bromoaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a pink solid (287 mg, 68%): δH (400 MHz, d6-DMSO) 10.07 (s, 1H, SO2NH), 8.60 (d, 1H, Ar, J = 8.4 Hz), 8.41 (d, 1H, Ar, J = 8.4 Hz), 8.31 (s, 1H, Ar), 7.78 (t, 1H, Ar, J = 7.4 Hz), 7.69 (t, 1H, Ar, J = 7.4 Hz), 7.48 (d, 1H, Ar, J = 7.6 Hz), 7.23 (t, 1H, Ar, J = 7.4 Hz), 7.15 (d, 1H, Ar, J = 8.0 Hz), 7.09 (t, 1H, Ar, J = 7.4 Hz);δC (100 MHz, d6-DMSO) 172.2, 165.2, 135.4, 133.5, 131.7, 131.1, 130.2, 128.9, 128.8, 128.7, 128.6, 127.2, 125.8, 125.6, 124.4, 120.9, 105.3; Calcd (M+): Calcd (M+): 421.0, Found: 420.2 ([M-H]-); tR = 12.0 min (98.6 %, II).
4-(N-(3-Bromophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3n)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 3-bromoaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a light pink solid (300 mg, 71%): δH (400 MHz, d6-DMSO) 11.86 (s, 1H, SO2NH), 8.61 (d, 1H, Ar, J = 8.8 Hz), 8.51 (s, 1H, Ar), 8.38 (d, 1H, Ar, J = 8.4 Hz), 7.87 (t, 1H, Ar, J = 7.4 Hz), 7.69 (t, 1H, Ar, J = 7.4 Hz), 7.18 (s, 1H, Ar), 7.14 – 7.00 (m, 3H, Ar); δC (100 MHz, d6-DMSO) 172.1, 165.5, 139.7, 131.6, 131.5, 131.4, 131.2, 127.4, 126.5, 125.6, 125.5, 124.7, 123.5, 122.2, 121.4, 117.8, 105.4; Calcd (M+): 421.0, Found: 420.2 ([M-H]-); tR = 12.3 min (98.5%, II).
4-(N-(2,4-Dibromophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3o)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2,4-dibromoaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a dark beige solid (291 mg, 58%): δH (400 MHz, d6-DMSO) 10.15 (s, 1H, SO2NH), 8.56 (d, 1H, Ar, J = 8.4 Hz), 8.41 (d, 1H, Ar, J = 8.4 Hz), 8.30 (s, 1H, Ar), 7.80 – 7.75 (m, 2H, Ar), 7.69 (t, 1H, Ar, J = 7.4 Hz), 7.50 (dd, 1H, Ar, J = 8.8, 1.6 Hz), 7.09 (d, 1H, Ar, J = 8.4 Hz); δC (100 MHz, d6-DMSO) 172.2, 165.3, 135.4, 135.1, 131.8, 131.6, 131.2, 130.3, 129.8, 127.3, 125.6, 125.5, 125.4, 124.5, 121.8, 120.0, 105.3; Calcd (M+): 498.9, Found: 498.1 ([M-H]-); tR = 18.2 min (97.0%, II).
1-Hydroxy-4-(N-(naphthalen-1-yl)sulfamoyl)-2-naphthoic acid (3p)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 1-naphthylamine according to General Procedure A on a 1 mmol scale to yield the title compound as a dark purple solid (283 mg, 72%): δH (400 MHz, d6-DMSO) 10.46 (s, 1H, SO2NH), 8.74 (d, 1H, Ar, J = 8.8 Hz), 8.38 (d, 1H, Ar, J = 8.8 Hz), 8.26 (s, 1H, Ar), 7.96 (d, 1H, Ar, J = 8.8 Hz), 7.85 – 7.83 (m, 2H, Ar), 7.73 – 7.68 (m, 2H, Ar), 7.41 (t, 1H, Ar, J = 7.6 Hz), 7.34 (t, 1H, Ar, J = 7.8 Hz), 7.28 (t, 1H, Ar, J = 7.4 Hz), 7.12 (d, 1H, Ar, J = 8.0 Hz); δC (100 MHz, d6-DMSO) 171.8, 164.6, 134.7, 133.8, 132.2, 131.3, 130.9, 130.0, 129.4, 128.5, 128.0, 126.9, 126.7, 126.2, 125.9, 125.6, 125.0, 124.9, 124.1, 123.0, 104.8; Calcd (M+): 393.1, Found: 416.2 ([M+Na]+); tR = 9.7 min (100%, II).
1-Hydroxy-4-(N-(naphthalen-2-yl)sulfamoyl)-2-naphthoic acid (3q)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2-naphthylamine according to General Procedure A on a 1 mmol scale to yield the title compound as a grey-purple solid (303 mg, 77%): δH (400 MHz, d6-DMSO) 10.83 (s, 1H, SO2NH), 8.72 (d, 1H, Ar, J = 7.6 Hz), 8.57 (s, 1H, Ar), 8.35 (d, 1H, Ar, J = 8.4 Hz), 7.87 (t, 1H, Ar, J = 8.0 Hz), 7.72 – 7.65 (m, 4H, Ar), 7.47 (s, 1H, Ar), 7.38 (t, 1H, J = 7.0 Hz), 7.32 (t, 1H, Ar, J = 7.0 Hz), 7.22 (d, 1H, Ar, J = 8.8 Hz); δC (100 MHz, d6-DMSO) 171.8, 164.9, 135.3, 133.2, 131.2, 131.0, 130.9, 129.8, 129.1, 127.5, 127.0, 126.9, 126.8, 125.2, 124.9, 124.6, 124.3, 123.7, 119.7, 115.0, 105.0; Calcd (M+): 393.1, Found: 392.2 ([M-H]-); tR = 12.7 min (94.5%, II).
4-(N-([1,1′-Biphenyl]-2-yl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3r)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2-aminobiphenyl according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (252 mg, 60%): δH (400 MHz, d6-DMSO) 9.60 (s, 1H), 8.33-8.28 (m, 2H), 8.07 (s, 1H), 7.62-7.58 (m, 2H), 7.23 (t, J= 4.8 Hz, 2H), 7.11 (t, J= 4.8 Hz, 1H), 7.03-7.00 (m, 6H). δC (100 MHz, d6-DMSO) 172.3, 164.9, 139.9, 138.6, 133.5, 131.4, 131.3, 130.9, 129.5, 129.1, 128.5, 127.8, 127.7, 126.9, 126.8, 126.4, 125.5, 125.4, 124.3, 105.1; Calcd (M+): 419.1, Found: 442.2 ([M+Na]+); tR = 14.6 min (98.7%, II).
4-(N-([1,1′-Biphenyl]-3-yl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3s)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 3-aminobiphenyl according to General Procedure A on a 1 mmol scale to yield the title compound as a dark brown solid (239 mg, 58%): δH (400 MHz, d6-DMSO) 10.45 (s, 1H), 8.61 (s,1H), 8.53 (d, J=8.8 Hz, 1H), 8.32 (d, J=8.8 Hz, 1H), 7.68 (t, J=8 Hz, 1H), 7.49 (t, J= 7.6 Hz, 1H), 7.40 (m, 4H), 7.34 (m, 1H), 7.28 (s, 1H), 7.20 (t, J=7.6 Hz, 1H), 7.13 (d, J=8 Hz, 1H), 6.95 (d, J= 8 Hz, 1H). δC (100 MHz, d6-DMSO) 171.1, 141.3, 140.2, 139.2, 134.1, 131.6, 130.1, 129.9, 129.4, 128.0, 126.9, 125.4, 125.2, 124.6, 121.5, 117.7, 116.6, 107.1; Calcd (M+): 419.1, Found: 418.3 ([M-H]-); tR = 13.9 min (100%, II).
4-(N-([1,1′-Biphenyl]-4-yl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3t)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-aminobiphenyl according to General Procedure A on a 1 mmol scale to yield the title compound as a brown solid (260 mg, 62%): δH (400 MHz, d6-DMSO) 10.37 (s, 1H), 8.54 (s, 1H), 8.47 (d, J=8 Hz, 1H), 8.31 (d, J=7.6 Hz, 1H), 7.62 (t, J=8 Hz, 1H), 7.51 (d, J= 8 Hz, 2H), 7.44 (d, J=8.4 Hz, 3H), 7.35 (t, J= 8 Hz, 2H), 7.25 (t, J=7.6 Hz, 1H), 7.05 (d, J = 7.6 Hz, 2H). δC (100 MHz, d6-DMSO) 172.5, 170.8, 139.9, 138.2, 134.8, 134.3, 131.6, 129.6, 129.2, 128.7, 127.6, 127.3, 126.6, 125.3, 124.8, 124.5, 118.8, 116.3, 107.9; Calcd (M+): 419.5, Found: 418.3 ([M-H]-); tR = 13.9 min (98.3%, II).
1-Hydroxy-4-(N-(2-(trifluoromethyl)phenyl)sulfamoyl)-2-naphthoic acid (3u)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 2-trifluoromethylaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a dark pink solid (222 mg, 54%): δH (400 MHz, d6-DMSO) 10.06 (s, 1H, SO2NH), 8.62 (d, 1H, Ar, J = 8.0 Hz), 8.44 (d, 1H, Ar, J = 8.8 Hz), 8.32 (s, 1H, Ar), 7.80 (t, 1H, Ar, J = 7.4 Hz), 7.71 – 7.66 (m, 2H, Ar), 7.52 (t, 1H, Ar, J = 7.2 Hz), 7.41 (t, 1H, Ar, J = 7.0 Hz), 6.99 (d, 1H, Ar, J = 7.6 Hz);δC (100 MHz, d6-DMSO) 172.1, 166.2, 134.5, 133.8, 133.7, 131.5, 131.0, 130.6, 129.5, 127.8, 127.4, 126.9, 126.4, 126.2 (q, 2JCF = 40 Hz), 125.5 (q, 1JCF = 108 Hz), 124.9, 124.6, 105.6; Calcd (M+): 411.0, Found: 410.2 ([M-H]-); tR = 10.7 min (96.7%, II).
4-(N-(4-Chlorophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3v)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-chloroaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a light brown solid (189 mg, 50%): δH (400 MHz, d6-DMSO) 10.74 (s, 1 H, NH), 8.62 (d, J = 8.8, 1 H, Ar), 8.46 (s, 1 H, Ar), 8.39 (d, J = 8.8, 1 H, Ar), 7.86 (t, J = 8.8, 1 H, Ar), 7.71 (t, J = 8.8, 1 H, Ar), 7.22 (d, J = 8.4, 2 H, Ar), 7.00 (d, J = 8.4, 2 H, Ar); δC (100 MHz, d6-DMSO) 172.2, 165.0, 136.9, 131.7, 131.2, 131.1, 129.5, 128.1, 127.4, 125.4, 124.9, 124.6, 124.2, 121.1, 105.3; Calcd (M+): 377.0, Found: 376.2 ([M-H]-); tR = 12.2 min (95.7%, II).
1-Hydroxy-4-(N-(4-(trifluoromethyl)phenyl)sulfamoyl)-2-naphthoic acid (3w)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-trifluoromethylaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a light pink solid (218 mg, 53%): δH (400 MHz, d6-DMSO) 11.13 (s, 1 H, NH), 8.60 (d, J = 8.8, 1 H, Ar), 8.57 (s, 1 H, Ar), 8.39 (d, J = 8.8, 1 H, Ar), 7.85 (t, J = 8.8, 1 H, Ar), 7.68 (t, J = 8.8, 1 H, Ar), 7.55 (d, J = 8.8, 2 H, Ar), 7.19 (d, J = 8.8, 2 H, Ar); δC (100 MHz, d6-DMSO) 172.0, 165.7, 141.8, 131.7, 131.5, 131.2, 127.4, 127.0, 125.9, 125.7, 124.8, 124.7, 123.6, 118.2, 105.5; Calcd (M+): 411.0, Found: 434.2 ([M+Na]+); tR = 15.6 min (95.4%, II).
1-Hydroxy-4-(N-(p-tolyl)sulfamoyl)-2-naphthoic acid (3x)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to p-toluidine according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (225 mg, 63%): δH (400 MHz, d6-DMSO) 10.39 (s, 1 H, NH), 8.65 (d, J = 8.8, 1 H, Ar), 8.43 (s, 1 H, Ar), 8.38 (d, J = 8.8, 1 H, Ar), 7.86 (t, J = 8.8, 1 H, Ar), 7.70 (t, J = 8.8, 1 H, Ar), 6.95 (d, J = 8.0, 2 H, Ar), 6.87 (d, J = 8.0, 2 H, Ar), 2.11 (s, 3 H, CH3); δC (100 MHz, d6-DMSO) 172.2, 165.0, 135.2, 133.4, 131.5, 131.4, 131.0, 129.9, 127.3, 125.4, 125.1, 124.5, 120.2, 105.3, 20.6; Calcd (M+): 357.1, Found: 380.2 ([M+Na]+); tR = 9.0 min (95.5%, II).
1-Hydroxy-4-(N-(4-isopropylphenyl)sulfamoyl)-2-naphthoic acid (3y)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to p-isopropylaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (235 mg, 61%): δH (400 MHz, d6-DMSO) 10.42 (s, 1 H, NH), 8.63 (d, J = 8.4, 1 H, Ar), 8.46 (s, 1 H, Ar), 8.39 (d, J = 8.4, 1 H, Ar), 7.84 (t, J = 8.4, 1 H, Ar), 7.70 (t, J = 8.4, 1 H, Ar), 7.02 (d, J = 8.4, 2 H, Ar), 6.90 (d, J = 8.0, 2 H, Ar), 2.71 (sept, J = 7.2, 1 H, CH), 1.06 (d, J = 7.2, 6 H, 2 × CH3); δC (100 MHz, d6-DMSO) 172.2, 164.8, 144.3, 135.5, 131.5, 131.4, 130.8, 127.3, 125.3, 125.1, 124.5, 120.0, 105.2, 33.1, 24.2; Calcd (M+): 385.1, Found: 408.3 ([M+Na]+); tR = 15.3 min (95.9%, II).
1-Hydroxy-4-(N-(4-methoxyphenyl)sulfamoyl)-2-naphthoic acid (3z)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to p-anisidine according to General Procedure A on a 1 mmol scale to yield the title compound as a light brown solid (281 mg, 75%): δH (400 MHz, d6-DMSO) 10.18 (s, 1H, SO2NH), 8.64 (d, 1H, Ar, J = 8.8 Hz), 8.38 (d, 1H, Ar, J = 8.4 Hz), 8.36 (s, 1H, Ar), 7.86 (t, 1H, Ar, J = 7.4 Hz), 7.71 (t, 2H, Ar, J = 7.6 Hz), 6.85 (d, 2H, Ar, J = 9.2 Hz), 6.71 (d, 2H, Ar, J = 8.8 Hz), 3.59 (s, 3H, OMe);δC (100 MHz, d6–DMSO): 172.2, 164.7, 156.8, 131.5, 131.4, 130.7, 130.3, 127.3, 125.3, 125.2, 124.8, 124.5, 123.3, 114.7, 105.2, 55.5; Calcd (M+): 373.1, Found: 372.3 ([M-H-); tR = 8.7 min (99.6%, III).
1-Hydroxy-4-(N-(4-isopropoxyphenyl)sulfamoyl)-2-naphthoic acid (3aa)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-isopropoxyaniline according to General Procedure A on a 1 mmol scale to yield the title compound as a white solid (289 mg, 72%): δH (400 MHz, d6-DMSO) 10.15 (s, 1H, SO2NH), 8.63 (d, 1H, Ar, J = 8.8 Hz), 8.39 (d, 1H, Ar, J = 8.0 Hz), 8.35 (s, 1H, Ar), 7.85 (t, 1H, Ar, J = 7.4 Hz), 7.71 (t, 1H, Ar, J = 7.8 Hz), 6.83 (d, 2H, Ar, J = 9.6 Hz), 6.69 (d, 2H, Ar, J = 8.8 Hz), 4.41 (hep, 1H, CH(CH3)2, J = 5.8 Hz), 1.14 (d, 6H, CH(CH3)2, J = 5.2 Hz); δC (100 MHz, d6-DMSO) 172.2, 164.7, 155.0, 131.5, 131.4, 130.7, 130.1, 127.3, 125.3, 125.2, 124.9, 124.5, 123.4, 116.5, 105.2, 69.7, 22.1; Calcd (M+): 401.1, Found: 402.0 ([M+H]+); tR = 10.2 min (98.6%, II).
4-(N-(4-Cyanophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3ab)
Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-aminobenzonitrile according to General Procedure A on a 1 mmol scale to yield the title compound as a light pink solid (254 mg, 69%): δH (400 MHz, d6-DMSO) 11.32 (s, 1H, SO2NH), 8.60 – 8.57 (m, 2H, Ar), 8.39 (d, 1H, Ar, J = 8.8 Hz), 7.87 (t, 1H, Ar, J = 7.4 Hz), 7.69 (t, 1H, Ar, J = 7.8 Hz), 7.63 (d, 2H, Ar, J = 8.4 Hz), 7.15 (d, 2H, Ar, J = 8.8 Hz);δC (100 MHz, d6-DMSO) 172.0, 166.1, 142.5, 134.1, 131.8, 131.7, 131.1, 127.3, 125.8, 124.8, 124.6, 122.9, 119.1, 118.1, 105.9, 105.3; Calcd (M+): 368.1, Found: 367.1 ([M-H]-); tR = 5.5 min (98.3%, II).
1-Hydroxy-4-(N-(4-nitrophenyl)sulfamoyl)-2-naphthoic acid (3ac)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-nitroaniline according to General Procedure A on a 2 mmol scale to yield the title compound as a dark yellow solid (249 mg, 64%): δH (400 MHz, d6-DMSO) 11.54 (s, 1H, SO2NH), 8.61 – 8.58 (m, 2H, Ar), 8.38 (d, 1H, Ar, J = 8.0 Hz), 8.07 (d, 2H, Ar, J = 8.4 Hz), 7.86 (t, 1H, Ar, J = 7.6 Hz), 7.68 (t, 1H, Ar, J = 7.6 Hz), 7.20 (d, 2H, Ar, J = 9.2 Hz); δC (100 MHz, d6-DMSO) 171.5, 166.2, 144.1, 142.2, 131.8, 131.3, 130.8, 126.9, 125.6, 125.5, 124.5, 124.1, 121.9, 117.2, 105.4; Calcd (M+): 388.0, Found: 387.3 ([M-H]-); tR = 4.8 min (97.4%, I).
4-(N-(3-Cyanophenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3ad)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 3-aminobenzonitrile according to General Procedure A on a 1 mmol scale to yield the title compound as a reddish-purple solid (188 mg, 61%): δH (400 MHz, d6-DMSO) 10.71 (s, 1 H, NH), 8.54 (s, 1 H, Ar), 8.40 (d, J = 8.4, 1 H, Ar), 8.32 (d, J = 8.4, 1 H, Ar), 7.63 (t, J = 8.4, 1 H, Ar), 7.44 (t, J = 8.4, 1 H, Ar), 7.39-7.27 (m, 4 H, Ar); δC (100 MHz, d6-DMSO) 173.3, 170.5, 139.9, 134.7, 131.5, 130.9, 129.7, 129.0, 126.5, 125.5, 124.9, 124.2, 122.7, 120.7, 118.8, 114.8, 112.1, 107.8; Calcd (M+): 368.1, Found: 367.1 ([M-H]-); tR = 7.4 min (97.2%, II).
1-Hydroxy-4-(N-isobutyl-N-(4-isopropoxyphenyl)sulfamoyl)-2-naphthoic acid (3ba)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-isopropoxyaniline (8a) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (350 mg, 69%): δH (400 MHz, d6-DMSO) 8.32 (d, 1H, Ar, J = 8.4 Hz), 8.30 (s, 1H, Ar), 8.10 (d, 1H, Ar, J = 7.6 Hz), 7.49 – 8.39 (m, 2H, Ar), 6.96 (d, 2H, Ph, J = 8.8 Hz), 6.77 (d, 2H, Ph, J = 8.8 Hz), 4.56 – 4.50 (m, 1H, OCH), 3.25 (d, 2H, CH2CH, J = 6.8 Hz), 1.39 – 1.34 (m, 1H, CH2CH), 1.22 (d, 6H, OCH(CH3)2, J = 6.4 Hz), 0.73 (d, 6H, CH2CH(CH3)2, J = 6.8 Hz); δC (100 MHz, d6-DMSO) 172.4, 170.8, 156.9, 134.7, 132.3, 131.9, 130.2, 128.9, 128.6, 125.1, 125.0 124.6, 115.9, 115.4, 108.0, 69.7, 57.5, 26.7, 22.2, 20.1; Calcd (M+): 457.2, Found: 456.2 ([M-H]-); tR = 16.7 min (99.5%, III).
1-Hydroxy-4-(N-(4-phenoxyphenyl)sulfamoyl)-2-naphthoic acid (3bb)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-phenoxyaniline (8b) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (340 mg, 78%): δH (400 MHz, d6-DMSO) 10.41 (s, 1H, SO2NH), 8.63 (d, 1H, Ar, J = 8.8 Hz), 8.40 – 8.38 (m, 2H, Ar), 7.85 (t, 1H, Ar, J = 7.6 Hz), 7.71 (t, 1H, Ar, J = 7.6 Hz), 7.31 (t, 2H, Ar, J = 8.0 Hz), 7.06 (t, 1H, Ar, J = 7.4 Hz), 6.96 (d, 2H, Ar, J = 8.8 Hz), 6.84 – 6.81 (m, 4H, Ar); δC (100 MHz, d6-DMSO) 172.2, 165.1, 158.2, 157.4, 133.5, 131.5, 131.0, 130.4, 127.3, 125.4, 125.1, 124.6, 123.5, 122.8, 120.2, 118.3, 116.0, 114.9, 105.3; Calcd (M+): 435.1, Found: 436.0 ([M+H]+); tR = 14.4 min (100%, II).
1-Hydroxy-4-(N-(4-(p-tolyloxy)phenyl)sulfamoyl)-2-naphthoic acid (3bc)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(p-tolyloxy)aniline (8c) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (342 mg, 76%): δH (400 MHz, d6-DMSO) 10.37 (s, 1H, SO2NH), 8.63 (d, 1H, Ar, J = 8.8 Hz), 8.41 – 8.39 (m, 2H, Ar), 7.85 (t, 1H, Ar, J = 7.4 Hz), 7.71 (t, 1H, Ar, J = 7.8 Hz), 7.11 (d, 2H, Ar, J = 8.8 Hz), 6.94 (d, 2H, Ar, J = 8.4 Hz), 6.78 (d, 2H, Ar, J = 8.8 Hz), 6.74 (d, 1H, Ar, J = 8.0 Hz), 2.24 (s, 3H, CH3); δC (100 MHz, d6-DMSO) 172.2, 164.5, 154.9, 153.9, 133.0, 132.7, 131.5, 130.9, 130.7, 127.3, 125.3, 125.1, 124.6, 122.8, 119.6, 118.6, 105.2, 20.6; Calcd (M+): 449.1, Found: 472.3 ([M+Na]+); tR = 9.3 min (100%, III).
1-Hydroxy-4-(N-(4-(naphthalen-1-yloxy)phenyl)sulfamoyl)-2-naphthoic acid (3bd)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(naphthalen-1-yloxy)aniline62 (8d) according to General Procedure A on a 1 mmol scale to yield the title compound as a light purple solid (351 mg, 70%): δH (400 MHz, d6-DMSO) 10.42 (s, 1H, SO2NH), 8.63 (d, 1H, Ar, J = 8.8 Hz), 8.42 – 8.39 (m, 2H, Ar), 7.99 (d, 1H, Ar, J = 8.8 Hz), 7.94 (d, 1H, Ar, J = 8.0 Hz), 7.86 (t, 1H, Ar, J = 7.4 Hz), 7.72 (t, 1H, Ar, J = 7.8 Hz), 7.66 (d, 1H, Ar, J = 8.8 Hz), 7.55 (t, 1H, Ar, J = 7.4 Hz), 7.49 (t, 1H, Ar, J = 7.0 Hz), 7.38 (t, 1H, Ar, J = 7.8 Hz), 6.98, 6.88 (ABq, 4H, Ar, JAB = 8.4Hz), 6.73 (d, 1H, Ar, J = 8.0 Hz); δC (100 MHz, d6-DMSO) 172.2, 165.2, 153.9, 153.0, 147.4, 134.9, 133.5, 131.4, 131.3, 131.0, 128.3, 127.3, 127.2, 126.6, 126.5, 125.5, 125.1, 124.6, 124.3, 123.5, 122.8, 121.7, 119.8, 113.1, 105.3; Calcd (M+): 485.1, Found: 508.3 ([M+Na]+); tR = 11.2 min (100%, III).
4-(N-(4-(3-Bromophenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3be)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(3-bromophenoxy)aniline63 (8e) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (334 mg, 65%): δH (400 MHz, d6-DMSO) 10.45 (s, 1H, SO2NH), 8.62 (d, 1H, Ar, J = 8.8 Hz), 8.43 (s, 1H, Ar), 8.40 (d, 1H, Ar, J = 8.8 Hz), 7.86 (t, 1H, Ar, J = 7.4 Hz), 7.71 (t, 1H, Ar, J = 7.4 Hz), 7.26 – 7.25 (m, 2H, Ar), 7.01 – 6.99 (m, 3H, Ar), 6.90 (d, 2H, Ar, J = 8.4 Hz), 6.83 – 6.80 (m, 1H, Ar);δC (100 MHz, d6-DMSO) 172.2, 165.2, 158.7, 152.2, 134.2, 132.1, 131.4, 131.0, 127.3, 126.2, 125.6, 125.1, 124.6, 122.6, 122.5, 120.9, 120.8, 117.0, 115.9, 112.5, 105.3; Calcd (M+): 513.0, Found: 513.9 ([M+H]+); tR = 10.7 min (99.6%, III).
4-(N-(4-(3,5-Dimethylphenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3bf)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(3,5-dimethylphenoxy)aniline (8f) according to General Procedure A on a 1 mmol scale to yield the title compound as a cream solid (306 mg, 66%): δH (400 MHz, d6-DMSO) 10.40 (s, 1H, SO2NH), 8.62 (d, 1H, Ar, J = 8.8 Hz), 8.41 – 8.39 (m, 3H, Ar), 7.85 (t, 1H, Ar, J = 7.4 Hz), 7.71 (t, 1H, Ar, J = 7.4 Hz), 6.95, 6.79 (ABq, 4H, Ar, JAB = 9.0 Hz), 6.70 (s, 1H, Ar), 6.43 (s, 2H, Ar), 2.18 (s, 6H, 2×CH3);δC (100 MHz, d6-DMSO) 172.2, 157.4, 153.4, 139.7, 133.3, 131.6, 131.4, 130.9, 129.9, 127.4, 127.3, 125.5, 125.1, 124.6, 124.5, 122.7, 120.1, 116.1, 105.3, 21.3; Calcd (M+): 463.1, Found: 463.91 ([M+H]+); tR = 19.8 min (98.9%, II).
4-(N-(4-(2,4-Dichlorophenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3bg)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(2,4-dichlorophenoxy)aniline64 (8g) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (297 mg, 66%): δH (400 MHz, d6-DMSO) 10.46 (s, 1H, SO2NH), 8.62 (d, 1H, Ar, J = 8.8 Hz), 8.41 – 8.39 (m, 2H, Ar), 7.85 (t, 1H, Ar, J = 7.4 Hz), 7.73 – 7.70 (m, 2H, Ar), 7.34 (dd, 1H, Ar, J = 8.8, 2.4 Hz), 6.97 (d, 2H, Ar, J = 8.8 Hz), 6.88 – 6.82 (m, 3H, Ar); δC (100 MHz, d6-DMSO) 172.1, 165.3, 151.6, 148.8, 133.9, 131.4, 131.0, 130.5, 129.1, 127.4, 127.3, 125.5, 125.0, 124.2, 124.1, 122.6, 121.6, 119.3, 105.3; Calcd (M+): 503.0, Found: 503.9 ([M+H]+); tR = 12.3 min (100%, III).
4-(N-(4-(4-Chlorophenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3bh)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(4-chlorophenoxy)aniline (8h) according to General Procedure A on a 1 mmol scale to yield the title compound as a beige solid (291 mg, 62%): δH (400 MHz, d6-DMSO) 10.45 (s, 1H, SO2NH), 8.63 (d, 1H, Ar, J = 8.8 Hz), 8.41 – 8.39 (m, 2H, Ar), 7.86 (t, 1H, Ar, J = 7.4 Hz), 7.72 (t, 1H, Ar, J = 7.4 Hz), 7.35 (d, 1H, Ar, J = 9.2 Hz), 6.98 (d, 1H, Ar, J = 8.4 Hz), 6.85 (app t, 4H, Ar, J = 8.6 Hz);δC (100 MHz, d6-DMSO) 172.2, 165.0, 156.4, 152.8, 133.9, 131.5, 131.4, 130.9, 130.2, 127.4, 127.2, 125.4, 125.1, 124.6, 124.5, 122.7, 120.4, 119.9, 105.2; Calcd (M+): 469.0, Found: 469.91 ([M+H]+); tR = 19.5 min (98.8%, II).
4-(N-(4-(4-Chloro-3,5-dimethylphenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3bi)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(4-chloro-3,5-dimethylphenoxy)aniline (8i) according to General Procedure A on a 1 mmol scale to yield the title compound as an off-white solid (374 mg, 75%): δH (400 MHz, d6-DMSO, TMS) δH 10.41 (s, 1H, NH), 8.61 (d, 1H, Ar-H, J = 8.8 Hz), 8.42 (s, 1H, Ar-H), 8.39 (d, 1H, Ar-H, J = 8.4 Hz), 7.84 (t, 1H, Ar-H, J = 7.6 Hz), 7.69 (t, 1H, Ar-H, J = 7.6 Hz), 6.96 (d, 2H, Ar-H, J = 8.8 Hz), 6.82 (d, 2H, Ar-H, J = 8.8 Hz), 6.70 (s, 2H, Ar-H), 2.24 (s, 6H, Ar-CH3). δC (100 MHz, d6-DMSO) 172.2, 164.8, 155.2, 153.2, 137.6, 133.4, 131.5, 130.8, 128.2, 127.3, 125.3, 125.1, 124.7, 124.5, 122.5, 119.9, 118.6, 105.2, 20.7; Calcd (M+): 497.1, Found: 496.2 ([M-H]-); tR = 4.0 min (95.0%, IV).
4-(N-(4-(4-Chloro-3,5-dimethylphenoxy)phenyl)-N-isobutylsulfamoyl)-1-hydroxy-2-naphthoic acid (3bj)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(4-chloro-3,5-dimethylphenoxy)-N-isobutylaniline (9a) according to General Procedure A on a 1 mmol scale to yield the title compound as a cream solid (399 mg, 72%): δH (400 MHz, d6-DMSO, TMS) δH 8.37 (d, 1H, Ar-H, J = 8 Hz), 8.27 (s, 1H, Ar-H), 8.14 (d, 1H, Ar-H, J = 8 Hz), 7.62 (t, 1H, Ar-H, J = 7.4 Hz), 7.56 (t, 1H, Ar-H, J = 7.4 Hz), 7.06 (d, 2H, Ar-H, J= 8.8 Hz), 6.86 (s, 2H, Ar-H), 6.85 (d, 2H, Ar-H, J = 8.8 Hz), 3.32 (d, 2H, NCH2, J = 7.2 Hz), 2.31 (s, 6H, Ar-CH3), 1.42 (m, 1H, CH2CH(CH3)2), 0.78 (d, 6H, CH(CH3)2, J = 6.4 Hz) δC (100 MHz, d6-DMSO) 156.3, 154.3, 137.9, 134.2, 132.9, 132.2, 130.6, 130.2, 128.9, 126.7, 126.1, 125.2, 124.7, 119.7, 118.7, 57.3, 26.8, 20.7, 20.2; Calcd (M+): 553.1, Found: 552.2 ([M-H]-); tR = 8.0 min (96.8%, IV).
4-(N-(4-(4-Chloro-3,5-dimethylphenoxy)phenyl)-N-cyclopentylsulfamoyl)-1-hydroxy-2-naphthoic acid (3bk)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(4-chloro-3,5-dimethylphenoxy)-N-isobutylaniline (9b) according to General Procedure A on a 1 mmol scale to yield the title compound as a white solid (396 mg, 70%): δH (400 MHz, d6-DMSO, TMS) δH 8.44 (app. t, 2H, Ar-H, J = 7.2 Hz), 8.27 (s, 1H, Ar-H), 7.90 (t, 1H, Ar-H, J = 7.8 Hz), 7.74 (t, 1H, Ar-H, J = 7.6 Hz), 6.94 (d, 2H, Ar-H, J = 8.8 Hz), 6.88 (d, 4H, Ar-H, J = 8.4 Hz), 4.54 (m, 1H, NCH(CH2)2, J = 8.2 Hz), 2.30 (s, 6H, Ar-CH3), 1.80-1.68 (m, 2H, (CHCH2CH2)), 1.50-1.17 (m, 6H, 3(CHCH2CH2)) δC (100 MHz, d6-DMSO) 172.2, 165, 157.3, 153.9, 138, 134, 131.6, 131.5, 130.2, 129.2, 127.3, 125.4, 125.2, 124.6, 124.4, 119.9, 118.3, 105.3, 59.9, 30.3, 22.4, 20.7; Calcd (M+): 565.1, Found: 564.3 ([M-H]-); tR = 8.5 min (95.4%, IV).
4-(N-Benzyl-N-(4-(4-chloro-3,5-dimethylphenoxy)phenyl)sulfamoyl)-1-hydroxy-2-naphthoic acid (3bl)
4-Chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-(4-chloro-3,5-dimethylphenoxy)-N-isobutylaniline (9c) according to General Procedure A on a 1 mmol scale to yield the title compound as a white solid (423 mg, 72%): δH (400 MHz, d6-DMSO, TMS) δH 8.43 (d, 1H, Ar-H, J = 8 Hz), 8.39 (s, 1H, Ar-H), 8.26 (d, 1H, Ar-H, J = 8 Hz), 7.73 (m, 2H, Ar-H, J = 7.2 Hz), 7.24-7.17 (m, 5H, Ph), 7.02 (d, 2H, Ar-H, J = 8.8 Hz), 6.77 (m, 3H, Ar-H), 4.78 (s, 2H, NCH2), 2.28 (s, 6H, (Ar-CH3)2) δC (100 MHz, d6-DMSO) 172.2, 165.2, 156.3, 154.2, 137.9, 136.5, 133.6, 132, 131.9, 131.2, 130.9, 128.9, 128.6, 127.8, 127.2, 125.5, 125.4, 124.5, 123.1, 119.6, 118.5, 105.5, 53.8, 20.7; Calcd (M+): 587.1, Found: 586.2 ([M-H]-); tR = 5.7 min (95.9%, IV).
4-(N-(4-Bromophenyl)-N-isobutylsulfamoyl)-1-hydroxy-2-naphthoic acid (3ca)
4-chlorosulfonyl-1-hydroxy-2-naphthoic acid (5) was coupled to 4-bromo-N-isobutylaniline (8j) according to General Procedure A on a 6 mmol scale to yield the title compound as a light pink solid (393 mg, 68%): δH (400 MHz, d6-DMSO, TMS) δH 8.41 – 8.39 (m, 1H, Ar), 8.29 (s, 1H, Ar), 8.15 – 8.13 (m, 1H, Ar), 7.68 – 7.65 (m, 2H, Ar), 7.48 (d, 2H, Ph, J = 8.8 Hz), 7.07 (d, 2H, Ph, J = 8.4 Hz), 3.36 (d, 2H, NCH2, J = 7.2 Hz), 1.44 – 1.37 (m, 1H, CH2CH), 0.77 (d, 6H, CH(CH3)2, J = 6 Hz) δC (100 MHz, d6-DMSO) 172.0, 165.8, 138.5, 132.4, 132.1, 131.9, 131.1, 130.8, 127.1, 125.7, 125.2, 124.5, 122.8, 120.9, 105.6, 57.1, 26.9, 19.9; Calcd (M+): 477.0, Found: 476.0 ([M-H]-); tR = 16.3 min (99.5%, III).
Methyl 4-(N-(4-bromophenyl)-N-isobutylsulfamoyl)-1-hydroxy-2-naphthoate (3cb) and Methyl 4-(N-(4-bromophenyl)-N-isobutylsulfamoyl)-1-methoxy-2-naphthoate (3cc)
Compound 3ca (752 mg, 1.3 mmol, 1 eq) was treated with MeI (2.2 eq) and K2CO3 (3 eq) in DMF (13 mL) at RT. After 16 h, the reaction was incomplete, yielding two products. Water (150 mL) was added to the reaction mixture, which was subsequently extracted with EtOAc (30 mL × 3). The EtOAc extractions were combined, washed with water (100 mL × 3), brine, dried (Na2SO4), filtered and concentrated. The residue was adsorbed onto silica gel and purified by flash column chromatography, eluting with a gradient of EtOAc in hexanes to deliver first 3cc (395 mg, 60%) and second 3cb (211 mg, 33%) as cream-coloured solids. 3cb: δH (400 MHz, d6-DMSO) 12.28 (s, 1H, OH), 8.40 (d, 2H, Ar, J = 8.4 Hz), 8.27 (s, 1H, Ar), 8.11 (d, 2H, Ar, J = 8.4 Hz), 7.70 – 7.45 (m, 2H, Ar), 7.46 (d, 2H, Ar, J = 8.4 Hz), 7.05 (d, 2H, Ar, J = 8.4 Hz), 3.96 (s, 3H, CO2CH3), 3.34 (d, 1H, CH2CH(CH3)2), J = 7.6 Hz), 1.38 (hep, 1H, CH(CH3)2, J = 6.9 Hz), 0.75 (d, 6H, CH(CH3)2, J = 6.0 Hz); δC (100 MHz, d6-DMSO) 174.3, 168.2, 143.1, 137.2, 136.5, 136.3, 135.7, 135.6, 132.5, 130.0, 129.9, 129.3, 129.2, 125.9, 109.8, 61.9, 58.4, 31.6, 24.7; Calcd (M+): 491.0, Found: 514.0 ([M+Na]+); tR = 9.5 min (95.1%, IV). 3cc: δH (400 MHz, d6-DMSO) 8.37 (d, 1H, Ar, J = 7.6 Hz), 8.31 (s, 1H, Ar), 8.15 (d, 1H, Ar, J = 8.8 Hz), 7.75 (t, 1H, Ar, J = 7.6 Hz), 7.67 (t, 1H, Ar, J = 7.4 Hz), 7.50 (d, 2H, Ar, J = 8.4 Hz), 7.08 (d, 2H, Ar, J = 8.4 Hz), 4.05 (s, 3H, CH3), 3.93 (s, 3H, CH3), 3.39 (d, 1H, CH2CH(CH3)2), J = 6.8 Hz), 1.43 (hep, 1H, CH(CH3)2, J = 6.7 Hz), 0.79 (d, 6H, CH(CH3)2, J = 5.2 Hz); δC (100 MHz, d6-DMSO) 165.1, 162.1, 138.2, 132.5, 132.2, 131.3, 130.9, 130.7, 129.3, 128.8, 128.3, 125.3, 124.7, 121.2, 117.1, 64.2, 57.2, 53.2, 26.9, 19.9; Calcd (M+): 505.1, Found: 528.1 ([M+Na]+).
4-(N-(4-Bromophenyl)-N-isobutylsulfamoyl)-1-methoxy-2-naphthoic acid (3cd)
Compound 3cc was saponified using LiOH.H2O (3eq) in a mixture of THF/H2O/MeOH (3:1:1) on a 0.2 mmol scale to deliver the title compound as a white solid (79 mg, 80%): δH (400 MHz, d6-DMSO) 13.5 (br s, 1H, CO2H), 8.36 (d, 1H, Ar, J = 8.4 Hz), 8.32 (s, 1H, Ar), 8.15 (d, 1H, Ar, J = 8.0 Hz), 7.73 (t, 1H, Ar, J = 7.8 Hz), 7.65 (t, 1H, Ar, J = 7.6 Hz), 7.49 (d, 2H, Ar, J = 8.4 Hz), 7.07 (d, 2H, Ar, J = 8.4 Hz), 4.06 (s, 3H, OMe), 3.40 (d, 2H, CH2CH(CH3)2), J = 6.8 Hz), 1.43 (hep, 1H, CH(CH3)2, J = 6.8 Hz), 0.80 (d, 6H, CH(CH3)2, J = 6.4 Hz); δC (100 MHz, d6-DMSO) 166.0, 161.6, 137.9, 132.5, 132.1, 130.8, 130.6, 130.0, 129.0, 128.2, 127.8, 124.9, 124.3, 120.8, 117.9, 63.6, 56.8, 26.5, 19.6; Calcd (M+): 491.0, Found: 490.0 ([M-H]-); tR = 15.3 min (98.5%, III).
Methyl 1-hydroxy-4-(N-isobutyl-N-(4-isopropoxyphenyl)sulfamoyl)-2-naphthoate (13)
3ba (458 mg, 1 mmol, 1 eq) was dissolved in MeOH (4 mL) at 0 °C. Thionyl chloride (1 mL, 12 mmol, 12 eq) was added dropwise to the stirred solution. The reaction was allowed to warm to room temperature, overnight. TLC (CH2Cl2/MeOH/AcOH, 92:7:1) indicated that reaction was complete, and was concentrated to dryness. The residue was azeotroped with CHCl3 (4 × 100 mL) to remove residual thionyl chloride. The resulting compound yielded the title compound as an off-white solid in quantitative yield, 472 mg: δH (400 MHz, CDCl3) 12.46 (s, 1H, OH), 8.46-8.42 (m, 2H Ar), 8.32-8.30 (m, 2H, Ar), 7.57-7.55 (m, 2H, Ar), 6.91 (d, 2H, Ph, J = 8.8 Hz), 6.67 (d, 2H, Ph, J = 8.4 Hz), 4.49-4.43 (m, 1H, OCH), 3.98 (s, 3H, MeO), 3.36 (d, 2H, CH2CH, J = 6.8 Hz), 1.60-1.53 (m, 1H, CH2CH), 1.29 (d, 6H, OCH(CH3)2, J = 6 Hz), 0.86 (d, 6H, CH2CH (CH3)2, J = 7.2 Hz); δC (100 MHz, CDCl3) 170.6, 164.4, 157.2, 132.4, 131.1, 130.8, 130.7, 130.2, 126.5, 125.7, 125.1, 125.0, 124.3, 115.9, 104.0, 70.0, 58.1, 52.7, 29.7, 26.7, 21.9, 19.9; Calcd (M+): 471.17, Found: 472.0 ([M+H]+); tR = 11.98 min (100%; IV).
Acetoxymethyl 1-hydroxy-4-(N-isobutyl-N-(4-isopropoxyphenyl)sulfamoyl)-2-naphthoate (14)
3ba (200 mg, 0.437 mmol, 1 eq) and K2CO3 (66.5 mg, 0.481 mmol, 1.1 eq) were dissolved in acetonitrile (4.4 mL) at 0 °C and stirred for 1 h. Bromomethyl acetate (43 μL, 0.437 mmol, 1 eq) was then added dropwise. The reaction was allowed to warm to RT and stirred overnight. The next day, TLC indicated the reaction was incomplete, so another 1.1 eq of K2CO3 and 1 eq of bromomethyl acetate were added and the reaction was stirred at RT for another 16 h. The reaction was quenched with water and extracted with EtOAc (3 × 25 mL). The organics were collected washed with brine, dried with Na2SO4, and concentrated to dryness. The crude material was purified by flash chromatography over silica gel using an eluent of Hex/EtOAc 3:1 to produce the title compound as a pale pink solid (116 mg, 50%): δH (400 MHz, CDCl3) 12.14 (s, 1H, OH), 8.48-8.46 (m, 1H, Ar), 8.43 (s, 1H, Ar), 8.33-8.30 (m, 1H, Ar), 7.59-7.57 (m, 2H, Ar), 6.93 (d, 2H, Ph, J = 8.8 Hz), 6.68 (d, 2H, Ph, J = 8.4 Hz), 6.04 (s, 2H, OCH2O), 4.49-4.44 (m, 1H, OCH), 3.37 (d, 2H, CH2CH, J = 6.8 Hz), 2.17 (s, 3H, CH3C=O), 1.61-1.54 (m, 1H, CH2CH), 1.30 (d, 6H, OCH(CH3)2, J = 6.4 Hz), 0.87 (d, 6H, CH2CH(CH3)2, J = 6 Hz); δC (100 MHz, CDCl3) 169.1, 165.0, 157.3, 132.6, 131.1, 131.0, 130.3, 130.1, 126.8, 125.8, 125.6, 125.1, 124.4, 115.9, 103.2, 79.9, 70.0, 58.1, 31.6, 26.7, 21.9, 20.7, 19.9; Calcd (M+): 529.18, Found: 530.0 ([M+H]+); tR = 26.99 min (100%; V).
Biology
General
All chemical reagents were ACS grade or higher unless otherwise indicated. All buffers were passed through Chelex-100 (Bio-Rad, Hercules, CA) to remove trace metals. The D2O, d6-DMSO, 15NH4Cl, and 13C-labeled glucose were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA).
Protein Production
A His6-MBP tagged recombinant human Mcl-1 residues 172 to 327 was produced in E. coli in either LB or minimal media supplemented with 15NH4Cl to produce unlabeled or 15N-labeled Mcl-1. The tagged protein was initially purified from the crude cell lysate by IMAC chromatography (GE Healthcare Life Sciences), and after dialysis to remove the imidazole the affinity tag was cleaved using PreScission Protease (GE Healthcare Life Sciences). A Sephacryl S-200 size exclusion column was used as a final purification step before the protein was concentrated with a 10,000 MWCO centifugal filter concentrator (Millipore). The protein purity was shown to be >98% by Coomassie Brilliant Blue (Bio-Rad) stained SDS-PAGE gel and the final concentration was determined using the Bradford protein assay (Bio-Rad) with BSA standards (Pierce).
Peptide
A 6-aminohexanoic acid linker was conjugated to the N-terminus of the Bak BH3 peptide (GQVGRQLAIIGDDINR), capped with fluorescein (on the amino group of the linker), and the peptide was amidated on the C-terminus to give FITC-Ahx-GQVGRQLAIIGDDINR-CONH2, hereafter referred to as “FITC-Bak” (synthesized by Neo BioScience in >95% purity).
Fluorescence polarization experiments
Fluorescence polarization experiments were conducted using a BMG PHERAstar FS multimode microplate reader equipped with two PMTs for simultaneous measurements of the perpendicular and parallel fluorescence emission. The assays were performed in black polypropylene 384-well microplate (Costar) with a final volume of 20 μL. Initially the affinity (KD) of the FITC-Bak peptide was determined by titrating Mcl-1172-327 into 10 nM FITC-Bak peptide in 20 mM HEPES, pH 6.8, 50 mM NaCl, 3 mM DTT, 0.01% Triton X-100 and 5% DMSO at room temperature while monitoring the perpendicular and parallel fluorescence emission with a 485 nm excitation and 520 nm emission filters. The fluorescence polarization competition assay (FPCA) was performed using 100 nM Mcl-1172-327 in the same buffer (thus, 15 nM FITC-Bak) with varying concentrations of either unlabeled peptide or experimental. Regression analysis was carried out using Origin (OriginLab, Northampton, MA) to fit the data to the Hill equation (1) to determine the initial binding affinity (KD) and the IC50 in the FPCA. For the fluorescence polarization competition titrations, an equation derived by Nikolovska-Coleska et al. was used to calculate the Ki from the IC50 data.49 The affinity of FITC-Bak for Mcl-1172-327 was determined to be 33.8 ± 0.50 nM in the assay conditions used.
Nuclear magnetic resonance spectroscopy
NMR spectra was collected at 25 °C with a Bruker AVANCE 800 NMR spectrometer (800.27 MHz for protons) equipped with pulsed-field gradients, four frequency channels, and triple resonance, z-axis gradient cryogenic probes. A one-second relaxation delay was used, and quadrature detection in the indirect dimensions was obtained with states-TPPI phase cycling; initial delays in the indirect dimensions were set to give zero- and first-order phase corrections of 90° and −180°, respectively.65,66 Data were processed using the processing program nmrPipe on Linux workstations.67 All proton chemical shifts are reported with respect to the H2O or HDO signal, taken to be 4.658 ppm relative to external TSP (0.0 ppm) at 37 °C. The 15N chemical shifts were indirectly referenced using the zero-point frequency at 37°C of 0.10132905 for 15N-1H, as previously described.68,69
Uniformly 15N-labeled Mcl-1 was used to collect two-dimensional 1H,15N-fast HSQC (heteronuclear single quantum coherence) spectra of Mcl-1 with and without compound to detect changes in the backbone 15N and 1H resonances of Mcl-1 due to the direct interaction with the compound.70 All compounds used for NMR were initially suspended in 100% d6-DMSO. The NMR samples contained 131 μM 15N-labeled Mcl-1, 182 μM compound, 20 mM HEPES, pH 6.8, 50 mM NaCl, 3 mM DTT, 20% D2O, and 5% d6-DMSO.
Cell titer-blue viability assay
A375 cells were plated at 5,000 cells/well seeding density in a 96 well black walled plate (Corning). Cells were cultured in 100μl DMEM (Gibco) plus 10% HIFBS and PenStrep overnight. Using a separate plate, serial dilutions were made of each compound in DMEM. 10μl of each serial dilution was transferred into the experimental plate so that the final compound concentrations were 4.69, 9.38, 18.75, 37.5, 75, 150, or 300 μM and 1% DMSO solvent. Untreated control cells also received DMSO vehicle at 1% final concentration. Cells were cultured with and without test compounds for 48 hours and then 20μl of Cell titer-blue reagent (Promega) was added to each well and to control wells containing media only to account for background. Cells were then incubated for 2 hours and fluorescence was read using Molecular Devices Spectra Max M5 (560Ex/590Em). Results were recorded as percent viability determined by dividing the experimental values by the untreated controls and subtracting the background from cell-free wells.
Supplementary Material
Scheme 5.
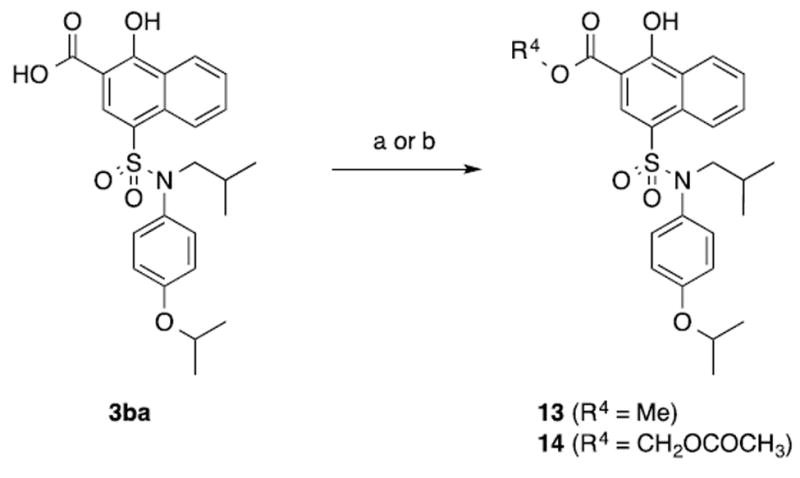
(a) SOCl2, MeOH, rt, 48 h; (b) BrCH2OCOCH3, K2CO3, MeCN, 0 °C, 16 h.
Acknowledgments
We thank the National Institutes of Health (T32GM066706 (MEL) and R43GM109635 (ADM)), the University of Maryland School of Pharmacy (SF), the University of Maryland Computer-Aided Drug Design Center and the Samuel Waxman Cancer Research Foundation (both to ADM) for financially supporting this research.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Complete computational analysis, including calculation of LGFE values. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Adams JM, Cory S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youle RJ, Strasser A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 3.Wang S. The Promise of Cancer Therapeutics Targeting the TNF-Related Apoptosis-Inducing Ligand and TRAIL Receptor Pathway. Oncogene. 2008;27:6207–6215. doi: 10.1038/onc.2008.298. [DOI] [PubMed] [Google Scholar]
- 4.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 5.Vogler M, Dinsdale D, Dyer MJS, Cohen GM. Bcl-2 Inhibitors: Small Molecules with a Big Impact on Cancer Therapy. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 6.Billard C. BH3 Mimetics: Status of the Field and New Developments. Mol Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 7.Lessene G, Czabotar PE, Colman PM. BCL-2 Family Antagonists for Cancer Therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 8.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, Rosenberg SH, Krivoshik AP, Enschede SH, Humerickhouse RA. Navitoclax, a Targeted High-Affinity Inhibitor of BCL-2, in Lymphoid Malignancies: a Phase 1 Dose-Escalation Study of Safety, Pharmacokinetics, Pharmacodynamics, and Antitumour Activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, Dive C, Enschede SH, Nolan C, Chiu YL, Busman T, Xiong H, Krivoshik AP, Humerickhouse R, Shapiro GI, Rudin CM. Phase I Study of Navitoclax (ABT-263), a Novel Bcl-2 Family Inhibitor, in Patients with Small-Cell Lung Cancer and Other Solid Tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, Chyla BJ, Dowell BL, Chakravartty A, Nolan CE, Rudersdorf N, Busman TA, Mabry MH, Krivoshik AP, Humerickhouse RA, Shapiro GI, Gandhi L. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients with Relapsed or Refractory Disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warr MR, Shore GC. Unique Biology of Mcl-1: Therapeutic Opportunities in Cancer. Curr Mol Med. 2008;8:138–147. doi: 10.2174/156652408783769580. [DOI] [PubMed] [Google Scholar]
- 13.Backus HH, van Riel JM, van Groeningen CJ, Vos W, Dukers DF, Bloemena E, Wouters D, Pinedo HM, Peters GJ. Rb, Mcl-1 and P53 Expression Correlate with Clinical Outcome in Patients with Liver Metastases From Colorectal Cancer. Ann Oncol. 2001;12:779–785. doi: 10.1023/a:1011112227044. [DOI] [PubMed] [Google Scholar]
- 14.Song L, Coppola D, Livingston S, Cress WD, Haura EB. Mcl-1 Regulates Survival and Sensitivity to Diverse Apoptotic Stimuli in Human Non-Small Cell Lung Cancer Cells. Cancer Biology & Therapy. 2004;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 15.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 Regulates Survival and Sensitivity to Diverse Apoptotic Stimuli in Human Non-Small Cell Lung Cancer Cells. Cancer Biology & Therapy. 2005;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 16.Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid Cell Leukemia-1 Inversely Correlates with Glycogen Synthase Kinase-3beta Activity and Associates with Poor Prognosis in Human Breast Cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 17.Y M, R H, M W, JU L, T K, K F, S T, S N, R D, M K, Y S, M I. Immunohistochemical Analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 Expression in Pancreatic Cancers. Oncology. 1998;56:73–82. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 18.Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F. The Expression of Mcl-1 in Human Cervical Cancer and Its Clinical Significance. Med Oncol. 2012;29:1985–1991. doi: 10.1007/s12032-011-0005-y. [DOI] [PubMed] [Google Scholar]
- 19.Brotin E, Meryet-Figuière M, Simonin K, Duval RE, Villedieu M, Leroy-Dudal J, Saison-Behmoaras E, Gauduchon P, Denoyelle C, Poulain L. Bcl-XL and MCL-1 Constitute Pertinent Targets in Ovarian Carcinoma and Their Concomitant Inhibition Is Sufficient to Induce Apoptosis. Int J Cancer. 2010;126:885–895. doi: 10.1002/ijc.24787. [DOI] [PubMed] [Google Scholar]
- 20.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A. Anti-Apoptotic Mcl-1 Is Essential for the Development and Sustained Growth of Acute Myeloid Leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, Deeb G, Dörken B, Thome M, Lenz P, Dirnhofer S, Hernandez-Ilizaliturri FJ, Tzankov A, Lenz G. MCL1 Is Deregulated in Subgroups of Diffuse Large B-Cell Lymphoma. Leukemia. 2013;27:1381–1390. doi: 10.1038/leu.2012.367. [DOI] [PubMed] [Google Scholar]
- 22.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of Apoptosis Sensitivity and Resistance to the BH3 Mimetic ABT-737 in Acute Myeloid Leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 23.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DCS. The BH3 Mimetic ABT-737 Targets Selective Bcl-2 Proteins and Efficiently Induces Apoptosis via Bak/Bax if Mcl-1 Is Neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Influence of Bcl-2 Family Members on the Cellular Response of Small-Cell Lung Cancer Cell Lines to ABT-737. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 Down-Regulation Potentiates ABT-737 Lethality by Cooperatively Inducing Bak Activation and Bax Translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 26.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJC, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers AJ. Potent and Selective Small-Molecule MCL-1 Inhibitors Demonstrate on-Target Cancer Cell Killing Activity as Single Agents and in Combination with ABT-263 (Navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei S-H, Dong K, Lin F, Wang X, Li B, Shen J-J, Zhang Q, Wang R, Zhang H-Z. Inducing Apoptosis and Enhancing Chemosensitivity to Gemcitabine via RNA Interference Targeting Mcl-1 Gene in Pancreatic Carcinoma Cell. Cancer Chemother Pharmacol. 2008;62:1055–1064. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 28.Friberg A, Vigil D, Zhao Bin, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. J Med Chem. 2012;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, Opferman JT, Walensky LD. A Competitive Stapled Peptide Screen Identifies a Selective Small Molecule That Overcomes MCL-1-Dependent Leukemia Cell Survival. Chem Biol. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap JL, Cao X, Vanommeslaeghe K, Jung K-Y, Peddaboina C, Wilder PT, Nan A, MacKerell AD, Smythe WR, Fletcher S. Relaxation of the Rigid Backbone of an Oligoamide-Foldamer-Based A-Helix Mimetic: Identification of Potent Bcl-xL Inhibitors. Org Biomol Chem. 2012;10:2928–2933. doi: 10.1039/c2ob07125h. [DOI] [PubMed] [Google Scholar]
- 31.Cao X, Yap JL, Newell-Rogers MK, Peddaboina C, Jiang W, Papaconstantinou HT, Jupitor D, Rai A, Jung KY, Tubin RP, Yu W, Vanommeslaeghe K, Wilder PT, MacKerell AD, Fletcher S, Smythe RW. The Novel BH3 A-Helix Mimetic JY-1-106 Induces Apoptosis in a Subset of Cancer Cells (Lung Cancer, Colon Cancer and Mesothelioma) by Disrupting Bcl-xL and Mcl-1 Protein-Protein Interactions with Bak. Mol Cancer. 2013;12:42. doi: 10.1186/1476-4598-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung K-Y, Vanommeslaeghe K, Lanning ME, Yap JL, Gordon C, Wilder PT, MacKerell AD, Fletcher S. Amphipathic A-Helix Mimetics Based on a 1,2-Diphenylacetylene Scaffold. Org Lett. 2013;15:3234–3237. doi: 10.1021/ol401197n. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z, Liu C, Li X, Song T, Wu Z, Liang X, Zhao Y, Shen X, Chen H. Fragment-Based Design, Synthesis, and Biological Evaluation of N-Substituted-5-(4-Isopropylthiophenol)-2-Hydroxynicotinamide Derivatives as Novel Mcl-1 Inhibitors. Eur J Med Chem. 2013;60:410–420. doi: 10.1016/j.ejmech.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z, Song T, Li X, Wu Z, Feng Y, Xie F, Liu C, Qin J, Chen H. Novel Soluble Myeloid Cell Leukemia Sequence 1 (Mcl-1) Inhibitor (E,E)-2-(Benzylaminocarbonyl)-3-Styrylacrylonitrile (4g) Developed Using a Fragment-Based Approach. Eur J Med Chem. 2013;59:141–149. doi: 10.1016/j.ejmech.2012.10.050. [DOI] [PubMed] [Google Scholar]
- 35.Ding X, Li Y, Lv L, Zhou M, Han L, Zhang Z, Ba Q, Li J, Wang H, Liu H, Wang R. De Novo Design, Synthesis and Evaluation of Benzylpiperazine Derivatives as Highly Selective Binders of Mcl-1. ChemMedChem. 2013;8:1986–2014. doi: 10.1002/cmdc.201300316. [DOI] [PubMed] [Google Scholar]
- 36.Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, Keller CE, Koenig M, He Y, Minond D, Mishra J, Cameron M, Spicer T, Hodder P, Cardone MH. Hydroxyquinoline-Derived Compounds and Analoguing of Selective Mcl-1 Inhibitors Using a Functional Biomarker. Bioorg Med Chem. 2013;21:6642–6649. doi: 10.1016/j.bmc.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petros AM, Swann SL, Song D, Swinger K, Park C, Zhang H, Wendt MD, Kunzer AR, Souers AJ, Sun C. Fragment-Based Discovery of Potent Inhibitors of the Anti-Apoptotic MCL-1 Protein. Bioorg & Med Chem Lett. 2014;24:1484–1488. doi: 10.1016/j.bmcl.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HDH, Nikolovska-Coleska Z. 3-Substituted-N-(4-Hydroxynaphthalen-1-Yl)Arylsulfonamides as a Novel Class of Selective Mcl-1 Inhibitors: Structure-Based Design, Synthesis, SAR, and Biological Evaluation. J Med Chem. 2014;57:4111–4133. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Lanning ME, Fletcher S. Small-Molecule Inhibitors of the Mcl-1 Oncoprotein. Austin J Anal Pharm Chem. 2015;1:1015. [Google Scholar]
- 40.Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, Erickson S, Fidanze S, Fry E, Hasvold L, Jenkins GJ, Jin S, Judge RA, Kovar PJ, Madar D, Nimmer P, Park C, Petros AM, Rosenberg SH, Smith ML, Song X, Sun C, Tao ZF, Wang X, Xiao Y, Zhang H, Tse C, Leverson JD, Elmore SW, Souers AJ. Structure-Guided Design of a Series of MCL-1 Inhibitors with High Affinity and Selectivity. J Med Chem. 2015;58:2180–2194. doi: 10.1021/jm501258m. [DOI] [PubMed] [Google Scholar]
- 41.Murayama R, Sato K, Kawakami S. Polymerizable Monomer, Polymeric Compound, Polymerizable Monomer, Polymeric Compound …. 2012 [Google Scholar]
- 42.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady ASA, Gulappa T, Cierpicki T, Owens S, Zhang T, Sun D, Stuckey JA, Mohammad RM, Nikolovska-Coleska Z. A Novel Small-Molecule Inhibitor of Mcl-1 Blocks Pancreatic Cancer Growth in Vitro and in Vivo. Mol Cancer Ther. 2014;13:565–575. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guvench O, MacKerell AD. Computational Fragment-Based Binding Site Identification by Ligand Competitive Saturation. PLoS Comput Biol. 2009;5:e1000435. doi: 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raman EP, Yu W, Guvench O, MacKerell AD. Reproducing Crystal Binding Modes of Ligand Functional Groups Using Site-Identification by Ligand Competitive Saturation (SILCS) Simulations. J Chem Inf Model. 2011;51:877–896. doi: 10.1021/ci100462t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raman EP, Yu W, Lakkaraju SK, MacKerell AD. Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. J Chem Inf Model. 2013;53:3384–3398. doi: 10.1021/ci4005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu W, Lakkaraju SK, Raman EP, MacKerell AD. Site-Identification by Ligand Competitive Saturation (SILCS) Assisted Pharmacophore Modeling. J Comput Aided Mol Des. 2014;28:491–507. doi: 10.1007/s10822-014-9728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu W, Lakkaraju SK, Raman EP, Fang L, MacKerell AD. Pharmacophore Modeling Using Site-Identification by Ligand Competitive Saturation (SILCS) with Multiple Probe Molecules. J Chem Inf Model. 2015;55:407–420. doi: 10.1021/ci500691p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raman EP, Yu W, Lakkaraju SK, MacKerell AD., Jr Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. J Chem Inf Model. 2013;53:3384–3398. doi: 10.1021/ci4005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Nimmer P, Rosenberg SH, Ng SC, Joseph M. Development of a High-Throughput Fluorescence Polarization Assay for Bcl-X(L) Anal Biochem. 2002;307:70–75. doi: 10.1016/s0003-2697(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 50.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 51.Pearlman DA, Charifson PS. Are Free Energy Calculations Useful in Practice? a Comparison with Rapid Scoring Functions for the P38 MAP Kinase Protein System. J Med Chem. 2001;44:3417–3423. doi: 10.1021/jm0100279. [DOI] [PubMed] [Google Scholar]
- 52.Yang C-Y, Wang S. Analysis of Flexibility and Hotspots in Bcl-xL and Mcl-1 Proteins for the Design of Selective Small-Molecule Inhibitors. ACS Med Chem Lett. 2012;3:308–312. doi: 10.1021/ml200301w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKee CS, Hill DS, Redfern CP, Armstrong JL, Lovat PE. Oncogenic BRAF Signalling Increases Mcl-1 Expression in Cutaneous Metastatic Melanoma. Exp Dermatol. 2013;22:767–769. doi: 10.1111/exd.12254. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Chauhan J, Hu A, Pendleton K, Yap JL, Sabato PE, Jones JW, Perri M, Yu J, Cione E, Kane MA, Fletcher S, Prochownik EV. Disruption of Myc - Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecuel 10074-G5. Oncotarget. 2013;4:936–947. doi: 10.18632/oncotarget.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 56.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, Flexible, and Free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 57.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone Φ, Ψ and Side-Chain X(1) and X(2) Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD. CHARMM General Force Field: a Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu W, He X, Vanommeslaeghe K, MacKerell AD. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J Comput Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. The Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 61.Molecular Operating Environment (MOE), Version 2012.10. Chemical Computing Group Inc; Montreal, Canada: 2012. [Google Scholar]
- 62.Percec V, Okita S, Wang JH. Synthesis of Aromatic Polyethers by Scholl Reaction. VI. Aromatic Polyethers by Cation-Radical Polymerization of 4,4′-, 3,3′-, and 2,2′-Bis(1-naphthoxy)biphenyls and of 1,3-Bis(1-naphthoxy)benzene. Macromolecules. 1992;25:64–74. [Google Scholar]
- 63.Szainman SH, Yan W, Bailey BN, Docampo R, Elhalem E, Rodriguez JB. Design and Synthesis of Aryloxyethyl Thiocyanate Derivatives as Potent Inhibitors of Trypanosoma cruzi Proliferation. J Med Chem. 2000;43:1826–1840. doi: 10.1021/jm9905007. [DOI] [PubMed] [Google Scholar]
- 64.Raiford LC, Thiessen GW, Wernert IJ. Some Derivatives of Diphenyl Ether. J Am Chem Soc. 1930;52:1205–1209. [Google Scholar]
- 65.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the Overlap Problem in the Assignment of 1H NMR Spectra of Larger Proteins by Use of Three-Dimensional Heteronuclear 1H-15N Hartmann-Hahn-Multiple Quantum Coherence and Nuclear Overhauser-Multiple Quantum Coherence Spectroscopy: Application to Interleukin 1 Beta. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 66.Bax A, Ikura M. An Efficient 3D NMR Technique for Correlating the Proton and 15N Backbone Amide Resonances with the Alpha-Carbon of the Preceding Residue in Uniformly 15N/13C Enriched Proteins. J Biomol NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 67.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 68.Edison AS, Abildgaard F, Westler WM, Mooberry ES, Markley JL. Practical Introduction to Theory and Implementation of Multinuclear, Multidimensional Nuclear Magnetic Resonance Experiments. Meth Enzymol. 1994;239:3–79. doi: 10.1016/s0076-6879(94)39003-7. [DOI] [PubMed] [Google Scholar]
- 69.Spera S, Ikura M, Bax A. Measurement of the Exchange Rates of Rapidly Exchanging Amide Protons: Application to the Study of Calmodulin and Its Complex with a Myosin Light Chain Kinase Fragment. J Biomol NMR. 1991;1:155–165. doi: 10.1007/BF01877227. [DOI] [PubMed] [Google Scholar]
- 70.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved Sensitivity of HSQC Spectra of Exchanging Protons at Short Interscan Delays Using a New Fast HSQC (FHSQC) Detection Scheme That Avoids Water Saturation. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.