

Abstract
Introduction
Multiple myeloma is a clonal malignancy of plasma B cells. While recent advances have improved overall prognosis, virtually all myeloma patients still succumb to relapsing disease. Therefore, novel therapies to treat this disease remain urgently needed. We have recently demonstrated that treatment of human multiple myeloma cells with an oncolytic virus known as myxoma results in rapid cell death even in the absence of viral replication; however, the specific mechanisms and pathways involved remain unknown.
Materials and Methods
To determine how Myxoma virus eliminates human multiple myeloma cells, we queried the apoptotic pathways which were activated following viral infection using immunoblot and other cell biology approaches.
Results
Our results indicate that myxoma virus infection initiates apoptosis in multiple myeloma cells through activation of the extrinsic initiator caspase-8. Caspase-8 activation subsequently results in cleavage of Bid and loss of mitochondrial membrane potential causing secondary activation of caspase-9. Activation of caspase-8 appears to be independent of extrinsic death ligands and instead correlates with depletion of cellular inhibitors of apoptosis. We hypothesize that this depletion results from virally mediated host-protein shutoff since a myxoma construct which overexpresses the viral decapping enzymes displays improved oncolytic potential.
Conclusion
Taken together, these results suggest that myxoma virus eliminates human multiple myeloma cells through a pathway unique to oncolytic poxviruses making it an excellent therapeutic option for the treatment of relapsed or refractory patients.
Keywords: oncolytics, caspase-8, host-protein shutoff, cIAPs, resistance
Introduction
Multiple myeloma (MM) is a clonal malignancy of plasma cells which is diagnosed in ~24,000 new patients each year 1, 2. Recent advances in the treatment of MM, including the development of novel chemotherapeutic agents such as bortezomib 3-5, has significantly improved initial prognosis; however, the disease displays a high degree of genetic heterogeneity and virtually all patients eventually succumb to relapse. Therefore, even with the best available treatments, the 5-year survival rate for newly diagnosed MM patients remains only 45%, indicating that novel treatment modalities are urgently needed 2.
One treatment option which has recently shown promise is the use of oncolytic viruses 6-10. Treatment with these viruses, including: reovirus 11, 12, recombinant measles 13-15, vesicular stomatitis virus 16-18, and adenovirus 19, 20, is highly cancer specific, reducing the chances of off target toxicities. Additionally, oncolytic viruses often eliminate infected cancer cells through virally distinct mechanisms, which makes them particularly attractive for the treatment of relapsed and/or refractory MM patients. Indeed, a recent, well-published human trial demonstrated that recombinant, oncolytic measles virus could induce complete remission even in late-stage, chemotherapy-refractive relapsed MM patients 21.
Our lab is interested in the clinical potential of an oncolytic poxvirus known as myxoma (MYXV). MYXV has several advantages as an oncolytic agent. First, infection is highly restricted to lagomorphs (rabbits) 22. No instance of MYXV infection of viral disease has ever been noted in any non-rabbit species. This provides MYXV with an excellent safety profile as well as eliminating the possibility of patients presenting with preexisting humoral immunity to the virus. Second, the virus is highly lytic and generally replicates to excellent titers in vitro suggesting feasible clinical translation.
We have previously demonstrated that treatment with MYXV can effectively kill human MM cell-lines as well as primary MM cells found in patient bone marrow samples. Elimination of MM cells was highly efficient and treatment of MM contaminated autologous transplant samples with MYXV prior to transplantation was sufficient to prevent disease relapse in animal models 23. Interestingly, while most oncolytic viruses eliminate infected cells through direct viral lysis, elimination of malignant MM cells by MYXV was independent of viral replication and instead appeared to occur through virally mediated induction of programmed cell death 23. The specific pathways involved in the induction of this programmed cell death, however, remained unknown.
Since resistance to treatment is a major clinical challenge in MM, and the development of this resistance depends on the specific pathways through which a treatment eliminates malignant cells, we sought to characterize the molecular pathways through which MYXV induces programmed cell death in infected MM cells with the goal of identifying how these pathways might influence treatment of relapsed or refractory patients.
Methods
Cells and reagents
U266 and MM1.S cells were purchased from ATCC (Manassas, VA, USA), RPMI-8226 cells were obtained from Dr. Bei Lu at the Medical University of South Carolina. Cells were cultured in RPMI-1640 supplemented with 20% fetal bovine serum and Penicillin/Streptomycin/Glutamine (Mediatech, Manassas, VA, USA). Cells were maintained between 0.2 – 0.8×106 cells/mL with no more than 10×106 cells/T175 flask. z-VAD-fmk, z-DEVD-fmk, z-LEHD-fmk, and z-IETD-fmk (BD Biosciences, San Jose, CA, USA) were used at a final concentration of 20µM. GSK2606414 (Calbiochem, Billerica, Massachusetts, USA) was used at a final concentration of 10nM. The following antibodies were used in these studies: Casp-8 (12F5) (Enzo Life Sciences Inc., Farmingdale, NY, USA); BID (2002), Casp-2 (2224), Casp-9 (9502), Casp-10 (9752), Mcl-1 (5453), PARP (9542), Survivin (2808), TNFR1 (3736), FAS (8023), DR5 (8074), and XIAP (2045) (Cell Signaling Technology, Beverly, MA, USA); FLIPS/L (sc5276), Actin (sc1615), eIF2α (sc11386), and p-eIF2α (sc101670) (Santa Cruz Biotechnology, Dallas, Texas, USA).
Virus and Viral infections
vMYX-GFP was a kind gift from Dr. Grant McFadden and was grown and purified as previously described 24. A viral construct overexpressing the viral decapping enzymes M084 and M085 (vMYX-ΔM083) was constructed in our lab and is described elsewhere 25. Unless otherwise noted, infections were done by concentrating cells to 10×106 cells/ml and then infecting at a multiplicity of Infection (MOI)=10 for 30 minutes at 37°C. After infection, cells were dilute d to a concentration of <1×106 cells/ml with fresh media. All drug treatments were done by pretreating cells with drug for >30 minutes prior to infection and subsequently diluting cells in complete media containing additional fresh drug.
MM Patient Samples
Bone marrow aspirates from patients with various stages of MM were obtained through the Medical University of South Carolina biorepository in accordance with institutional IRB guidelines. Red blood cells were removed through ACK lysis and then samples were infected with vMYXV-GFP at an MOI=10 as detailed above.
Western Blots
Cell lysates were generated at a concentration of 1×106 cells/100ul of Laemlli sample buffer. Samples were separated on SDS-PAGE gels, and subsequently transferred to PVDF. Membranes were blocked for 30 minutes with 5% non-fat dry milk (Mix’n Drink, SACO, Middleton, WI, USA ) in TBS-T (25mM Tris, 150mM Nacl, 2mM KCl, 0.1% Tween20 pH 7.4), and then incubated overnight at 4°C with primary antibody diluted in 1% non-fat dry milk in TBS-T. Appropriate secondary antibody incubations were done at 4°C for one hour, and blots were treat ed with chemiluminescent substrate (Pierce Biotechnology, Rockford, IL, USA) before exposure to film.
Immune Precipitations
2×107 cells were used per condition. Cells were lysed with 1ml cold precipitation buffer (20mM Tris pH 7.4, 150mM NaCl, 2mM EDTA, 5% Glycerol, 1% NP40, Protease Inhibitor Cocktail) for 20 minutes at 4°C. Samples were then centrifuged at 15,000rpm in a pre-chilled centrifuge for 10min to remove large cell debris. The supernatant was placed into a fresh tube containing 30µl of equilibrated Protein A/G beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and rocked on ice for 30 minutes. Samples were centrifuged and the precleared supernatant was transferred to a fresh tube containing 10μl of primary antibody. Samples were incubated for 45 minutes on ice. 30μl of equilibrated Protein A/G beads were then added and samples were rocked for an additional 45 minutes. Bead beds were washed five times with 500μl cold precipitation buffer, and then eluted in Laemmli Sample Buffer.
Flow cytometry
For analysis of mitochondrial membrane potential, U266 cells were labeled with 3μM Tetramethylrhodamine, methyl ester, perchlorate (TMRM) (Life Technologies, Grand Island, NY, USA) in complete media for 30min at 37°C. Cells were then pelleted and resuspended in complete media containing 1μM TMRM. At the indicated times samples were pelleted, resuspended in 2% paraformaldehyde in phosphate buffered saline (PBS), and analyzed using flow cytometer. For analysis of cell surface phosphoserine exposure, U266 cells were infected with vMYX-GFP as indicated above for six hours. Cells were then pelleted, resuspended in binding buffer (100mM HEPES pH 7.4, 140mM NaCl, 25mM CaCl2), and labeled with APC-Annexin V (BD Biosciences, San Jose, CA, USA) for 30 minutes at 4°C. Cells were then washed with PBS, stained with near-IR LIVE/DEAD Vital Dye (Life Technologies, Grand Island NY, USA) and analyzed using flow cytometry. All flow cytometry analysis was conducted using a FACSCalibur machine (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJoX (FlowJo, LLC, Ashland, OR, USA).
Other Assays
Enzymatic caspase activity was measured using Caspase-Glo kits (Promega, Madison, WI, USA) according to the manufacturer instructions. In brief, MYXV infected cells were collected over time and lysed in the caspase activity reagent, and samples were read on a FLUOstar Optima Plate reader (BMG LABTECH Inc, Cary, NC, USA). Cell viability experiments were performed using CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) (Promega, Madison, WI, USA) according to the manufacturer instructions and read on a FLUOstar Optima Plate reader.
Results
MYXV induces apoptosis in a variety of MM cell lines
Our previous results had suggested that MYXV eliminated infected human U266 MM cells by inducing a rapid apoptotic response 23. However, whether this mechanism occurred in other infected MM cells remained unknown. We therefore asked whether MYXV induced phenotypic markers of apoptosis in three different human MM cell lines. U266, MM1.S, or RPMI-8226 human MM cells were mock-treated or infected with MYXV at an MOI=10. Two hours post infection, cells were harvested and exposure of phosphotidyl serine to the exterior side of the plasma membrane was determined by staining with annexin-V. Early apoptotic cells were further identified from dead and/or necrotic cells by staining cells with a membrane impermeable vital dye (Figure 1A). Consistent with MYXV inducing apoptosis, we observed that viral treatment induced a rapid increase in annexin-V+/Vital Dye− cells in all three cell lines. In contrast, at this early time point, the numbers of dead and/or necrotic cells remained largely constant. To further determine whether all infected MM cells progressed through complete apoptosis, we next asked whether viral treatment induced cleavage of the late stage apoptotic marker PARP. U266, MM1.S, or RPMI-8226 human MM cells were mock-treated or infected with MYXV at an MOI=10. Six hours post infection, cells were harvested and the cleavage status of PARP was analyzed using immunoblot (Figure 1B). In mock-treated cells, virtually all cellular PARP existed as a full length protein. In contrast, infection with MYXV induced robust cleavage of PARP in all three tested cell lines. Taken together, these data suggest that MYXV induced MM cell apoptosis is a general phenomenon and is not unique to individual MM clones.
Figure 1. MYXV induces apoptosis in infected MM cells.

(A) Human U266, RPMI-8226, or MM.1S cells were either mock-treated or infected with MYXV at MOI=10. Two hours post-infection, cells were stained with annexin-V and Live/Dead vital dye to identify apoptotic cells. (B) Human U266, RPMI-8226, or MM.1S cells were either mock-treated or infected with MYXV at MOI=10. 24 hours post-infection, cells were harvested and samples were separated on a SDS-PAGE gel. The presence and cleavage status of PARP was then analyzed by immunoblot.
MYXV induces apoptosis in primary MM cells
Our previous results indicated that MYXV infection induced apoptosis in established MM cell lines (Figure 1 and 23). We next wished to determine whether MYXV also induced apoptosis in primary MM cells. It has been previously reported that primary MM cells undergoing apoptosis rapidly lose surface expression of CD138 26. Consistent with these results, we observed that human U266 MM cells treated with either UV light or infected with vMYX-GFP displayed a significant reduction in CD138 surface expression as early as six hours after treatment (Figure 2A). To assess the induction of apoptosis in primary MM cells, we therefore infected bone marrow aspirates from MM patients with vMYX-GFP at an MOI = 10 and assayed the surface expression of CD138 after six hours. In mock treated samples, we observed two distinct populations of CD138+ cells. CD138hi cells, identified as viable malignant MM cells, and CD138lo cells, identified as normal plasma B cells (Figure 2B). In MYXV treated samples, the population of CD138lo normal plasma cells remained readily identifiable and the number of these cells and their CD138 expression appeared similar to those observed in mock treated samples. In contrast, MYXV samples displayed a loss of virtually all CD138hi cells and the gain of a new population of infected (GFP+) CD138lo cells (Figure 2B). This result, which is consistent with primary MM cells undergoing rapid apoptosis following infection with MYXV, was observed in 100% (8/8) of tested patient samples (Figure 2C).
Figure 2. MYXV indices apoptosis in primary MM cells.
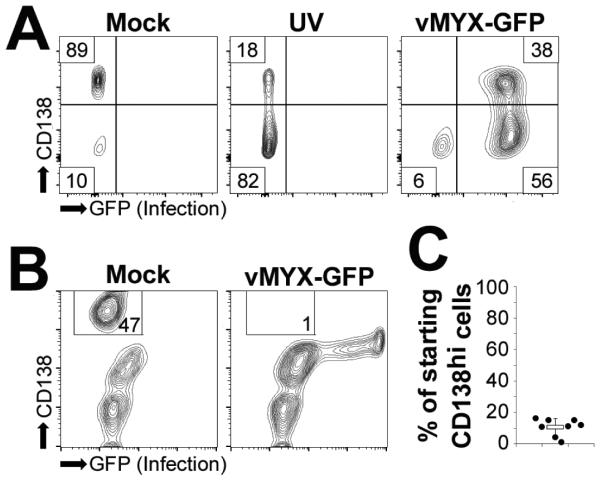
(A) Human U266 MM cells were either mock-treated, subjected to UV light for one hour, or infected with vMYX-GFP at an MOI=10. Six hours after treatment, cell surface expression of CD138 was analyzed using flowcytometry. (B) Human bone marrow aspirates depleted of red blood cells were either mock-treated or infected with vMYX-GFP at an MOI=10. Six hours post-infection cell surface expression of CD138 was analyzed using flowcytometry. (C) Quantitation of ‘B’. Data presented as percent of starting CD138hi cells which was calculated by comparing the number of CD138hi cells in mock and vMYX-GFP infected samples.
MYXV induces multiple apoptotic pathways
Apoptosis is an incredibly complex form of cell death which can occur through a variety of distinct pathways. While these pathways all converge during the late stages of apoptosis, they are initiated by a relatively small number of distinct caspases 27 including: Casp-2 (DNA damage induced apoptosis 28, 29), Casp-8 (extrinsic apoptosis 30), Casp-9 (intrinsic apoptosis 31) and potentially Casp-10 32, 33. Each of these caspases exists as an inactive, full-length precursor which is activated through proteolytic cleavage during apoptotic initiation. In order to determine which apoptotic pathways might be involved in the MYXV-induced death of MM cells, we therefore asked which of these initiator caspases was cleaved early after viral infection. Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. Six hours post-infection, cells were harvested and cleavage of each initiator caspase was determined using immunoblot (Figure 3). In mock-treated cells, all four initiator caspases existed as inactive full length precursors. Six hours after infection with MYXV, however, cleavage products of both Casp-8 and Casp-9 could be readily observed. In contrast, no cleavage of Casp-2 or Casp-10 was seen at this time point. These data suggest that MYXV induces both intrinsic and extrinsic apoptosis in infected MM cells.
Figure 3. MYXV activates both intrinsic and extrinsic apoptosis.
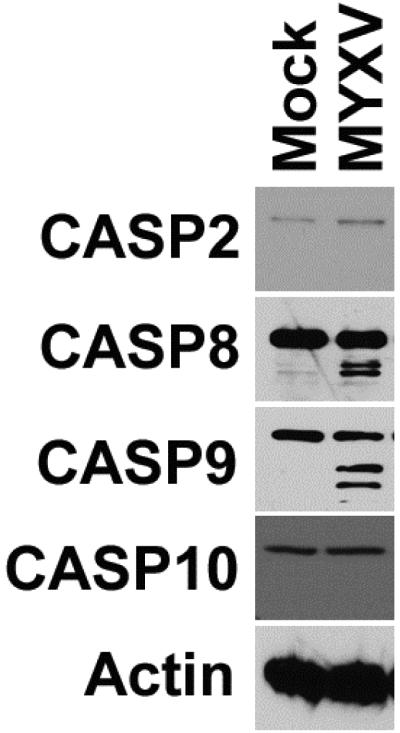
Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. 24 hours post-infection, cells were harvested and samples were separated on a SDS-PAGE gel. The presence and cleavage status of the initiator caspases Casp-2, Casp-8, Casp-9, and Casp-10 was determined using immunoblot.
MYXV initiates apoptosis through activation of Casp-8
Our previous data suggested that MYXV infection resulted in activation of both intrinsic and extrinsic apoptosis in infected human MM cells. Previous work, however, has demonstrated that these two pathways can overlap through various feedback and cross-talk mechanisms 34-38. We therefore wished to determine whether MYXV initiated activation of intrinsic and extrinsic apoptosis through distinct mechanisms or if primary activation of one pathway induced secondary activation of the other pathway. To address this question, we initially queried the kinetics of activation of both Casp-8 and Casp-9. Human U266 MM cells were either mock-treated or infected with MYXV at MOI=10. At the indicated times post-infection, cells were harvested and the enzymatic activity of both Casp-8 and Casp-9 were determined (Figure 4A). The results demonstrated that enzymatic activity of Casp-8 began to increase in MYXV infected cells between 3 and 3.5 hours post infection. In contrast, enzymatic activity of Casp-9 did not begin to increase until 4 hours post-infection suggesting that Casp-8 activation occurred prior to activation of Casp-9. To determine whether this difference was merely due to slower activation kinetics of Casp-9 or truly represented sequential activation of each pathway, we next asked how inhibition of either Casp-8 or Casp-9 affected the cleavage of the other. Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10 in the presence of various small molecular caspase inhibitors, including: zVAD-fmk (pan-caspase inhibitor), v-DEVD-fmk (Casp-3 inhibitor), v-IETD-fmk (Casp-8 inhibitor), or v-LEHD-fmk (Casp-9 inhibitor). Six hours after infection, cells were harvested and the cleavage of both Casp-8 and Casp-9 were assayed using immunoblot (Figure 4B). The results indicated that inhibition of either, all caspases or Casp-8 specifically, was sufficient to completely block cleavage of Casp-9. In contrast, inhibition of all caspases or Casp-9 specifically, had no effect on the cleavage of Casp-8. Inhibition of the downstream effector Casp-3 had no effect on cleavage of either Casp-8 or Casp-9 indicating that these results were not due to non-specific defects in apoptosis following caspase inhibition. These results suggested a two part mechanism in which MYXV infection initially induces extrinsic apoptosis through activation of Casp-8 and that this activation subsequently induces secondary activation of intrinsic apoptosis and cleavage of Casp-9.
Figure 4. MYXV initiates apoptosis through activation of Casp-8.
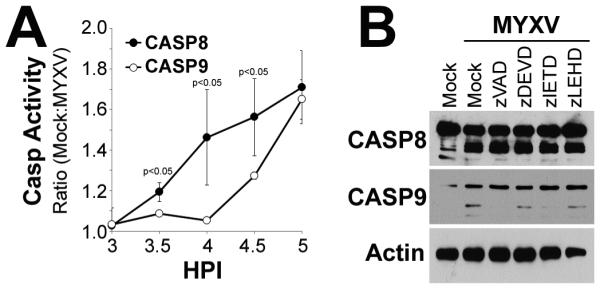
(A) Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were lysed and the activity of both Casp-8 and Casp-9 were measured using Caspase-Glo™ analysis. Data is presented as the ratio of signal from mock-treated and infected cells. (B) Human U266 MM cells were pretreated with the indicated caspase inhibitors for 30 minutes and then either mock-treated or infected with MYXV at an MOI=10. Six hours post-infection, cells were harvested and samples were separated on a SDS-PAGE gel. The presence and cleavage status of both Casp-8 and Casp-9 was then determined using immunoblot.
Virally activated Casp-8 mediates loss of mitochondrial membrane potential through cleavage of BID, but this is not required for completion of apoptosis
One of the major mechanisms of cross-talk between the intrinsic and extrinsic apoptotic pathways is through Casp-8 mediated cleavage of BID 34, 35, 39. Once cleaved, BID translocates to the mitochondrial where it induces loss of mitochondrial membrane potential and activation of Casp-9. To determine whether this mechanism might be responsible for the secondary activation of intrinsic apoptosis following MYXV infection, we asked whether virally infected cells displayed evidence of caspase mediated cleavage of BID. Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10 in the presence of the pan-caspase inhibitor z-VAD-fmk. At the indicated times post-infection, cells were harvested and the expression of full-length BID was assayed by immunoblot (Figure 5A). We observed that MYXV infected cells displayed reduced expression of full-length BID beginning approximately four hours-post infection. This loss of BID was not observed in cells pretreated with z-VAD-fmk indicating that it was caused by caspase mediated cleavage. To confirm whether cleavage of BID played a role in activation of intrinsic apoptosis, we next asked how mitochondrial membrane potential was altered by MYXV infection in the presence and absence of caspase activity. Human U266 MM cells were labeled with the mitochondrial membrane potential dye TMRM for 30 minutes in either the presence of absence of z-VAD-fmk. Cells were then mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were harvested and their mitochondrial membrane potential was analyzed by detecting TMRM fluorescence using flowcytometry (Figure 5B). Consistent with MYXV inducing secondary intrinsic apoptosis, we observed that virally infected cells began to lose TMRM fluorescence around four hours post-infection. This loss of TMRM fluorescence, however, was greatly reduced in the presence of z-VAD-fmk. Taken together, these data suggest that Casp-8 mediates secondary activation of intrinsic apoptosis through proteolytic cleavage of BID resulting in loss of mitochondrial membrane potential.
Figure 5. MYXV activates secondary intrinsic apoptosis through cleavage of BID.

(A) Human U266 MM cells were either untreated or pretreated with z-VAD-fmk for 30 minutes and then either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection cells were harvested and samples were separated on a SDS-PAGE gel. The presence of full length BID was then determined using immunoblot. (B) Human U266 MM cells were pre-labeled with TMRM for 30 minutes in either the presence or absence of z-VAD-fmk. Cells were then mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection cells were harvested and mitochondrial membrane potential analyzed by detecting TMRMhi cells using flowcytometry. (C) Human U266 MM cells were pretreated with the indicated caspase inhibitors for 30 minutes and then either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were harvested and samples were separated on a SDS-PAGE gel. The presence and cleavage status of PARP was then determined using immunoblot.
Our data suggested that viral activation of Casp-8 was required to induce apoptosis in infected MM cells. We next wanted to determine whether this activation was also sufficient to induce apoptosis or if secondary activation of Casp-9 was also required. We therefore asked how specific inhibition of Casp-8 or Casp-9 impacted the completion of the apoptotic cascades in infected cells. Human U266 MM cells were pretreated with either z-IETD-fmk (Casp-8 inhibitor) or z-LEHD-fmk (Casp-9 inhibitor) for 30 minutes. Cells were then either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were harvested and completion of the apoptotic cascade was determined by assaying the cleavage of the effector molecule PARP (Figure 5C). Consistent with our previous work (Figures 1, 4, and 23) MYXV infection induced cleavage of PARP around 6-8 hours post-infection. This cleavage was largely prevented by pretreatment of cells with the Casp-8 inhibitor, v-IETD-fmk. In contrast, pretreatment of cells with the Casp-9 inhibitor z-LEHD-fmk had virtually no effect on PARP cleavage. Taken together, these data suggest that viral activation of the extrinsic apoptotic pathway is both necessary and sufficient to induce complete apoptosis in infected human MM cells.
MYXV activates a variety of extrinsic death receptors by depleting cellular inhibitors of apoptosis
Extrinsic apoptosis is initiated through activation of any one of a number of cellular death receptors including: TNFR1 and TNFR2, FasR, DR3 (tweak receptor), DR4/5 (TRAIL receptors), and DR6 40-46. Typically these receptors are activated through ligation of their cognate ligands, which induces receptor trimerization and recruitment of Casp-8. Following recruitment and activation, Casp-8, along with the adaptor molecules FADD or TRADD, then disassociate from the activated death receptor forming the functional death-inducing signaling complex (DISC). In order to determine the mechanism through which MYXV might activate extrinsic apoptosis in infected MM cells, we therefore asked which death receptors displayed association with Casp-8 following viral infection. Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. After three hours, cells were lysed and Casp-8 was immunoprecipitated. Association of various death receptors with Casp-8 was then determined by immunoblotting precipitated samples for TNFR1, FasR, DR5 (Trail receptor), and DR3 (Tweak receptor) (Figure 6A). We observed that, in mock-treated samples, Casp-8 was associated at low levels with TNFR1, FasR, and DR5. The association of all three of these receptors with Casp-8, however, was greatly increased in cells infected with MYXV. Association of Casp-8 with DR3 was not observed, however, this receptor was not detected in mock-treated or virally infected whole cell lysates suggesting it is not expressed in U266 MM cells (data not shown). To further determine whether initial association of Casp-8 with each death receptor resulted in functional activation, we next asked whether Casp-8 remained associated with each receptor over time or whether it disassociated into the DISC. Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. After six hours, cells were lysed and the indicated death receptors were immunoprecipitated. Association of each death receptors with Casp-8 was then determined by immunoblotting precipitated samples for Casp-8 (Figure 6B). In contrast to our results at three hours, we observed that at six hours post-infection, virtually no Casp-8 remained associated with TNFR1, FasR, or DR5. Since this disassociation correlated kinetically with the appearance of enzymatically active Casp-8 (Figure 4A), these data suggest that MYXV infection initiates extrinsic apoptosis by activating multiple cellular death receptors.
Figure 6. MYXV activates multiple extrinsic pathways.

(A) Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. Three hours post-infection, cells were lysed and Casp-8 was immunoprecipitated. Immunoprecipitated samples were then separated on a SDS-PAGE gel and association of Casp-8 with the indicated death receptors was analyzed using immunoblot. (B) Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. Six hours post-infection, cells were lysed and the indicated death receptors were immunoprecipitated. Immunoprecipitated samples were then separated on a SDS-PAGE gel and association of each death receptor with Casp-8 was analyzed using immunoblot. (C) Human U266 MM cells were either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were lysed and samples separated on a SDS-PAGE gel. The expression of the indicated cIAPs was then analyzed using immunoblot.
Each death receptor is normally activated through ligation of a distinct ligand. However, it seemed unlikely that MYXV infection would provide distinct ligands for three separate death receptors. We therefore hypothesized that a ligand-independent mechanism might be involved. Typically, extrinsic apoptosis is tightly controlled by a series of cellular inhibitors of apoptosis (cIAPs), such as XIAP, survivin, and cFLIP. Depletion of these cIAP’s can significantly enhance induction of extrinsic apoptosis and is even sufficient to directly induce activation of Casp-8 in some models 47-50. Additionally, we have previously demonstrated that MYXV infection inhibits expression of the cIAP Mcl1 in the context of an activated unfolded protein response (UPR) 51. We therefore asked whether viral infection might also reduce expression of other cIAPs. Human U266 MM cells were either mock-treated or infected at an MOI=10. At the indicated times post-infection, cells were harvested and the expression of the indicated cIAP’s were analyzed using immunoblot (Figure 6C). The results indicated that Mcl1, XIAP, and cFLIP were all rapidly depleted in MYXV infected cells. In contrast, levels of survivin remained largely stable over the course of eight hours. These results indicate that depletion of cIAPs correlates with the viral activation of Casp-8 and suggest a possible mechanism through which MYXV might induce ligand independent extrinsic apoptosis.
MYXV does not deplete cIAPs through the UPR
Many cIAPs are short lived proteins which can be depleted during prolonged periods of translational arrest 52-54. Our previous work has demonstrated that MYXV infection induces translational arrest during an active UPR by causing PERK mediated phosphorylation of eIF2α 51, 55. We therefore asked whether depletion of cIAPs might be caused by viral activation of PERK. Human U266 MM cells were pretreated with the PERK specific inhibitor GSK2606414 and then either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were harvested and the phosphorylation status of eIF2α as well as expression of the cIAP Mcl1 was analyzed using immunoblot (Figure 7A). Consistent with previous results, treatment of U266 cells with GSK2606414 prevented virally induced phosphorylation of eIF2α. Surprisingly, however, drug treatment did not prevent general depletion of cIAP levels. Additionally, GSK2606414 failed to prevent cleavage of Casp-8, Casp-9, or Casp-3 (data not shown) and did not prevent loss of MM cell viability (Figure 6B) suggesting that MYXV-based activation of PERK was not responsible for depletion of cIAPs or the induction of extrinsic apoptosis.
Figure 7. Viral deletion of cIAPs is not mediated by activation of the UPR.

(A) Human U266 MM cells were pretreated with GSK2606414 for 30 minutes and then either mock-treated or infected with MYXV at an MOI=10. At the indicated times post-infection, cells were harvested and the levels of the indicated proteins was analyzed using immunoblot. (B) Human U266 MM cells were pretreated with GSK2606414 for 30 minutes and then either mock-treated or infected with MYXV at an MOI=10. Cellular viability was analyzed after 24 hours.
A MYXV construct with enhanced host-protein shut-off displays increased depletion of cIAPs and improved oncolytic capacity
A second mechanism through which MYXV might deplete cIAPs is by globally preventing protein translation through a well characterized phenomenon known as host-protein shutoff 56. In MYXV, host-protein shutoff is mediated by the two poxviral decapping enzymes, M084 and M085 57. These enzymes remove the 5’ cap from viral and cellular mRNA’s thus preventing their translation 58, 59. We have recently characterized a MYXV mutant (vMYX-ΔM083) which has enhanced expression of M084 and M085 due to the local insertion of a strong poxviral promoter 25. In order to assess the role of host-protein shutoff in depletion of cIAPs we therefore asked how expression of these proteins was altered in cells infected by either ‘normal’ MYXV (vMYX-GFP) or vMYX-ΔM083. Human U266 MM cells were either mock-treated or infected with vMYX-GFP or vMYX-ΔM083 at inceasing MOI’s. Two hours post-infection, cells were harvested and expression of the indicated proteins was determined using immunoblot (Figure 8A). The results indicated that, at this early time point, vMYX-GFP did not induce significant depletion of cIAPs at any MOI. In contrast, infection with vMYX-ΔM083 resulted in significantly lower levels of expression of various cIAPs at MOI’s of either 1 or 3. To further determine whether this enhanced depletion of cIAPs altered the killing of MM cells, we infected human U266 MM cells with either vMYX-GFP or vMYX-ΔM083 at various MOI’s and measured the resulting effects on cellular viability after 24 hours (Figure 8B). Consistent with our previous results 23, vMYX-GFP caused a dramatic reduction in MM cell viability in a dose dependent manner. Treatment with vMYX-ΔM083, however, caused an even more pronounced reduction in cellular viability. Consistent with the depletion of cIAPs, this difference became even more pronounced at lower MOI’s. This difference in MM cell killing was not due to altered infection of MM cells between the two viruses since both viruses displayed identical rates of infection (measured by the numbers of cells displaying evidence of GFP) at all tested MOI’s (Figure 8C). Taken together, these data suggest that MYXV eliminates cIAPs through a host-protein shutoff mediated mechanism and that altering the efficacy of this process can enhance virally mediated killing of MM cells.
Figure 8. Enhancing viral host-protein shut-off improves MYXV’s oncolytic efficacy.
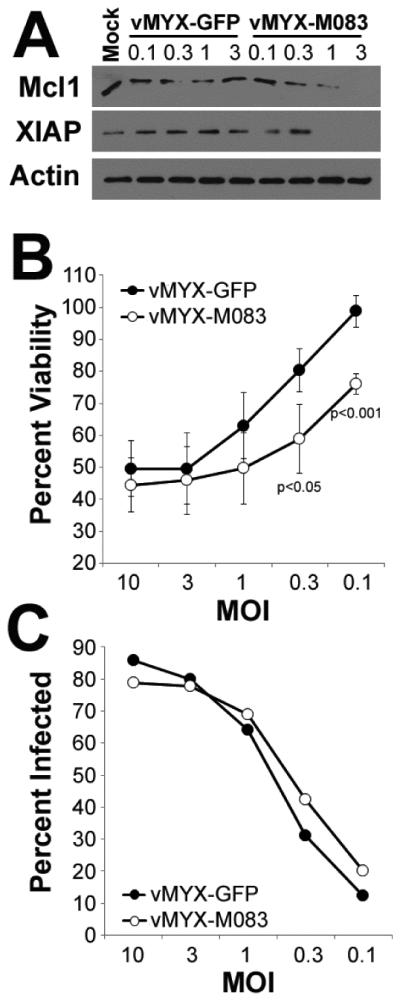
(A) Human U266 MM cells were either mock-treated or infected with vMYX-GFP or vMYX-ΔM083 at the indicated MOIs. Two hours post-infection, cells were harvested and the expression of the indicated proteins was analyzed using immunoblot. Human U266 MM cells were infected with either vMYX-GFP or vMYX-ΔM083 at the indicated MOIs. (B) Cellular viability or (C) GFP expression was analyzed after 24 hours.
Discussion
Our previous work established that MYXV effectively killed human MM cells by inducing a rapid apoptotic response 23. Here we provide evidence that this apoptotic response is initiated through the activation of Casp-8 and suggest that this activation is the result of depletion of cIAPs due to poxviral host-protein shut-off.
Interestingly, while our data strongly indicates that MM cell apoptosis following MYXV treated is initiated through activation of Casp-8, the complete mechanism involved in this activation is less clear. For example, our data demonstrates that MYXV depletes cIAPs prior to activation of Casp-8 and a viral construct which is more efficient at depleting these proteins is also more efficient at killing MM cells. These data strongly suggest that depletion of cIAPs plays a key role in the induction of apoptosis in infected MM cells. However, while depletion of cIAPs has been shown to be sufficient to activate apoptotic pathways in other models, in our hands depletion of these molecules in the absence of viral infection, using the translational inhibitor cyclohexamide, was insufficient to activate Casp-8 (our unpublished observations). Thus, additional apoptotic activating stimuli might be provided by MYXV.
We have observed that, while MYXV infection induces apoptosis in virtually every tested MM cell (Figure 1 and 23, 60), the kinetics of this induction can vary from cell line to cell line. Since our data suggests that depletion of cIAPs plays a major role in the induction of this apoptotic response, it is attractive to hypothesize that the kinetics of apoptotic induction are related to the initial expression of any apoptotic proteins within a given myeloma. Since upregulation of various cIAPs is a major mechanism through which MM cells develop resistance 61 we would predict that MYXV treatment might display differential kinetics in primary versus relapsed patients. It should be noted, however, that the mechanisms of poxviral host-protein shut-off are predicated on decapping of cellular mRNA’s. To our knowledge, this is a unique viral mechanism which is not utilized by any small molecular chemotherapeutics. Therefore, cross-treatment resistance seems unlikely, a hypothesis which is supported by our observation that MYXV induced apoptosis in 100% of tested primary MM patient samples (Figure 2C). On a similar note, while treatment with caspase inhibitors prevents the molecular completion of apoptosis (Figures 4 and 5) it largely fails to prevent virally induced loss of MM cell viability (our unpublished observations). Indeed, despite repeated attempts to block multiple programmed cell death pathways, including simultaneous blockade of: apoptosis, necroptosis, autophagy, and viral replication, we were unable to identify any method to prevent MYXV from rapidly killing every MM cell it infected. This is consistent with our previous results indicating that MYXV treatment eliminates primary MM cells from patient bone marrow samples with equal efficacy regardless of relapse or resistance status 23. While this suggests resistance to MYXV treatment is unlikely to develop, its clinical significance remains unclear. The primary draw-back of MYXV as a therapy for MM is that the virus fails to complete its replication cycle in infected MM cells due to the speed with which these cells die 23. Therefore, the development of MM cell ‘resistance’ to MYXV might slow cell death sufficiently to allow for amplification of the viral agent thus actually increasing overall efficacy. Further studies on how modulation of host protein shut-off impacts overall efficacy of viral treatment in preclinical models is needed to address these question.
Conclusions
We demonstrate that MYXV eliminates MM cells by depletion of cIAPs due to host-protein shutoff mediated translational arrest. This mechanism is fundamental to cellular biology and unique to poxviruses, suggests that MYXV represents an excellent tool for the treatment of relapsed or refractory MM patients.
Micro Abstract: We have previously shown that the myxoma virus kills human myeloma cells. Here we demonstrate that this killing occurs due to the ligand independent activation of caspase-8. We hypothesize that this activation is the result of viral host-protein shutoff depleting a variety of cellular inhibitors of apoptosis.
Clinical Practice Points: It has been previously demonstrated that an oncolytic poxvirus known as myxoma can rapidly eliminate infected multiple myeloma cells thus preventing disease relapse in models of autologous stem cell transplant. Interestingly, elimination of infected cells occurred even in the complete absence of viral replication suggesting that it might be due to some form of programmed cells death. The mechanisms mediating induction of this cell death, however, remained unknown.
The current manuscript advances these previous findings in two ways. First we demonstrate that myxoma virus treatment induces caspase-8 mediated apoptosis in infected multiple myeloma cells. This apoptosis is ligand independent and instead correlates with depletion of cellular inhibitors of apoptosis. Second, we demonstrate that a viral construct which enhances depletion of these inhibitors, through overexpression of the viral mRNA decapping enzymes, displays an enhanced ability to eliminate multiple myeloma cells, particularly at low multiplicities of infection.
This work could impact clinical practice in two ways. First it provides a mechanistic explanation for the observed killing of multiple myeloma cells by myxoma virus thus alleviating some of the hesitancy to use a live replicating virus as a therapeutic while also suggesting potential methods to screen patients for potential responses to treatment. Second, it demonstrates that genetic manipulation of the myxoma genome can enhance the efficacy of viral treatment and suggests a possible second generation virus which might be more therapeutically efficacious.
Acknowledgements
Dr. Bartee is supported by a grant from the NIAID (1K22AI095372-01A1), a grant from the American Cancer Society (#IRG-97-219-14), and startup funding from the Medical University of South Carolina. This work was also supported in part by the Hollings Cancer Center’s Cancer Center Support Grant P30 CA138313, the South Carolina Clinical and Translational Research (SCTR) Institute NIH grant UL1TR000062, and the MUSC center for Oral Health Research through NIH Grant 5P30GM103331
Abbreviations
- MYXV
Myxoma virus
- MM
Multiple Myeloma
- MOI
multiplicity of Infection
- TMRM
Tetramethylrhodamine, methyl ester, perchlorate
- cIAPs
cellular inhibitors of apoptosis
- UPR
unfolded protein response
- DISC
death-inducing signaling complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.society L. l. Disease Information & Support: myeloma. 2012.
- 3.Alexanian R, Haut A, Khan AU, Lane M, McKelvey EM, Migliore PJ, Stuckey WJ, Jr., Wilson HE. Treatment for multiple myeloma. Combination chemotherapy with different melphalan dose regimens. JAMA : the journal of the American Medical Association. 1969;208:1680–1685. doi: 10.1001/jama.208.9.1680. [DOI] [PubMed] [Google Scholar]
- 4.Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood. 2008;111:2521–2526. doi: 10.1182/blood-2007-08-104984. [DOI] [PubMed] [Google Scholar]
- 5.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, Greipp PR, Kyle RA, Gertz MA. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bais S, Bartee E, Rahman MM, McFadden G, Cogle CR. Oncolytic virotherapy for hematological malignancies. Advances in virology. 2012;2012:186512. doi: 10.1155/2012/186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 8.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nature clinical practice. Oncology. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 9.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Bahlis N, Neri P, Pho M, Stewart D, Mansoor A, Morris DG. Reovirus as a successful ex vivo purging modality for multiple myeloma. Bone marrow transplantation. 2014;49:80–86. doi: 10.1038/bmt.2013.130. [DOI] [PubMed] [Google Scholar]
- 12.Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, Neri P, Pho M, Stewart D, Mansoor A, Morris DG. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4962–4972. doi: 10.1158/1078-0432.CCR-11-3085. [DOI] [PubMed] [Google Scholar]
- 13.Dingli D, Peng KW, Harvey ME, Greipp PR, O'Connor MK, Cattaneo R, Morris JC, Russell SJ. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 14.Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:1155–1164. doi: 10.1038/mt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell SJ, Peng KW. Measles virus for cancer therapy. Current topics in microbiology and immunology. 2009;330:213–241. doi: 10.1007/978-3-540-70617-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarde DN, Nace RA, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti-myeloma activity in vivo. Experimental hematology. 2013;41:1038–1049. doi: 10.1016/j.exphem.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yarde DN, Naik S, Nace RA, Peng KW, Federspiel MJ, Russell SJ. Meningeal myeloma deposits adversely impact the therapeutic index of an oncolytic VSV. Cancer gene therapy. 2013;20:616–621. doi: 10.1038/cgt.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naik S, Nace R, Barber GN, Russell SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer gene therapy. 2012;19:443–450. doi: 10.1038/cgt.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senac JS, Doronin K, Russell SJ, Jelinek DF, Greipp PR, Barry MA. Infection and killing of multiple myeloma by adenoviruses. Human gene therapy. 2010;21:179–190. doi: 10.1089/hum.2009.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raus S, Coin S, Monsurro V. Adenovirus as a new agent for multiple myeloma therapies: Opportunities and restrictions. The Korean journal of hematology. 2011;46:229–238. doi: 10.5045/kjh.2011.46.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, Lowe V, O'Connor MK, Kyle RA, Leung N, Buadi FK, Rajkumar SV, Gertz MA, Lacy MQ, Dispenzieri A. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic proceedings. 2014;89:926–933. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fenner F, Ratcliffe FN. Myxomatosis. Cambridge University Press; Cambridge, UK: 1965. [Google Scholar]
- 23.Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2012;18:1540–1551. doi: 10.1016/j.bbmt.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Current protocols in microbiology. 2010 doi: 10.1002/9780471729259.mc14a01s17. Chapter 14:Unit 14A 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunlap KM, Wolfe M, Bartee M, Bartee E. Myxoma Virus M083L is a virulence factor which mediates systemic dissemination by enhancing infection of activated T cells. Journal of virology. Submitted;Submitted. doi: 10.1128/JVI.02186-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jourdan M, Ferlin M, Legouffe E, Horvathova M, Liautard J, Rossi JF, Wijdenes J, Brochier J, Klein B. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. British journal of haematology. 1998;100:637–646. doi: 10.1046/j.1365-2141.1998.00623.x. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Wang J. Initiator caspases in apoptosis signaling pathways. Apoptosis : an international journal on programmed cell death. 2002;7:313–319. doi: 10.1023/a:1016167228059. [DOI] [PubMed] [Google Scholar]
- 28.Bouchier-Hayes L. The role of caspase-2 in stress-induced apoptosis. Journal of cellular and molecular medicine. 2010;14:1212–1224. doi: 10.1111/j.1582-4934.2010.01037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochemical and biophysical research communications. 2005;331:859–867. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 30.Salvesen GS. Caspase 8: igniting the death machine. Structure. 1999;7:R225–229. doi: 10.1016/s0969-2126(00)80048-9. [DOI] [PubMed] [Google Scholar]
- 31.Kuida K. Caspase-9. The international journal of biochemistry & cell biology. 2000;32:121–124. doi: 10.1016/s1357-2725(99)00024-2. [DOI] [PubMed] [Google Scholar]
- 32.Krug HF. Caspase-10 is the key initiator caspase involved in tributyltin-mediated apoptosis in human immune cells. Journal of toxicology. 2012;2012:395482. doi: 10.1155/2012/395482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 35.Esposti MD. The roles of Bid. Apoptosis : an international journal on programmed cell death. 2002;7:433–440. doi: 10.1023/a:1020035124855. [DOI] [PubMed] [Google Scholar]
- 36.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nature reviews. Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 37.Roy S, Nicholson DW. Cross-talk in cell death signaling. The Journal of experimental medicine. 2000;192:F21–25. [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreira KS, Kreutz C, Macnelly S, Neubert K, Haber A, Bogyo M, Timmer J, Borner C. Caspase-3 feeds back on caspase-8, Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apoptosis : an international journal on programmed cell death. 2012;17:503–515. doi: 10.1007/s10495-011-0691-0. [DOI] [PubMed] [Google Scholar]
- 39.Yamada H, Tada-Oikawa S, Uchida A, Kawanishi S. TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochemical and biophysical research communications. 1999;265:130–133. doi: 10.1006/bbrc.1999.1641. [DOI] [PubMed] [Google Scholar]
- 40.Debatin KM, Goldmann CK, Bamford R, Waldmann TA, Krammer PH. Monoclonal-antibody-mediated apoptosis in adult T-cell leukaemia. Lancet. 1990;335:497–500. doi: 10.1016/0140-6736(90)90735-n. [DOI] [PubMed] [Google Scholar]
- 41.Nakayama M, Harada N, Okumura K, Yagita H. Characterization of murine TWEAK and its receptor (Fn14) by monoclonal antibodies. Biochemical and biophysical research communications. 2003;306:819–825. doi: 10.1016/s0006-291x(03)01051-9. [DOI] [PubMed] [Google Scholar]
- 42.Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine & growth factor reviews. 2003;14:241–249. doi: 10.1016/s1359-6101(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 43.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 45.Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS letters. 1998;431:351–356. doi: 10.1016/s0014-5793(98)00791-1. [DOI] [PubMed] [Google Scholar]
- 46.Moller P, Henne C, Schmidt A, Eichelmann A, Leithauser F, Bruderlein S, Dhein J, Krammer PH. [Expression of APO-1, a cell surface molecule mediating apoptosis, during normal B cell ontogeny and in B cell tumors. Co-expression and coregulation of APO-1 and ICAM-1 (CD54) in germinal central cells] Verhandlungen der Deutschen Gesellschaft fur Pathologie. 1992;76:237–242. [PubMed] [Google Scholar]
- 47.Ramakrishnan V, Painuly U, Kimlinger T, Haug J, Rajkumar SV, Kumar S. Inhibitor of apoptosis proteins as therapeutic targets in multiple myeloma. Leukemia. 2014;28:1519–1528. doi: 10.1038/leu.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desplanques G, Giuliani N, Delsignore R, Rizzoli V, Bataille R, Barille-Nion S. Impact of XIAP protein levels on the survival of myeloma cells. Haematologica. 2009;94:87–93. doi: 10.3324/haematol.13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romagnoli M, Trichet V, David C, Clement M, Moreau P, Bataille R, Barille-Nion S. Significant impact of survivin on myeloma cell growth. Leukemia. 2007;21:1070–1078. doi: 10.1038/sj.leu.2404602. [DOI] [PubMed] [Google Scholar]
- 50.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 51.Dunlap KM, Bartee MY, Bartee E. Myxoma virus attenuates expression of activating transcription factor 4 (ATF4) which has implications for the treatment of proteasome inhibitor– resistant multiple myeloma. Oncolytic Virotherapy. 2015;4:1–11. doi: 10.2147/OV.S72372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. Journal of cell science. 2000;113:4363–4371. doi: 10.1242/jcs.113.23.4363. Pt 23. [DOI] [PubMed] [Google Scholar]
- 53.Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Molecular and cellular biology. 2010;30:3099–3110. doi: 10.1128/MCB.01266-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE. Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail. The Journal of biological chemistry. 2005;280:27345–27355. doi: 10.1074/jbc.M504019200. [DOI] [PubMed] [Google Scholar]
- 55.Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virgin HW, McFadden G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nature immunology. 2004;5:1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 56.Knipe D, Howley P. Fields Virology. 6th Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 57.Cameron C, Hota-Mitchell S, Chen L, Barrett J, Cao JX, Macaulay C, Willer D, Evans D, McFadden G. The complete DNA sequence of myxoma virus. Virology. 1999;264:298–318. doi: 10.1006/viro.1999.0001. [DOI] [PubMed] [Google Scholar]
- 58.Liu SW, Wyatt LS, Orandle MS, Minai M, Moss B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. Journal of virology. 2014;88:202–211. doi: 10.1128/JVI.02426-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parrish S, Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. Journal of virology. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan WM, Bartee EC, Moreb JS, Dower K, Connor JH, McFadden G. Myxoma and vaccinia viruses bind differentially to human leukocytes. Journal of virology. 2013;87:4445–4460. doi: 10.1128/JVI.03488-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu J, Dang N, Menu E, De Bryune E, Xu D, Van Camp B, Van Valckenborgh E, Vanderkerken K. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012;119:826–837. doi: 10.1182/blood-2011-07-366492. [DOI] [PubMed] [Google Scholar]