

Abstract
Non-small cell lung cancer (NSCLC) accounts for 85% of all types of lung cancer and is the leading cause of world-wide cancer-associated mortalities. Radiation therapy has long been regarded as a fundamental therapeutic treatment strategy for NSCLC. However, alternative therapies for NSCLC remain insufficient, with the majority of cancers developing a high incidence of radioresistance. MicroRNAs (miRNAs/miRs) are endogenous oligonucleotide RNAs that serve an important role in carcinogenesis and tumor progression. In the present study, a novel function of miR-133b that is associated with the radiosensitivity of lung cancer cells is reported. miR-133 was downregulated in radioresistant lung cancer cells, which exhibited an elevated glycolysis rate when compared with radiosensitive cells. Additionally, it was observed that pyruvate kinase isoform M2 (PKM2) is a target of miR-133b, and that the expression of PKM2 is positively correlated with radioresistance. Finally, it was demonstrated that overexpression of miR-133b resensitizes radioresistant lung cancer cells through the inhibition of PKM2-mediated glycolysis. The current study may indicate a novel function of miR-133b, potentially aiding the development of anticancer therapeutics.
Keywords: microRNA, miR-133, non-small cell lung cancer, radiation resistance, glycolysis, pyruvate kinase isoform M2
Introduction
Lung cancer presents with a particularly high mortality rate and is understood to be the leading cause of worldwide cancer-associated mortalities. Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers (1). To date, the treatment options that are available for patients with lung cancer consist of combinations of chemotherapy and radiotherapy (RT) (2). Despite decades of research, alternative NSCLC therapies remain insufficient, and the majority of lung cancers demonstrate a high incidence of relapse due to the development of resistance (3). RT is a treatment that utilizes high-energy rays or particles to destroy lung cancer cells. However, the efficacy of RT is limited due to the survival of lung cancer cells following the treatment, subsequently resulting in recurrence (4). Thus, the identification of novel therapeutic drugs to mitigate possible chemo- or radioresistance is critical to allow the development of successful lung cancer treatments.
The majority of cancer cells exhibit elevated glycolysis and depressed mitochondrial oxidative phosphorylation, allowing the generation of ATP to function as their energy supply. This phenomenon is known as the Warburg effect (5). Furthermore, this switch in energy metabolism, and the associated increased expression of glycolytic enzymes, provides a survival advantage for the cancer cells (6). It is reported that dysregulated glycolysis is closely associated with chemo- and radioresistance in cancer (7), suggesting that the inhibition of glycolysis may be an effective method to incorporate during the development of optimal combination regimens for the treatment of cancer.
MicroRNA (miRNA/miR) is non-coding, single-stranded RNA of ~22 nucleotides in length. miRNA regulates gene expression at a post-transcription level by binding onto specific mRNA molecules at the 3′-untranslated region (3′-UTR), and performs essential roles in a variety of biological processes (8). miRNAs are associated with proliferation, differentiation, migration, the cell cycle and apoptosis (9). Furthermore, previous studies have demonstrated that miRNAs have important functions within cancer development, malignant transformation and drug resistance (10), and that they may serve as potential oncogenes or tumor suppressors (9–11). Recently, the role of miRNA-21 within NSCLC radioresistance has been identified, indicating that miRNAs may be selected as a possible therapeutic target (12). Additionally, miR-133b in particular has been reported to function as a tumor suppressor in multiple types of cancer (13–15). However, the precise role of miR-133b within radiosensitivity is not yet clear. In the present study, the role of miR-133b in NSCLC radiosensitivity is investigated, with the association between dysregulated glycolysis and radiosensitivity also examined. The subsequent results demonstrate that miR-133b may function as a therapeutic target for the treatment of radioresistant lung cancer.
Materials and methods
Cell culture ionizing radiation treatment
NSCLC A549 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and were cultured in Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin. Cell cultures were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Ionizing radiation treatment of cells
Cell cultures containing 5×105 cells were exposed to different doses of radiation (0, 0.2, 0.4, 0.5, 0.6, 1.0, 2.0, 3.0, 4.0, 5.0, 6.0 Gy) at room temperature, with a Cs-137 irradiator (HWM D-2000; Siemens AG, Munich, Germany) at a dose rate of 2 Gy/min. Following irradiation, the cells were subjected to the following experiments, or were placed back in the incubator in a humidified atmosphere of 5% CO2 at 37°C.
miRNA transfection
The cells were seeded in 6-well plates at 1×105 cells/well and cultured overnight. The cells were subsequently transfected with 100 nM of the pre-miR-133b, inhibitors [anti-miR-133b (Shanghai GenePharma Co., Ltd., Shanghai, China)] or negative control mRNA (Shanghai GenePharma Co., Ltd.) using Lipofectamine 2000® and Opti-MEM I reduced serum medium (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocols. The precursor and antisense of miR-133b were chemically synthesized by Shanghai GenePharma Co., Ltd. Following a total of 48 h post-transfection, the cells were prepared for further analysis.
Plasmid DNA transfection
(Myc-DDK-tagged)-Human pyruvate kinase (PKM) transcript variant 2 was purchased from OriGene Technologies, Inc. (catalog no. RC219382; Rockville, MD, USA) and cloned into pCMV6 vector (catalog no. PS100001, OriGene Technologies, Inc.). pCMV6 empty vector was used as a control. Plasmid DNA transfection was performed using Lipofectamine 2000® and Opti-MEM I reduced serum medium (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocols. Following a total of 48 h post-transfection, the cells were prepared for further analysis.
Cell viability assay
A total of 1×105 cells for each well were seeded in 12-well plates overnight. The medium was replaced daily. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Absorbance was measured spectrophotometrically at 570 nm by the EL800 Universal Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA).
RNA extraction and quantitative polymerase chain reaction (qPCR)
miRNAs from the cultured cells were extracted and purified using the mirVana miRNA Isolation kit (Ambion; Thermo Fisher Scientific, Inc.), following the manufacturer's protocols. Total RNA concentration was adjusted to 2 ng/µl using a spectrophotometer. Total RNA (1 µg) was reverse transcribed with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). The cDNA reaction was diluted to 1:10 for use as a template for qPCR of mature miRNA.
miRNA expression patterns in the cultured cells and tissues were evaluated using TaqMan® OpenArray® Human MicroRNA Panels (Applied Biosystems; Thermo Fisher Scientific, Inc.).
miRNA from the samples was converted into cDNA using 156 specific stem-loop reverse transcription primers. Specific TaqMan® qPCR primers and probes were then used to conduct qPCR and determine the quantity of the miRNAs. qPCR was performed using the GeneAmp® Fast PCR Master mix and the 7900HT Fast Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The qPCR conditions were as follows: 95°C for 10 min; 40 cycles of 95°C for 15 sec; and 60°C for 60 sec. All reactions were performed in duplicate. Expression levels of the mature miRNAs were evaluated using the comparative quantification cycle (Cq) method of 2(−ΔΔCq). The ΔCq for PKM2 mRNA expression was calculated relative to the Cq of 18S ribosomal RNA. Relative mRNA expression was calculated using the formula 2(−ΔΔCq). The primers used for qPCR were: PKM2 forward primer, 5′-GAGGCCTCCTTCAAGTGCT-3′, and reverse primer, 5′-CCAGACTTGGTGAGGACGAT-3′. All reactions were performed at least twice in duplicate.
Measurements of glucose consumption
The glucose concentration in the diluted medium was measured using the Glucose (GO) assay kit (Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer's protocols. Glucose consumption was calculated by subtracting the concentration of glucose remaining in the medium at the indicated time from the concentration of glucose present in fresh cell culture medium. The results were normalized to the total amount of protein when compared with the control cells.
Measurements of lactate production
Lactate concentrations were determined using a lactate assay kit (BioVision, Inc., Milpitas, CA, USA), according to the manufacturer's protocols. Samples and lactate standard with lactate assay buffer was prepared in a 96-well plate. A total of 50 µl of the reaction mix, containing the lactate enzyme mix, was added to each well. The plate was then incubated for 30 min at room temperature. Optical density values at 570 nm were measured with a SpectraMax® M2e Multimode Microplate Reader (Molecular Devices, LLC, Sunnyvale, CA, USA). The results were normalized to the total amount of protein when compared with the control cells.
Western blot analysis
The cells were harvested and washed with ice-cold phosphate-buffered saline. Cell lysates were obtained by resuspending the cells in RIPA buffer [10 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100 and 1% Na-deoxycholate (Kanto Chemical Co., Ltd., Tokyo, Japan)], and 5 mM ethylenediaminetetraacetic acid that was supplemented with a protease inhibitor cocktail (Sigma-Aldrich). The protein concentration of the cell lysates was determined by using the Bradford protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of protein were loaded and subsequently separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis prior to being electrotransferred onto a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). The membranes were blocked and incubated overnight with primary antibodies at 1:1,000 dilution. The primary antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) as follows: Rabbit anti-human PKM2 monoclonal (dilution, 1:1,000; catalog no. 4053) and rabbit anti-human β-actin monoclonal (dilution, 1:1,000; catalog no. 4967) antibodies. The membranes were then washed and incubated at room temperature for 1 h with HRP-conjugated goat anti-rabbit immunoglobulin G (Nichirei Bioscience Inc., Tokyo, Japan). Protein bands were visualized with the Chemi-Lumi One L western blotting substrate (Nacalai Tesque, Kyoto, Japan).
Statistical analysis
Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used to calculate unpaired Student's t test for data analysis. All data are presented as the mean ± standard error. P<0.05 was considered to indicate a statistically significant difference.
Results
Establishment of radioresistant NSCLC cell line
To investigate the roles of miRNA-133b in the radiosensitivity of human NSCLC, a radioresistant cell line was established from A549 cells. A549 parental cells were exposed to increased intensities of radiation (1 to 5 Gy) and the surviving cells were selected. Following 1 month of consecutive selection, the surviving cell clones were pooled and subjected to resistance verification. Results of cell viability experiments are presented in Fig. 1A and B. The A549 radiosensitive cells demonstrated a significant inhibition of viability following irradiation with 0.5 to 6 Gy. By contrast, A549 resistant cells exhibited significantly increased viability following radiation exposure. The irradiation dosage for 50% cell viability inhibition in the radioresistant cells was 8 Gy, which was greater than that of the radiosensitive cells.
Figure 1.

Radiation-resistant cells exhibit elevated glucose metabolism. (A) Selection of radiation-resistant cells from the A549 parental cells. A549 radioresistant cells were pooled and cell viabilities were analyzed by treatment with radiation at 0, 0.5, 1, 2, 4 and 6 Gy. (B) Cell death and morphological changes in the A549 radiosensitive and -resistant cells in response to radiation treatments at 0, 1 and 4 Gy. (C) Glucose uptake (left) and lactate production (right) were measured in the A549 radiosensitive and radioresistant cells. Columns represent the mean of three independent experiments; bars represent the standard error. *P<0.05 and **P<0.01 vs. IR sensitive cells. IR, irradiation; -, no radiation; +, radiation at 1 Gy; ++, radiation at 4 Gy.
Radioresistant A549 cells have increased glucose metabolism
As aforementioned, dysregulated glucose metabolism is associated with chemo- and radioresistance in cancer cells (7). To investigate whether the glucose metabolic profile was altered by radiation treatment, the glucose uptake and lactate product of the A549 cells was measured following different dosages of radiation treatment. Notably, the results from the present study demonstrated that the glucose uptake and lactate product were induced by radiation treatment (Fig. 1C), indicating that there may be an association between glucose metabolism and radiosensitivity in NSCLC cells. As expected, the radioresistant A549 cells exhibited increased glucose metabolism when compared with the sensitive cells, suggesting that the upregulated glucose metabolism may contribute to radioresistance, and may be targeted to develop anti-radioresistance drugs.
miR-133b is negatively correlated with radioresistance
The present study subsequently investigated the mechanism underlying the upregulated glucose metabolism in radioresistant lung cancer cells. As it has been previously reported that miR-133b acts as a tumor suppressor in lung cancer, the expression levels of miR-133b in A549 cells were measured following radiation treatment (13–18). The expression of miR-133b was significantly reduced following radiation treatments of 0.2 to 0.6 Gy (Fig. 2A). Additionally, the expression of miR-133b in the radiation-resistant A549 cells was observed to be lower than that in the radiation sensitive cells (Fig. 2B), suggesting miR-133b may be involved in the regulation of radiation sensitivity.
Figure 2.

miR-133b is downregulated in radioresistant cancer cells. (A) The expression of miR-133b in A549 parental cells irradiated with 0, 0.2, 0.4 and 0.6 Gy. (B) The expression of miR-133b in A549 radiosensitive and radioresistant cells. Columns represent the mean of three independent experiments; bars represent standard error. *P<0.05, **P<0.01 and ***P<0.001 vs. IR sensitive cells. miR, microRNA; ctrl, control; IR, irradiation.
PKM2 is a target of miR-133b in NSCLC cells
The aforementioned results identified the correlation between dysregulated glycolysis, the expression of miR-133b and radiation resistance. To investigate the possible association between miR-133b and glycolysis, miRNA databases were searched for potential miR-133b targets that may contribute to the regulation of glycolysis. Results from miRBase (http://www.mirbase.org/) indicated that PKM2 may function as a target for miR-133b, and that the 3′-UTR of PKM2 contains a highly-conserved binding site for miR-133b. To determine whether PKM2 is the target gene of miR-133b, the protein expression level of PKM2 in the A549 cell line was analyzed in response to the overexpression or inhibition of miR-133b (Fig. 3A and B left). Reduced PKM2 expression in cells transfected with miR-133b was observed (Fig. 3A), as was increased PKM2 levels following the inhibition of miR-133b expression (Fig. 3B), indicating that miR-133b may suppress the glycolysis of lung cancer cells by targeting PKM2.
Figure 3.

PKM2 is a target of miR-133b in lung cancer cells. (A) A549 cells were transfected with 100 nM pre-miR-negative (control miR) or pre-miR-133b for 48 h (left). Cell lysates were prepared for western blotting analysis of the expression of PKM2 (right). β-actin was used as a loading control. (B) A549 cells were transfected with 100 nM anti-miR-negative (control miR) or antisense-miR-133b for 48 h (left). Cell lysates were prepared for western blotting analysis of the expression of PKM2 (right). β-actin was used as a loading control. Columns represent the mean of three independent experiments; bars represent standard error. **P<0.01 vs. control miR. miR, microRNA; PKM2, pyruvate kinase isoform M2.
Radioresistant A549 cells exhibit elevated PKM2 expression
To verify whether the inhibited PKM2 expression, caused by miR-133b, is the mechanism for the radiosensitivity observed in lung cancer cells, the expression of PKM2 at the protein and mRNA levels in radiosensitive and -resistant cells was measured. As expected, PKM2 was upregulated in radioresistant cells when compared with sensitive cells (Fig. 4A and B), suggesting that the upregulation of glycolysis in radioresistant cells is subsequent to the upregulation of PKM2.
Figure 4.

PKM2 is upregulated in radioresistant lung cancer cells, and miR-133b suppresses glucose metabolism. (A) A549 radiosensitive and -resistant cells were analyzed by western blotting and (B) quantitative polymerase chain reaction for the expression of PKM2. β-actin was used as a loading control. *P<0.01 and ***P<0.001 vs. radiation sensitive cells. (C) A549 cells were transfected with 100 nM pre-miR-negative (control miR) or pre-miR-133b for 48 h. Cell were prepared for the measurements of glucose uptake (right) and lactate product (right). Columns represent the mean of three independent experiments; bars represent standard error. *P<0.01 and ***P<0.001 vs. control miR. miR, microRNA; PKM2, pyruvate kinase isoform M2.
Overexpression of miR-133b resensitizes A549 radioresistant cells to irradiation through targeting on PKM2
The present study examined the glycolysis rate through the overexpression of miR-133b in the A549 cells. The results, presented in Fig. 4C, demonstrated that miR-133b negatively regulated glucose uptake and lactate product, thus the overexpression of miR-133b significantly suppressed glucose metabolism. To investigate whether the overexpression of miR-133b in lung cancer cells could resensitize radioresistant cells to irradiation through the inhibition of PKM2, pre-miR-133b was transfected with the overexpression vector containing wild-type PKM2 or a control vector and was added to the A549 radioresistant cells. The exogenous overexpression of PKM2 restored the PKM2 to the initial level (Fig. 5A), indicating that the activities of glycolysis may be recovered by the overexpression of PKM2 in miR-133b-overexpressing cells. Overexpression of miR-133b significantly sensitized the radioresistant cells to radiation (Fig. 5B), indicating that miR-133b may serve as a therapeutic target aiding the the development of drugs. Additionally, restoration of the activity of glycolysis by the overexpression of PKM2 in the miR-133b overexpressing cells led to a significant resistance to irradiation when compared with the transfection of the control vector in radioresistant cells (Fig. 5B), suggesting that the overexpression of miR-133b sensitized lung cancer radioresistant cells to irradiation by the inhibition of PKM2-mediated glycolysis.
Figure 5.
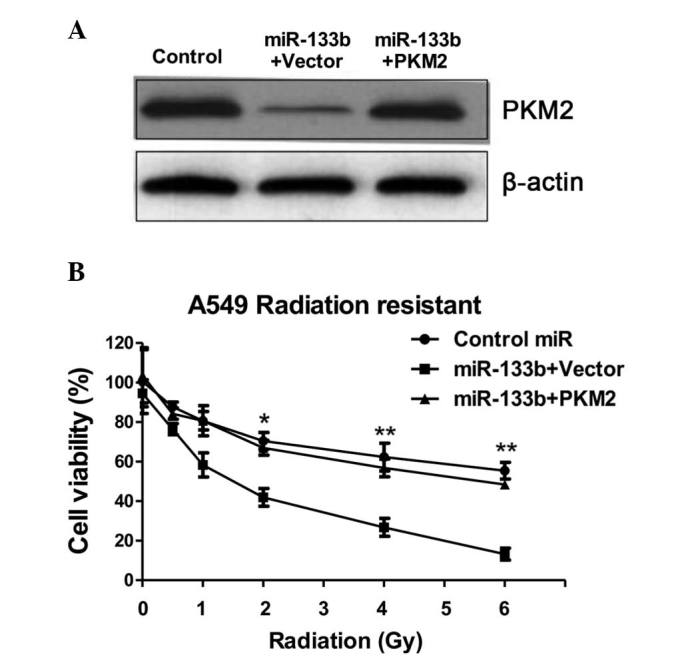
Overexpression of miR-133b resensitizes radioresistant lung cancer cells through the inhibition of glucose metabolism. (A) A549 radioresistant cells were transfected with 100 nM pre-miR-negative (control miR) or cotransfected with pre-miR-133b + vector control or cotransfected with pre-miR-133b + PKM2 for 48 h. Cell lysates were prepared for western blotting analysis of the expression of PKM2. Western blotting experiments demonstrated that the overexpression of PKM2 in the miR-133b-overexpressing A549 radioresistant cells restored the expression of PKM2 to a similar level to that observed in the control cells. (B) A540 radioresistant cells were transfected with control miR, miR-133b + vector and miR-133b + PKM2 for 48 h, and subsequently, cell viabilities were analyzed following radiation treatments of 0, 0.5, 1, 2, 4 and 6 Gy. Columns represent the mean of three independent experiments; bars represent the standard error. *P<0.05 and **P<0.01 vs. control miR. miR, microRNA; PKM2, pyruvate kinase isoform M2.
Discussion
miR-133b has been reported as a tumor suppressor functioning in numerous types of cancer that demonstrate low miR-133b expression, as noted in colorectal (13), gastric (14), bladder (15), lung (16), esophageal (17), ovarian (18) and breast (19) cancer. However, the role of miR-133b within radiosensitivity remains under investigation. During the present study, a significant downregulation of miR-133b in radioresistant NSCLC cells was observed. Additionally, it was demonstrated that miR-133b suppresses glycolysis within lung cancer cells by targeting PKM2, which is an essential enzyme involved in glycolysis. As aforementioned, cancer cells exhibit an upregulated rate of glycolysis when compared with normal cells. Together, the results of the present study are consistent with those of previous studies reporting that miR-133b functions as a tumor suppressor.
Pyruvate kinase catalyzes the production of pyruvate from phosphoenol pyruvate through a glycolytic cascade (20). Pyruvate kinase has 4 different isoforms (L, R, M1 and M2), with each isoform expressed in a tissue-specific manner (21). Additionally, PKM2 upregulation has been observed in numerous types of cancer (21). The current study reported that PKM2 is a target of miR-133b. Notably, the expression of PKM2 was correlated with radioresistance in the assessed lung cancer cells. Radioresistant lung cancer cells demonstrated upregulated PKM2 expression, indicating that PKM2 may serve as a target to overcome radiation resistance.
The altered energy metabolism of cancer cells has recently been studied and recognized as a novel hallmark of cancer (7). Glucose metabolism in cancer cells is primarily characterized by two major biochemical events: i) The increased uptake of glucose and ii) the increased production of lactate. To the best of our knowledge, the present study is the first to demonstrate that miR-133b significantly suppresses glucose metabolism within lung cancer cells, identifying a novel mechanism of miR-133b as a tumor suppressor. It has been reported that the increased accumulation of lactate may act as an anti-oxidant in cancer cells and contribute to radioresistance (22). The results of the current study demonstrated that radioresistant lung cancer cells have increased levels of lactate product, which may be the mechanism underlying radioresistance. However, further understanding is required with regard to the signaling pathway and molecular mechanisms. In summary, a novel function of miR-133b associated with the radiosensitivity of lung cancer cells was identified. Furthermore, it was observed that PKM2 serves as a target of miR-133b, and the glucose metabolism of lung cancer cells was positively correlated with radioresistance. Overexpression of miR-133b resensitized radioresistant lung cancer cells through the inhibition of PKM2-mediated glycolysis. The present study provides further insight into the cellular and molecular mechanisms that may be involved in the exhibition of radiation resistance in NSCLC.
Acknowledgements
The authors would like to thank Dr Yi Li for providing editorial assistance (Department of Internal Medicine, Tianjin Huanhu Hospital, Tianjin, China).
References
- 1.Sechler M, Cizmic AD, Avasarala S, Van Scoyk M, Brzezinski C, Kelley N, Bikkavilli RK, Winn RA. Non-small-cell lung cancer: Molecular targeted therapy and personalized medicine - drug resistance, mechanisms, and strategies. Pharmgenomics Pers Med. 2013;6:25–36. doi: 10.2147/PGPM.S26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71:3–10. doi: 10.1016/j.lungcan.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Willers H, Azzoli CG, Santivasi WL, Xia F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013;19:200–207. doi: 10.1097/PPO.0b013e318292e4e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimura T, Noma N, Sano Y, Ochiai Y, Oikawa T, Fukumoto M, Kunugita N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother Oncol. 2014;112:302–307. doi: 10.1016/j.radonc.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 9.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng T, Wang J, Chen X, Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126:2–10. doi: 10.1002/ijc.24782. [DOI] [PubMed] [Google Scholar]
- 11.Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17:523–531. doi: 10.1038/cgt.2010.18. [DOI] [PubMed] [Google Scholar]
- 12.Gwak HS, Kim TH, Jo GH, Kim YJ, Kwak HJ, Kim JH, Yin J, Yoo H, Lee SH, Park JB. Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS One. 2012;7:e47449. doi: 10.1371/journal.pone.0047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin CW, Li XR, Zhang Y, Hu G, Guo YH, Zhou JY, Du J, Lv L, Gao K, Zhang Y, Deng H. TAp63 suppress metastasis via miR-133b in colon cancer cells. Br J Cancer. 2014;110:2310–2320. doi: 10.1038/bjc.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Huang J, Zhang L, Qu Y, Li J, Yu B, Yan M, Yu Y, Liu B, Zhu Z. MiR-133b is frequently decreased in gastric cancer and its overexpression reduces the metastatic potential of gastric cancer cells. BMC Cancer. 2014;14:34. doi: 10.1186/1471-2407-14-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen XN, Wang KF, Xu ZQ, Li SJ, Liu Q, Fu DH, Wang X, Wu B. MiR-133b regulates bladder cancer cell proliferation and apoptosis by targeting Bcl-w and Akt1. Cancer Cell Int. 2014;14:70. doi: 10.1186/s12935-014-0070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford M, Batte K, Yu L, Wu X, Nuovo GJ, Marsh CB, Otterson GA, Nana-Sinkam SP. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem Biophys Res Commun. 2009;388:483–489. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kano M, Seki N, Kikkawa N, Fujimura L, Hoshino I, Akutsu Y, Chiyomaru T, Enokida H, Nakagawa M, Matsubara H. miR-145, miR-133a and miR-133b: Tumor-suppressive miRNAs target FSCN1 in esophageal squamous cell carcinoma. Int J Cancer. 2010;127:2804–2814. doi: 10.1002/ijc.25284. [DOI] [PubMed] [Google Scholar]
- 18.Dai A, Sun H, Fang T, Zhang Q, Wu S, Jiang Y, Ding L, Yan G, Hu Y. MicroRNA-133b stimulates ovarian estradiol synthesis by targeting Foxl2. FEBS Lett. 2013;587:2474–2482. doi: 10.1016/j.febslet.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 19.Shen L, Li J, Xu L, Ma J, Li H, Xiao X, Zhao J, Fang L. miR-497 induces apoptosis of breast cancer cells by targeting Bcl-w. Exp Ther Med. 2012;3:475–480. doi: 10.3892/etm.2011.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015;356:184–191. doi: 10.1016/j.canlet.2014.01.031. [DOI] [PubMed] [Google Scholar]
- 21.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi: 10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koukourakis MI, Giatromanolaki A, Panteliadou M, Pouliliou SE, Chondrou PS, Mavropoulou S, Sivridis E. Lactate dehydrogenase 5 isoenzyme overexpression defines resistance of prostate cancer to radiotherapy. Br J Cancer. 2014;110:2217–2223. doi: 10.1038/bjc.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]