

Abstract
Sleep homeostasis reflects a centrally mediated drive for sleep, which increases during waking and resolves during subsequent sleep. Here we demonstrate that mice deficient for glial adenosine kinase (AdK), the primary metabolizing enzyme for adenosine (Ado), exhibit enhanced expression of this homeostatic drive by three independent measures: (1) increased rebound of slow-wave activity; (2) increased consolidation of slow-wave sleep; and (3) increased time constant of slow-wave activity decay during an average slow-wave sleep episode, proposed and validated here as a new index for homeostatic sleep drive. Conversely, mice deficient for the neuronal adenosine A1 receptor exhibit significantly decreased sleep drive as judged by these same indices. Neuronal knock-out of AdK did not influence homeostatic sleep need. Together, these findings implicate a glial-neuronal circuit mediated by intercellular Ado, controlling expression of homeostatic sleep drive. Because AdK is tightly regulated by glial metabolic state, our findings suggest a functional link between cellular metabolism and sleep homeostasis.
SIGNIFICANCE STATEMENT The work presented here provides evidence for an adenosine-mediated regulation of sleep in response to waking (i.e., homeostatic sleep need), requiring activation of neuronal adenosine A1 receptors and controlled by glial adenosine kinase. Adenosine kinase acts as a highly sensitive and important metabolic sensor of the glial ATP/ADP and AMP ratio directly controlling intracellular adenosine concentration. Glial equilibrative adenosine transporters reflect the intracellular concentration to the extracellular milieu to activate neuronal adenosine receptors. Thus, adenosine mediates a glial-neuronal circuit linking glial metabolic state to neural-expressed sleep homeostasis. This indicates a metabolically related function(s) for this glial-neuronal circuit in the buildup and resolution of our need to sleep and suggests potential therapeutic targets more directly related to sleep function.
Keywords: adenosine, adenosine kinase, delta power, glia, SWS, sleep
Introduction
While the function(s) of sleep remain unknown, the importance of sleep is underscored by CNS deficits resulting from sleep deprivation (SD) in flies, rodents, and humans. Acute SD is associated with CNS dysfunction in memory consolidation (Walker et al., 2003), synaptic plasticity and related protein kinase A signaling pathways (Frank et al., 2001; Ganguly-Fitzgerald et al., 2006; Vecsey et al., 2009; Tononi and Cirelli, 2014), and executive function (Kuriyama et al., 2008; Bjorness et al., 2009). Chronic SD, either voluntarily or experimentally induced, is associated with disrupted neuroendocrine control of metabolism (Knutson et al., 2007) and neural degeneration (Shaw et al., 2002; Kang et al., 2009).
Further evidence for the importance of sleep derives from the homeostatic nature of sleep modulation. Sleep homeostasis reflects both a buildup of sleep drive during waking and its decay during slow-wave sleep (SWS). Previous waking time has a direct association with the magnitude of neuronal-generated slow-wave activity (SWA) recorded from the surface EEG during ensuing SWS episodes (Borbély et al., 1982; Tobler et al., 1986; Franken et al., 2001). The magnitude of SWA is quantified as the spectral power of EEG in the 0.5–4.5 Hz range (also called δ power). It is commonly characterized as the SWA rebound response and provides a quantifiable measure for the sleep drive buildup during waking.
Activation of neuronal A1 receptors (AdoRA1) by adenosine (Ado) is necessary for the expression of the rebound SWA response to SD (Bjorness et al., 2009). AdoRA1s enhance the amplitude of SWA in cortical and thalamocortical local circuits by facilitation of SWA-like oscillations (Pape, 1992), an effect that may contribute to synchronization of SWA in regional cortical and thalamic circuits. In addition, Ado can facilitate SWA by reducing global cholinergic tone (Rainnie et al., 1994; Materi et al., 2000). Finally, the increase in Ado is activity-dependent (Mitchell et al., 1993; Manzoni et al., 1994; Huston et al., 1996), and glutamatergic synaptic activity is high during waking (Evarts et al., 1962; Evarts, 1964), consistent with the observed increase of CNS extracellular Ado with prolonged waking (Porkka-Heiskanen et al., 1997, 2000) promoting increased rebound SWA. Conversely, reducing extracellular Ado tone by knocking out 5′ectonucleotidase, an enzyme that converts AMP to Ado or by reducing glial release of ATP, which may be converted to Ado, reduces homeostatic sleep rebound (Halassa et al., 2009; Zielinksi et al., 2012).
Factors that increase metabolic demand (e.g., with increased neuronal activity) or decreased metabolite availability increase Ado (McIlwain and Pull, 1972), suggesting that Ado mediates negative feedback for homeostatic control of metabolic state and neuronal excitability (Greene and Haas, 1991). At the local circuit level, synaptic glutamate can activate NMDA receptors to increase extracellular Ado concentration (Brambilla et al., 2005). This Ado, in turn, mediates a negative feedback on excitatory glutamatergic synaptic activity through activation of inhibitory AdoRA1s on presynaptic glutamate terminals, closing the local circuit homeostatic loop (Brambilla et al., 2005). Thus, AdoRA1 activation effects on the local circuit level are consistent with the global effects to enhance homeostatic SWA drive related to prior waking.
However, the question remains as to the relationship of the Ado system to the biological basis of homeostatic sleep drive, particularly with regard to its resolution during SWS, as reflected by SWS-SWA decay. We used two conditional knock-outs affecting the Ado-system: one a neuron specific AdoRA1 knock-out, and the other, a glial targeted knockdown of AdK to examine this system's role in the control of sleep homeostatic SWA. AdK is the CNS enzyme most responsible for controlling Ado levels under nonpathological conditions (for review, see Boison, 2013). Full knock-out of AdK is lethal (Boison et al., 2002), whereas a rare missense mutation in humans results in AdK deficiency and a severe disruption of the methionine cycle (Bjursell et al., 2011); therefore, we used a conditionally inducible knockdown model to modestly and selectively reduce AdK in postmaturational glia within the CNS. Our findings implicate a metabolically related, glial-neuronal circuit, mediated by Ado, in the expression of global sleep need.
Materials and Methods
Subjects
Adult male mice were single housed throughout experimental procedures with a 12:12 light/dark cycle (8 A.M./8 P.M.) in temperature- and humidity-controlled animal rooms. Mice had ad libitum access to food and water at all times. All experiments were approved by the Dallas Veterans Administration Medical Center Institutional Animal Care and Use Committee or the University of Texas Southwestern Institutional Animal Care and Use Committee (based on location of specific experimental procedures).
Conditional AdoRA1 deletion (fAdoRA1;CaMKII:Cre, n = 10).
For a more detailed description, see Bjorness et al. (2009). Briefly, mice with the Adora1 gene flanked by loxP sites (Scammell et al., 2003) were crossed with the T50 line of mice in which the CaMKII promoter drove Cre recombinase expression (Tsien et al., 1996). This resulted in deletion of the AdoRA1 from excitatory neurons (primarily glutamatergic neurons) in many sleep-related regions of the brain, including the forebrain, parietal neocortex, hypothalamus, and thalamus (except n. reticularis), and dorsal pontine tegmentum beginning at P10 and reaching full expression before P60. Data from a subset of these animals have been reported previously (Bjorness et al., 2009). fAdoRA1 mice (n = 5) were used as a genotype control.
Tamoxifen-inducible adenosine kinase deletion (fAdK;GFAP:CreER, n = 52).
To make a fAdK;GFAP:CreER double conditional knockdown, alleles for AdK were replaced by knock-in of loxP sequencing flanking an internal exon (10) encoding an Asp residue critical to AdK enzymatic activity for both long and short isoforms (developed by J.A.B., A.A.F., R.W.G.), and these mutants were crossed with those harboring a GFAP:CreER transgene (Hirrlinger et al., 2006). PCR was performed using a set of primers able to distinguish homozygous, heterozygous, and wild-type AdK and another set of primers able to distinguish the presence of the GFAP:CreER transgene. Mice homozygous for floxed AdK with the presence of the GFAP:CreER transgene were used for experiments. In the adult, expression of CreER is limited to glia and a few neuronal progenitor cells. Exposure to tamoxifen (Tam) allows access of the CreER to the nucleus, thus restricting Cre-mediated recombination of the floxed alleles to glia and the few adult neuronal progenitors still present. This is important because AdK expression switches from neuronal to mostly astrocytic by P14 (Studer et al., 2006) with only a small subset of neurons retaining expression of AdK into adulthood such that Tam exposure in adulthood alters primarily glial expression of AdK. Mice were injected with either Tam (fAdK;GFAP:CreER_Tam; n = 26) or vehicle (fAdK;GFAP:CreER_Veh; n = 26) (for details, see below). Furthermore, because there is a small neuronal population expressing AdK into adulthood, the role of glial versus neuronal AdK was addressed by use of a conditional neuronal AdK knock-out in which floxed AdK mice were crossed with CaMKII:Cre mice resulting in a neuronal knock-out of AdK (fAdK;CaMKII:Cre; n = 5). Floxed AdK mice treated with Tam (fAdK_Tam; n = 6) were used as a control for the fAdK;CaMKII:Cre mice.
Wild-type controls.
C57BL/6 mice were treated with Tam (Charles River; BL6_Tam; n = 4 and University of Texas Southwestern breeding core; C57BL/6_Tam; n = 3), vehicle (University of Texas Southwestern breeding core; C57BL/6_Veh; n = 4), or untreated (University of Texas Southwestern breeding core; BL6; n = 4).
Tamoxifen treatment.
Tamoxifen (Sigma-Aldrich) was dissolved in 10% ethanol/90% sunflower oil and administered intraperitoneally for 5 d at a dose of 180 mg/kg. Vehicle (Veh) control consisted of a similar volume of 10% ethanol/90% sunflower oil. The Tam injections occurred at least 18 days before the beginning of baseline recording or tissue collection for AdK, Ado, and in vitro electrophysiology measurements. For mice undergoing polysomnography, Tam injections followed recovery from surgical and acclimation to recording cables (see below). The dose, number, and time period between injection and recording were based on previous studies showing that these parameters were well tolerated and resulted in significant gene knockdown (Hirrlinger et al., 2006).
Quantitative PCR analysis.
In a subset of fAdK;GFAP:CreER_Tam and fAdK;GFAP:CreER_Veh mice (n = 5/group), mRNA was extracted from cortical tissue using RNA STAT-60, reverse-transcribed using Superscript2 (Invitrogen), and quantitative PCR was performed with Sybr Green (Tm 60C, primer concentration: 900 nm each, ABI), using the following primers aatttggaagcgtgtgtgtg and ccccaagtccatcagatgtc. Values were normalized with cyclophilin-B.
Western blot analysis.
In a separate subset of fAdK;GFAP:CreER_Tam and fAdK;GFAP:CreER_Veh mice (n = 4/group), Western blot analysis was performed as described previously (Sahin et al., 2004) using a laboratory-generated rabbit polyclonal antibody (see below). Briefly, tissue was frozen in liquid nitrogen and then homogenized in boiling SDS lysis buffer. Protein concentrations were defined by BCA against a BSA standard curve. Equal amounts of protein from each sample was subjected to 12% SDS-PAGE and transferred to PVDF membranes. Coomassie staining of the membranes verified equal protein loading (see Fig. 4A). Western blot analysis of GFAP was performed using a commercial murine GFAP probe (GA5, Cell Signaling Technology).
Figure 4.
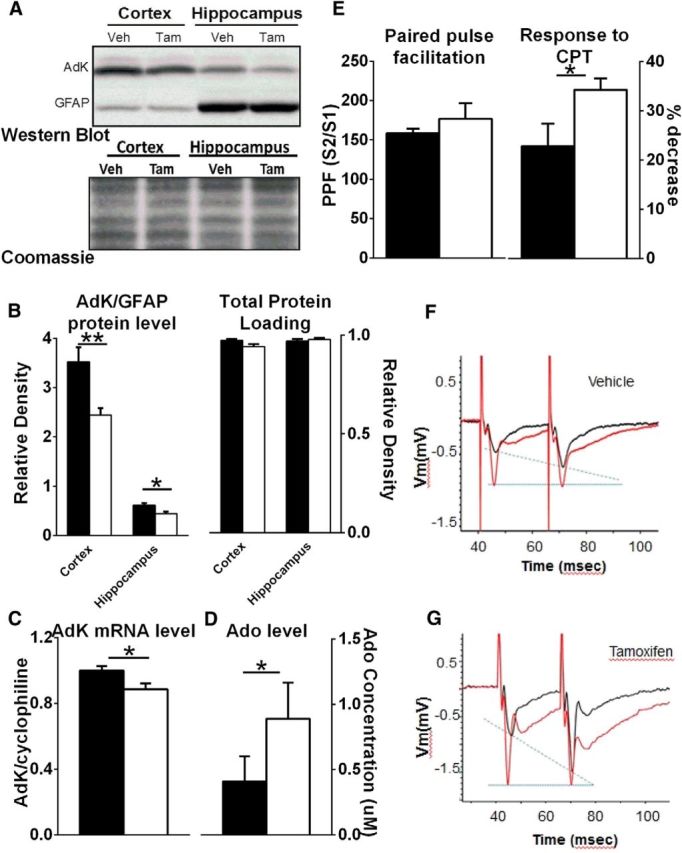
Characterization of AdK knockdown. A, Top, Representative examples of Western blots using AdK (top row) and GFAP (bottom row) antibodies from cortical and hippocampal tissue in one Veh- and one Tam-treated fAdK;GFAP:CreER mouse. Negative images and resized plots are shown. Light upper band represents a nonspecific cross reactive band that is typical for antibodies that detect protein in the 50 kDa range. Dark lower band represents both the short and long AdK isoforms that do not easily resolve (43.5 and 45 kDa, respectively). Bottom, Coomassie stain of total protein from the membrane used for the Western blots shown in top blot. This shows equivalent relative densities of Coomassie stain between genotypes and between regions and demonstrates the loading of equal amounts of protein. Thus, the similarity in GFAP expression between genotypes within the same region and the difference in GFAP expression over different regions is not attributable to differential protein loading (top and bottom blots). B, Tamoxifen decreases AdK protein as measured by quantitative immunoblotting using a monoclonal antibody against AdK (open symbols indicate Tam; closed symbols indicate Veh). AdK level is presented relative to glia-specific GFAP level because AdK is primarily expressed in glia in adults. C, Tamoxifen treatment decreases AdK mRNA as measured by quantitative PCR of fAdK;GFAP:CreER mice (open bars indicate Tam; closed bars indicate Veh). AdK values are presented relative to cyclophilin. D, Knockdown of AdK increases extracellular Ado concentration. Biosensors sensitive to the presence of Ado were used to measure Ado concentration from acute hippocampal slices taken from Veh-treated (filled bar) and Tam-treated (open bar) fAdK;GFAP:CreER animals. Tam-treated mice showed significantly higher Ado levels. E, Paired pulse facilitation, evoked by stimulation of the Schaffer collaterals and recorded as fEPSPs in the stratum radiatum of CA1, was measured in acute hippocampal slices from Veh-treated (filled bar) or Tam-treated (open bar) fAdK;GFAP:CreER animals. The ratios of the evoked response (measured by the slope of the fEPSP) of the second stimulus (S2) to the first stimulus (S1) were compared under baseline conditions (E, left axis; F, G, black traces). PPF indicating presynaptic inhibition is increased in slices obtained from Tam-treated mice. CPT, an AdoRA1 antagonist, blocks PPF (1 μm; F, G, gray traces). The Tam-treated group showed a greater sensitivity of the PPF to CPT, indicating a greater AdoRA1-mediated, tonic, presynaptic inhibition (E, right axis).
Generation of AdK antibody
A rabbit polyclonal antibody was generated against the long and short forms of recombinant mouse AdK (mAdK1 and mAdK2, respectively). Following the isolation of 10–15 ml of preimmune serum, each of three New Zealand white rabbits was immunized by subcutaneous injection of 1 ml of a 1:1 emulsified mixture of Freund's complete adjuvant and 1 mg of recombinant AdK (equal parts mAdK1 and mAdK2) diluted in 10 mm PBS, pH 7.4. Each animal received a booster injection 2 weeks after immunization, followed by additional booster injections at 4 week intervals. Booster injections were identical in volume and composition to the initial injection, except Freund's complete adjuvant was replaced with Freund's incomplete adjuvant. A test bleed (10–15 ml) was obtained from each animal 2 weeks after the first booster. A larger production bleed (20–30 ml) was obtained 2 weeks after each subsequent booster. Animals were given a total of 10 booster injections before being killed. Collected blood was incubated at 37°C for 1 h and placed at 4°C overnight. Serum was isolated from clotted blood by centrifugation at 200 × g for 10 min. Immunoblot analysis was performed to evaluate all antisera (1:200) for the ability to detect recombinant mAdK1 (50 ng) and endogenous AdK in mouse brain lysates (100 μg of total protein). Polyclonal antibodies for mouse AdK were purified from high-titer antisera by affinity-column chromatography using an AminoLink resin (Pierce) conjugated to recombinant mouse AK. Peak elution fractions were dialyzed in 10 mm MOPS, pH 7.5, and 154 mm NaCl, and used at a dilution of 1:1000 for immunoblot analysis, or 1:500 for immunohistochemistry.
Immunoblot analysis of recombinant mAdK1 and total protein from mouse brain homogenates showed that the affinity-purified antibody was highly selective for a doublet corresponding to the two isoforms of AdK in the brain, whereas immunohistochemical detection of AdK was apparent in ubiquitously distributed discrete cell bodies, with little or no staining of cellular processes or white matter tracts (data not shown). Dense populations of AdK-positive cells were located in the striatum, olfactory bulb, and granular layer of the cerebellum, whereas lower numbers of AdK-positive cells were present in the cerebral cortex and hippocampus. The punctate immunofluorescence staining pattern observed in these tissues was absent from control sections stained with secondary antibody alone (data not shown).
Adenosine measurements.
In a distinct subset of fAdK;GFAP:CreER_Tam and fAdK;GFAP:CreER_Veh (n = 4 group), mice were killed under isoflurane anesthesia, and acute hippocampal slices (400 μm) were made. Ado biosensors (Warwick Biosensors Group, University of Warwick, Coventry, United Kingdom) were used to determine Ado level as described previously (Frenguelli et al., 2003). Briefly, biosensors were calibrated to known Ado concentrations, and extracellular measurements were determined based on the differential signal between a pair of Ado biosensors and inosine biosensors (both biosensors are coated with nucleoside phosphorylase and xanthine oxidase, whereas only the Ado biosensor is coated with Ado deaminase making the inosine sensor impervious to Ado).
In vitro electrophysiology.
In the same subset of mouse hippocampal slices that were used with Ado biosensors, local field recordings were made to obtain paired-pulse facilitation (PPF, n = 5, fAdK;GFAP:CreER_Tam; n = 4, fAdK;GFAP:CreER_Veh). Additionally, PPF was measured in hippocampal slices from Tam-treated (n = 3) and Veh-treated (n = 4) C57BL/6 mice to ensure that Tam does not alter basal inhibitory tone. The hippocampus was used because it is the best characterized cortical region for Ado/AdoRA1 physiology (Dunwiddie and Masino, 2001; Fredholm et al., 2005). This is in part due to its well-known amenability to in vitro electrophysiological population responses, especially the CA3-CA1 synapse (Johnston and Wu, 1995). There is no evidence to indicate any qualitative difference in regard to AdoRA1 physiology to any other cortical region thus far examined. Hippocampal slices were prepared and stored in a submersion chamber in normal aCSF (in mm) as follows: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2-6H2O, 2 CaCl2-2H2O, 25 NaHCO3, and 25 dextrose, up to 6 h constantly saturated with 95% O2 5% CO2. Electrophysiological recordings were performed in a submersion chamber with slices constantly perfused (at a rate of 2–3 ml/min) with normal aCSF saturated with 95% O2 5% CO2 at 30°C for the duration of recording (up to 6 h) supplemented with drugs as indicated (the AdoRA1 antagonist, cyclopentyltheophylline) for experiments. A recording electrode was filled with normal aCSF and placed in stratum radiatum of CA1 and a stimulating electrode, placed within 200–300 μm in stratum radiatum to stimulate Schaffer collaterals. Stimulation was set to deliver 2 consecutive pulses 25 ms apart (paired pulses). The ratio of these pulses (PPR; P2/P1) was used to indicate any change in presynaptic release. PPF was also probed in the presence of 8-cyclopentyl-theophylline (1.0 μm), a selective A1 Ado receptor antagonist.
Surgery and recovery protocol.
Animals were prepared for surgery and fitted with frontal and parietal EEG electrodes (insulated wire tipped with a gold amphenol pin [Plastics One], soldered to a self-tapping screw) and dorsal nuchal muscle EMG electrodes, as previously described (Bjorness et al., 2009). Following a 2 week recovery from surgical procedures, mice were acclimated to a recording tether (Plastics One) for 1 week, given another week to recover from the tether acclimation, and then retethered and acclimated to the treadmill apparatus (TM) with the TM motionless (TMoff) for 1 d before baseline data collection as previously described (Bjorness et al., 2009). Baseline activity was recorded for 24 h (fAdoRA1;CaMKII:Cre) or 48 h (all other groups) immediately before acute or chronic, partial SD.
EEG and EMG recording, and sleep/waking analysis.
EEG and EMG signals were recorded using a Grass model 15LT amplifier system (Grass Technologies). Signals were sampled at 256 Hz. Following acquisition, data files were scored in 10 s epochs for sleep/waking state using a custom MATLAB (The MathWorks) based, sleep-scorer program. Epochs were manually checked to ensure they were assigned the correct state and flagged for removal (for power analyses) if they contained artifact. Standard criteria were used for state assignment; waking was characterized by low-amplitude, high-frequency activity in the EEG and modulation in the EMG, SWS was characterized by high-amplitude, low-frequency activity in the EEG and little modulation in the EMG, and rapid eye movement sleep (REM) was characterized by low-amplitude, high-frequency activity in the EEG and a lack of muscle tone with occasional muscle twitches in the EMG. State episodes began with 30 consecutive seconds of a single state and were ended by 30 consecutive seconds of another state. Power spectrum values were calculated in a 2 s window with a 1 s overlap and a Hamming window using the mean squared spectrum function in MATLAB.
SD.
For acute SD, the TM belt was on for 4 or 6 h/d for 7 or 4 consecutive days, respectively, to provide an adequate number of SWS episodes of recovery sleep. Data for recovery sleep were gathered from 0 to 3.5 h after SD (ZT 4–7.5 h, ZT 6–9.5 h for 4 and 6 h SD, respectively) based on the approximate return to baseline of the SWS-SWA SD-induced rebound (matched circadian time, data not shown).
For chronic SD, a TMon-TMoff cycle of 4 h on (SD), then off for 2 h (recovery), for a total of 8 cycles over 48 h was used. SWA values from the TMon (SD) and TMoff (recovery) condition were compared with baseline values taken from the same circadian time for each animal.
Sleep need parameters.
Four parameters were used to assess sleep need, three of which have been used previously to assess homeostatic sleep need (SWS-SWA power, SWS consolidation, decay across the circadian inactive phase) and a novel parameter (decay of an averaged SWS episode), which is described and validated here for the first time.
SWS-SWA power.
Power in the SWA band (0.5–4.5 Hz) was normalized to gamma power (30–50 Hz).
SWS episode consolidation.
A cumulative histogram was used to determine the relative contribution of SWS episode lengths. The first bin was 8 (10 s) epochs; then each successive bin increased by 4 epochs. The cumulative number of episodes in each successive bin length was expressed as a percentage of the total number of episodes.
SWS-SWA decay analysis
Decay across the circadian inactive phase.
SWS-SWA was normalized to the 24 h SWS-SWA, and the percentage change between the first and last consolidated sleep episode for each baseline day for each mouse was calculated as previously described (Nelson et al., 2013). The baseline day with the clearest consolidation periods at the beginning and end of the light phase was used for statistical analysis.
Decay of an averaged SWS episode.
A quantitative assessment of SWA decay within an averaged SWS episode provides a directly measured SWS-SWA decay response with fewer uncontrolled variables than more traditional, indirect measures. The assessment of decay from the beginning to the end of the circadian inactive phase involves many SWS episodes of variable duration (SWA wanes during each SWS episode) interspersed with waking episodes of variable duration (waxing of sleep need occurs during each waking episode).
It is important to consider the large variability of SWA expression that occurs during SWS.
A major source of SWS-SWA decay variance is peak amplitude of SWS-SWA because it is a function of the varying, preceding time in waking. The exponential decay rate is a function of the instantaneous SWA amplitude and will accordingly vary with preceding waking duration. However, the time constant for decay is independent of the amplitude and can be used to characterize the kinetics of the decay independently of SWA amplitude.
SWS episodes of at least 5 min in duration were collected for each animal, and SWA per epoch was normalized to average gamma power or average waking SWA. The normalization factor was chosen based on the power versus frequency distribution such that a band with the same relative power distribution was used.
SWS episodes of ≥5 min duration were selected for analysis because peak SWA was not obtained until 3–4 min following the transition from waking to SWS at t = 0. This constraint has previously been used when modeling SWA decay across SWS episodes (Franken et al., 2001). Using Excel (Microsoft), each SWS episode meeting the 5 min duration criteria was transcribed to a separate column. Each column represents a single SWS episode, and each row represents a consecutive 10 s epoch, starting with t = 0 (the transition from waking to SWS) to whenever the episode spontaneously terminates (e.g., a 10 min episode terminates at t = 60 or row 60). Each cell of the spreadsheet reflects the normalized SWA recorded for that particular epoch in that particular episode.
Next, using Prism (GraphPad Software), a natural log transform was used to linearize the SWA to time relationship. The linearized SWA to time curves for all episodes were averaged to obtain the average slope for each animal. Group averages were then created by averaging the average episode for each member of the group, after which values were retransformed by taking the exponential to provide a group average SWA power versus time curve.
Threshold criteria for determination of a tau for SWA decay of an averaged SWS episode were used as follows: (1) each animal must express a minimum of 5 episodes of SWS of 15 min or longer (5× >15 min) in length (with a maximum of 52 SWS episodes/animal due to Prism software limitation); and (2) the group average must consist of at least 5 animals. The goodness of fit assessed by the absolute sum of squares was <1.0 for all groups having a determination of tau. If <5× >15 min episodes and/or <5 animals/group were used, the variability of the decay toward the end of the average episode became too large for useful determination of the time constant. Two mice from the fAdK;GFAP:CreER_Veh group were excluded and the fAdoA1R;CaMKII:Cre group was excluded by these SWS episode duration-related criteria. Even if the criterion of 5× episodes of >15 min was relaxed for the fAdoA1R;CaMKII:Cre mice to 5× >12 min (n = 7 mice in this group met this relaxed criterion), the tau was indeterminate because there was little to no decay (see Fig. 3D).
Figure 3.

Neuronal AdoRA1s are necessary for SWA decay across light phase, during a SWS episode and for SWS consolidation. A, Integrated SWA/10 s epoch during SWS declines over the course of the 12 h light phase (ZT0–12 on x-axis) for a typical control wild-type mouse (fAdoRA1). B, An example of SWS-SWA/epoch across the 12 h light phase from an AdoRA1 conditional knock-out mutant mouse (fAdoRA1;CamkII:Cre) shows a lack of SWS-SWA decay. C, Pooled data for percentage decrease in SWS-SWA from the first to last hour of the light phase. Filled bar represents fAdoRA1. Open bar represents fAdoRA1;CaMKII. D, Loss of AdoRA1 attenuates both expression of SWA during SWS and abolishes the decay of SWA during an average SWS episode. The SWS-SWA amplitude is expressed as fold increase over the 24 h average waking SWA. E, Loss of AdoRA1 fragments SWS episodes, as demonstrated by cumulative distribution of SWS episode duration for fAdoRA1;CaMKII:Cre (open circles) and fAdoRA1 (filled circles) animals.
There was one exception to the 5× >15 min and 5 animals/group criteria made for the acute and chronic, partial SD groups. The animals in these two groups had too few 15 min episodes within their restricted period of recording (SWS episodes were obtained only for the 3.5 h period following acute SD or for 2 h recovery periods between successive SD bouts of chronic, partial SD). Thus, the 5× >15 min episode criterion was relaxed to 5× >12 min episode length, and the number of animals/group was reduced to at least 4 animals. Despite the relaxed constraint, 4 consecutive days of acute 6 h SD and 7 d of 4 h SD were still required to obtain 5× >12 min duration SWS episodes for each experimental subject. The acute and chronic SD, mice from the wild-type phenotype groups (BL6_Tam, fAdK_Tam, fAdK;GFAP:CreER_Veh) were pooled into a single control group because many mice did not reach the threshold of 5× >12 min SWS (n = 4 BL6_Tam, n = 1 fAdK_Tam, n = 3 fAdK;GFAP:CreER_Veh excluded). This pooled SD group fit had an absolute sum of squares of <1.0. These groups were also combined into a pooled control group for comparison of decay of an averaged SWS-SWA episode under baseline conditions. There was no significant difference between BL6_Tam, fAdK;GFAPCreER_Veh, and fAdK_Tam for normalized SWA power, SWS-SWA rebound following chronic, partial SD, and average SWS episode duration (one-way ANOVA; data not shown). Additionally, this lack of difference in sleep need parameters between Veh-treated and Tam-treated controls indicates that Tam treatment itself does not influence sleep behavior.
Analysis of the decay rate.
To determine the best fit for a single exponential decay rate, a fragment of the decay curve (t = ∼3.0 to t = ∼12 min) was fit using a single-phase nonlinear regression analysis, which can be represented as SWAt = (SWA0 − plt) × exp(−kt) + plt, where SWA0 is the value of SWA at time = 0 when SWA is maximal, plt (plateau) is the value of SWA as time approaches infinity when SWA is minimal, k is the rate constant of decay, and t is time [τ, the time constant of decay = 1/k]. Plateau does not equal 0 but is close to the SWA level upon transition from SWS to waking at the end of the average SWS episode. The fragment began at t = ∼3 min to exclude the initial rising phase of SWA. This allowed the most accurate estimation of the SWA0 and plt values by extrapolation. The fragment captured the least variable part of the decay, and the size of the fragment was at least as long as the expected τ (for baseline) based on preliminary analysis. Once determined, the plt was subtracted and the span (SWA0 − plt) was divided out, allowing direct comparison of the decay from SWAt/SWA0 = 1 to SWAt/SWA∞ = ∼0. This gives the equation, (SWAt − plt)/(SWA0 − plt) = exp(−kt). Transformation by natural log yields: ln(SWAt − plt)/(SWA0 − plt) = −kt, where the negative slope = k or 1/τ for every episode regardless of the SWA0 of that particular episode. Transformations and curve fitting were done using GraphPad Prism (GraphPad Software); averaging within and across animals was done in Excel (Microsoft).
Statistical analyses
Statistical analyses were performed using Prism (GraphPad Software). Comparisons were made between baseline and SD conditions and across groups (fAdK;GFAP:CreER_Tam vs fAdK;GFAP:CreER_Veh, fAdK;CaMKII:Cre vs fAdK_Tam). For SWA decay analysis, a comparison of fits was done using an F test of the k values of the decay. For average episode duration, SWA, and time in state analyses, an unpaired t test was used for unpaired data and a paired t test was used for paired data unless otherwise noted, whereas for SWA or time in state across the circadian period, a two-way ANOVA was used. For the percentage change in SWS-SWA from the beginning of the light cycle to the end of the light cycle, a one-way ANOVA was used. Statistical significance was set at p < 0.05, and average values ± SEM are provided, except where ± SD is explicitly noted.
Results
Humans and other primates have highly consolidated sleep, with a single sleep period, typically at night, in which sleep predominates and waking time is relatively brief. Rodents show sleep/waking behavior that is quite similar to humans, with the exception of having comparatively fragmented sleep, with average sleep episode durations of less <10 min interspersed with waking episodes in which sleep need and SWA power can reaccumulate. We sought an assessment of SWA decay within an SWS episode to quantify the resolution of sleep need and SWA power during sleep. However, there is considerable variability of SWA expression during SWS as it is affected by inevitable changes from sensory input or the internal state of the animal, even if not of sufficient strength to induce a transition to waking (Dijk and Beersma, 1989). This uncontrolled variability necessitates an average of the SWA decay over many individual SWS episodes (for complete details, see Materials and Methods). We first measured SWA decay under baseline and SD recovery conditions in a set of wild-type animals, after which we investigated the influence of neuronal AdoRA1 receptors and glial AdK expression on this SWA decay, along with standard homeostatic sleep need measures, including changes in SWA power and SWS episode duration.
Time constant of SWS-SWA decay increases with prolonged waking
The time constant, τ, of SWS-SWA decay within an averaged SWS episode under baseline, unrestricted conditions was compared with that from SWS episodes following acute 4 h SD and acute 6 h SD control animals. τ increased from 9.6 min under baseline conditions to 13.6 min and 17.4 min after 4 and 6 h SD, respectively (Fig. 1A–C,F; p < 0.0001 comparison of fits test, baseline vs 4 h SD, baseline vs 6 h SD, 4 h SD vs 6 h SD).
Figure 1.

SWA decay time constant correlates with sleep need in wild-type mice. The time constant of SWA decay (τ) during an average SWS episode following differing amounts of SD is determined by a single phase exponential, regression fit in wild-type mice. The conditions for C57BL/6 mice include baseline, 4 and 6 h acute SD (A–C, respectively). D, E, The pooled group of genetic control strains (BL6_Tam, fAdK_Tam, fAdK;GFAP:CreER_Veh) experienced chronic SD (4 h SD with 2 h recovery for 8 consecutive cycles). Plots represent time of the SWS episode (plotted for each 10 s epoch; x-axis) versus normalized SWA (y-axis). F, Histogram of SWA decay during an average SWS episode determined for each SD condition shows a graded slowing in proportion to previous enforced W duration. “BL+/−St. Dev.” is a pooled control strain used for chronic SD. The τ determined under baseline conditions during either the phase of minimal sleep need (6 h, early part of the active phase, ZT12–18_BL) compared with the phase of high sleep need (6 h, early part of the inactive period, ZT0–6_BL) shows a similar increase in duration with sleep need.
In a separate group of control animals, τ was assessed under chronic, partial SD conditions, consisting of cycles of 4 h SD followed by 2 h of recovery, repeated over 48 h. τ increased from 9.4 min to 17.6 min from baseline to chronic, partial SD condition (Fig. 1D–F; p < 0.0001 comparison of fits test, baseline vs SD).
Next, τ during a period of relatively low sleep need (first half of active dark phase, ZT12–18, immediately following a period of high sleeping time) was compared with a period of relatively high sleep need (first half of inactive light phase, ZT0–6, immediately following a period of low sleeping time) under baseline conditions. In control animals, τ was 9 min, early in the dark phase, compared with 11.6 min, early in the light phase (Fig. 1F; p = 0.0002; n = 18 light phase, n = 9 dark phase). Together, these results show that the time constant of SWS-SWA decay within an average SWS episode is proportional to previous time spent awake under both experimentally enforced (SD) and freely behaving undisturbed (active phase vs inactive phase) conditions.
Time course of initial buildup of SWA within an SWS episode matches gamma decay
SWS-SWA decays were determined based on a fragment of the average SWS episode such that the initial period of the average SWS episode was excluded due to the rise in SWA power that follows the transition from waking to SWS. This is necessary because peak SWA occurs minutes after the start of SWS. The time constant of SWA increase from t = 0 to t = ∼3 min corresponded to the time constant of gamma power decrease following the waking to SWS transition (Fig. 2). The amplitude of the gamma power has been associated with monoaminergic and cholinergic arousal center activity (Borbély et al., 1982; Franken et al., 2001). Accordingly, the decrease of the gamma power is consistent with the reported gradual decrease of monoaminergic arousal center activity accompanying this transition (Tobler and Borbély, 1986; Pape, 1992).
Figure 2.

SWS-SWA for an averaged SWS episode. A, SWA decay is shown during an averaged SWS episode from a wild-type strain (C57BL/6_Tam, n = 5). B, C, On transition from W to SWS, SWA increases at a similar rate (τ = ∼0.57 min) as gamma power decreases (τ = 0.58 min). B, Inset, Full averaged SWS episode shown in A with the SWA rising phase marked in red.
Necessity of neuronal AdoRA1 in the expression of homeostatic sleep need
We previously showed that mice lacking neuronal AdoRA1s do not have the typical increase in SWS-SWA power following SD (i.e., rebound SWA) (Bjorness et al., 2009). This loss of prolonged, waking-induced increased SWA could likely affect the SWS-SWA decay. Accordingly, decay in conditional AdoRA1 knock-out mice under freely behaving undisturbed and experimentally enforced waking conditions was examined. First, it was measured across the inactive, sleep phase of the circadian period from ZT0–12 under baseline conditions in fAdoRA1;CaMKII:Cre (AdoRA1 KO) and fAdoRA1 (control) mice. The mean SWA from the first consolidated SWS episode to the last episode of the circadian sleep phase significantly decreased in fAdoRA1 mice by 17.5 ± 3.2% (p < 0.03, Wilcoxon matched pairs; n = 5; Fig. 3A,C). In contrast, AdoRA1 mutant mice (Cre+) showed little or no decrease in mean SWA across the circadian inactive phase (4.7 ± 5.8%; n = 6; Fig. 3B,C). Next, the SWA decay was compared within an averaged SWS episode in fAdoRA1;CaMKII:Cre and fAdoRA1. The time constant of decay in the control fAdoRA1 mice was 10 min, similar to the time constant found in other wild-type controls (Fig. 3D). In contrast, SWA decay was absent in the Cre+ mice (τ could not be determined; Fig. 3D). Both decays across the circadian inactive phase and within an averaged SWS episode were dependent on neuronal AdoRA1s.
Another index of homeostatic sleep drive is SWS episode consolidation, which is usually measured as the average SWS episode duration. Consolidation of SWS episodes marks the buildup of sleep drive during waking in mammals (Trachsel et al., 1991; Tobler et al., 1997; Franken et al., 1999; Leemburg et al., 2010; Gvilia et al., 2011) and flies (Huber et al., 2004). To fully characterize the role of AdoRA1 in sleep drive expression, we compared average SWS duration in fAdoRA1;CaMKII:Cre and fAdoRA1 to mice. AdoRA1 knock-out mice had shorter average SWS episode durations (unpaired t test, p = 0.0002, data not shown) indicative of fragmentation (decreased consolidation) of sleep episodes under baseline conditions, as demonstrated by the leftward shift in the cumulative distribution of SWS episode durations (Fig. 3E). Together, the expression of sleep drive, as assessed by three sleep-drive indices (SWA rebound, SWA decay across and within SWS episodes, and sleep consolidation), was attenuated or absent in the neuronal AdoRA1 knock-out mice.
Glial AdK reduction increases Ado levels and inhibitory tone
Treatment of AdK;GFAP:CreER mice with Tam caused 20% reduction of AdK protein in neocortex and archicortex tissue compared with Veh treatment, as assessed by Western blotting (hippocampal tissue: p = 0.014, cortex tissue: p = 0.029, Mann–Whitney t test). The AdK protein was normalized to glial-specific GFAP protein because AdK is expressed almost exclusively in glia in adults (Studer et al., 2006) (Fig. 4A,B). A 10%–15% reduction of AdK mRNA relative to cyclophilin mRNA was also observed in cortical tissue as measured by qPCR (p = 0.029, Mann–Whitney t test; Fig. 4C). These relatively modest decreases in AdK mRNA and protein abundance are consistent with previously reported efficiency of Tam-induced recombination of cortical glia (∼28%) using the same GFAP:CreER construct crossed with a reporter transgene (Hirrlinger et al., 2006).
Cortical extracellular Ado concentration, measured using Ado biosensors in acutely prepared hippocampal slices from fAdK;GFAP:CreER_Tam mice, was significantly greater than Ado concentration in slices from fAdK;GFAP:CreER_Veh mice (p = 0.035, Mann–Whitney t test; Fig. 4D).
Presynaptic, inhibitory Ado tone was assessed in vitro using AdoRA1 antagonist-sensitive PPF of cortical Schaffer collateral synapses. The tone, as measured by the percentage decrease in response to CPT (1 μm), was significantly greater in slices from fAdK;GFAP:CreER_Tam mice (p = 0.04, unpaired t test; Fig. 4E–G). However, there was no significant effect of Tam alone on PPF response to CPT (p = 0.49, unpaired t test of C57BL/6_Veh vs C57BL/6_Tam; data not shown). These data provide convergent evidence for the control of extracellular Ado concentration and AdoRA1 inhibitory tone by glial AdK.
Reduced glial AdK increases SWA power
It has previously been shown that a reduction of extracellular Ado resulting from transgenic overexpression of AdK in all cell types decreases SWA in SWS and waking (Palchykova et al., 2010), whereas administration of an AdK inhibitor increases SWA (Radek et al., 2004). However, the effect of decreased expression of glial AdK on SWA has not been previously described. SWA was significantly increased in fAdK;GFAP:CreER_Tam mice during both SWS and waking compared with fAdK;GFAP:CreER_Veh mice (p = 0.024, Mann–Whitney test; Fig. 5A,C).
Figure 5.

Glial adenosine kinase knockdown increases SWA. A, Average, normalized SWA during SWS and waking (left y-axis). Under baseline conditions, fAdK;GFAP:CreER_Tam mice (open bars) show a significant increase in SWA during SWS and waking compared with Veh-treated mice (filled bars). Tam-treated mice spent the same amount of time in SWS compared with fAdK;GFAP:CreER_Veh mice (right y-axis). B, The circadian distribution of SWS-SWA is illustrated by the percentage SWA in 2 h bins of the average 48 h baseline SWS-SWA for each animal and is similar between groups. C–E, Power spectrum distribution in Tam- and Veh-treated fAdK;GFAP:CreER mice (open symbols indicate Tam; closed symbols indicate Veh) during waking (C), SWS (D), and REM (E). F, SWA across states is shown over 48 h of chronic, partial SD with 4 h/2 h cycles of SD and recovery. SWA is greater during both SD (TMon) and during the recovery period (TMoff) in the Tam-treated compared with the control mice. G, Tam-treated mice (open bars) show a greater percentage increase of SWA from enforced wake to SD recovery sleep (left y-axis, TMon to TMoff) compared with Veh-treated mice (filled bars). The same two genotypes were also compared for the percentage increase from baseline SWS time to SD recovery SWS time (right y-axis, base to TMoff). Unlike the greater increase in SWA in Tam-treated mice, both groups show similar increases in SWS time following SD.
The Tam-induced increase in average SWA per 2 h bin was apparent throughout the circadian cycle (p = 0.045, two-way ANOVA, F(1,24) = 4.49; data not shown). However, the circadian distribution of SWA was not significantly altered across genotypes (Fig. 5B). There was only a slight change in the state-specific EEG power distribution across the EEG frequency spectrum between Tam- and Veh-treated mice (a small increase of power in the 2–4 Hz range compensated for by a nonsignificant decrease spread over the 10–30 Hz range; Fig. 5C–E).
Reduction of glial AdK slows SWS-SWA decay and increases SWS consolidation
The effect of glial AdK on sleep drive expression was assessed using three indices that directly correlate with prior waking experience, including the following: (1) rebound SWA; (2) SWA decay across and within SWS episodes; and (3) SWS episode consolidation.
Tam- and Veh-treated fAdK;GFAP:CreER mice were subjected to chronic, partial SD via a slowly moving TM for 4 h SD/2 h recovery repeated 8 times over 48 h (Fig. 5F,G) to examine SWA rebound. The ratio of SWA during recovery sleep to the SWA recorded during SD (TMon compared with TMoff) was used to quantitatively compare rebound SWA between genotypes. The reduction of glial AdK in the Tam-treated mice resulted in a larger SWA rebound ratio (p = 0.038, Mann–Whitney test; Fig. 5F,G). Remarkably, there was no significant difference in average time spent in SWS (Fig. 5G) or waking (difference in medians = 2.6%; p = 0.22, Mann–Whitney test; data not shown) between genotypes. Compared with the large effect on rebound SWA, the insignificant effect on SWS or waking time indicates a dissociation of the need for sleep from sleep time.
The decay of SWS-SWA across episodes was assessed over the circadian inactive phase (ZT0–12) by comparing the mean SWA of the first consolidated SWS episode of the inactive phase with the last. The fAdK;GFAP:CreER_Tam mice showed a similar percentage decrease in SWA across the light phase to the Veh-treated and pooled control animals (Veh = −10.3 ± 3.4%, Tam = −13.5 ± 2.9%, data not shown). However, the Tam-treated group's SWS-SWA was greater at both the start (ZT0) and end (ZT12) of the inactive period consistent with a slowed rate constant for SWS-SWA decay within SWS episodes. To test this, the decay of SWA within an averaged SWS episode was examined in both Tam- and Veh-treated fAdK;GFAP:CreER mice. The decay time constant in Tam-treated mice was more than doubled at τ = 22.5 min compared with the τ = 8.6 min of Veh-treated controls (p < 0.0001, comparison of fits test, Fig. 6A,B). Thus, the effect of reduced Ado metabolism by glial AdK reduction was a marked slowing of the SWS-SWA decay time constant showing the glial control of SWS-SWA decay.
Figure 6.

Reduction of glial, but not neuronal, AdK slows SWA decay during an SWS episode and induces increased SWS consolidation. A, B, Tam-treated mice have a significantly slowed decay rate (τ = 22.5 min) compared with Veh-treated mice (τ = 8.6 min). C, Cumulative distribution of SWS episode durations is shifted to the right (more consolidated) in Tam-treated fAdK;GFAP:CreERVeh-treated mice. D, E, The time constant of SWA decay between control fAdK_Tam and neuronal AdK knock-out animals, fAdK;CaMKII:Cre (τ = 11.1 vs 10.1 min) was similar. F, Mice lacking neuronal AdK (fAdK;CaMKII:Cre) have a similar SWS episode duration distribution as fAdK_Tam mice.
Reduction of glial AdK caused a significant shift to the right of the cumulative histogram of SWS episode durations compared with control Veh-treated mice, with an evenly distributed increase of sleep episode duration or consolidation (average episode duration for Tam-treated = 6.86 ± 0.51 min, Veh-treated mice = 5.63 ± 0.46 min; p = 0.036, Mann–Whitney test; Fig. 6C). Thus, animals with reduced AdK glial metabolism of Ado show increased sleep drive expression based on the indices of increased SWS, increased rebound SWA in response to SD, and slowed decay of SWS-SWA.
AdK expression in adult neurons is negligible; thus, a selective loss of neuronal AdK-mediated Ado metabolism would not be expected to affect sleep drive indices. This was confirmed by a conditional knock-out of AdK induced by a cross of the floxed AdK mouse with aCaMKII:Cre mouse expressing Cre recombinase primarily in excitatory neurons of the forebrain. Neither SWS episode consolidation nor the decay of SWA during SWS was affected in the neuronal AdK knock-out, compared with genetic control mice (Fig. 6D–F). Moreover, in contrast to the glial loss of AdK, neuronal AdK loss did not impact total SWS-SWA (fAdK;CaMKII:Cre = 12.2 ± 1.3; fAdK_Tam = 14.3 ± 1.4; data not shown). Because the AdoRA1 affecting the sleep drive indices in the fAdoRA1;CaMKII:Cre mice are exclusively neuronal (Fig. 3) and the primary metabolism of the extracellular Ado that activates neuronal AdoRA1 is glial, a neuronal-glial circuit controlling expression of sleep drive is indicated (Fig. 7).
Figure 7.

The decay time constant for SWA during the SWS episode is influenced by the genes coding for AdoRA and glial AdK. A, Histogram of the time constants for decay (τ) of SWA during an average SWS episode under baseline conditions for different genotypes. Loss of neuronal AdoRA1s (fAdoRA1;CaMKII:Cre) results in loss of decay. Reduction of glial AdK expression (fAdK;GFAP:CreER_Tam) increases τ by >10× the SD (SD = 1.1 min). B, Diagram depicting systems-level relationships between waking, which is facilitated by arousal and increases sleep need, and sleep function, which decreases sleep need and presumably enhances arousal. Increased waking results in increased Ado tone that increases sleep need indices. C, At the local circuit level, the expression of sleep need is mediated by Ado acting on neuronal AdoRA1 to facilitate rebound SWA in response to prolonged enforced waking. During SWS, Ado flows down its concentration gradient into glia by glial equilibrative transporters. There it is metabolized by the low-capacity but high-affinity glial enzyme, AdK, thus reducing the activation of neuronal AdoRA1 and thereby controlling the rate of SWA decay.
Discussion
Sleep disruption is part of the symptomatology of all major psychiatric and neurodegenerative disorders. However, the relationship of this sleep-related pathology to sleep homeostasis and function remains unclear because the biological substrate for sleep homeostasis has not been defined. The indices of sleep homeostasis reflecting the drive for sleep include rebound SWA expressed during SWS, SWS consolidation, the decrease in SWA across the inactive period, and the averaged SWS-SWA episode decay. All of these indices are controlled by CNS extracellular Ado, which acts at neuronal AdoRA1 to enhance homeostatic SWA. The Ado is transported into glia where it is metabolized by AdK to reduce homeostatic SWA. Thus, a glial-neuronal circuit mediated by Ado can control sleep homeostasis.
However, not all of SWA correlates with sleep drive. Modulation of SWA is controlled in at least two distinct ways: (1) by the level of arousal related to cholinergic and monoaminergic modulatory neurotransmitter activation; and (2) through sleep homeostatic influence mediated by the AdoRA1-AdK system. With respect to arousal, on transition from waking to SWS, there is an increase in SWS-SWA over waking SWA, even in the absence of neuronal AdoRA1 activation (Bjorness et al., 2009). This SWA is associated with the waking-to-SWS transitional decrease of cholinergic (Steriade et al., 1990) and monoaminergic (Hobson et al., 1975; Aston-Jones and Bloom, 1981) activity, and it occurs independently of sleep drive. A further increase in SWS-SWA correlates with previous waking duration (Franken et al., 2001) whether waking is driven by circadian phase or experimentally induced and is characterized as the homeostatic sleep response. The results presented here demonstrate that the homeostatic response requires neuronal AdoRA1 and is under the dynamic control of glial AdK.
The Ado-mediated control of SWS-SWA can occur at the local circuit level. Its release is controlled by local neuronal activity (Mitchell et al., 1993; Manzoni et al., 1994). The source of this Ado is probably both neuronal and astrocytic (Wall and Dale, 2013) with the greater part from neurons. Once released, Ado acts on AdoRA1s at both presynaptic sites to reduce presynaptic glutamate release (Scanziani et al., 1992; Brambilla et al., 2005) and postsynaptic sites to increase inwardly rectifying potassium conductance (Trussell et al., 1985; Gerber et al., 1989; Greene and Haas, 1991; Rainnie et al., 1994) and reduce Ih conductance (Pape, 1992; Rainnie et al., 1994). These AdoRA1 effects can act together to destabilize cortical up-states (Steriade et al., 2001) increasing the frequency of transitions between up and down states, and it is these transitions that are most responsible for the generation of EEG SWA (Steriade et al., 2001; Destexhe et al., 2007; Haider et al., 2009).
The increase in extracellular Ado associated with SD (Porkka-Heiskanen et al., 1997) increases activation of neuronal AdoRA1 responsible for the SWS-SWA rebound. In rodents, the SWA rebound then resolves over a series of SWS episodes (Franken et al., 2001) as the extracellular Ado concentration declines (Porkka-Heiskanen et al., 1997). The SWS episodes are interspersed with waking or REM episodes when Ado may reaccumulate but after SD or during the circadian active phase the SWS episodes are relatively consolidated such that SWS-SWA decay predominates.
Within an SWS episode, glial AdK controls the rate of SWA's resolution. AdK is the highest-affinity CNS enzyme for the metabolism of Ado. It is an evolutionarily ancient enzyme that binds ATP and Ado (Km = 0.2–2 μm) and releases AMP and ADP by an ordered Bi-Bi mechanism, subject to substrate (Ado) inhibition (Arch and Newsholme, 1978; Mimouni et al., 1994; Mathews et al., 1998). In the adult mammalian CNS, extracellular Ado diffuses down its concentration gradient into glia by equilibrative transporters (Peng et al., 2005). Glia are the primary source of AdK in adult rodent CNS (Studer et al., 2006); and under physiological conditions, AdK maintains the Ado diffusion gradient from the extracellular space into the glia. A reduction in AdK activity increases extracellular Ado and the activation of neuronal AdoRA1 by slowing or potentially reversing the glial influx of Ado (Pak et al., 1994; Arrigoni et al., 2001). An increase in expression of AdK, induced by gliosis, may cause epileptogenesis through increased Ado metabolism and concomitantly reduced AdoRA1 activation (Boison, 2006). Furthermore, experimental overexpression of AdK throughout the CNS is not only epileptogenic (Masino et al., 2011) but also reduces SWA (Palchykova et al., 2010). Thus, glial AdK has a critical role in regulating extracellular Ado and its activation of neuronal AdoRA1. The results presented here suggest that the Ado regulated by glial AdK is responsible for the homeostatic part of SWS-SWA and, furthermore, support the potential of AdK as a therapeutic target of Ado-related pathological conditions (Boison, 2013) that may be extended to include insomnia.
The rate of AdK-mediated metabolism of Ado may be affected by the following: (1) its limited capacity, which may result in a shift toward zero order kinetics as Ado levels increase beyond AdK's capacity; (2) substrate inhibition of AdK by Ado (Mathews et al., 1998) consistent with a step-like decrease in AdK velocity as Ado levels increase; and (3) the ratio of [ATP]/[AMP × ADP] (Mimouni et al., 1994), consistent with a function of glial AdK as a metabolic sensor with Ado levels as a readout. Of these three, only the AdK capacity is expected to be directly reduced by the Tam-induced reduction in glial AdK. The reduced capacity was sufficient to slow the average time constant of decay, τ, of SWA decay during an SWS episode by twofold. A similar slowing of τ was observed in response to both acute and chronic, partial SD (Fig. 7A). The net result is that the sleep homeostatic response of rebound SWS-SWA is greatly enhanced in conditions where AdK capacity is likely to be exceeded, including SD.
The τ determined from the SWA decay during an average SWS episode shows little variability under baseline conditions across several genetic control strains. In contrast, mutations in Ado system genes resulted in a changes of ∼10 units of SD of this τ. In particular, τ for SWS-SWA decay is indeterminate in the absence of neuronal AdoRA1, probably because rebound SWA is for the most part absent in the AdoRA1 mutant (Bjorness et al., 2009). The τ is more than doubled in a genetic strain with a reduction of Ado's primary metabolizing enzyme in the CNS (fAdK;GFAP:CreER_Tam). The Z-score derived from the sample population means is >10 for the Ado system mutants, demonstrating the critical role of the Ado system in this sleep homeostatic parameter. Thus, τ for SWS-SWA decay is a robust phenotypic marker for sleep homeostasis and under control by the Ado system (Fig. 7B,C). One potential confound is that sleep fragmentation itself can influence SWS-SWA decay, given the 5× >15 min duration criteria, such that longer recordings may be necessary in animals with phenotypically short SWS episode duration. Additionally, given that a group average SWS episode was used for SWA decay analysis, instances of higher variability within a group in regards to SWS duration or normalized SWA power could adversely impact the ability to fit the “average SWS episode” using a single-phase exponential decay.
The Ado-mediated effects are relatively selective for SWA and SWS consolidation compared with time spent in SWS, suggesting dissociation between SWS time and sleep drive (Bjorness et al., 2009; Suzuki et al., 2013). A similar selective effect on sleep time was observed in the Shaker null in Drosophila (Cirelli et al., 2005). A gene deletion of the mammalian homolog, Kcna1, in mice also decreases sleep time with little effect on sleep drive (Douglas et al., 2007) an effect consistent with increased arousal rather than loss of sleep drive. Indeed, increased arousal is typically used experimentally to prevent sleep in the face of increased drive for sleep. This is an important consideration for phenotypic characterization of sleep genes because time spent asleep appears most sensitive to level of arousal. Together with our findings, SWS-SWA rebound and decay controlled by the Ado system are more sensitive to homeostatic sleep drive.
These observations support a model of homeostatic sleep drive controlled by extracellular levels of Ado locally within the forebrain CNS. The degree of Ado-mediated activation of neuronal forebrain AdoRA1 can determine the amount of SD-evoked rebound SWS-SWA, SWS consolidation, and SWS-SWA decay. By using neuronal and glial targeted conditional knock-out/knockdown mice, we show that glial, rather than neuronal, AdK determines the rate of SWA decay that resolves the homeostatic sleep drive response to prior waking. AdK expression shifts from neuronal to mainly glial at P14 (Studer et al., 2006), which is the developmental period in which slow-wave expression shifts from mainly active sleep to quiet sleep (Frank and Heller, 1997). Homeostatic regulation of sleep is developmentally regulated with SWA rebound occurring after SWS consolidation (Frank et al., 1998), whereas aged animals show SWA rebound without SWS consolidation (Shiromani et al., 2000a). Although not tested here, it is likely that the decay of SWS-SWA is also influenced by development.
These findings implicate a glial-neuronal circuit, mediated by Ado, neuronal AdoRA1, and glial AdK that can modulate sleep homeostasis in a manner influenced by glial metabolic state. This glial-neuronal interaction may subserve a metabolic functional balance between neurons and glia needed to avoid adverse consequences of SD. Further, these findings suggest that the loss of the SWA sleep homeostatic response, even in the absence of major changes in time spent asleep, may provide clinically relevant indicators of suboptimal brain metabolism and function.
Footnotes
This work was supported by the National Institutes of Health Grants MH 06777 and NS075545, the Department of Veterans Affairs, Dallas Veterans Administration Medical Center Grants MH79710, MH083711, and DA016672 to R.W.G. and to J.A.B., a sponsored agreement between University of Texas Southwestern Medical Center to J.A.B., A.A.F., and R.W.G., Intra-Cellular Therapies, Inc., and the Japan Society for the Promotion of Science through the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST) to M.Y. We thank Dr. Frank Kirchhoff for the generous gift of GFAP:CreER mice; Dr. Amelia Eisch for help with the tamoxifen injection protocol; Dr. Detlev Boison for help with preliminary Western analysis of AdK knockdown; Dr. Ayako Suzuki for the qPCR analysis of AdK expression; and Lehong Nguyen for technical assistance.
The authors declare no competing financial interests.
References
- Arch JR, Newsholme EA. Activities and some properties of 5′-nucleotidase, adenosine kinase and adenosine deaminase in tissues from vertebrates and invertebrates in relation to the control of the concentration and the physiological role of adenosine. Biochem J. 1978;174:965–977. doi: 10.1042/bj1740965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Rainnie DG, McCarley RW, Greene RW. Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci. 2001;21:1076–1085. doi: 10.1523/JNEUROSCI.21-03-01076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci. 2009;29:1267–1276. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, Smith D, Struys E, von Döbeln U, Gustafsson CM, Lundeberg J, Wedell A. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet. 2011;89:507–515. doi: 10.1016/j.ajhg.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci U S A. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Brambilla D, Chapman D, Greene R. Adenosine mediation of presynaptic feedback inhibition of glutamate release. Neuron. 2005;46:275–283. doi: 10.1016/j.neuron.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Hughes SW, Rudolph M, Crunelli V. Are corticothalamic ‘up’ states fragments of wakefulness? Trends Neurosci. 2007;30:334–342. doi: 10.1016/j.tins.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk DJ, Beersma DG. Effects of SWS deprivation on subsequent EEG power density and spontaneous sleep duration. Electroencephalogr Clin Neurophysiol. 1989;72:312–320. doi: 10.1016/0013-4694(89)90067-9. [DOI] [PubMed] [Google Scholar]
- Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G, Cirelli C. Sleep in Kcna2 knockout mice. BMC Biol. 2007;5:42. doi: 10.1186/1741-7007-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Evarts EV. Temporal patterns of discharge of pyramidal tract neurons during sleep and waking in the monkey. J Neurophysiol. 1964;27:152–171. doi: 10.1152/jn.1964.27.2.152. [DOI] [PubMed] [Google Scholar]
- Evarts EV, Bental E, Bihari B, Huttenlocher PR. Spontaneous discharge of single neurons during sleep and waking. Science. 1962;135:726–728. doi: 10.1126/science.135.3505.726. [DOI] [PubMed] [Google Scholar]
- Frank MG, Heller HC. Development of REM and slow wave sleep in the rat. J Physiol. 1997;272:R1792–R1799. doi: 10.1152/ajpregu.1997.272.6.R1792. [DOI] [PubMed] [Google Scholar]
- Frank MG, Morrissette R, Heller HC. Effects of sleep deprivation in neonatal rats. J Physiol. 1998;275:R148–R157. doi: 10.1152/ajpregu.1998.275.1.R148. [DOI] [PubMed] [Google Scholar]
- Frank MG, Issa NP, Stryker MP. Sleep enhances plasticity in the developing visual cortex. Neuron. 2001;30:275–287. doi: 10.1016/S0896-6273(01)00279-3. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–2621. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Llaudet E, Dale N. High-resolution real-time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. J Neurochem. 2003;86:1506–1515. doi: 10.1046/j.1471-4159.2003.01957.x. [DOI] [PubMed] [Google Scholar]
- Ganguly-Fitzgerald I, Donlea J, Shaw PJ. Waking experience affects sleep need in Drosophila. Science. 2006;313:1775–1781. doi: 10.1126/science.1130408. [DOI] [PubMed] [Google Scholar]
- Gerber U, Greene RW, Haas HL, Stevens DR. Characterization of inhibition mediated by adenosine in the hippocampus of the rat in vitro. J Physiol. 1989;417:567–578. doi: 10.1113/jphysiol.1989.sp017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RW, Haas HL. The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-L. [DOI] [PubMed] [Google Scholar]
- Gvilia I, Suntsova N, Angara B, McGinty D, Szymusiak R. Maturation of sleep homeostasis in developing rats: a role for preoptic area neurons. Am J Physiol Regul Integr Comp Physiol. 2011;300:R885–R894. doi: 10.1152/ajpregu.00727.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, McCormick DA. Rapid neocortical dynamics: cellular and network mechanisms. Neuron. 2009;62:171–189. doi: 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirrlinger PG, Scheller A, Braun C, Hirrlinger J, Kirchhoff F. Temporal control of gene recombination in astrocytes by transgenic expression of the tamoxifen-inducible DNA recombinase variant CreERT2. Glia. 2006;54:11–20. doi: 10.1002/glia.20342. [DOI] [PubMed] [Google Scholar]
- Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- Huber R, Hill SL, Holladay C, Biesiadecki M, Tononi G, Cirelli C. Sleep homeostasis in Drosophila melanogaster. Sleep. 2004;27:628–639. doi: 10.1093/sleep/27.4.628. [DOI] [PubMed] [Google Scholar]
- Huston JP, Haas HL, Boix F, Pfister M, Decking U, Schrader J, Schwarting RK. Extracellular adenosine levels in neostriatum and hippocampus during rest and activity periods of rats. Neuroscience. 1996;73:99–107. doi: 10.1016/0306-4522(96)00021-8. [DOI] [PubMed] [Google Scholar]
- Johnston D, Wu SM. Foundations of cellular neurophysiology. Cambridge, MA: Massachusetts Institute of Technology; 1995. [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–178. doi: 10.1016/j.smrv.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama K, Mishima K, Suzuki H, Aritake S, Uchiyama M. Sleep accelerates the improvement in working memory performance. J Neurosci. 2008;28:10145–10150. doi: 10.1523/JNEUROSCI.2039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemburg S, Vyazovskiy VV, Olcese U, Bassetti CL, Tononi G, Cirelli C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc Natl Acad Sci U S A. 2010;107:15939–15944. doi: 10.1073/pnas.1002570107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J Clin Invest. 2011;121:2679–2683. doi: 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materi LM, Rasmusson DD, Semba K. Inhibition of synaptically evoked cortical acetylcholine release by adenosine: an in vivo microdialysis study in the rat. Neuroscience. 2000;97:219–226. doi: 10.1016/S0306-4522(00)00075-0. [DOI] [PubMed] [Google Scholar]
- Mathews II, Erion MD, Ealick SE. Structure of human adenosine kinase at 1.5 A resolution. Biochemistry. 1998;37:15607–15620. doi: 10.1021/bi9815445. [DOI] [PubMed] [Google Scholar]
- McIlwain H, Pull I. Release of adenine derivatives on electrical stimulation of superfused tissues from the brain. J Physiol. 1972;221:9P–10P. [PubMed] [Google Scholar]
- Mimouni M, Bontemps F, Van den Berghe G. Kinetic studies of rat liver adenosine kinase: explanation of exchange reaction between adenosine and AMP. J Biol Chem. 1994;269:17820–17825. [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV. Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J Neurosci. 1993;13:3439–3447. doi: 10.1523/JNEUROSCI.13-08-03439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Faraguna U, Zoltan JT, Tononi G, Cirelli C. Sleep patterns and homeostatic mechanisms in adolescent mice. Brain Sci. 2013;3:318–343. doi: 10.3390/brainsci3010318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak MA, Haas HL, Decking UK, Schrader J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology. 1994;33:1049–1053. doi: 10.1016/0028-3908(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Palchykova S, Winsky-Sommerer R, Shen HY, Boison D, Gerling A, Tobler I. Manipulation of adenosine kinase affects sleep regulation in mice. J Neurosci. 2010;30:13157–13165. doi: 10.1523/JNEUROSCI.1359-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Adenosine promotes burst activity in guinea-pig geniculocortical neurones through two different ionic mechanisms. J Physiol. 1992;447:729–753. doi: 10.1113/jphysiol.1992.sp019026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Huang R, Yu AC, Fung KY, Rathbone MP, Hertz L. Nucleoside transporter expression and function in cultured mouse astrocytes. Glia. 2005;52:25–35. doi: 10.1002/glia.20216. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 2000;99:507–517. doi: 10.1016/S0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Radek RJ, Decker MW, Jarvis MF. The adenosine kinase inhibitor ABT-702 augments EEG slow waves in rats. Brain Res. 2004;1026:74–83. doi: 10.1016/j.brainres.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin B, Kansy JW, Nairn AC, Spychala J, Ealick SE, Fienberg AA, Greene RW, Bibb JA. Molecular characterization of recombinant mouse adenosine kinase and evaluation as a target for protein phosphorylation. Eur J Biochem. 2004;271:3547–3555. doi: 10.1111/j.1432-1033.2004.04291.x. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW. Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci. 2003;23:5762–5770. doi: 10.1523/JNEUROSCI.23-13-05762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature. 2002;417:287–291. doi: 10.1038/417287a. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Lu J, Wagner D, Thakkar J, Greco MA, Basheer R, Thakkar M. Compensatory sleep response to 12 h wakefulness in young and old rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R125–R133. doi: 10.1152/ajpregu.2000.278.1.R125. [DOI] [PubMed] [Google Scholar]
- Steriade M, Datta S, Paré D, Oakson G, Curró Dossi RC. Neuronal activities in brain-stem cholinergic nuclei related to tonic activation processes in thalamocortical systems. J Neurosci. 1990;10:2541–2559. doi: 10.1523/JNEUROSCI.10-08-02541.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience. 2006;142:125–137. doi: 10.1016/j.neuroscience.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Sinton CM, Greene RW, Yanagisawa M. Behavioral and biochemical dissociation of arousal and homeostatic sleep need influenced by prior wakeful experience in mice. Proc Natl Acad Sci U S A. 2013;110:10288–10293. doi: 10.1073/pnas.1308295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler I, Borbély AA. Sleep EEG in the rat as a function of prior waking. Electroencephalogr Clin Neurophysiol. 1986;64:74–76. doi: 10.1016/0013-4694(86)90044-1. [DOI] [PubMed] [Google Scholar]
- Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachsel L, Tobler I, Achermann P, Borbély AA. Sleep continuity and the REM-nonREM cycle in the rat under baseline conditions and after sleep deprivation. Physiol Behav. 1991;49:575–580. doi: 10.1016/0031-9384(91)90283-T. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Jackson MB. Adenosine-activated potassium conductance in cultured striatal neurons. Proc Natl Acad Sci U S A. 1985;82:4857–4861. doi: 10.1073/pnas.82.14.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/S0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, Kim SS, Chen T, Shang YZ, Zhuo M, Houslay MD, Abel T. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature. 2009;461:1122–1125. doi: 10.1038/nature08488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MP, Brakefield T, Hobson JA, Stickgold R. Dissociable stages of human memory consolidation and reconsolidation. Nature. 2003;425:616–620. doi: 10.1038/nature01930. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Dale N. Neuronal transporter and astrocytic ATP exocytosis underlie activity-dependent adenosine release in the hippocampus. J Physiol. 2013;591:3853–3871. doi: 10.1113/jphysiol.2013.253450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Taishi P, Clinton JM, Krueger JM. 5′-Ectonucleotidase-knockout mice lack non-REM sleep responses to sleep deprivation. Eur J Neurosci. 2012;35:1789–1798. doi: 10.1111/j.1460-9568.2012.08112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]