

Abstract
In Alzheimer's disease (AD), the accumulation and deposition of amyloid-β (Aβ) peptides in the brain is a central event. Aβ is cleaved from amyloid precursor protein (APP) by β-secretase and γ-secretase mainly in neurons. Although mutations in APP, PS1, or PS2 cause early-onset familial AD, ABCA7 encoding ATP-binding cassette transporter A7 is one of the susceptibility genes for late-onset AD (LOAD), in which its loss-of-function variants increase the disease risk. ABCA7 is homologous to a major lipid transporter ABCA1 and is highly expressed in neurons and microglia in the brain. Here, we show that ABCA7 deficiency altered brain lipid profile and impaired memory in ABCA7 knock-out (Abca7−/−) mice. When bred to amyloid model APP/PS1 mice, plaque burden was exacerbated by ABCA7 deficit. In vivo microdialysis studies indicated that the clearance rate of Aβ was unaltered. Interestingly, ABCA7 deletion facilitated the processing of APP to Aβ by increasing the levels of β-site APP cleaving enzyme 1 (BACE1) and sterol regulatory element-binding protein 2 (SREBP2) in primary neurons and mouse brains. Knock-down of ABCA7 expression in neurons caused endoplasmic reticulum stress highlighted by increased level of protein kinase R-like endoplasmic reticulum kinase (PERK) and increased phosphorylation of eukaryotic initiation factor 2α (eIF2α). In the brains of APP/PS1;Abca7−/− mice, the level of phosphorylated extracellular regulated kinase (ERK) was also significantly elevated. Together, our results reveal novel pathways underlying the association of ABCA7 dysfunction and LOAD pathogenesis.
SIGNIFICANCE STATEMENT Gene variants in ABCA7 encoding ATP-binding cassette transporter A7 are associated with the increased risk for late-onset Alzheimer's disease (AD). Importantly, we found the altered brain lipid profile and impaired memory in ABCA7 knock-out mice. The accumulation of amyloid-β (Aβ) peptides cleaved from amyloid precursor protein (APP) in the brain is a key event in AD pathogenesis and we also found that ABCA7 deficit exacerbated brain Aβ deposition in amyloid AD model APP/PS1 mice. Mechanistically, we found that ABCA7 deletion facilitated the processing of APP and Aβ production by increasing the levels of β-secretase 1 (BACE1) in primary neurons and mouse brains without affecting the Aβ clearance rate in APP/PS1 mice. Our study demonstrates a novel mechanism underlying how dysfunctions of ABCA7 contribute to the risk for AD.
Keywords: ABCA7, APP, BACE1, cognitive function, lipid homeostasis, neuron
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease and is the leading cause of dementia in the elderly (Alzheimer's Association, 2015). Cleavage of amyloid-β (Aβ) peptides from amyloid precursor protein (APP) by β- and γ-secretase, and their subsequent accumulation, aggregation, and deposition in the brain likely trigger the pathogenic cascade in AD (Hardy and Selkoe, 2002). Although carrying mutations in APP, PSEN1, or PSEN2 results in early-onset familial AD, the majority of cases are sporadic late-onset AD (LOAD) (Alzheimer's Association, 2015). Importantly, accumulating evidence demonstrates that the onset of LOAD is also strongly affected by genetic factors (Bertram et al., 2010). In addition to APOE, recent genome-wide studies have identified nine novel LOAD susceptibility loci: ABCA7, CLU, BIN1, PICALM, CR1, CD2AP, EPHA1, MS4A4A/MS4A6E (Hollingworth et al., 2011, Naj et al., 2011), and TREM2 (Guerreiro et al., 2013, Jonsson et al., 2013). Among them, a meta-analysis in African-American participants showed that variants in ABCA7 coding ATP-binding cassette (ABC) transporter A7 (ABCA7) were strongly associated with the increased risk for LOAD [odds ratio (OR): 1.79], which was comparable to the increased risk associated with APOE4 (OR: 2.31) in that cohort (Reitz et al., 2013). ABCA7 rs3764650C is also shown to exacerbate cognitive decline in subjects with a final diagnosis of mild cognitive impairment or LOAD (Carrasquillo et al., 2015). Furthermore, the loss-of-function variants in ABCA7 have been demonstrated to significantly increase AD risk (OR: 1.73–2.12) (Cuyvers et al., 2015, Steinberg et al., 2015). Conversely, carrying ABCA7 rs3764650T is protective against AD, likely by increasing its expression level in the brain (Vasquez et al., 2013). Although the roles of ABCA7 in the brain are not fully revealed, the suppression of ABCA7 levels/functions is predicted to contribute to LOAD progression. Therefore, a better understanding of the functional aspects for ABCA7 in the brain is critical to exploring the complex pathogenesis of LOAD.
ABCA7 is abundantly expressed in the brain, as well as in the spleen, lungs, and adrenal glands, whereas its expression level is moderate in the peripheral macrophages and lymphocytes and low in the liver (Wang et al., 2003, Abe-Dohmae et al., 2006). RNA sequencing shows that the ABCA7 level is the highest in neurons followed by microglia in the mouse brain (Zhang et al., 2014). ABCA7 is a member of the subfamily of ABC transporters that predominantly function in distributing lipids and other lipophilic molecules across membranes (Kim et al., 2008, Tanaka et al., 2010). Although the roles of ABCA7 in macrophages are actively studied because of its contribution to phagocytosis (Iwamoto et al., 2006, Jehle et al., 2006), the function of neuronal ABCA7 has received less attention despite its high expression level in neurons. Here, using in vitro cellular and in vivo animal models, we show that ABCA7 deficiency affects brain lipid homeostasis, disturbs cognitive functions, and accelerates Aβ production. Furthermore, ABCA7 deficiency induces endoplasmic reticulum (ER) stress and activates the extracellular regulated kinase (ERK) pathway in neurons, which may influence AD pathology. Our study provides novel insights into the contribution of ABCA7 dysfunctions in neurons toward developing AD through multiple pathways.
Materials and Methods
Animals.
APPswe/PS1dE9 (APP/PS1) mice (Jankowsky et al., 2004) were purchased from the Jackson Laboratory. Abca7−/− mice (Kim et al., 2005) were crossed to APP/PS1 mice to generate APP/PS1;Abca7−/− mice. Both male and female mice with a C57/B6J background were used in this study. All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee and were in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
Shotgun lipidomic analysis of brain lipids.
Shotgun lipidomics was performed as described previously (Yang et al., 2009, Yang and Han, 2011) in the hemi-forebrains of the male mice (n = 5/group) at 20–22 months of age. Lipids were extracted by the modified Bligh and Dyer method. A triple-quadrupole mass spectrometer equipped with a NanoMate device and Xcalibur system was used to analyze lipids. All mass spectrometry (MS) analyses, including tandem MS, were acquired automatically by a customized sequence operated under Xcalibur software. Internal standards for quantification of individual molecular species of other lipid classes were added to each brain tissue sample at the start of extraction procedure.
Behavioral tests.
For behavioral test, 23 control (male; n = 10, female; n = 13) and 30 Abca7−/− (male; n = 15, female; n = 15) mice were analyzed at 20–22 months of age. Spatial reference memory was investigated using Morris water maze (MWM) test. To assess anxiety or motor performance deficits, the open-field test was performed described below. Mice were monitored with an overhead camera and their behaviors were evaluated using Any Maze software.
MWM test.
The maze consisted of a 122-cm-diameter pool filled with water made opaque with nontoxic white tempera paint at a temperature of 20–21°C during testing. For both visible and hidden platform training, the pool was surrounded by a hanging white curtain with four distinct extra maze cues suspended high on the curtain. Before hidden platform training, all mice underwent one session of six pretraining trials in which a visible marker was placed on the platform. When mice did not mount the platform within 60 s, they were guided to the platform by the experimenter and allowed to sit for 10 s before being removed by experimenter. For hidden platform training, the platform was submerged 1.5 cm below the surface and was always located in the northwest quadrant of the pool. Training sessions composed of 6 trials per day were performed on 3 consecutive days. Each day, all mice were moved from the housing room to the training room, where they were allowed to acclimate for 1 h before training began. After training, all mice were returned to the housing room. On each trial, mice were held by the tail and gently lowered into the pool at the wall with the release site for each of the six trials varying randomly every training day. All mice were allowed to sit on the platform for 10 s after mounting it successfully. The maximum time allowed per trial was 60 s. If mice did not mount the platform within 60 s, they were guided to the platform by the experimenter and allowed to sit on it for 10 s before being removed; for latency analysis, these mice were excluded. For the probe test, the platform was removed and each mouse was allowed to swim for 60 s. The starting location for the probe test was at the southeast edge of the pool 180° from the northwest platform location used during training. All behaviors were recorded with a video tracking system (AnyMaze; Stoelting). Escape latencies, distance traveled, swim paths, swim speeds, and platform entries were recorded automatically for subsequent analysis.
Open-field test.
For open-field analysis, after acclimation to the testing room for 1 h, mice were tested for 15 min in a clear plastic chamber (41 × 41 × 30 cm) with two 16 × 16 photo-beam arrays detecting horizontal and vertical movements. Activity in the open field was monitored by an overhead camera to track movement with AnyMaze software. Mice were analyzed for multiple measures, including total distance traveled, average speed, and the time spent in the center of chamber (digitally designed by an 8 × 8 cm region) compared with the perimeter.
In vivo microdialysis.
In vivo microdialysis in APP/PS1 and APP/PS1;Abca7−/− littermates was performed essentially as described previously (Cirrito et al., 2003, 2011). Briefly, under isoflurane volatile anesthetic, guide cannula (BR style; Bioanalytical Systems) were cemented into the hippocampus (3.1 mm behind bregma, 2.5 mm lateral to midline, and 1.2 mm below dura at a 12° angle). A microdialysis probe (38 kDa molecular weight cutoff membrane; Bioanalytical Systems) was inserted through the guide cannula into the brain. Microdialysis perfusion buffer consisted of 4% bovine serum albumin (Sigma-Aldrich) in artificial CSF. The basal level of ISF Aβ40 was defined as the mean concentration of Aβ over 5 h before drug administration using a constant flow rate of 1.0 μl/min. To measure Aβ elimination half-life, mice were administered a γ-secretase inhibitor, LY411575 (5 mg/kg, i.p.), to block Aβ production rapidly. Microdialysis samples were collected every 60 min for 6 h and assayed for Aβ40 or Aβ42 by sandwich ELISA similar to methods described previously (Cirrito et al., 2011). mHJ2 (mouse-anti-Aβ35-40) or mHJ7.4 (mouse-anti-Aβ37-42) was used as the capture antibody and biotinylated mHJ5.1 (mouse-anti-Aβ13-28) as the detection antibody. The half-life of ISF Aβ was calculated from the slope of the semilog plot of percentage change in Aβ versus time (Cirrito et al., 2003).
Immunohistochemical staining and analysis.
Paraffin-embedded sections were immunostained using pan-Ab (Ab 33.1.1; human Ab 1–16 specific) and anti- Iba-1 (WAKO) antibodies. Immunohistochemically stained sections were scanned using the ImageScope XT image scanner (Aperio Technologies) and analyzed using ImageScope software. The intensity of Aβ or Iba1 was calculated using the positive pixel count program available with the ImageScope software (Aperio Technologies). The numbers of Iba1-positive microglia were counted manually by two investigators if their cell body or processes were overlapped with Congo-red-stained plaques. At least three amyloid plaques per mouse brain were analyzed. All of the above analyses were performed in a blinded manner.
Tissue collection.
Animals were anesthetized with isoflurane and transcardially perfused with cold PBS. The brain was bisected and one hemisphere was fixed in 10% neutralized formalin in phosphate buffer. For biochemical assays, the brain was homogenized in RIPA buffer containing protease inhibitor mixture (Thermo Scientific) and phosphatase inhibitor (Sigma-Aldrich, T1892).
Cell cultures.
Mouse neuroblastoma N2a cells and N2a-APP cells were maintained in DMEM/Opti-MEM (1:1) containing 5% in fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen) or DMEM medium supplemented with 5% FBS and 1% penicillin/streptomycin (Invitrogen) and grown at 37°C in 5% CO2 incubator. These cells were transiently transfected with the indicated human ABCA7 cDNA or mouse ABCA7 siRNA (Invitrogen) using Lipofectamine 2000 following the manufacturer's instructions. Primary cultures of neurons were prepared using cerebral cortices of embryonic day 16 (E16) mice. Cortical brain tissues were dissociated on ice and then incubated in papain dilution solution composed of 5 mm l-cysteine, 1 mm EDTA, pH 8.0, 10 mm HEPES-NaOH, pH 7.3, 100 μg/ml BSA in PBS (Invitrogen, CM free) supplemented with 10 U/ml papain (Worthington) and 100 μg/ml DNase I (Sigma-Aldrich) for 10 min. After aspiration of the supernatant, the tissues were triturated in FBS. After the pellets were washed twice with DMEM containing 10% FBS and 1% P/S, they were resuspended in Neurobasal medium (Invitrogen) supplemented with B27 and Glutamax (Invitrogen) and then filtered with a cell strainer (40 μm; Falcon). The cells at were plated on poly-d-lysine (BD Biosciences)-coated 12-well plates (0.3 × 106 cells/well) or 6-well plates (0.75 × 106 cells/well) and maintained in Neurobasal medium (Invitrogen) supplemented with B-27 and Glutamax (Invitrogen) in a 5% CO2 incubator.
Lentivirus preparation.
Lentiviral vectors carrying control nontargeting shRNA and shRNA targeting Abca7 were obtained through partnership agreement between Sigma-Aldrich and the Mayo Clinic RNA Interference Shared Resource. The virapower lentiviral expression kit (Invitrogen) was used to produce lentiviral particles into HEK293FT cells using Lipofectamine 2000 as per the manufacturer's instructions. Viral particles in the medium were concentrated by centrifugation through a 20% sucrose cushion for 2 h at ∼72,000 × g. The pellets of viral particles were resuspended in PBS and filtered through 0.22 μm centrifugal filters (Millipore). Titers of the viral preparations were measured using the Lenti-X qRT-PCR Titration kit from Clontech. The purified viruses with titers ranging from 1 × 109 to 1 × 1011 viral particles/ml were used. Primary mouse cortical neurons were infected with lentivirus carrying nontarget control shRNA or each specific-shRNA at 7–9 d in vitro (DIV), and used for Western blotting or quantitative PCR at 17–24 DIV.
Aβ measurements.
After 4–8 d, levels of Aβ40 and Aβ42 in the medium were quantified by a multiplex microsphere-based immunoassay through xMAP technology using INNO-BIA (Innogenetics). To measure Aβ levels in APP/PS1 mouse brains, the tissues were homogenized in RIPA buffer and centrifuged at 100,000 × g for 60 min at 4°C. After the supernatant (RIPA-soluble fraction) was isolated, pellets were resuspended in 5 m GDN-HCl, rotated at room temperature overnight and centrifuged at 16,000 × g for 30 min. The supernatant was used as GDN-soluble fraction. Aβ levels in both fractions were determined by ELISA using end-specific mAbs 2.1.3 (human Aβx-42 specific) or mAb 13.1.1 (human Aβx-40 specific) as the capturing antibody and biotin-conjugated mAb 266 (human Aβ15-24 specific) as the detection antibody (Bard et al., 2003, Shinohara et al., 2013).
Western blot.
Protein concentrations of cell lysates or brain extracts were measured using the BCA method (Pierce). Samples with equal amounts of protein were separated by 4–20% Tris-glycine SDS-PAGE (Invitrogen) gels or 4–12% NuPAGE Bis-Tris gel (Life Technologies), followed by transfer to nitrocellulose membranes. After blocking with 5% milk diluted in Tris-buffered saline for 1 h, membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used; anti-β-site APP cleaving enzyme 1 (anti-BACE1; Thermo Scientific), anti-APP C-terminal antibody (IBL), anti-82E1 antibody (IBL), anti-SREBP2 antibody (Cayman Chemical), anti-protein kinase R-like endoplasmic reticulum kinase (anti-PERK; Cell Signaling Technology), rabbit anti-phospho ERK1/2 (T202/Y204; Cell Signaling Technology, 4370), anti-ERK (Cell Signaling Technology), anti-phosphorylated eukaryotic initiation factor 2 (anti-eIF2; Cell Signaling Technology), anti-eIF2 (Cell Signaling Technology), anti-phospho-JNK (Cell Signaling Technology), anti-phospho-p38 (Cell Signaling Technology), anti-PS1 (Cell Signaling Technology), anti-GFAP antibody (Sigma-Aldrich), anti Iba1 antibody (WAKO), and anti-GAPDH (Sigma-Aldrich). After incubation with horseradish peroxidase-conjugated goat anti-rabbit or mouse antibodies, the chemiluminescent bands were visualized using the ECL system (Pierce) and quantified densitometrically.
BACE1 activity assay.
BACE1 activity assay was performed spectrofluorometrically using commercial assay kits from Millipore in homogenates of forebrains from control and Abca7−/− mice following the manufacturer's instructions. Fluorescence intensity was measured with a microplate reader (SpectraMax) at an excitation wavelength of 345 nm and an emission wavelength of 505 nm.
RNA isolation, reverse transcription, quantitative PCR.
Total RNA was isolated by using small-scale phenol-free total RNA isolation kit (RNAqueous; Ambion). For real-time PCR analysis, total RNA from each sample was used for first-strand cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA from each sample was amplified via real-time PCR using assays on demand premixed TaqMan primer/probe set for mouse and normalized against the Geomean of HPRT, TFRC, and GAPDH using a 7900HT sequence analyzer (Applied Biosystems). The following ABI probes were used: Mm00497010_m1 for detection of mouse Abca7 mRNA, Mm00478664_m1 for detection of mouse Bace1 mRNA, Mm00446968_m1 for detection of mouse Hprt mRNA, Mm00441941_m1 for detection of mouse Tfrc mRNA, and Mm99999915_g1 for detection of mouse Gapdh mRNA. The mRNA levels for each experimental group were quantified using the comparative CT method. The statistical analysis was performed on dCT ± SEM for each target gene.
Statistical analysis.
Results are expressed as mean ± SEM. For all experiments, values that were not within the predetermined criterion of 2 SDs from the mean were considered statistical outliers and were excluded from analysis. Student's t test (two-tailed) was used for single comparison of means. For latency and distance in the hidden platform of the MWM test, one-way ANOVA with Fisher's LSD post hoc test was used to evaluate statistical significance. p < 0.05 was considered statistically significant.
Results
ABCA7 deficiency alters lipid profile in mouse brains
Because ABCA7 belongs to the ABC transporter family, it has been postulated that it is also involved in the regulation of lipid metabolism (Kim et al., 2008). Therefore, we assessed lipid profiles in the brains of the control and ABCA7 knock-out (Abca7−/−) mice (males, n = 5/group) at 20–22 months of age by unbiased shotgun lipidomic analysis for 275 lipid species (Yang et al., 2009, Yang and Han, 2011). The lipids were grouped into phosphatidylethanolamine (PE; 41 subspecies), phosphatidylinositol (PI; 20 subspecies), phosphatidylserine (PS; 25 subspecies), phosphatidylglycerol (PG; 18 subspecies), phosphatidylcholine (PC; 24 subspecies), lysophosphatidylcholine (LPC; 14 subspecies), sphingomyelin (SM; 14 subspecies), ceramide (CER; 4 subspecies), phosphatidic acid (PA; 12 subspecies), lysocardiolipin (LCL; 19 subspecies), cardiolipin (CL; 42 subspecies), sulfatide (ST; 12 subspecies), carnitine (CAR; 9 subspecies), cerebroside (CBS; 20 subspecies), and cholesterol (FCHL). Although we did not detect differences in the total amount of lipid species in each class, the levels of 24 specific subspecies were significantly modulated by ABCA7 deficiency; 12 subspecies in PE, 3 in PG, 1 in LPC, and 2 in SM were significantly reduced in the brains of aged Abca7−/− mice compared with those of control mice, although 3 subspecies in PC, 1 in CER, 3 in ST, and 1 in CBS were increased (Table 1). These results indicate that ABCA7 contributes to lipid metabolism in the brain, which might affect diverse brain functions.
Table 1.
Lipidomic analysis for forebrain samples from Abca7−/− mice
| Lipid subspecies (nmol/mg protein) | Control (n = 5) | Abca7−/− (n = 5) | p-value |
|---|---|---|---|
| PE | |||
| P16:0–18:2 | 0.3235 ± 0.0168 | 0.2219 ± 0.0133 | 0.0015 |
| D16:0-18:2/D16:1-18:1 | 0.4339 ± 0.0130 | 0.3498 ± 0.0171 | 0.0045 |
| P14:0-22:6 | 0.2206 ± 0.0169 | 0.0925 ± 0.0255 | 0.0031 |
| A18:0-18:1 | 0.4889 ± 0.0231 | 0.4019 ± 0.0245 | 0.0324 |
| D16:1-20:4 | 0.0722 ± 0.0179 | 0.0000 ± 0.0000 | 0.0038 |
| D18:1-18:2/D16:0-20:3 | 0.3837 ± 0.0160 | 0.2911 ± 0.0248 | 0.0138 |
| P20:0-18:0/P18:0-20:0 | 0.1580 ± 0.0301 | 0.0080 ± 0.0040 | 0.0011 |
| D16:1-22:6 | 0.2423 ± 0.0124 | 0.0933 ± 0.0265 | 0.0009 |
| P18:2-22:6/D18:1-20:1 | 0.4684 ± 0.0247 | 0.3202 ± 0.0506 | 0.0300 |
| A20:0-20:4/P18:0-22:3 | 0.4435 ± 0.0279 | 0.3492 ± 0.0251 | 0.0362 |
| P18:0-22:2 | 0.3079 ± 0.0189 | 0.1565 ± 0.0176 | 0.0004 |
| D20:0-20:1/D18:0-22:1 | 0.1823 ± 0.0257 | 0.0721 ± 0.0241 | 0.0141 |
| PG | |||
| 20:4-20:4/18:2-22:6 | 0.1064 ± 0.0057 | 0.0845 ± 0.0031 | 0.0097 |
| 20:4-22:6 | 0.2131 ± 0.0150 | 0.1687 ± 0.0116 | 0.0474 |
| 20:3-22:6/20:4-22:5 | 0.0119 ± 0.0031 | 0.0026 ± 0.0026 | 0.0490 |
| PC | |||
| A16:0-18:0 | 0.1838 ± 0.0198 | 0.2696 ± 0.0258 | 0.0298 |
| D18:0-20:3 | 0.3522 ± 0.0114 | 0.4338 ± 0.0177 | 0.0047 |
| D18:1-22:6/D18:2-22:5 | 1.4960 ± 0.0613 | 1.7113 ± 0.0550 | 0.0309 |
| LPC | |||
| 16:1 | 0.2648 ± 0.0106 | 0.2230 ± 0.0105 | 0.0233 |
| SM | |||
| N16:0 | 0.1541 ± 0.0063 | 0.1208 ± 0.0071 | 0.0080 |
| N20:0 | 0.2171 ± 0.0091 | 0.1647 ± 0.0065 | 0.0016 |
| CER | |||
| N24:1 | 0.4716 ± 0.0328 | 0.5742 ± 0.0190 | 0.0269 |
| ST | |||
| N18:0 | 0.1373 ± 0.0049 | 0.1757 ± 0.0150 | 0.0416 |
| OH_N22:0 | 1.0494 ± 0.0474 | 1.3249 ± 0.0971 | 0.0342 |
| N25:0/OH_N24:1 | 1.5221 ± 0.0666 | 1.8540 ± 0.1178 | 0.0398 |
| CBS | |||
| OH_N22:0 | 2.9963 ± 0.0498 | 3.3674 ± 0.1224 | 0.0229 |
The subset of lipids significantly modulated by ABCA7 deficiency is shown. The first number in each pair refers to the number of carbon atoms in the fatty acid chain. The number after the colon refers to the number of double bonds. The two fatty acid chain designations are separated by a hyphen. The prefix “D” indicates a diacyl chain linkage in the phospholipids. The prefix “P” indicates a vinyl ether plasmenyl chain linkage. The prefix “A” indicates an alkyl ether chain linkage. The prefix “N” indicates that the fatty acid is in an amide linkage with the sphingosine. The prefix “OH” indicates an additional hydroxyl group in them. Values are mean ± SEM. Student's t test was used for the statistical analysis.
ABCA7 deficiency causes spatial memory deficit in aged mice
To investigate whether ABCA7 deficiency affects the memory, the MWM test was conducted in Abca7−/− and control wild-type mice at 20–22 months of age. The mice underwent 3 d of hidden platform training (6 sessions per day) after visible platform training at day 0. Throughout the training trials with the visible platform condition, there were no statistical differences in latencies to find the platform between the genotypes (Fig. 1A). After the completion of training, a probe trial was performed in which the platform was removed and putative indices of spatial memory were measured. During training sessions, Abca7−/− mice exhibited longer time in escape latency (Fig. 1B). In addition, Abca7−/− mice crossed the platform location fewer times in probe tests compared with control mice (Fig. 1C). Swim speeds during the probe test were similar among the groups (Fig. 1D). We also tested anxiety-related behavior in the open-field test and did not observe significant differences in anxiety-related behaviors between control and Abca7−/− mice (Fig. 1E). These results indicate that ABCA7 deficiency compromises spatial memory in 20- to 22-month-old mice.
Figure 1.

Spatial learning abilities are disturbed in Abca7−/− mice. Control (n = 23) and Abca7−/− (n = 30) mice were tested through MWM tests at 20–22 months of age. A, Mice received 6 training sessions in the MWM test with the visible platform condition at training day 0. B, Escape latency and distance to platform in the MWM test with the hidden platform condition were plotted against the training days. C, D, Crossings of target and equivalent nontarget locations for 60 s (C) and swim speed (D) were also assessed during the probe test of the MWM test. E, Control (n = 23) and Abca7−/− (n = 30) mice were tested through the open-field test at 20–22 months of age. Total distance traveled, average speed, and time spent in center in the open field were shown. Values are mean ± SEM. *p < 0.05. N.S., Not significant. One-way ANOVA (A), one-way ANOVA with post hoc Fisher's LSD t test (B), and Student's t test (C–E) were used for the statistical analysis.
Deletion of ABCA7 promotes APP processing and Aβ deposition by increasing BACE1 level in APP/PS1 mice
To further determine the effects of ABCA7 deficiency on amyloid pathology in vivo, we bred Abca7−/− mice with APPswe/PS1dE9 (APP/PS1) amyloid model mice (Jankowsky et al., 2004). When the plaque burden in APP/PS1;Abca7−/− mice and control APP/PS1 mice were compared by immunostaining for Aβ, amyloid plaque loads in the cortex and hippocampus were significantly increased in APP/PS1;Abca7−/− mice at 7 months of age (Fig. 2A–C). Consistently, ELISA showed higher levels of radioimmunoprecipitation assay (RIPA) buffer-soluble Aβ42 (Fig. 2D) and RIPA-insoluble Aβ42 levels in the guanidine (GDN) fraction in the hippocampus of APP/PS1;Abca7−/− mice than in control APP/PS1 mice (Fig. 2E). RIPA-insoluble Aβ40 level was also increased in APP/PS1;Abca7−/− mice (Fig. 2F), although it was not significant by Student's t test (p = 0.07). When analyzed by two-way ANOVA (genotype and sex), significant main effects of ABCA7 deficiency on increased levels of insoluble Aβ40 (F(3,15) = 10.57, p < 0.05) were detected.
Figure 2.
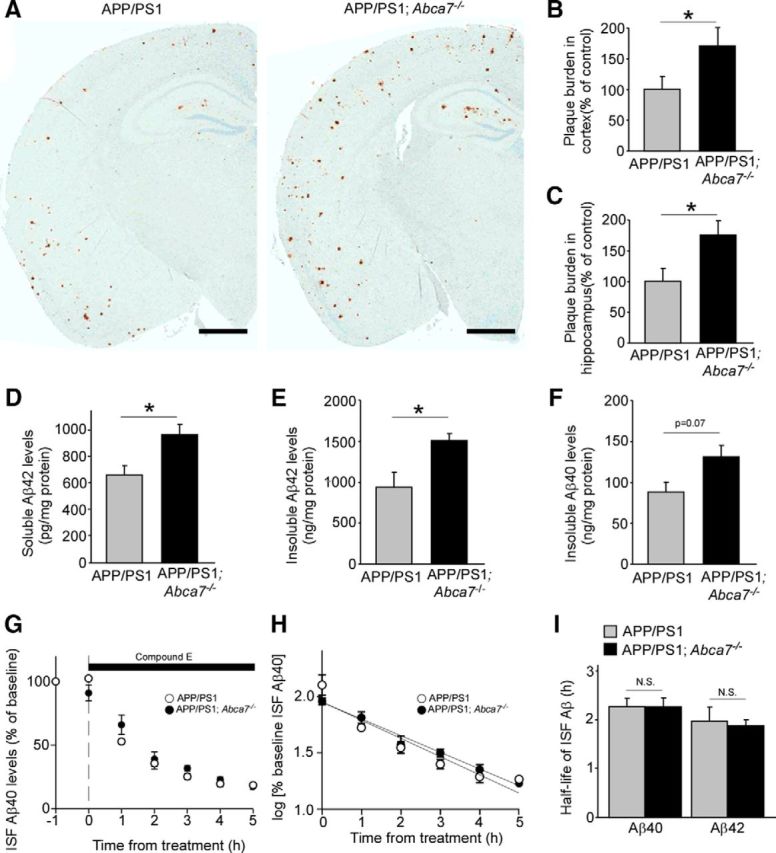
ABCA7 deficiency exacerbates Aβ pathology in APP/PS1 mice without affecting Aβ clearance. A, Brain sections from control APP/PS1 and APP/PS1;Abca7−/− mice were immunostained for Aβ at 7 months of age. Scale bar, 1 mm. B, C, Area for Aβ plaque burden was quantified in the cortex (B) and hippocampus (C) from these mice (n = 7–8 per group). D–F, Aβ42 levels in the RIPA-soluble fraction (D) and the insoluble GDN fraction (E), and Aβ40 level (F) in the insoluble fraction of hippocampus from APP/PS1 and APP/PS1;Abca7−/− mice were measured by ELISA at 7 months of age (n = 6–9 per group). G–I, ISF Aβ40 clearance was assessed using in vivo microdialysis in APP/PS1 and APP/PS1;ABCA7−/− mice at the age of 3 months (n = 3–4 per group). G, Mice were treated with a γ-secretase inhibitor and the cortical ISF levels of Aβ40 were monitored for 6 h after a baseline of cortical ISF Aβ40 levels was achieved through 5 h monitoring. H, Common logarithms of percentage baseline ISF Aβ40 concentrations versus time were plotted. The slope from the individual linear regressions from log [% ISFAβ40] versus time for each mouse was used to calculate the mean half-life (t1/2) of elimination for Aβ40 from the ISF. I, Similarly, the mean half-life of Aβ42 elimination was also calculated. Values are mean ± SEM. *p < 0.05. N.S., Not significant. Student's t test was used for the statistical analysis.
Next, we investigated Aβ clearance rate in the interstitial fluid (ISF) using in vivo microdialysis (Cirrito et al., 2003) in the hippocampus of control APP/PS1 and APP/PS1;Abca7−/− mice at 3 months of age. The elimination rate of Aβ from the ISF was measured by infusing a γ-secretase inhibitor directly into the hippocampus of APP/PS1 and APP/PS1;Abca7−/− mice to block Aβ production (Fig. 2G). Although a gradual decrease of ISF Aβ levels was observed after treatment with γ-secretase inhibitor, there was no significant difference between APP/PS1 and APP/PS1;Abca7−/− mice (Fig. 2H). The half-lives of elimination from the ISF for Aβ40 were calculated as 2.28 h and 2.27 h in APP/PS1 mice and APP/PS1;Abca7−/− mice, respectively (Fig. 2I). Similarly, the half-lives of ISF Aβ42 were determined as 1.99 h for APP/PS1 mice and 1.89 h for APP/PS1;Abca7−/− mice (Fig. 2I). These results indicate that deletion of ABCA7 does not influence the elimination rate of soluble Aβ from the ISF.
Therefore, to assess the effects of ABCA7 deficiency on APP cleavage, we analyzed levels of APP derivatives in the brains of these mice by Western blotting. We found that the levels of APP C-terminal fragment-β (CTF-β) were significantly increased in the cortex of APP/PS1;Abca7−/− mice, whereas CTFα levels and total APP were not significantly changed when analyzed using the APP C-terminal specific antibody (Fig. 3A,B). The increased CTF-β level was also confirmed by CTF-β N-terminal specific antibody (82E1) in these mouse brains (Fig. 3C,E). Furthermore, elevated BACE1 2006, Sun et al., 2012), sterol regulatory element-binding protein 2 (SREBP2) is known as one of the transcription factors for BACE1 (Mastrocola et al., 2011). Therefore, we analyzed the level of SREBP2 by Western blotting in these mice and found that it was significantly increased in the cortex of APP/PS1;Abca7−/− mice compared with control mice (Fig. 3G,H). BACE1 activity was also significantly increased in the brains of Abca7−/− mice compared with those from control mice (Fig. 3I). These results suggest that ABCA7 regulates SREBP2-BACE1 activity in the mouse brain and that the disturbance of the pathway results in exacerbated APP processing and Aβ deposition.
Figure 3.

Deletion of ABCA7 accelerates APP processing in APP/PS1 mice. The levels of APP, CTF-α, and CTF-β were analyzed by Western blot using APP C-terminal specific antibody (A, B) and CTF-β N-terminal specific antibody (82E1) (C, E) in the cortex of control APP/PS1 and APP/PS1;Abca7−/− mice at 7 months of age. The levels of BACE1 (D, F) and SREBP2 (G, H) were analyzed in the cortex of these mice by Western blot. Values are mean ± SEM (n = 3–6 per group). I, BACE1 activity was also analyzed in the cortex of control and Abca7−/− mice at 20–22 months of age. Values are mean ± SEM (n = 6 per group). *p < 0.05. N.S., Not significant. Student's t test was used for the statistical analysis.
ABCA7 deficiency facilitates Aβ production in neurons
To further assess the effects of ABCA7 deficiency on APP processing, ABCA7 expression was suppressed by infection of a lentivirus carrying its specific shRNA in mouse primary cultures of neurons (Fig. 4A). Although suppression of ABCA7 did not affect the levels of full-length APP (Fig. 4B) and PS1 (Fig. 4C), the BACE1 level was significantly increased upon ABCA7 knock-down, which is consistent with results from APP/PS1;Abca7−/− mice (Fig. 4D). The increased mRNA level of Bace1 was also detected by RT-PCR (Fig. 4F). As predicted, we also found increased levels of Aβ40 and Aβ42 in the conditioned medium from primary neurons upon ABCA7 knock-down (Fig. 4E). Furthermore, we found a significant increase of SREBP2 in ABCA7 knocked-down neurons compared with control (Fig. 4G), suggesting that ABCA7 deficiency promotes BACE1 transcription by increasing SREBP2, resulting in accelerated Aβ production.
Figure 4.

ABCA7 regulates APP processing in neuronal cells. A–D, Expression levels of APP (B), BACE1 (C), and PS1 (D) were assessed by Western blot in primary neurons with or without knock-down of ABCA7 using lentivirus carrying the specific shRNA (n = 12 per group). E, BACE1 mRNA levels were quantified by real-time PCR in control and ABCA7 KD neurons (n = 6). F, Mouse endogenous levels of Aβ40 and Aβ42 in the culture medium of control and ABCA7 KD neurons were evaluated by ELISA (n = 9 per group). G, Expression levels of SREBP2 were assessed in control and ABCA7 KD neurons by Western blot (n = 8–9). H, Levels of full-length APP and APP-CTFs cells were examined by Western blot in mouse neuroblastoma N2a cells with or without knock-down of ABCA7 through its specific siRNA (n = 6 per group). I, N2a cells stably expressing human APP were transfected with either control or human ABCA7 vector. The levels of full-length APP and APP-CTF cells were examined by Western blot (n = 3 per group). Values are mean ± SEM. *p < 0.05; **p < 0.01. N.S., Not significant. Student's t test was used for the statistical analysis.
To confirm the effects of ABCA7 deficiency on APP processing, we further investigated the effects of modulating ABCA7 expression on APP processing in mouse neuroblastoma N2a cells. First, we performed gene silencing for ABCA7 using specific siRNA and found that ABCA7 knock-down led to increased levels of APP-CTFs without affecting those of full-length APP (Fig. 4H). Conversely, overexpression of human ABCA7 in N2a cells stably expressing human APP significantly reduced the levels of CTF-α and CTF-β without affecting those of full-length APP (Fig. 4I). Together, these results indicate that ABCA7 deficiency increases Aβ production by regulating APP processing in neurons.
ABCA7 deficiency induces ER stress in neurons
To further investigate the alternative mechanisms underlying ABCA7-related neuronal pathology, we investigated effects of ABCA7 deficiency on ER stress in neurons because increased ER stress has been shown in AD brains (O'Connor et al., 2008, Yoon et al., 2012). When the levels of ER stress sensors PERK and eIF2α were analyzed by Western blotting, we found that there were significantly increased levels in ABCA7 knock-down neurons (Fig. 5A). These results suggest the potential involvement of ABCA7 in the regulation of the neuronal PERK-eIF2a pathway during the ER stress response. We did not observe any significant difference in brain levels of PERK and p-eIF2α between control and Abca7−/− mice at 20–22 months of age (Fig. 5B), likely due to the presence of other brain cell types that are not relevant to such ABCA7 function.
Figure 5.

ABCA7 deletion leads to ER stress in primary neurons. A, Levels of PERK and eIF2α phosphorylation were examined by Western blot in control and ABCA7 KD neurons (n = 9 per group). B, Levels of PERK and phosphorylated eIF2α in the cortex of control and Abca7−/− mice were also analyzed at 20–22 months of age (n = 10 per group). Values are mean ± SEM. *p < 0.05; **p < 0.01. Student's t test was used for the statistical analysis.
Deletion of ABCA7 activates ERK pathway in the brain of APP/PS1 mice
Aβ and soluble APP have been shown to activate ERK pathway in neurons (Rohe et al., 2008, Serrano et al., 2009). In addition, the mitogen-activated protein kinase (MAPK)–ERK pathway contributes to the stability of memory (Davis and Laroche, 2006). Therefore, we examined the effect of ABCA7 deficiency on this signaling pathway in vivo. In APP/PS1;Abca7−/− mice, immunostaining demonstrated that basal levels of phosphorylated ERK (p-ERK) were higher in the cortex at 7 months of age than those in control APP/PS1 mice, in which p-ERK was mainly colocalized with the neuronal marker NeuN (Fig. 6A). Western blot also confirmed the increased levels of p-ERK in the cortex of APP/PS1;Abca7−/− mice compared with control mice (Fig. 6B). These results suggest that ABCA7 deficiency activates the ERK pathway specifically in neurons. When brain levels of p-p38 and p-JNK, the other major components of MAPK cascades (Kim and Choi, 2010), were analyzed by Western blotting, there were no significant differences in these signaling molecules between ABCA7-deficient and control mice (Fig. 6C). Next, we evaluated p-ERK levels in the brains from control and Abca7−/− mice at 20–22 months of age and found no significant difference between the groups (Fig. 6D). These results indicate that ABCA7 regulates MAPK/ERK pathways in the brain in the presence of Aβ pathology.
Figure 6.
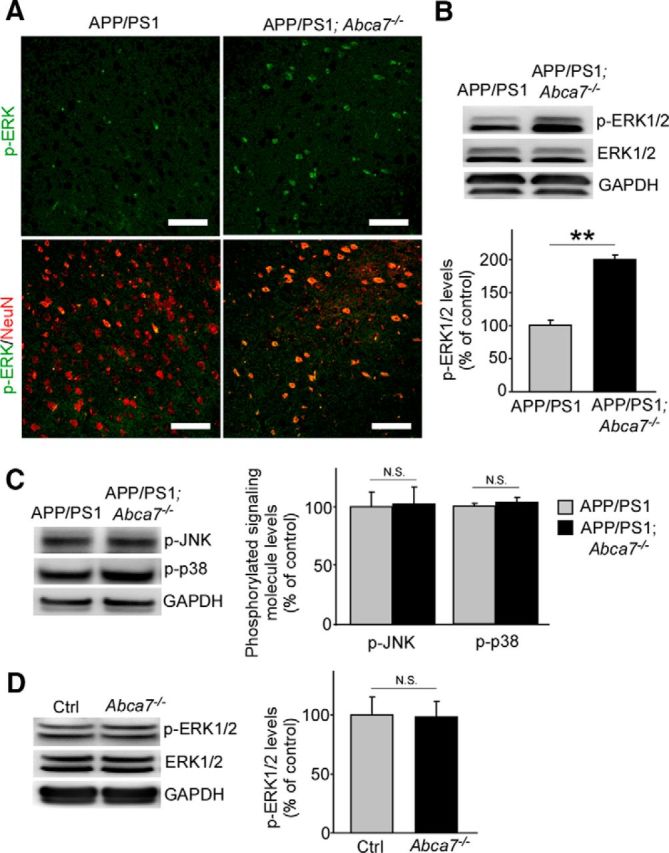
ABCA7 deficiency upregulates p-ERK levels in the brains of APP/PS1 mice. A, Brain sections from control APP/PS1 and APP/PS1;Abca7−/− mice were immunostained for p-ERK (green) and NeuN (red) at the age of 7 months. Scale bar, 100 μm. B, Levels of p-ERK were examined by Western blot in the cortex of the mice. C, Similarly, levels of phosphorylated JNK and p38 were also analyzed. Values are mean ± SEM (n = 6 per group). D, Levels of p-ERK in the cortex of control and Abca7−/− mice were analyzed at 20–22 months of age. Values are mean ± SEM (n = 7 per group). **p < 0.01. N.S., Not significant. Student's t test was used for the statistical analysis.
ABCA7 deficient does not enhance plaque associated microglia in APP/PS1 mice
Because ABCA7 is expressed in microglia and neurons in the brain (Kim et al., 2013, Zhang et al., 2014), we investigated the phenotypes of microglia in APP/PS1;Abca7−/− mouse brains through immunostaining for the microglia marker Iba1. Although Iba1-positive microglia were abundant, in particular around amyloid plaques, in the cortex (Fig. 7A,B) and hippocampus (Fig. 7C,D) in both control APP/PS1 and APP/PS1;Abca7−/− mice, there was no significant difference in the Iba-1 densities between the genotypes (Fig. 7B,D). Consistent with these results, Western blotting also revealed that the levels of Iba1 (Fig. 7E) in cortex were not altered by ABCA7 deficiency in APP/PS1 mice. In addition to Iba1 levels, levels of the astrocyte marker GFAP were also not affected by the genotypes (Fig. 7F).When Iba1-positive microglia around Congo-red positive amyloid plaques were counted (Fig. 7G,H), we did not detect significance differences in the cortex (Fig. 7H) and hippocampus (Fig. 7I) of the mice. These results suggest that ABCA7 deficiency does not exacerbate microglia activation status in APP/PS1 mice.
Figure 7.

Microglia activation is not altered in APP/PS1;Abca7−/− mice. A–D, Brain sections from control APP/PS1 and APP/PS1;Abca7−/− mice were immunostained for a microglia marker Iba1 at the age of 7 months. Scale bar, 100 μm. Iba1 immunoreactivity was quantified in cortex (A, B) and hippocampus (C, D). E, F, Levels of Iba1 (E) and the astrocyte marker GFAP (F) were also analyzed by Western blot in the cortex. G–I, After costaining with Iba1 and Congo red, the numbers of Iba1-positive microglia around Congo red-positive amyloid plaques were counted in their cortex (H) and hippocampus (I). Scale bar, 20 μm. Values are mean ± SEM (n = 6–7 per group). N.S., Not significant. Student's t test was used for the statistical analysis.
Discussion
A recent meta-analysis for AD susceptibility loci has confirmed the genome-wide associations of the nine already known genes, including ABCA7. Furthermore, 11 new genes (CASS4, CELF1, FERMT2, HLA-DRB5/HLA-DRB1, INPP5D, MEF2C, NME8, PTK2B, SLC24A4/RIN3, SORL1, and ZCWPW1) have been identified as LOAD-related genes (Lambert et al., 2013). Functionally, their coding proteins contribute to the pathways for lipid metabolism, innate immunity, and/or endocytosis, although some of them are poorly characterized (Hardy et al., 2014, Medway and Morgan, 2014). Although the pathophysiological mechanisms underlying the association of these genes with AD risk remain to be elucidated, loss-of-function variants in ABCA7 were recently reported to be associated with the increased risk for AD (Cuyvers et al., 2015, Steinberg et al., 2015). Consistently, our results demonstrate that deficiency of ABCA7 facilities APP processing and Aβ production in neurons. We also found increased Aβ levels and plaque deposition in APP/PS1 mice in Abca7−/− background compared with control APP/PS1 mice.
ABCA7 belongs to ABC transporter subfamily A, which is a transmembrane protein homologous to ABCA1 that regulates the generation of high-density lipoprotein (HDL) and the translocation of lipids (Tanaka et al., 2011). Our lipidomic analysis revealed that ABCA7 deficiency does not influence brain cholesterol levels in aged mice, which is consistent with previous in vitro experiments showing that endogenous ABCA7 has less effects on HDL biogenesis (Ikeda et al., 2003, Kim et al., 2008). Nonetheless, we found that some of the lipid subspecies, in particular those that belong to PE and SM classes, were modified in the brains of Abca7−/− mice. PE and SM are also major components of lipid rafts (Pike et al., 2005), which is a dynamic platform for signal transduction. More importantly, BACE1 is known to cleavage APP in lipid rafts (Vetrivel and Thinakaran, 2010, Fabelo et al., 2014). Although further studies are needed, the modification of lipid raft function/components due to ABCA7 deficiency may facilitate the interaction between APP and BACE1, resulting in accelerating Aβ production in AD brains. In addition, we also found that increased BACE1 levels were accompanied by elevated SREBP2 levels in ABCA7-suppressed neurons and in APP/PS1;Abca7−/− mouse brains. Consistently, when the active truncated form of SREBP2 was constitutively overexpressed, β-secretase activity was significantly enhanced, resulting in neurodegeneration in APP/PS1 mice (Barbero-Camps et al., 2013). Although SREBP2 regulates the transcription of the genes involved in lipid synthesis and cellular uptake (Bengoechea-Alonso and Ericsson, 2007), exposure to high cholesterol also activates the SREBP2 pathway, resulting in an increase of BACE1 expression (Mastrocola et al., 2011). Therefore, it is tempting to speculate that ABCA7 deficiency increases the levels of SREBP2, which leads to downstream upregulation of BACE1 expression. Given that SREBP2 is also a transcription factor regulating ABCA7 (Iwamoto et al., 2006), the increased SREBP2 level by ABCA7 deficiency may be due to a compensative mechanism, although there is a possibility that altered lipid profiles in the brains due to ABCA7 deficiency activate the SREBP2-BACE1 pathway. Another study reported that ABCA7 deletion facilitates APP endocytosis, which promotes APP cleavage (Satoh et al., 2015). Therefore, ABCA7 dysfunctions likely accelerate Aβ production through multiple pathways to facilitate APP processing.
Furthermore, we found that aged Abca7−/− mice have impaired spatial learning memory without Aβ pathology when assessed with the MWM test. Another study conflictingly showed that ABCA7 plays only a minor role in cognitive domains with a subtle sex-specific effect at the age of 5 months when analyzed by cheeseboard test and noble object recognition test (Logge et al., 2012), suggesting that cognitive decline associated with ABCA7 deficiency may be age dependent. Interestingly, ABCA7 knock-down induced ER stress, as represented by elevated PERK levels, which induces activation of eIF2α in primary neurons. Indeed, phosphorylated eIF2α levels are elevated in the brains of LOAD patients and AD model mice (O'Connor et al., 2008, Yoon et al., 2012, Ma et al., 2013). Deletion of neuronal PERK has also been shown to ameliorate synaptic dysfunctions and memory deficits in APP/PS1 mice (Ma et al., 2013). Therefore, the cognitive decline observed in aged Abca7−/− mice may be partially related to ER stress, in particular activation of the PERK-eIF2α pathway. Because we did not observe significant effects of ABCA7 deficiency on these molecules in mouse cortex, ABCA7 deficiency may induce ER stress specifically in neurons, although further studies are required. Conversely, SREBP2 levels have been shown to be upregulated under ER stress conditions (Barbero-Camps et al., 2014). Phosphorylation of eIF2α also can increase BACE1 levels directly and promote APP processing (O'Connor et al., 2008). Because Aβ induces ER stress through the phosphorylation of PERK/eIF2α (Lee et al., 2010), ABCA7 dysfunctions may trigger a vicious cycle between ER stress and Aβ pathology by activating both the SREBP2-BACE1and PERK-eIF2α pathways.
We also found significantly elevated levels of phosphorylated ERK in neurons in APP/PS1;Abca7−/−mice compared with control APP/PS1 mice. Interestingly, a pathological study of AD patients revealed that the p-ERK level is upregulated in the projecting neurons in transentorhinal cortex at the initial stage of neurofibrillary degeneration before Aβ deposition in AD patients, which further extends into other brain regions coincident with the progressive sequence of neurofibrillary changes (Pei et al., 2002). In addition, the administration of a MEK inhibitor has been reported to rescue early memory impairment by suppressing ERK activation in 3xTg AD model mice at 6 months of age (Feld et al., 2014). Therefore, the abnormal activation of the ERK pathway likely induces detrimental effects on synaptic integrity and cognition, whereas ERKs are essential for synaptic plasticity related to learning and memory formation (Davis and Laroche, 2006, Samuels et al., 2009). Our findings suggest that ABCA7 dysfunctions are associated with excess activation of the ERK pathway, which may be one of the crucial endophenotypes in early stage of LOAD. Because we did not observe a significant increase of p-ERK levels in Abca7−/− mice without the APP/PS1 background, ABCA7 likely plays a role in regulating the ERK pathway activated by Aβ and/or other factors in AD.
In summary, our findings provide new evidence that ABCA7 dysfunctions in neurons contribute to AD pathogenesis, likely by affecting brain lipid homeostasis, APP processing, ER stress, and ERK activity (Fig. 8). Although ABCA7 is highly expressed in microglia, as well as in neurons in the brains (Zhang et al., 2014), we found that ABCA7 deficiency did not influence microglia activation in APP/PS1 mice, which is consistent with a previous report (Kim et al., 2013). ABCA7 deletion likely exacerbates brain Aβ accumulation by suppressing microglia/macrophage-mediated phagocytosis in J20 AD model mice (Kim et al., 2013); however, we did not observe significant differences in ISF Aβ40 clearance between APP/PS1 and APP/PS1;Abca7−/− mice when analyzed by in vivo microdialysis. Therefore, our results indicate that ABCA7 dysfunctions contribute to AD pathogenesis by modifying brain lipid profile and facilitating Aβ production in neurons through an activated SREBP2-BACE1 pathway, rather than influencing Aβ clearance. Furthermore, ER stress is induced by ABCA7 deletion in neurons, which activates the PERK-eIF2α pathway, causing cognitive decline and further activation of the SREBP2-BACE1 pathway (Fig. 8).Cholesterol and phospholipids have been shown to modulate the activity of BACE1 and γ-secretase (Di Paolo and Kim, 2011). ER function is also affected by lipid composition and lipid biosynthetic enzymes (Lagace and Ridgway, 2013). Therefore, future studies should define how alterations of brain lipid homeostasis due to ABCA7 loss of function affect AD pathogenesis. Our study has identified novel mechanisms underlying the association between ABCA7 dysfunction and risk for LOAD. Investigating further functional contribution of genetic and/or environmental factors determined through unbiased approaches to LOAD pathogenesis should allow us to refine common diagnostic and therapeutic targets for the disease.
Figure 8.

Possible pathways for ABCA7-related LOAD pathogenesis. ABCA7 dysfunctions likely facilitate Aβ production in neurons by activating the SREBP2-BACE1 pathway directly and/or by modifying the brain lipid profile indirectly. Furthermore, ER stress is also induced by ABCA7 loss of function in neurons, which activates the PERK-eIF2α pathway, causing cognitive declines and further activation of the SREBP2-BACE1 pathway. Excessive activation of the ERK signaling pathway caused by ABCA7 dysfunctions may also contribute directly to AD pathogenesis.
Footnotes
This work was supported by the Robert and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program (S.G.Y.), the National Institutes of Health (Grants R01AG18023 to S.G.Y.; Grants R01AG035355, R01AG027924, R01AG046205, RF1AG051504 01, P01NS074969, and P50AG016574 to G.B.; and Grants R01HL074136 and R01HL101274 to M.L.F.), the Alzheimer's Association (T.K.), and the Cure Alzheimer's Fund (G.B. and T.K.). We thank Sumiko Abe-Dohmae at Nagoya City University for the gift of human and mouse ABCA7 antibodies; Kazumitsu Ueda at Kyoto University for the gift of human ABCA7 plasmid constructs; Dennis W. Dickson, Melissa E. Murray, Monica Castanedes Casey, and Linda Rousseau at Mayo Clinic Jacksonville for histology and immunohistochemical analyses; Tynickwa Mims and Tammee M. Parson in the Younkin laboratory for animal care; Mary D. Davis for careful reading of this manuscript; and Christopher Janus at the University of Florida and Michelle Mielke at Mayo Clinic Rochester for critical comments.
The authors declare no competing financial interests.
References
- Abe-Dohmae S, Ueda K, Yokoyama S. ABCA7, a molecule with unknown function. FEBS Lett. 2006;580:1178–1182. doi: 10.1016/j.febslet.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 2015;11:332–384. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Barbero-Camps E, Fernández A, Martínez L, Fernández-Checa JC, Colell A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet. 2013;22:3460–3476. doi: 10.1093/hmg/ddt201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero-Camps E, Fernández A, Baulies A, Martínez L, Fernández-Checa JC, Colell A. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol. 2014;184:2066–2081. doi: 10.1016/j.ajpath.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, et al. Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100:2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19:215–222. doi: 10.1016/j.ceb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Carrasquillo MM, Crook JE, Pedraza O, Thomas CS, Pankratz VS, Allen M, Nguyen T, Malphrus KG, Ma L, Bisceglio GD, Roberts RO, Lucas JA, Smith GE, Ivnik RJ, Machulda MM, Graff-Radford NR, Petersen RC, Younkin SG, Ertekin-Taner N. Late-onset Alzheimer's risk variants in memory decline, incident mild cognitive impairment, and Alzheimer's disease. Neurobiol Aging. 2015;36:60–67. doi: 10.1016/j.neurobiolaging.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Disabato BM, Restivo JL, Verges DK, Goebel WD, Sathyan A, Hayreh D, D'Angelo G, Benzinger T, Yoon H, Kim J, Morris JC, Mintun MA, Sheline YI. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A. 2011;108:14968–14973. doi: 10.1073/pnas.1107411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuyvers E, De Roeck A, Van den Bossche T, Van Cauwenberghe C, Bettens K, Vermeulen S, Mattheijssens M, Peeters K, Engelborghs S, Vandenbulcke M, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Sleegers K. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. The Lancet Neurology. 2015;14:814–822. doi: 10.1016/S1474-4422(15)00133-7. [DOI] [PubMed] [Google Scholar]
- Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5:61–72. doi: 10.1111/j.1601-183X.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabelo N, Martín V, Marín R, Moreno D, Ferrer I, Díaz M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer's disease and facilitates APP/BACE1 interactions. Neurobiol Aging. 2014;35:1801–1812. doi: 10.1016/j.neurobiolaging.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Feld M, Krawczyk MC, Sol Fustiñana M, Blake MG, Baratti CM, Romano A, Boccia MM. Decrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2014;40:69–82. doi: 10.3233/JAD-131076. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy J, Bogdanovic N, Winblad B, Portelius E, Andreasen N, Cedazo-Minguez A, Zetterberg H. Pathways to Alzheimer's disease. J Intern Med. 2014;275:296–303. doi: 10.1111/joim.12192. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Abe-Dohmae S, Munehira Y, Aoki R, Kawamoto S, Furuya A, Shitara K, Amachi T, Kioka N, Matsuo M, Yokoyama S, Ueda K. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem Biophys Res Comm. 2003;311:313–318. doi: 10.1016/j.bbrc.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Iwamoto N, Abe-Dohmae S, Sato R, Yokoyama S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J Lipid Res. 2006;47:1915–1927. doi: 10.1194/jlr.M600127-JLR200. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006;174:547–556. doi: 10.1083/jcb.200601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Kim WS, Fitzgerald ML, Kang K, Okuhira K, Bell SA, Manning JJ, Koehn SL, Lu N, Moore KJ, Freeman MW. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J Biol Chem. 2005;280:3989–3995. doi: 10.1074/jbc.M412602200. [DOI] [PubMed] [Google Scholar]
- Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–1166. doi: 10.1111/j.1471-4159.2007.05099.x. [DOI] [PubMed] [Google Scholar]
- Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK, Cheng D, Karl T, Garner B. Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer's disease. J Neurosci. 2013;33:4387–4394. doi: 10.1523/JNEUROSCI.4165-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace TA, Ridgway ND. The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2499–2510. doi: 10.1016/j.bbamcr.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee do Y, Lee KS, Lee HJ, Kim do H, Noh YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC, Jeong Y, Kim DK, Lee WB, Kim SS. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS One. 2010;5:e10489. doi: 10.1371/journal.pone.0010489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logge W, Cheng D, Chesworth R, Bhatia S, Garner B, Kim WS, Karl T. Role of Abca7 in mouse behaviours relevant to neurodegenerative diseases. PLoS One. 2012;7:e45959. doi: 10.1371/journal.pone.0045959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola R, Guglielmotto M, Medana C, Catalano MG, Cutrupi S, Borghi R, Tamagno E, Boccuzzi G, Aragno M. Dysregulation of SREBP2 induces BACE1 expression. Neurobiol Dis. 2011;44:116–124. doi: 10.1016/j.nbd.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Medway C, Morgan K. Review: The genetics of Alzheimer's disease; putting flesh on the bones. Neuropathol Appl Neurobiol. 2014;40:97–105. doi: 10.1111/nan.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer's disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/S0169-328X(02)00488-6. [DOI] [PubMed] [Google Scholar]
- Pike LJ, Han X, Gross RW. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: a shotgun lipidomics study. J Biol Chem. 2005;280:26796–26804. doi: 10.1074/jbc.M503805200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue M, Baldwin CT, Green RC, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309:1483–1492. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohe M, Carlo AS, Breyhan H, Sporbert A, Militz D, Schmidt V, Wozny C, Harmeier A, Erdmann B, Bales KR, Wolf S, Kempermann G, Paul SM, Schmitz D, Bayer TA, Willnow TE, Andersen OM. Sortilin-related receptor with A-type repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J Biol Chem. 2008;283:14826–14834. doi: 10.1074/jbc.M710574200. [DOI] [PubMed] [Google Scholar]
- Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression–implications for Alzheimer's disease. Prog Neurobiol. 2006;79:95–111. doi: 10.1016/j.pneurobio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Saitta SC, Landreth GE. MAP'ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Abe-Dohmae S, Yokoyama S, St George-Hyslop P, Fraser PE. ABCA7 Loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290:24152–24165. doi: 10.1074/jbc.M115.655076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano F, Chang A, Hernandez C, Pautler RG, Sweatt JD, Klann E. NADPH oxidase mediates beta-amyloid peptide-induced activation of ERK in hippocampal organotypic cultures. Mol Brain. 2009;2:31. doi: 10.1186/1756-6606-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Petersen RC, Dickson DW, Bu G. Brain regional correlation of amyloid-beta with synapses and apolipoprotein E in non-demented individuals: potential mechanisms underlying regional vulnerability to amyloid-beta accumulation. Acta Neuropathol. 2013;125:535–547. doi: 10.1007/s00401-013-1086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, Kong A, Helisalmi S, Soininen H, Lah JJ, Aarsland D, Fladby T, Ulstein ID, Djurovic S, Sando SB, White LR, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47:445–447. doi: 10.1038/ng.3246. [DOI] [PubMed] [Google Scholar]
- Sun X, Bromley-Brits K, Song W. Regulation of beta-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer's disease. J Neurochem. 2012;120:62–70. doi: 10.1111/j.1471-4159.2011.07515.x. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Abe-Dohmae S, Iwamoto N, Fitzgerald ML, Yokoyama S. Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J Lipid Res. 2010;51:2591–2599. doi: 10.1194/jlr.M006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Abe-Dohmae S, Iwamoto N, Yokoyama S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb. 2011;18:274–281. doi: 10.5551/jat.6726. [DOI] [PubMed] [Google Scholar]
- Vasquez JB, Fardo DW, Estus S. ABCA7 expression is associated with Alzheimer's disease polymorphism and disease status. Neurosci Lett. 2013;556:58–62. doi: 10.1016/j.neulet.2013.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer's disease beta-amyloid production. Biochim Biophys Acta. 2010;1801:860–867. doi: 10.1016/j.bbalip.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Lan D, Gerbod-Giannone M, Linsel-Nitschke P, Jehle AW, Chen W, Martínez LO, Tall AR. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278:42906–42912. doi: 10.1074/jbc.M307831200. [DOI] [PubMed] [Google Scholar]
- Yang K, Han X. Accurate quantification of lipid species by electrospray ionization mass spectrometry meet a key challenge in lipidomics. Metabolites. 2011;1:21–40. doi: 10.3390/metabo1010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, Nagahara AH, Lu KP, Nelson RJ, Tuszynski MH, Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron. 2012;75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]