

Abstract
Glioblastoma is the most common and most aggressive brain tumor in adults. The introduction of temozolomide (TMZ) has advanced chemotherapy for malignant gliomas, but it is not curative. The difficulties in treating glioblastoma may be as a result of the presence of glioma stem cells (GSCs), which are a source of relapse and chemoresistance. Another reason may be that endogenous Akt kinase activity may be activated in response to clinically relevant concentrations of TMZ. Akt activation is correlated with the increased tumorigenicity, invasiveness and stemness of cancer cells and overexpression of an active form of Akt increases glioma cell resistance to TMZ. Mounting evidence has demonstrated that cancer stem cells are preferentially sensitive to an inhibitor of Akt and down-regulation of the PI3K/Akt pathway may enhance the cytotoxicity of TMZ. Metformin (MET), the first-line drug for treating diabetes, it has been proved that it reduces AKT activation and selectively kills cancer stem cells, but whether it can potentiate the cytotoxicity of TMZ for GSCs remains unknown. In the present study, the GSCs isolated from human glioma cell line U87 and Rat glioma cell line C6, in vitro treatment with TMZ either alone or with MET. The present study demonstrates that MET acts synergistically with TMZ in inhibiting GSCs proliferation and generating the highest apoptotic rates when compared to either drug alone. These findings implicate that GSCs cytotoxicity mediated by TMZ may be stimulated by MET, have a synergistic effect, but the definite mechanisms remain elusive.
Keywords: temozolomide, metformin, glioma, cancer stem cell, Akt
Introduction
Glioblastoma is the most common and devastating primary malignant intracranial tumor in adults (1). The current standard of care (SOC) for newly diagnosed glioblastomas is surgical resection followed by radiotherapy (RT) plus concomitant and adjuvant temozolomide (TMZ) (2). The prognosis remains relatively poor with a median overall survival of only 14.6 months, median progression free survival of 6.9 months and 5 year survival rate of only 9.8% following diagnosis (2,3). The fast recurrence and multi-drug resistance are some of the key challenges in combating brain tumors. Glioma stem cells (GSCs) are considered to be a major source of relapse and chemoresistance (4), and are therefore an important therapeutic target for glioblastoma.
Accumulating evidence has also demonstrated that glioblastoma frequently display hyperactivation of the phosphatidylinositol-3-kinase (PI3K)-Akt pathway (5–7) and endogenous Akt kinase activity may be activated in response to clinically relevant concentrations of TMZ (8,9). Akt activation is correlated with the increased tumorigenicity, invasiveness and stemness (6) and overexpression of an active form of Akt increases glioma cell resistance to TMZ (5,8). It has demonstrated that cancer stem cells are preferentially sensitive to an inhibitor of Akt (7,10) and down-regulation of the PI3K/Akt pathway enhances the cytotoxicity of TMZ (11). At present, the most effective drug for the treatment of glioblastoma is TMZ, the primary path leading to glioma cell death is formation of O-6-methylguanine and apoptotic signalling triggered by O-6-methyl G:T mispairs, but apoptotic signalling goes through a step mediated by AMPK (12). However, there are obvious disadvantages as described above.
Metformin (MET), a first-line treatment for type-2 diabetes, can reduce cancer incidence (13) and mortality (14), and increases the number of breast carcinoma patients obtaining complete response to neo-adjuvant therapy (15). Previous studies have demonstrated that MET can selectively kill cancer stem cells (16–19) with minor adverse effects (20). Its mechanism of antiproliferative action is considered to be activation of AMP-activated protein kinase (AMPK) (17,18) and/or inhibition of Akt activity (19,21). On the basis of this evidence, a number of clinical trials are underway (22,23). Soritau et al (24) proved that the tumor cells isolated from patients with high-grade gliomas who were treated with TMZ and MET exhibited a significant decrease in proliferation rate when compared to those treated with TMZ alone. But whether MET may potentiate the cytotoxicity of TMZ for GSCs is unknown.
In the present study, GSCs isolated from human glioma cell line U87 and Rat glioma cell line C6, were treated with TMZ either alone or with MET. The combination index (CI) value was analyzed and calculated by the Chou-Talalay method (25). The present study demonstrated that MET acts synergistically with TMZ in inhibiting GSCs proliferation and expansion, showed a significant apoptosis compared to either agent alone and reinforced the effect on cell cycle arrest. The present study provides a rationale for why the combination of MET and TMZ may improve treatment of patients with glioblastoma.
Materials and methods
Cell and gliosphere culture
The human glioma cell line U87 and Rat glioma cell line C6 were purchased from Nanjing KGI Biotechnology Company (Nanjing, China). The cells were cultured in Gibco DMEM media (Thermo Fisher Scientific, Inc., Waltham, USA), supplemented with Hyclone 10% fetal bovine serum (Thermo Fisher Scientific, Inc.), Hyclone Penicillin-Streptomycin (100 U/ml), Hyclone glutamine (2 M) in a humidified atmosphere of 5% CO2 at 37°C. The cells were dissociated using 0.25% trypsin (Gibco; Thermo Fisher Scientific, Inc.) and 0.02% EDTA solution and subcultured once in 3–5 days. To generate gliospheres, U87 and C6 glioblastoma cells were dissociated from DMEM cultures using trypsin-EDTA solution and cultured in Gibco Neurobasal medium (NBM) supplemented with Gibco N2 (1x), B27 (1x), glutaMAX (1x), heparin (2 ug/ml), recombinant human FGF-basic (b-FGF, 20 ng/ml; PeproTech China, Suzhou, China), recombinant human epidermal growth factor (EGF, 20 ng/ml; PeproTech), Hyclone Penicillin-Streptomycin (100 U/ml). The gliospheres were cultured in 6-well plates in 5% CO2 incubator at 37°C with a medium change every 2–3 days. After gliospheres formed and reached 100–200 cells/sphere, within 10 days, gliospheres were dissociated by accutase (Sigma-Aldrich, St. Louis, MO, USA) and reseeded at a ratio of 1:2-3.
GSCs identification (immunofluorescence staining)
Gliospheres were plated onto poly-L-lysine (Sigma-Aldrich) coated glass cover slips in DMEM with 10% FBS for 8 h. The Gliospheres were washed with cold PBS (0.01 M), fixed with 4% paraformaldehyde (Gibco; Thermo Fisher Scientific, Inc.) for 30 min, permeabilized with 0.1% Triton X-100 for 15 min and blocked in 5% BSA (Sigma-Aldrich) for 1 h at room temperature. Then the gliospheres were immunostained with rabbit anti-human polyclonal CD133 (cat. no. ZA-0426; 1:100; ZSGB-BIO; OriGene Technologies, Inc., Beijing, China), mouse anti-human monoclonal nestin (cat. no. ZM-0323; 1:40; ZSGB-BIO; OriGene Technologies, Inc.), mouse anti-human monoclonal β-tubulin III (cat. no. TA500047; 1:100; ZSGB-BIO; OriGene Technologies, Inc.) and rabbit anit-human monoclonal glial fibrillary acidic protein (GFAP; cat. no. ZA-0529; 1:100, ZSGB-BIO; OriGene Technologies, Inc.) antibodies at 4°C overnight. Subsequent visualization was performed with fluorescein-conjugated AffiniPure goat anti-rabbit IgG (cat. no. ZF-0311; 1:100) and tetramethylrhodamine-conjugated AffiniPure goat anti-mouse IgG (cat. no. ZF-0313; 1:100) secondary antibodies (ZSGB-BIO; OriGene Technologies, Inc.) for 30 min at room temperature in darkness, and the nuclei were counterstained with DAPI (0.5 µg/ml; Institute of Biotechnology, Haimen, China).
For immunostaining of differentiated tumor cells, gliospheres were transferred to DMEM with 10% FBS for another 7 days and immunocytochemistry was performed as described above. The fluorescent signals were detected and images were captured with a fluorescence microscope (Olympus IX51, Olympus Corporation, Tokyo, Japan).
Gliosphere formation and expansion assay
To test the effect of two agents (TMZ and/or MET) on gliosphere formation, after primary sphere formation was observed, gliospheres were dissociated and plated in 96-well plates (5×103/ml/well) in NBM with B27, N2, glutaMAX, 2 µg/ml heparin and 20 ng/ml EGF+bFGF in the absence or presence of TMZ, MET or TMZ plus MET. Cultures were fed 0.02 ml of NBM every 2 days and images were captured (x200 magnification) after 7 days using IX51 Olympus microscope. To determine the gliosphere counts, U87 and C6 gliospheres were dissociated and plated in 96-well plates (5×103/ml/well) in NBM with B27, N2, glutaMAX, 2 µg/ml heparin and 20 ng/ml EGF+bFGF in the presence of TMZ, MET or TMZ plus MET for 7 days and the number of gliospheres counted under IX51 Olympus microscope.
Combination proliferation assay
The cytotoxic effect of TMZ or/and MET on GSCs was measured by cell counting kit-8 (CCK-8) assay. Briefly, U87 and C6 gliospheres were cultured in 96-well tissue culture plates (5×103 per 100 µl per well) in NBM with B27, N2, glutaMAX, 2 µg/ml heparin and 20 ng/ml EGF+bFGF at 37°C for 24 h, then different concentrations of TMZ (0–3.2 mmol, Sigma-Aldrich) or MET (0–160 mmol, Sigma-Aldrich) were added and compared with the DMSO-treated control. The CCK-8 reagent (10 µl/well) was added at 93 h, and the OD was measured at 450 nm after 96 h using a Synergy HTX Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA). In order to determine the combination cytotoxic effect on GSCs, parallel studies were performed in cells treated with TMZ (0–3.2 mmol) plus MET (0–160 mmol) (1:50, TMZ:MET). The combination index (CI) value was analyzed by CalcuSyn software (version 2; Biosoft, Cambridge, UK) and calculated by the Chou-Talalay method (25). CI values <1.0, =1, and >1.0 indicates synergistic interaction (more than additive), summation (additive), and antagonistic interaction (less than additive), respectively.
Cell cycle analysis
To determine the effect of TMZ in combination with MET on cell cycle progression, U87 and C6 gliospheres were cultured in NBM with B27, N2, glutaMAX, 2 µg/ml heparin and 20 ng/ml EGF+bFGF in the absence or presence of 0.4 mmol TMZ, 20 mmol MET or 0.4 mmol TMZ plus 20 mmol MET in 5% CO2 incubator at 37°C. After 48 h, the GSCs were dissociated by accutase (Sigma-Aldrich), fixed and permeabilized with 500 µl of 70% cold ethanol at −20°C overnight. After washing in PBS, the cells were treated with 100 µg/ml of RNase (Sigma-Aldrich) and incubated at 37°C for 30 min. Then the cells stained with 100 µg/ml of propidium iodide (PI, Sigma-Aldrich) at 4°C for another 30 min in darkness. The percentage of cells at different cell cycle stages (G0/G1, G2/M and S phase) was determined on the basis of DNA content by flow cytometer using ModFit LT 3.3 software (Verity Software House, Inc., Topsham, ME, USA).
Apoptosis assay
To determine the effect of TMZ or/and MET on GSCs apoptosis, U87 and C6 gliospheres were cultured in NBM with B27, N2, glutaMAX, 2 µg/ml heparin and 20 ng/ml EGF+bFGF in the absence or presence of 0.4 mmol TMZ, 20 mmol MET or 0.4 mmol TMZ plus 20 mmol MET in 5% CO2 incubator at 37°C. After 48 h, the GSCs were collected. Apoptotic and necrotic cell death was analyzed by double staining with fluorescein-isothiocyanate (FITC)-conjugated annexin V and PI, in which annexin V bound to the early and late apoptotic cells with membrane-exposed phosphatidylserine, while PI labeled only the late apoptotic/necrotic cells with membrane damage. Staining was performed using the FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA), according to the manufacturer's instructions. The stained cells were analyzed with a BD FACSAria III flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The numbers of viable (annexin V-/PI-), apoptotic (annexin V+/PI-), and necrotic (annexin V+/PI+) cells were calculated with the FACSDiva software, version 6.2 (BD Biosciences).
Western blot analysis
To investigate possible molecular determinants of MET effects, U87 and C6 gliospheres were treated with TMZ (400 µmol), MET (20 mmol) or TMZ+MET (400 µmol+20 mmol) for 72 h were collected and lysed in RIPA buffer. Equal amounts of proteins (30 µg) were separated by SDS-PAGE gels and were transferred to a PVDF membrane (Pierce; Thermo Fisher Scientific, Inc.) and blocked for 2 h in 5% nonfat dry milk in PBS-T. The blots were incubated with primary antibodies, including rabbit anti-human monoclonal phospho-Akt (cat. no. 13038P; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-human monoclonal Akt (cat. no. 4691P; 1:1,000; Cell Signaling Technology, Inc.) and mouse anti-human monoclonal β-actin (cat. no. A1978; 1:1,000; Sigma-Aldrich), at 4°C for 12 h. The membranes were then washed with PBS-T and incubated with HRP-conjugated goat anti-rabbit IgG (cat. no. ZB-2301; 1:5,000) or HRP-conjugated goat anti-mouse IgG (cat. no. ZB-2305; 1:5,000) secondary antibodies (ZSGB-BIO; OriGene Technologies, Inc.) for 2 h. After subsequent washes, specific signals were showed using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc.) and detected by the western blot automated chemiluminescence image analysis system (Tanon5200; Shanghai-Day Technology Co., Ltd., Shanghai, China).
Statistical analysis
The experiments were performed ≥3 times and data were expressed as the mean ± standard deviation. Statistical comparisons were assessed using the Student's t test. Statistical analysis was performed with SPSS software, version 19.0 (IBM SPSS< Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Isolation, characterization and differentiation of GSCs isolated from U87 and C6 glioma cells
U87 and C6 gliospheres cultures were characterized for recognized GSC signatures: Self-renewal, neural stem cell marker expression and differentiation. The cells within the sphere were positive for neural stem cell markers CD133 (Fig. 1A) and nestin (Fig. 1B), and lack of immunoreactivity for markers of differentiated neural cell types such as GFAP for astrocytes and β-tubulin III for neurons. The assay of multi-lineage differentiation capacity of cells within the sphere was demonstrated by culturing the cells in differentiation-inducing culture medium (DMEN+10% FBS) for 7 days. These cells lost expression of CD133 and nestin when subjected to differentiating conditions, showed typical morphological differentiation towards neuronal and astrocytic lineages, identified as β-tubulin-III positive for neurons and GFAP positive for astrocytes (Fig. 1C). The self-renewing capacity of the tumor spheres was assayed by dissociation of primary tumor spheres. When the self-renewal capacity was compared among tumor subtypes at a plating density of 5×103 cells/well, U87 were observed to generate a greater mean number of secondary tumor spheres (151±5) compared with C6 (123±4) (Fig. 2).
Figure 1.

Identification, characterization and differentiation U87-GSCs and C6-GSCs. U87 and C6 gliospheres were cultured for 7 days, immunocytochemical staining of (A) the cancer stem cell marker, CD133, and (B) the neural progenitor marker, nestin, in U87-GSC and C6-GSC cells. (C) Following differentiation, the U87 and C6 glioma cells were immunostained with β-tubulin-III and GFAP. The fluorescent signals were detected and images were captured at x200 magnification. GSCs, glioma stem cells; GFAP, glial fibrillary acidic protein.
Figure 2.
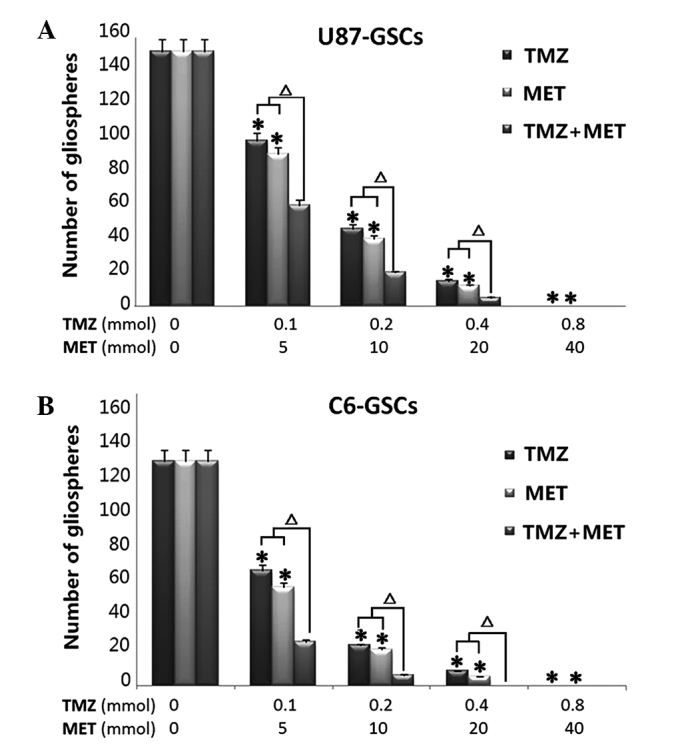
Inhibition of gliosphere formation and expansion by TMZ and MET. (A) U87-GSCs and (B) C6-GSCs were cultured in the absence or presence of TMZ, MET or TMZ and MET for 7 days and the number of gliospheres counted under a microscope. The results are representative of 3 independent experiments *P<0.05, compared with control group; ∆P<0.01, compared with single agents. GSCs, glioma stem cells; TMZ, temozolomid; MET, metformin.
TMZ plus MET contribute more effectively to inhibit gliospheres formation and expansion
To determine whether TMZ plus MET is more effective in the inhibition of U87 and C6 gliosphere formation and expansion compared with either drug alone, culture conditions were established to generate gliospheres in vitro. U87 and C6 glioma cells were cultured in NBM with B27, N2, glutaMAX, 2 ug/ml heparin and 20 ng/ml EGF+bFGF, promotes gliosphere formation in 5–7 days and increases in size in 7–10 days. In vitro treatment with TMZ or MET resulted in a marked reduction in the number and size of gliospheres generated from U87 and C6 cells (P<0.05). Notably, when the cells were treated with MTZ plus MET, a significant decrease was observed in the number and size of gliospheres compare to either agent alone (P<0.01). Further analyses demonstrated that in vitro treatment with TMZ, MET or TMZ plus MET resulted in a dose-dependent decrease in the number and size of gliospheres (Fig. 2). These results indicate that TMZ plus MET combination treatment is more effective at inhibiting U87 and C6 gliosphere formation and expansion compared with single agent treatment.
TMZ plus MET acts synergistically in inhibiting GSCs proliferation
In order to determine whether the MET, which is currently in clinical trials for cancer treatment, augments the cell proliferation inhibitory effects of TMZ in GSCs, U87 and C6 GSCs were plated with 0–3.2 mmol TMZ either alone or with 0–160 mmol MET for 72 h and the cells were assessed using the CCK-8 assay. Both U87-GSCs and C6-GSCs exhibited reduced proliferation levels as a dose-dependent response to TMZ and MET. The combination of TMZ and MET (1:50, TMZ:MET) resulted in a significant shift in the proliferation inhibition curve compared with treatment with either drug alone(Fig. 3). The statistical combination index (CI) was determined for the dual therapy to determine whether combination therapy was synergistic, additive, or antagonistic. As shown in Fig. 3 and Table I, MET acted synergistically (CI<1.0) with TMZ to inhibit U87-GSCs and C6-GSCs growth at all combination doses tested. As the concentration of TMZ in combination with MET was increased, the trend in CI value was reduced, suggesting that increasing the dosage of TMZ and MET increases the synergistic effect.
Figure 3.

MET augments the effects of TMZ in GSCs. (A) U87-GSCs and (C) C6-GSCs were treated with TMZ alone (0–160 mmol), MET (0–3.2 mmol) or the combination of TMZ and MET (1:50, TMZ:MET). Calcusyn software was used to ascertain the combination index (CI) value for each combination ratio to determine whether combination treatments have an additive or synergistic effect on (B) U87-GSCs or (D) C6-GSCs. CI values <1.0, =1, and >1.0 indicates synergistic interaction (more than additive), summation (additive), and antagonistic interaction (less than additive), respectively. Results are representative of 3 independent experiments. GSCs, glioma stem cells; TMZ, temozolomid; MET, metformin; CI, combination index.
Table I.
Combination treatments with TMZ and MET synergistically inhibit cell growth in U87-GSCs and C6-GSCs.
| TMZ | MET | Combination | |||||
|---|---|---|---|---|---|---|---|
| Cell line | Dose (mmol) | Growth inhibition (%) | Dose (mmol) | Growth inhibition (%) | Ratio | Growth inhibition (%) | CI value |
| U87-GSCs | 0.05 | 15.9±0.83 | 2.5 | 14.0±0.75 | 1:50 | 24.8±0.87 | 0.602 |
| 0.1 | 16.9±1.22 | 5 | 18.9±1.24 | 1:50 | 32.6±2.65 | 0.691 | |
| 0.2 | 26.4±1.15 | 10 | 22.7±1.32 | 1:50 | 43.8±1.39 | 0.693 | |
| 0.4 | 32.8±1.85 | 20 | 29.4±1.58 | 1:50 | 57.6±2.12 | 0.619 | |
| 0.8 | 45.2±3.21 | 40 | 41.6±2.01 | 1:50 | 68.1±1.85 | 0.643 | |
| C6-GSCs | 0.05 | 26.1±0.95 | 2.5 | 32.5±0.98 | 1:50 | 46.3±2.34 | 0.582 |
| 0.1 | 38.2±1.30 | 5 | 44.2±2.09 | 1:50 | 58.2±1.08 | 0.493 | |
| 0.2 | 42.6±1.59 | 10 | 55.1±2.32 | 1:50 | 70.0±3.05 | 0.388 | |
| 0.4 | 51.6±2.03 | 20 | 62.3±1.68 | 1:50 | 79.7±2.71 | 0.308 | |
| 0.8 | 64.7±2.08 | 40 | 69.0±2.41 | 1:50 | 84.2±3.02 | 0.358 | |
Cells were treated with a 1:50 combination (ratio) of TMZ:MET at the indicated concentrations. Percentage growth inhibition of the combination treatments was calculated compared to untreated cells (DMSO). Calcusyn software was used to ascertain the combination index to determine whether combination treatments have an additive or synergistic effect. CI values <1.0, =1, and >1.0 indicates synergistic interaction (more than additive), summation (additive), and antagonistic interaction (less than additive), respectively. TMZ, temozolomid; MET, metformin; CI, Combination index.
TMZ and/or MET induced cell cycle arrest in GSCs
Whether the inhibition of proliferation by TMZ or MET was due to cell cycle arrest or apoptosis in GSCs was investigated using flow cytometry. U87-GSCs and C6-GSCs were treated with TMZ (0.4 mmol), MET (20 mmol) or TMZ (0.4 mmol) plus MET (20 mmol) for 48 h and the cell cycle phase was determined. As shown in Fig. 4 and Table II, U87-GSCs cultured in NBM with B27, N2, glutaMAX, heparin and EGF+bFGF showed 62.79% G0/G1 cells, 18.3% G2/M cells and 18.91% S-phase cells, addition of TMZ or MET resulted in a reduction in G0/G1 cells (45.19 and 59.2%, respectively), a marked increase in G2/M cells (34.49 and 22.19%, respectively) and an increase in S-phase cells (20.32 and 18.61%, respectively). However, when U87-GSCs were treated with TMZ plus MET, a greater reduction in G0/G1 cells (22.67%; P<0.05), and a greater increase in G2/M and S-phase cells was observed (48.89 and 28.43%, respectively; P<0.05). This result indicates that the U87-GSC cells were arrested at the G2/M phase by TMZ or MET treatment, but TMZ plus MET induced cell cycle arrest at both the G2/M phase and S-phase; the effect was markedly better than treatment with a single agent, particularly for the G2/M phase arrest. C6-GSCs, cultured in NBM with B27, N2, glutaMAX, heparin and EGF+bFGF, were analyzed by flow cytometry and the result demonstrated that 66.43% cells were in G0/G1, 8.53% were in G2/M and 25.04% were in S-phase. The addition of TMZ or MET resulted in a slight decrease in the percentage of G0/G1 cells (60.06 and 56.34%, respectively), an increase in the percentage of G2/M cells (22.64 and 25.02%, respectively) and a slight decrease in the percentage of S-phase cells (17.30 and 18.65%, respectively). When C6-GSCs were treated with TMZ plus MET resulted in a greater reduction in the percentage of G0/G1 cells (43.09%; P<0.05) and a marked increase in the percentage of cells in S-phase (31.92%; P<0.05). These results indicated that the C6-GSCs cycles were arrested at S phase following treatment with TMZ plus MET; however, little difference was observed in the percentage of C6-GSCs in G2/M arrest when treated with TMZ plus MET compared with either single agent. These results suggest that TMZ plus MET treatment regulates cell cycle progression of GSCs in culture and using the agents together enhances the effects.
Figure 4.

Effects of TMZ, MET or TMZ and MET on cell cycle distribution. (A) U87-GSCs and (B) C6-GSCs (2×105 cells) were cultured in the presence of 0.4 mmol TMZ, 20 mmol MET or TMZ and MET. The cells were harvested after 48 h and stained with propidium iodide (PI), and analyzed by flow cytometry. GSCs, glioma stem cells; TMZ, temozolomid; MET, metformin.
Table II.
Modulation of cell cycle and apoptosis by TMZ, MET or TMZ plus MET.
| Cell line | Sub-group | Mean (%) Sub G0 phase | Mean (%) G0/G1 phase | Mean (%) S phase | Mean (%) G2/M phase |
|---|---|---|---|---|---|
| U87-GSGS | Control | 0.08±0.02 | 62.79±3.10 | 18.91±2.30 | 18.30±1.65 |
| TMZ | 6.22±0.20 | 45.19±1.28 | 20.32±1.41 | 34.49±1.69 | |
| MET | 5.38±0.03 | 59.20±2.36 | 18.61±1.36 | 22.19±2.52 | |
| TMZ+MET | 14.77±1.09a | 22.67±3.21a | 28.43±2.84a | 48.89±3.21a | |
| C6-GSCs | Control | 0.05±0.01 | 66.43±2.89 | 25.04±1.05 | 8.53±0.39 |
| TMZ | 5.37±0.05 | 60.06±2.03 | 17.30±0.56 | 22.64±0.58 | |
| MET | 6.28±0.24 | 56.34±1.38 | 18.65±0.47 | 25.02±0.12 | |
| TMZ+MET | 19.61±0.31a | 43.09±0.45a | 31.92±0.90a | 24.99±0.14 |
The analysis of cell cycle distribution and apoptosis rate in U87-GSCs and C6-GSCs. Data is presented as the mean and are representative of three independent experiments
(P<0.05, compared with single agent). TMZ, temozolomid; MET, metformin; GSCs, glioma stem cells.
TMZ plus MET significantly induced apoptosis in GSCs
The present study also aimed to determine whether the TMZ or MET-induced reduction in cell viability is accompanied by cell apoptosis. Annexin V and PI staining and flow cytometry was used to detect apoptosis in U87-GSCs and C6-GSCs treated with TMZ (0.4 mmol), MET (20 mmol) or TMZ (0.4 mmol) plus MET (20 mmol) for 48 h. As shown in Fig. 5, U87-GSCs and C6-GSCs cultured without TMZ or MET exhibited low apoptotic cell death. Treatment with TMZ or MET induced cell apoptosis (P<0.05); however, treatment with TMZ plus MET resulted in a significant increase in apoptotic cells compared with single agents (P<0.05). In U87-GSCs, the percentage of apoptotic cells in the control group was 15.86%, while those of TMZ and MET groups were 31.84 and 28.23%, respectively, and was significantly increased to 61.73% in TMZ plus MET group (P<0.05). In C6-GSCs, the percentage of apoptotic cells in the control group was 11.21%, and those of the TMZ and MET groups were 36.96% and 41.59%, respectively; and was significantly increased to 62.73% in TMZ plus MET group (P<0.05). The above results demonstrated that TMZ or MET significantly induced apoptosis in GSCs compared with the control groups and MET treatment enhances TMZ-induced apoptosis.
Figure 5.

TMZ plus MET significantly induced apoptosis in GSCs. (A) U87-GSCs and (B) C6-GSCs (2×105 cells) were cultured in the presence of 0.4 mmol TMZ, 20 mmol MET or TMZ plus MET. The cells were harvested after 48 h and stained with Annexin V-FITC, propidium iodide, and analyzed by flow cytometry. (C) Quantitative analysis of the early and late apoptosis rate. Results are representative of three independent experiments (*P<0.05, compared with single agents). GSCs, glioma stem cells; TMZ, temozolomid; MET, metformin.
MET potentiates the cytotoxicity of TMZ for GSCs by inhibition of Akt activation
Fig. 6 demonstrates that when GSCs were treated with TMZ alone for 72 h, Akt phosphorylation is slightly enhanced. However, Akt phosphorylation was inhibited by MET, suggesting that Akt inhibition is required for MET antiproliferative activity in GSCs. Notably, the phosphorylated Akt (P-Akt) levels were markedly down-regulated compared with the control group and TMZ alone groups compared with treatment with TMZ and MET in combination. These results indicate that the potential mechanism of MET enhancing TMZ cytotoxicity may involve down-regulation of P-Akt.
Figure 6.

Western blot analysis of phosphorylated Akt protein expression. MET acts synergistically with TMZ in inhibiting GSCs proliferation and expansion by inhibition of Akt activation. GSCs, glioma stem cells; TMZ, temozolomid; MET, metformin
Discussion
Previous studies have indicated that glioblastomas originate from a pool of stem-like cells that share properties in common with neuronal stem cells. GSCs are a small population of cells that have self-renewal, transplantation, recurrence properties and multidrug resistance (26). The identification of stem-like cells in tumors and the proposed role for this subpopulation in drug resistance opened the search for targeted molecular therapies and novel drugs able to deplete this tumor cell subpopulation, either by activating differentiation or through selective toxicity that causes loss of self-renewal (19). However, in medicine, it is usually difficult to get agents with high target specificity; only a few of compounds have showed selective toxicity for cancer stem cells (27,28). However, certain drugs already used in the clinic that may have encouraging results, such as MET, may selectively kill cancer stem cells (16–19) with minor adverse events (20).
To the best of our knowledge, the present report demonstrates for the first time the ability of MET to function synergistically with TMZ to reduce U87-GSCs and C6-GSCs proliferation, although it has been proved previously that MET improves the survival of cisplatin-treated tumour cells (29).
TMZ is currently the most effective drug for the treatment of glioblastoma and one mechanism of TMZ-induced apoptosis involves an obligatory AMP-activated protein kinase (AMPK) activation step (12); however, this increases the activity of Akt which increases glioma cell resistance to TMZ (5,8) and may increase the tumorigenicity, invasiveness and stemness of tumor cells (6). The anticancer mechanism of MET is considered to be activation of AMPK (17,18) or inhibition of the PI3K-Akt pathway (19,21). Although the present study demonstrated that MET potentiates the cytotoxicity of TMZ for GSCs and has a synergistic effect, whether this is achieved through increased AMPK activity and/or inhibition of AKT activity in GSCs remains unknown.
The potential for MET to target GBM proliferation was proposed due to the hypothesized synergy with TMZ on AMPK activation (17); the present authors are sceptical of this hypothesis and it has not been confirmed. However, certain studies support this hypothesis. Liu et al (30), suggested that MET's growth inhibition in vitro does not require AMPK. Instead, AMPK activation may be a response mechanism to counter stress induced by anticancer agents (30). It is possible that the growth-inhibiting effects are a combination of the AMPK-independent mTOR-dependent cellular and systemic effects of MET (30). Gritti et al (16) determined that MET exhibits its selective antiproliferative effects in human GSCs via inhibition of the CLIC1-mediated ion current, although this requires further studies to confirm this observation.
It has been proposed that the Akt pathway represents a novel target for the sensitization of Akt overexpression gliomas to chemotherapeutic methylating agents such as TMZ (5), due to the ability of Akt overexpression to suppress TMZ-induced mitotic catastrophe as well as TMZ-induced senescence (5). It has been proved that TMZ combined with the PI3K inhibitor LY294002 inhibits melanoma cell growth, survival and invasion (31); and similar results were observed in glioma cells (11). Akt activation has been reported to suppress activation of the G2 checkpoint in human colon carcinoma cells exposed to radiation (32), Akt activation has also been proven to suppresses TMZ-induced G2 arrest (5). However, in our trial, MET, through the inhibition of Akt activation, selectively killed cancer stem cells and acted synergetic with TMZ, enhancing TMZ-induced G2 arrest for U87-GSCs; however, in C6-GSCs, they just reinforced each other and induced cell cycle arrest at S phase. Based on the experimental results of the present study, MET may enhance the cytotoxicity of TMZ by down-regulation of the PI3K/Akt pathway, these results warrant further in vivo exploration.
Although the present study presents evidence that MET acts synergistically with TMZ in inhibiting GSCs proliferation and generating the highest apoptotic rates when compared with either drug alone, but the exact mechanism of action remains unclear. Soritau et al (24) reported that tumor cells isolated from patients with high-grade gliomas who were treated with TMZ and MET exhibited a significant decrease in proliferation rate when compared to those treated with TMZ alone. In conclusion, future trials are required to explore the detailed mechanisms.
Acknowledgements
The present study was supported by the Young Scientists Fund of the National Natural Sciene Foundation of China (grant no. 21401072).
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Cancer stem cells in solid tumors. Semin Cancer Biol. 2010;20:77–84. doi: 10.1016/j.semcancer.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005;65:4861–4869. doi: 10.1158/0008-5472.CAN-04-2633. [DOI] [PubMed] [Google Scholar]
- 6.Molina JR, Hayashi Y, Stephens C, Georgescu MM. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12:453–463. doi: 10.1593/neo.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027–3036. doi: 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caporali S, Levati L, Starace G, Ragone G, Bonmassar E, Alvino E, D'Atri S. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol. 2008;74:173–183. doi: 10.1124/mol.107.044743. [DOI] [PubMed] [Google Scholar]
- 9.De Salvo M, Maresca G, D'Agnano I, Marchese R, Stigliano A, Gagliassi R, Brunetti E, Raza GH, De Paula U, Bucci B. Temozolomide induced c-Myc-mediated apoptosis via Akt signalling in MGMT expressing glioblastoma cells. Int J Radiat Biol. 2011;87:518–533. doi: 10.3109/09553002.2011.556173. [DOI] [PubMed] [Google Scholar]
- 10.Gallia GL, Tyler BM, Hann CL, Siu IM, Giranda VL, Vescovi AL, Brem H, Riggins GJ. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther. 2009;8:386–393. doi: 10.1158/1535-7163.MCT-08-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, Han L, Shi Z, Zhang K, Liu Y, Zheng Y, Jiang T, Pu P, Jiang C, Kang C. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol Med Rep. 2012;5:575–579. doi: 10.3892/mmr.2011.674. [DOI] [PubMed] [Google Scholar]
- 12.Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, Yang Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010;285:40461–40471. doi: 10.1074/jbc.M110.164046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 15.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gritti M, Wurth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E, Sirito R, Daga A, et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget. 2014;5:11252–11268. doi: 10.18632/oncotarget.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kast RE, Karpel-Massler G, Halatsch ME. Can the therapeutic effects of temozolomide be potentiated by stimulating AMP-activated protein kinase with olanzepine and metformin? Br J Pharmacol. 2011;164:1393–1396. doi: 10.1111/j.1476-5381.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, Narita Y, Shibui S, Kayama T, Kitanaka C. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem cells Transl Med. 2012;1:811–824. doi: 10.5966/sctm.2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12:145–156. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolen S, Feldman L, Vassy J, Wilson L, Yeh HC, Marinopoulos S, Wiley C, Selvin E, Wilson R, Bass EB, et al. Systematic review: Comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med. 2007;147:386–399. doi: 10.7326/0003-4819-147-6-200709180-00178. [DOI] [PubMed] [Google Scholar]
- 21.Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res. 2010;123:271–279. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
- 22.Rattan R, Ali Fehmi R, Munkarah A. Metformin: An emerging new therapeutic option for targeting cancer stem cells and metastasis. J Oncol. 2012;2012:928127. doi: 10.1155/2012/928127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24:469–480. doi: 10.1016/j.tem.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Soritau O, Tomuleasa C, Aldea M, Petrushev B, Susman S, Gheban D, Ioani H, Cosis A, Brie I, Irimie A, et al. Metformin plus temozolomide-based chemotherapy as adjuvant treatment for WHO grade III and IV malignant gliomas. J BUON. 2011;16:282–289. [PubMed] [Google Scholar]
- 25.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 27.Sachlos E, Risueño RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012;149:1284–1297. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 28.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janjetovic K, Vucicevic L, Misirkic M, Vilimanovich U, Tovilovic G, Zogovic N, Nikolic Z, Jovanovic S, Bumbasirevic V, Trajkovic V, et al. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651:41–50. doi: 10.1016/j.ejphar.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci USA. 2014;111:E435–E444. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meier FE, Lasithiotakis K, Schittek B, Sinnberg T, Garbe C. Temozolomide combined with the PI3K inhibitor LY294002 or the mTOR inhibitor rapamycin inhibits melanoma cell growth, survival and invasion. J Clin Oncol. 2007;25(Suppl 18):8559. [Google Scholar]
- 32.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cellular Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]