

Abstract
The aim of the current study was to observe the effects of simvastatin and taurine on delayed cerebral vasospasm (DCVS) following experimental subarachnoid hemorrhage (SAH) in rabbits. A total of 48 New Zealand white rabbits were allocated at random into four groups (control, SAH, SAH + simvastatin and SAH + taurine groups; n=12 each). The rabbit model of DCVS was established using a double hemorrhage method, which involved injecting autologous arterial blood into the cisterna magna in the SAH groups. The SAH + simvastatin group was administered oral simvastatin (5 mg/kg) daily between days 0–6. The SAH + taurine group was administered oral taurine (50 mg/kg) daily between days 0–6. Starch (50 mg/kg) was administered orally to the animals in the other two groups (control and SAH groups). The control group were not subjected to any other injections or treatment. The internal diameter and internal diameter/wall thickness of the basilar artery (BA) were measured. The expression levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 were determined using immunohistochemical and quantitative polymerase chain reaction methods following the sacrifice of all animals on day 7. The activity of nuclear factor (NF)-κB in the BA was also measured using an electrophoretic mobility shift assay. The BA walls in the SAH + simvastatin and SAH + taurine groups exhibited reduced narrowing and corrugation of the tunica elastica interna compared with the SAH group. At the protein and cDNA levels, it was found that cerebral vasospasm of the BA in the SAH + simvastatin and SAH + taurine groups was alleviated, as indicated by the reduced expression of TNF-α, IL-1β, IL-6 and NF-κB compared with the SAH group (P<0.05). In conclusion, simvastatin and taurine reduced DCVS following SAH in rabbits, which suggests that these compounds may exert anti-inflammatory effects.
Keywords: cerebral vasospasm, inflammation, simvastatin, subarachnoid hemorrhage, taurine
Introduction
Subarachnoid hemorrhage (SAH) is a severe, life-threatening type of stroke caused by bleeding into the space surrounding the brain. Up to 50% of all cases of SAH are fatal and 10–15% of stroke victims succumb to the condition prior to hospital admission. Patients that survive often exhibit neurological or cognitive impairment. A common and severe complication following aneurysmal SAH is delayed cerebral ischemia (DCI), which occurs in ~30% of patients surviving the ictus of the hemorrhage. DCI may be reversible, but in certain cases progresses to a cerebral infarction, which is associated with an increased risk of severe disability and mortality (1–3). Therefore, delayed cerebral vasospasm (DCVS) is a common and potentially fatal complication in patients that have survived an SAH (4–6). Cerebral vasospasm is the prolonged, intense vasoconstriction of the larger conducting arteries in the subarachnoid space, which is initially surrounded by a clot. During the first few days following aneurysmal rupture, significant narrowing of the cerebral vasculature develops gradually. This spasm usually reaches its maximum level within 1 week of the hemorrhagic event. Vasospasm is among the leading causes of mortality following an aneurysmal rupture, along with the effect of the initial hemorrhage and subsequent bleeding (7,8).
It has been reported that an inflammatory response may be crucial to the development of DCVS (9). Cerebral vasospasm following an aneurysmal SAH, which is considered to be caused by sustained contraction of smooth muscle cells of the major cerebral arteries, induces cerebral ischemia and affects subsequent mortality and morbidity (10). Currently, the most common drug in use for reducing the incidence of DCI and the poor outcome following an SAH is nimodipine (11). As the effects of nimodipine are relatively modest, considerable research efforts have focused on investigating and developing novel drugs for the prevention and treatment of this complication. Among the suggested therapeutic options is treatment with statins. Numerous intracellular signalling transduction pathways are considered to be implicated in the sustained contraction of smooth muscle cells during cerebral vasospasm. The critical event in this response is the recruitment of circulating leukocytes into the inflammatory site (12–14). Genes associated with inflammation, particularly certain cytokines, are highly expressed in spastic arteries, which suggests that the inflammatory response may cause sustained contraction of the cerebral arteries (4).
Cerebral vasospasm is among the most common and severe complications of SAH, and exhibits a complex pathogenesis. The initial processes that occur in SAH involve the activation of genes associated with angiogenesis, inflammation and extracellular matrix remodeling (15–17). Tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and nuclear factor (NF)-κB are the primary contributors to the inflammatory response (18–20). TNF-α (also known as cachexin or cachectin) is an adipokine that is involved in systemic inflammation, and is a member of a group of cytokines that stimulate the acute phase reaction. TNF-α is produced primarily by activated macrophages, as well as by a number of other cell types, such as CD4+ lymphocytes, natural killer cells and neurons.
The primary role of TNF-α is the regulation of immune cells. TNF-α is an endogenous pyrogen, which can induce fever, cachexia, apoptotic cell death and inflammation. In addition, it can inhibit tumorigenesis and viral replication, and respond to sepsis via IL-1β- and IL-6-producing cells. Furthermore, TNF-α is able to promote the activation of NF-κB, a heterodimeric transcription factor that translocates to the nucleus and mediates the transcription of a vast array of proteins involved in cell survival and proliferation, anti-apoptotic factors and inflammatory responses (21–23). These processes increase in the presence of proangiogenic factors and the expression of proinflammatory genes. In addition, multiple factors and mechanisms are considered to be active in the inflammatory response. Recently, simvastatin and taurine have been demonstrated to exert anti-inflammatory effects. The anti-inflammatory effects of simvastatin have been demonstrated in patients with chronic heart failure (24), while those of taurine have been shown in a rat model of stroke (25). In the current study, simvastatin was administered in a rabbit model of SAH to prevent DCVS, and the underlying molecular biological mechanisms were investigated with the aim of identifying a potential method for preventing DCVS.
Materials and methods
Animal treatment
In total, 48 New Zealand male white rabbits (weight range, 1.8–2.2 kg; mean age, 2 months) were randomly assigned to four groups (n=12 per group), as follows: SAH, SAH + simvastatin, SAH + taurin and control groups. In the SAH groups, a DCVS model was established using the double hemorrhage method by injecting autologous arterial blood into the cisterna magna (12). The SAH + simvastatin group was administered oral simvastatin (5 mg/kg) daily between days 0–6. The SAH + taurine group was administered oral taurine (50 mg/kg) daily between days 0–6. Starch (50 mg/kg) was administered orally to the animals in the other two groups (control and SAH groups). The control group mice were not subjected to any other injections or treatment.
Observation of structure using histochemistry
The rabbits were anesthetized by intramuscular injection of xylazine sailaqin (ml/kg; Jilin Huamu Animal Health Products Co., Ltd, Changchun, China), then rapidly sacrificed. The spastic vertebrobasilar arteries were rapidly removed for hematoxylin and eosin staining (Sigma-Aldrich, St. Louis, MO, USA). The internal diameter and internal diameter / wall thickness of the basilar artery (BA) were measured.
Immunohistochemistry
Paraffin-embedded artery specimens were cut into 4-µm sections and baked at 65°C for 30 min. The sections were deparaffinized with xylene, rehydrated, submerged in ethylenediaminetetraacetic acid (EDTA; pH 8.0), autoclaved for antigen retrieval, treated with 3% hydrogen peroxide, and then incubated with 1% fetal bovine serum. Primary goat anti-mouse polyclonal antibodies against TNF-α (sc-1349; 1:200), IL-1β (sc-1251; 1:500), IL-6 (sc-1265; 1:200; Santa Cruz Biotechnology, Inc., La Jolla, CA, USA) and mouse monoclonal NF-κB (N8523; 1:500; Sigma-Aldrich) were added and the sections were incubated overnight at 4°C. Next, a horseradish peroxidase-labeled secondary antibody was applied, and the sections were incubated for 30 min at room temperature, and then for 5 min at room temperature with diaminobenzidine. The sections were then counterstained with hematoxylin and eosin, and mounted with Permount mounting medium (Yansheng Biotechnology Company, Shanghai, China). Subsequently, the sections were visualized and photographed using a light microscope. The degree of immunostaining was scored separately by two independent investigators and the scores were determined based on the proportion of positively stained cells (17). The scores from the two investigators were averaged for further comparison of expression.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
TRIzol was purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). Primers were designed using Premier Primer 5, modified by Oligo 6 (Premier Biosoft International, Palo Alto, CA, USA) and synthesized by Invitrogen (Beijing, China). The PrimeScript RT-PCR kit, and SYBR Premix ExTaq (Perfect Real-Time) were purchased from Fermentas (Thermo Fisher Scientific, Waltham, MA, USA). The Stratagene Mx3000P qPCR system (Agilent Technologies, La Jolla, CA, USA) was used for qPCR, in order to analyze the TNF-α, IL-1β, IL-6, NF-κB and β-actin expression levels. Cerebral vasospasm scores of the BA were collected, and then total RNA was extracted using the TRIzol reagent and reverse-transcribed into cDNA according to the instructions provided with the PrimeScript RT-PCR kit. The primers used were as follows: β-actin forward, 5′-ATCGTGCGGGACATCAA-3′, and reverse, 5′-AGGAAGGAGGGCTGGAA-3′; TNF-α forward, 5′-AAACCCGCAAGTGGAG-3′, and reverse, 5′-AGAACCTGGGAGTAGATGAG-3′; IL-1β forward, 5′-GCAGGGTAGGTTTATCGTCTTT-3′, and reverse, 5′-GCAGGGTAGGTTTATCGTCTTT-3′; IL-6 forward, 5′-CTGGCGGAAGTCAATCTG-3′, and reverse, 5′-ATAGTGTCCTAACGCTCATC-3′; and NF-κB forward, 5′-CCCAGCCATTTGCACACCTCAC-3′, and reverse, 5′-TTCAGAATTGCCCGACCAGTTTTT-3′. The qPCR reaction was performed using a ROXII kit (Takara Bio, Dalian, China), in a 25-µl reaction mixture containing the following: Master mix (12.5 µl); ROXII dye (5 µM; 0.5 µl each), forward and reverse primers (10 nmol; 0.5 µl each); sample cDNA (1 µl); and MilliQ H2O (10 µl). The amplification conditions were as follows: For β-actin, 95°C for 5 min, then 40 cycles at 95°C for 30 sec, 56.9°C for 40 sec, and 72°C for 30 sec; for TNF-α, 95°C for 5 min, then 40 cycles at 95°C for 30 sec, 55°C for 40 sec, and 72°C for 30 sec; for IL-1β, 95°C for 5 min, then 40 cycles at 95°C for 35 sec, 55°C for 40 sec, and 72°C for 35 sec; for IL-6, 95°C for 5 min, then 40 cycles at 95°C for 40 sec, 53°C for 40 sec, and 72°C for 40 sec; and for NF-κB, 95°C for 5 min, then 40 cycles at 95°C for 30 sec, 55°C for 40 sec, and 72°C for 30 sec. The automatic dissociation curve conditions were added for all the samples. The relative mRNA expression levels of the genes were calculated from the cycle threshold value using the 2−∆∆Cq threshold method for quantification.
Electrophoretic mobility shift assay (EMSA)
NF-κB promoter was additionally analyzed using an EMSA kit (Pierce Biotechnology, Inc., Rockford, IL, USA), following a standard laboratory procedure (19). Briefly, end-labeled double-stranded oligonucleotide probes for the EMSA were prepared using T4 polynucleotide kinase and adenosine triphosphate γ-32P (Board of Radiation and Isotope Technology, Hyderabad, India). The DNA-protein binding was conducted in a 25-µl reaction mixture using 4 µg tissue extract in a binding buffer containing 20 mM HEPES (pH 7.5), 60 mM KCl, 0.2 mM EDTA, 10% glycerol, 1 mM DTT and 0.25 µl protease inhibition cocktail (all from Sigma-Aldrich). The nuclear extract from tissues was mixed with all the components, with the exception of an appropriate end-labeled probe, and incubated for 10 min. For competition experiments, a 50-fold excess of identical unlabeled probe was added 15 min prior to the addition of the end-labeled probe. In order to analyze the supershift, a mouse monoclonal anti-NF-κB antibody (1:500; N8523; Sigma-Aldrich) was added 15 min prior to the addition of the end-labeled probe. Subsequently, 50 fM end-labeled probe was added and incubated for 20 min. All incubation steps were performed on ice. Following incubation, DNA-protein complexes were resolved on a 5% non-denaturing polyacrylamide gel in 0.5X Tris-boric acid-EDTA buffer at 4°C and 30 mA. The oligo sequences for the NF-κB promoter used in the EMSA were 5′-AGTTGAGGGGACTTTCCCAGGC-3′ (forward) and 5′-TGAACTCCCCTGAAAGGGTCCG-3′ (reverse). A double-stranded probe was constructed by mixing an equal quantity of two oligos in Tris-HCl-EDTA (pH 8.0), followed by heating at 65°C for 10 min. The mixture was then gradually cooled to room temperature over 30 min.
Statistical analysis
Statistical analysis was performed using the SPSS software, version 20.0 (IBM SPSS, Armonk, NY, USA) for t-tests. For immunohistochemical analysis, an Automatic Analytic System, version 2.0 was used to record the percentage of positive cells and the Alpha Imager 1220 documentation and analysis system, version 5.50 (both from Emerald Green Biotech Co., Ltd., Hangzhou, China) was used to measure the integral optical density of NF-κB. P<0.01 was considered to indicate a statistically significant difference.
Results
Histochemistry
The results of histochemical analysis are presented in Table I and Fig. 1. The basilar artery (BA) walls in the SAH + simvastatin and SAH + taurine groups exhibited reduced narrowing and corrugation of the tunica elastica interna compared with the SAH group (P<0.05).
Table I.
Internal diameter and internal D/T of the basilar artery on day 7 after SAH.
| Group | Vessel diameter (µm) | D/T |
|---|---|---|
| SAH | 413.8±45.2 | 10.73±4.85 |
| SAH + simvastatin | 534.6±54.0 | 18.75±4.72 |
| SAH + taurine | 523.6±48.0 | 17.58±4.56 |
| Control | 588.9±58.0 | 25.37±6.46 |
D/T, diameter/wall thickness; SAH, subarachnoid hemorrhage.
Figure 1.
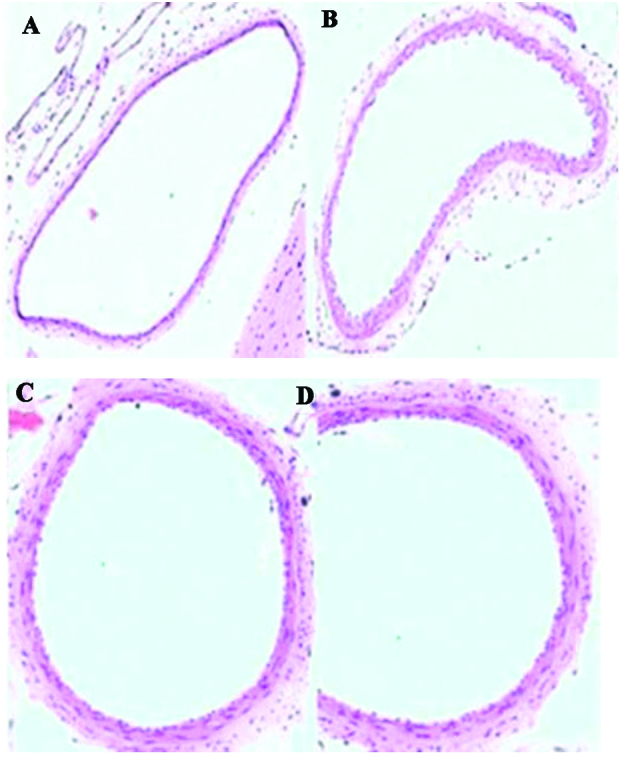
Representative histological structural differences in the arterial wall of the basilar artery (magnification, 100x stain, hematoxylin and eosin). (A) Control group. (B) Subarachnoid haemorrhage (SAH) group. (C) SAH + simvastatin group. (D) SAH + taurine group.
Immunohistochemistry
The expression of TNF-α, IL-1β and IL-6 was detected using immunohistochemistry after all animals were sacrificed on day 7. The immunohistochemistry results revealed that cerebral vasospasm of the BA in the SAH + simvastatin and SAH + taurine groups was alleviated, with reduced expression of TNF-α, IL-1β, IL-6 and NF-κB, compared with the SAH group (P<0.05; Table II). Furthermore, there was no statistically significant difference between the SAH + simvastatin and SAH + taurine groups (P>0.05; Fig. 2).
Table II.
Protein expression on day 7 after SAH as determined using immunohistochemistry.
| Group | TNF-α | IL-1β | IL-6 | NF-κB |
|---|---|---|---|---|
| SAH | 0.4109±0.0934 | 0.4396±0.0981 | 0.4020±0.0659 | 0.4430±0.0623 |
| SAH + simvastatin | 0.2531±0.0723a | 0.2362±0.1139a | 0.2769±0.0872a | 0.2238±0.0953a |
| SAH + taurine | 0.2621±0.0798a | 0.2351±0.1142a | 0.2745±0.0853a | 0.2312±0.0827a |
| Control | 0.2181±0.0870 | 0.1967±0.1056 | 0.2842±0.0528 | 0.2942±0.0645 |
P<0.01 vs. SAH group. SAH, subarachnoid hemorrhage; TNF-α, tumor necrosis factor-alpha; IL-1β, interleukin-1β; IL-6, interleukin-6; NF-κB, nuclear factor-κB.
Figure 2.

Photomicrographs of immunohistochemical staining for tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and nuclear factor (NF)-κB. Upper panel: TNF-α positive results for the (A) control, (B) subarachnoid haemorrhage (SAH) and (C) SAH + simvastatin (5 mg/kg) and (D) SAH + taurine groups. (E) NF-κB positive results of immunohistochemical stain (magnification, x20 and x40), (F) IL-1β positive results of immunohistochemical stain (magnification, x20 and x40), (G) IL-6 positive results of immunohistochemical stain (magnification, x20 and x40).
RT-qPCR
The expression levels of TNF-α, IL-1β and IL-6 mRNA were detected using RT-qPCR after all animals were sacrificed on day 7. The RT-qPCR results indicated that cerebral vasospasm of the BA in the SAH + simvastatin and SAH + taurine groups was alleviated, with reduced expression of TNF-α, IL-1β, IL-6 and NF-κB compared with the SAH group (P<0.05; Table III). There was no significant difference between the SAH + simvastatin and SAH + taurine groups. These results indicated that simvastatin or taurine exhibited an anti-inflammatory effect in treatment of SAH, and NF-κB was also involved.
Table III.
mRNA expression levels on day 7 after SAH by RT-qPCR.
| Group | TNF-α | IL-1β | IL-6 | NF-κB |
|---|---|---|---|---|
| SAH | 0.5431±0.2011 | 0.6324±0.1762 | 0.5002±0.1590 | 0.6542±0.1542 |
| SAH + simvastatin | 0.3526±0.1745a | 0.3816±0.0908a | 0.3224±0.1837a | 0.3513±0.1845a |
| SAH + taurine | 0.4050±0.1296a | 0.3125±0.1285a | 0.3671±0.1379a | 0.3565±0.1388a |
| Control | 0.3060±0.1672 | 0.3595±0.1261 | 0.2876±0.1535 | 0.2751±0.1452 |
P<0.01 vs SAH group. SAH, subarachnoid hemorrhage; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; TNF-α, tumor necrosis factor-alpha; IL-1β, interleukin-1β; IL-6, interleukin-6; NF-κB, nuclear factor-κB.
EMSA
The results of the EMSA are presented in Table IV. Following treatment with simvastatin or taurine, the NF-κB activity was significantly decreased compared with the SAH group (P<0.01), and the value of NF-κB activity was comparable to that in the control group (P>0.05). These results suggest that simavastin and/or taurine are able to alleviate the SAH via the regulation of NF-κB activity.
Table IV.
Activity of NF-κB as determined by EMSA (IOD).
| Group | NF-κB |
|---|---|
| SAH | 16,782±4,095 |
| SAH + simvastatin | 6,689±1,319a |
| SAH + taurine | 6,796±1,365a |
| Control | 5,927±1,945 |
P<0.01 vs SAH group. NF-κB, nuclear factor-κB; EMSA, electrophoretic mobility shift assay; IOD, integral optical density; SAH, subarachnoid hemorrhage.
Discussion
Vasospasm, in which the blood vessels constrict and restrict blood flow, is a severe complication of SAH and may result in ischemic brain injury (referred to as delayed ischemia) and permanent brain damage due to insufficient oxygen (26). Vasospasm may be fatal if severe. Delayed ischemia is characterized by new neurological symptoms and may be confirmed by transcranial Doppler or cerebral angiography. Among all the patients admitted with SAH, ~33% exhibit delayed ischemia, and 50% of these patients experience permanent neurological damage as a result (26–28). It is possible to screen for the development of vasospasm using a transcranial Doppler every 24–48 h. A blood flow velocity of >120 cm/sec is suggestive of vasospasm (28). The pathogenesis underlying vasospasm is complicated. The initial processes that lead to SAH involve the activation of genes involved in angiogenesis, inflammation and extracellular matrix remodeling. Modern pharmacology has demonstrated that taurine has sedating, anti-inflammatory and antioxidative stress properties (29).
Immune regulation and induction of various inflammatory and growth regulatory genes, including IL-1β, IL-6, TNF-α and granulocyte-macrophage colony-stimulating factor (GM-CSF), require the activation of transcription factors, such as NF-κB, activated transcription factor (ATF-2), c-Fos and cAMP response element-binding protein (CREB) (30). Previous studies have reported that taurine treatment significantly reduces the secretion of the aforementioned proinflammatory cytokines and the expression of the IL-1β, IL-6, TNF-α, GM-CSF, IL-12 and p40 genes (31,32). At concentrations of 2.5, 5 and 10 mg/ml, taurine inhibits the collagen matrix invasion of B16F-10 melanoma cells in a dose-dependent manner. A previous study demonstrated that taurine is able to inhibit matrix metalloproteinase production using zymographic analysis (33). Furthermore, the study revealed that the nuclear translocation of p65, p50, c-Rel subunits of NF-κB and other transcription factors, such as ATF-2, c-Fos and CREB, were inhibited by treatment with taurine (33). TNF-α, IL-1β, IL-6 and NF-κB are dominant factors in the regulation of inflammation, and the cytokines IL-6 and TNF-α are produced by various cells, including adipose tissue (34) and macrophages (35). Overexpression of TNF-α occurs in the adipose tissues of obese animals and humans (36). TNF-α may stimulate plasminogen activator inhibitor (PAI)-1 secretion in human adipose tissue fragments, and circulating plasma IL-6 is associated with PAI-1 plasma levels. Statins have been used in the prevention of vasospasm, on the basis of the hypothesis that the actions of statins may upregulate endothelial nitric oxide synthase (eNOS) (37). Impairment of eNOS, endothelium-dependent relaxation and cerebrovascular autoregulation all occur in vasospastic cerebral arteries following SAH. Statins improve endothelial function and increase the mRNA and protein expression of eNOS, and enzymatic activity three-fold. The results of previous animal models of SAH suggest that statins may ameliorate cerebral vasospasm (38,39). It has been hypothesized that patients chronically treated with statins may exhibit a decreased risk of symptomatic vasospasm after SAH. Rasmussen et al (40) studied a TNF-α-activated human umbilical vein endothelial cell model, in which fluvastatin inhibited the activation and release of the active p50 subunit of NF-κB and inhibited NF-κB activity. Dysregulation of NF-κB, phosphorylated-extracellular signal-regulated kinases and Bax may trigger apoptosis, cell cycle arrest and oxidative stress in vascular endothelium. Taurine reduces ischemic brain damage by suppressing the inflammation associated with poly(ADP-ribose) polymerase (PARP) and NF-κB in a rat model of stroke, and taurine significantly reduced the levels of TNF-α, IL-1β, inducible NOS and intracellular adhesion molecule-1 (25). An inflammatory reaction associated with the PARP and NF-κB-induced expression of inflammatory mediators may be a mechanism underlying the effects of taurine against ischemic stroke. Zelvyte et al (41) reported that pravastatin is able to effectively suppress NF-κB expression in human monocytes. In addition, numerous studies have indicated that statins may inhibit the expression of NF-κB (42,43).
In the current study, simvastatin and taurine were administered to rabbits in an SAH model to evaluate their preventative and therapeutic effects against DCVS. Furthermore, the mechanism by which statins reduce DCVS at the molecular level was investigated in an effort to identify a novel method for the treatment of DCVS. In conclusion, simvastatin and taurine were able to reduce DCVS following SAH in the rabbit model, which may be associated with the anti-inflammatory effect of statins.
Acknowledgements
This study was supported by a grant from the National Natural Science Fund (no. 81271313).
References
- 1.Kerz T, Victor A, Beyer C, Trapp I, Heid F, Reisch R. A case control study of statin and magnesium administration in patients after aneurysmal subarachnoid hemorrhage: Incidence of delayed cerebral ischemia and mortality. Neurol Res. 2008;30:893–897. doi: 10.1179/174313208X338034. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki H, Kanamaru K, Shiba M, Fujimoto M, Kawakita F, Imanaka-Yoshida K, Yoshida T, Taki W. Tenascin-C is a possible mediator between initial brain injury and vasospasm-related and -unrelated delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl. 2015;120:117–121. doi: 10.1007/978-3-319-04981-6_20. [DOI] [PubMed] [Google Scholar]
- 3.da Costa L, Fisher J, Mikulis DJ, Tymianski M, Fierstra J. Early identification of brain tissue at risk for delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl. 2015;120:105–109. doi: 10.1007/978-3-319-04981-6_18. [DOI] [PubMed] [Google Scholar]
- 4.Hirashima Y, Endo S, Kato R, Takaku A. Prevention of cerebrovasospasm following subarachnoid hemorrhage in rabbits by the platelet-activating factor antagonist, E5880. J Neurosurg. 1996;84:826–830. doi: 10.3171/jns.1996.84.5.0826. [DOI] [PubMed] [Google Scholar]
- 5.Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, Ley KF, Kassell NF, Lee KS. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003;53:123–133. doi: 10.1227/01.NEU.0000068863.37133.9E. discussion 133–125. [DOI] [PubMed] [Google Scholar]
- 6.Lin CL, Dumont AS, Calisaneller T, Kwan AL, Hwong SL, Lee KS. Monoclonal antibody against E selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg Neurol. 2005;64:201–205. doi: 10.1016/j.surneu.2005.04.038. discussion 205–206. [DOI] [PubMed] [Google Scholar]
- 7.Kim E, Kim HC, Park SY, Lim YJ, Ro SH, Cho WS, Jeon YT, Hwang JW, Park HP. Effect of red blood cell transfusion on unfavorable neurological outcome and symptomatic vasospasm in patients with cerebral aneurysmal rupture: Old versus fresh blood. World Neurosurg. 2015 Aug 28; doi: 10.1016/j.wneu.2015.08.024. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 8.Nickele C, Muro K, Getch CC, Walker MT, Bernstein RA. Severe reversible cerebral vasoconstriction syndrome mimicking aneurysmal rupture and vasospasm. Neurocrit Care. 2007;7:81–85. doi: 10.1007/s12028-007-0001-4. [DOI] [PubMed] [Google Scholar]
- 9.Salunke P, Patra DP, Mukherjee KK. Delayed cerebral vasospasm and systemic inflammatory response syndrome following intraoperative rupture of cerebral hydatid cyst. Acta Neurochir (Wien) 2014;156:613–614. doi: 10.1007/s00701-013-1675-3. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Zheng T, Altura BT, Altura BM. Antioxidants prevent elevation in [Ca(2+)](i) induced by low extracellular magnesium in cultured canine cerebral vascular smooth muscle cells: Possible relationship to Mg(2+) deficiency-induced vasospasm and stroke. Brain Res Bull. 2000;52:151–154. doi: 10.1016/S0361-9230(00)00260-4. [DOI] [PubMed] [Google Scholar]
- 11.Heffren J, McIntosh AM, Reiter PD. Nimodipine for the prevention of cerebral vasospasm after subarachnoid hemorrhage in 12 children. Pediatr Neurol. 2015;52:356–360. doi: 10.1016/j.pediatrneurol.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Munakata A, Ohkuma H, Shimamura N. Effect of a free radical scavenger, edaravone, on free radical reactions: Related signal transduction and cerebral vasospasm in the rabbit subarachnoid hemorrhage model. Acta Neurochir Suppl. 2011;110:17–22. doi: 10.1007/978-3-7091-0356-2_4. [DOI] [PubMed] [Google Scholar]
- 13.Nishziawa S. Roles of signal transduction mechanisms in cerebral vasospasm following subarachnoid hemorrhage: Overview. Acta Neurochir Suppl. 2011;110:27–30. doi: 10.1007/978-3-7091-0353-1_5. [DOI] [PubMed] [Google Scholar]
- 14.Zubkov AY, Nanda A, Zhang JH. Signal transduction pathways in cerebral vasospasm. Pathophysiology. 2003;9:47–61. doi: 10.1016/S0928-4680(02)00055-X. [DOI] [PubMed] [Google Scholar]
- 15.Jośko J, Hendryk S, Jedrzejowska-Szypułka H, Słowiński J, Gwóźdź B, Lange D, Snietura M, Zwirska-Korczala K, Jochem J. Cerebral angiogenesis after subarachnoid hemorrhage (SAH) and endothelin receptor blockage with BQ-123 antagonist in rats. J Physiol Pharmacol. 2001;52:237–248. [PubMed] [Google Scholar]
- 16.Zhou ML, Zhu L, Wang J, Hang CH, Shi JX. The inflammation in the gut after experimental subarachnoid hemorrhage. J Surg Res. 2007;137:103–108. doi: 10.1016/j.jss.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 17.Sarrafzadeh A, Copin JC, Bengualid DJ, Turck N, Vajkoczy P, Bijlenga P, Schaller K, Gasche Y. Matrix metalloproteinase-9 concentration in the cerebral extracellular fluid of patients during the acute phase of aneurysmal subarachnoid hemorrhage. Neurol Res. 2012;34:455–461. doi: 10.1179/1743132812Y.0000000018. [DOI] [PubMed] [Google Scholar]
- 18.De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20:E83–E88. doi: 10.1161/01.ATV.20.11.e83. [DOI] [PubMed] [Google Scholar]
- 19.Song YS, Lee YS, Chan PH. Oxidative stress transiently decreases the IKK complex (IKKalpha, beta, and gamma), an upstream component of NF-kappaB signaling, after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:1301–1311. doi: 10.1038/sj.jcbfm.9600123. [DOI] [PubMed] [Google Scholar]
- 20.Huang CY, Fujimura M, Noshita N, Chang YY, Chan PH. SOD1 down-regulates NF-kappaB and c-Myc expression in mice after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:163–173. doi: 10.1097/00004647-200102000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Guan Y, Zhao G, Fu XJ, Guo TZ, Liu YT, Ren XL, Wang W, Liu HR, Li YQ. Elevated IL-6 and TNF-α levels in cerebrospinal fluid of subarachnoid hemorrhage patients. Mol Neurobiol. 2015 Jun 11; doi: 10.1007/s12035-015-9268-1. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 22.Young AM, Karri SK, You W, Ogilvy CS. Specific TNF-alpha inhibition in cerebral aneurysm formation and subarachnoid hemorrhage. Curr Drug Saf. 2012;7:190–196. doi: 10.2174/157488612803251315. [DOI] [PubMed] [Google Scholar]
- 23.Hanafy KA, Grobelny B, Fernandez L, Kurtz P, Connolly ES, Mayer SA, Schindler C, Badjatia N. Brain interstitial fluid TNF-alpha after subarachnoid hemorrhage. J Neurol Sci. 2010;291:69–73. doi: 10.1016/j.jns.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinchuk TV, Fedulaev YN, Khairetdinova GA, Denisova NN, Chura OV, Logunova IY. Anti-inflammatory effects of simvastatin in patients with chronic heart failure. Bull Exp Biol Med. 2014;157:552–554. doi: 10.1007/s10517-014-2612-z. [DOI] [PubMed] [Google Scholar]
- 25.Sun M, Zhao Y, Gu Y, Xu C. Anti-inflammatory mechanism of taurine against ischemic stroke is related to down-regulation of PARP and NF-κB. Amino Acids. 2012;42:1735–1747. doi: 10.1007/s00726-011-0885-3. [DOI] [PubMed] [Google Scholar]
- 26.Seiyama A, Yoshikawa N, Imamura Y. Ischemic pretreatment delays ischemic brain vasospasm injury in gerbils. Adv Exp Med Biol. 2014;812:247–252. doi: 10.1007/978-1-4939-0620-8_33. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Zhang B, Li S, Liang C, Xu K, Li S. Whole brain CT perfusion combined with CT angiography in patients with subarachnoid hemorrhage and cerebral vasospasm. Clin Neurol Neurosurg. 2013;115:2496–2501. doi: 10.1016/j.clineuro.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. N Engl J Med. 2006;354:387–396. doi: 10.1056/NEJMra052732. [DOI] [PubMed] [Google Scholar]
- 29.Miyazaki T, Matsuzaki Y. Taurine and liver diseases: A focus on the heterogeneous protective properties of taurine. Amino Acids. 2014;46:101–110. doi: 10.1007/s00726-012-1381-0. [DOI] [PubMed] [Google Scholar]
- 30.Juvela S. Nonsteroidal anti-inflammatory drugs as risk factors for spontaneous intracerebral hemorrhage and aneurysmal subarachnoid hemorrhage. Stroke. 2003;34:e34–e36. doi: 10.1161/01.STR.0000075560.89823.DF. [DOI] [PubMed] [Google Scholar]
- 31.Joo K, Lee Y, Choi D, Han J, Hong S, Kim YM, Jung Y. An anti-inflammatory mechanism of taurine conjugated 5-aminosalicylic acid against experimental colitis: Taurine chloramine potentiates inhibitory effect of 5-aminosalicylic acid on IL-1beta-mediated NFkappaB activation. Eur J Pharmacol. 2009;618:91–97. doi: 10.1016/j.ejphar.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Marcinkiewicz J, Kurnyta M, Biedroń R, Bobek M, Kontny E, Maśliński W. Anti-inflammatory effects of taurine derivatives (taurine chloramine, taurine bromamine, and taurolidine) are mediated by different mechanisms. Adv Exp Med Biol. 2006;583:481–492. doi: 10.1007/978-0-387-33504-9_54. [DOI] [PubMed] [Google Scholar]
- 33.Quinn MR, Barua M, Liu Y, Serban V. Taurine chloramine inhibits production of inflammatory mediators and iNOS gene expression in alveolar macrophages; a tale of two pathways: Part I, NF-kappaB signaling. Adv Exp Med Biol. 2003;526:341–348. doi: 10.1007/978-1-4615-0077-3_42. [DOI] [PubMed] [Google Scholar]
- 34.Buechler C, Ullrich H, Aslanidis C, Bared SM, Lingenhel A, Ritter M, Schmitz G. Lipoprotein (a) downregulates lysosomal acid lipase and induces interleukin-6 in human blood monocytes. Biochim Biophys Acta. 2003;1642:25–31. doi: 10.1016/S0167-4889(03)00083-1. [DOI] [PubMed] [Google Scholar]
- 35.Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc. 2001;60:349–356. doi: 10.1079/PNS2001110. [DOI] [PubMed] [Google Scholar]
- 36.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asahi M, Huang Z, Thomas S, Yoshimura S, Sumii T, Mori T, Qiu J, Amin-Hanjani S, Huang PL, Liao JK, et al. Protective effects of statins involving both eNOS and tPA in focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:722–729. doi: 10.1038/sj.jcbfm.9600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer AH. Statins in the management of aneurysmal subarachnoid hemorrhage: An overview of animal research, observational studies, randomized controlled trials and meta-analyses. Acta Neurochir Suppl. 2011;110:193–201. doi: 10.1007/978-3-7091-0356-2_36. [DOI] [PubMed] [Google Scholar]
- 39.Sabri M, Macdonald RL. Statins: A potential therapeutic addition to treatment for aneurysmal subarachnoid hemorrhage? World Neurosurg. 2010;73:646–653. doi: 10.1016/j.wneu.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen LM, Hansen PR, Nabipour MT, Olesen P, Kristiansen MT, Ledet T. Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells. Biochem J. 2001;360:363–370. doi: 10.1042/0264-6021:3600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zelvyte I, Dominaitiene R, Crisby M, Janciauskiene S. Modulation of inflammatory mediators and PPARgamma and NFkappaB expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol Res. 2002;45:147–154. doi: 10.1006/phrs.2001.0922. [DOI] [PubMed] [Google Scholar]
- 42.Kaji H, Kanatani M, Sugimoto T, Chihara K. Statins modulate the levels of osteoprotegerin/receptor activator of NFkappaB ligand mRNA in mouse bone-cell cultures. Horm Metab Res. 2005;37:589–592. doi: 10.1055/s-2005-870538. [DOI] [PubMed] [Google Scholar]
- 43.Ahn KS, Sethi G, Aggarwal BB. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem Pharmacol. 2008;75:907–913. doi: 10.1016/j.bcp.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]