

Abstract
Mesenchymal stem cells (MSCs) have been considered to have potential as ideal carriers for the delivery of anticancer agents since the capacity for tumor-oriented migration and integration was identified. In contrast to DNA-based vectors, mRNA synthesized in vitro may be readily transfected and is mutagenesis-free. The present study was performed in order to investigate the effects of phosphatase and tensin homolog (PTEN) mRNA-engineered MSCs on human glioma U251 cells under indirect co-culture conditions. PTEN-bearing mRNA was generated by in vitro transcription and was transfected into MSCs. The expression of PTEN in transfected MSCs was detected by immunoblotting, and the migration ability of MSCs following PTEN-bearing mRNA transfection was verified using Transwell co-cultures. The indirect co-culture was used to determine the effects of PTEN-engineered MSCs on the viability of U251 glioma cells by luminescence and fluorescence microscopy. The synthesized PTEN mRNA was expressed in MSCs, and the expression was highest at 24 h subsequent to transfection. An enhanced migration rate was observed in MSCs transfected with PTEN mRNA compared with non-transfected MSCs (P<0.05). A significant inhibition of U251 cells was observed when the cells were cultured with conditioned medium from PTEN mRNA-engineered MSCs (P<0.05). The results suggested that anticancer gene-bearing mRNA synthesized in vitro is capable of being applied to a MSC-mediated anticancer strategy for the treatment of glioblastoma patients.
Keywords: mesenchymal stem cells, synthesized mRNA, phosphatase and tensin homolog, gene therapy, glioma cell
Introduction
Gliomas are recognized as highly malignant and intractable cancers and are the most common type of primary brain tumors throughout the world (1). The complex cellular composition, diffuse invasiveness and capacity to escape conventional therapies have been challenging and have hampered progress towards an effective treatment (2,3). A major obstacle limiting the effectiveness of conventional therapies for gliomas and other types of cancer, is the lack of tumor specificity (4,5). Therefore, exploring the therapeutic strategies that specifically target tumor tissues is critical.
The mesenchymal stem cell (MSC)-mediated strategy has demonstrated considerable potential for the development of clinically meaningful patient-tailored anticancer therapies (6–9). The tumor-directed migration and incorporation of MSCs have been demonstrated in a number of preclinical studies in vitro, using Transwell migration assays, and in vivo, using animal tumor models (10–12). Directed at the tumor microenvironment, engineered MSCs are able to express or release anticancer substances that may constantly act on the adjacent tumor cells (13). As a direct attacker, an anticancer gene that was pre-engineered on MSCs plays a critical role for the action in this system (14). Plasmid DNA transfection and viral vector transduction are widely used in gene therapy-associated studies (15–19). These methodologies have the potential hazards of genomic recombination or insertional mutagenesis, which is a significant problem when this technology is applied to gene therapy (20,21). Therefore, an approach that carries anticancer genes using MSCs, without genetic modification and without risks for clinical applications, may be desired for anticancer medicine. Recently, a novel stabilized mRNA construct has become a more attractive alternative to the most commonly used DNA-based plasmid (pDNA) (22). Synthesized mRNA in vitro has been widely used to generate induced pluripotent stem cells or induce cell reprogramming (23–26).
Phosphatase and tensin homolog (PTEN) functions as the important negative regulator of PI3K-AKT-mTOR pathway in controlling cells apoptosis (27–29). The activity of PTEN is lost by deletions, mutations or promoter methylation at a high frequency in numerous human cancers (30). Therefore, restoring PTEN function in cancer cells may break down the PTEN mutation-dependent cancer cell growth, termed oncogene addiction, and demonstrates considerable promise for cancer therapy. The present study describes an experimental strategy that inhibits malignant glioma U251 cells using a conditioned medium (CM) from PTEN-engineered MSCs through the use of in vitro synthesized PTEN mRNA. The strategy has initiated the application of advantageous PTEN mRNA in the field of cancer research. The novel synthetic modified PTEN mRNA possesses great potential for use as an effective therapeutic candidate for glioblastoma patients.
Materials and methods
Cells and culture conditions
MSCs were isolated from healthy human pancreatic ductal tissue and expanded ex vivo as previously described (31). Briefly, human pancreases were obtained with full informed consent from adult heart-beating cadaver organ donors through the organ procurement program at the British Columbia Transplant Society (Vancouver, Canada). A total of 5 pancreatic ductal tissue samples were collected between July 2011 and November 2011 at Vancouver General Hospital (Vancouver, Canada). The pancreatic ductal tissue taken from the Ricordi® Chamber (Biorep Technologies, Inc., Miami, FL, USA), which was used for islet isolation, was utilized as the starting material for MSC isolation. The human glioma U251 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) to be used as target cells in the present study. U251 cells were maintained as suggested by ATCC, and the culture conditions were kept consistent with those of the MSCs. In order to track the viability in a timely manner, the U251 cells were pre-labeled with luciferase using the pGL4.51[luc2/CMV/Neo] vector (Promega Corporation, Madison, WI, USA), according to the manufacturer's protocol.
In vitro synthesis of PTEN mRNA and MSC transfection
The in vitro transcription template construction and RNA synthesis is schematized in Fig. 1. The oligonucleotide sequences used in the construction of in vitro transcription templates are shown in Table I. The human 5′-UTR with Kozak and 3′-UTR sequences were synthesized commercially by Integrated DNA Technologies (Coralville, IA, USA), sub-cloned into a pcDNA3.3 plasmid and termed the pcDNA 3.3-TOPO TA vector. As previously described (23), plasmid inserts were excised using restriction enzyme digestion and used to template tail polymerase chain reaction (PCR). RNA was synthesized using the Ambion MEGAscript T7 kit (Thermo Fisher Scientific, Waltham, MA, USA). The ribonucleoside blend was composed of anti-reverse cap analogs (New England Biolabs, Inc., Ipswich, MA, USA) and adenosine triphosphate, guanosine triphosphate, 5-methylcytidine triphosphate and pseudouridine triphosphate (TriLink Biotechnologies, San Diego, CA, USA). Reactions were incubated for 5 h at 37°C and followed by DNase treatment, according to the manufacturer's protocol. Then, the reactions were treated with Antarctic Phosphatase (New England Biolabs, Inc.) for 2 h at 37°C in order to remove residual 5′-triphosphates. The synthesized RNA was purified with Ambion MEGAclear spin columns (Thermo Fisher Scientific) and quantified using NanoDrop (Thermo Fisher Scientific). RNA transfection was performed using TransIT-mRNA (Mirus Bio, Madison, WI, USA). RNA was diluted in Invitrogen Opti-MEM basal media (Thermo Fisher Scientific), and the Boost reagent (Mirus Bio LLC, Madison, WI, USA) and TransIT-mRNA were added sequentially according to the manufacturer's protocol. Subsequent to 2 min incubation at room temperature (RT), the RNA-lipid complexes were delivered to culture media in the culture plates. The plates were then returned to the incubator and PTEN expression was analyzed 12, 24 and 36 h later using western blot analysis.
Figure 1.
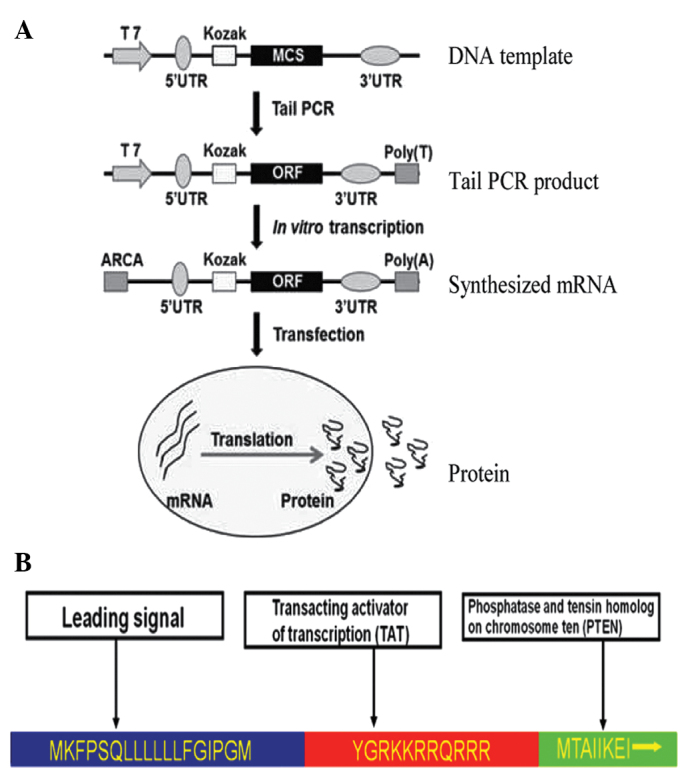
Flow charts of PTEN mRNA synthesis in vitro and the design of the stPTEN fusion protein. (A) Schematic diagram of RNA synthesis in vitro. (B) Design of the stPTEN fusion protein. PTEN, phosphatase and tensin homolog; stPTEN, secretory phosphatase and tensin homolog.
Table I.
Primers for in vitro synthesized PTEN mRNA.
| PTEN mRNA | Forward primer | Reverse primer |
|---|---|---|
| PTEN ORF | F1: ATGAAGTTTCCTTCTCAACTTC | R1: TCAGACTTTTGTAATTTGTGTATG |
| F2: CGCGGATCCGCCACCATGAAGTTTCC | R2: CCGCTCGAGTCAGACTTTTGTAATTT | |
| 5′ Splint | GTACTGCTCCTCGCCGTTCATGGTGGC | |
| TCTTATATTTCTTCTTACTC | ||
| 3′ Splint | CCCGCAGAAGGCAGCTTACTCATCGTG | |
| GTTCCTGCGGCC | ||
| 5′ UTR | TTGGACCCTCGTACAGAAGCTAATACG | |
| ACTCACTATAGGGAAATAAGAGAGAAA | ||
| AGAAGAGTAAGAAGAAATATAAGAGCC | ||
| ACC | ||
| 3′ UTR | GCTGCCTTCTGCGGGGCTTGCCTTCTG | |
| GCCATGCCCTTCTTCTCTCCCTTGCAC | ||
| CTGTACCTCTTGGTCTTTGAATAAAGC | ||
| CTGAGTAGGAAGTGAGGGTCTAGAACT | ||
| AGTGTCGACGC | ||
| Ligation product PCR | TTGGACCCTCGTACAGAAGCTAATACG | GCGTCGACACTAGTTCTAGACCCTCA |
| Tail PCR | TTGGACCCTCGTACAGAAGCTAATACG | T120CTTCCTACTCAGGCTTTATTCAAA |
PTEN, phosphatase and tensin homolog; mRNA, messenger RNA; ORF, open reading frame; UTR, untranslated region; PCR, polymerase chain reaction.
Viability assay of tumor cells in indirect co-culture
The cytotoxic effects of PTEN-engineered MSCs (MSCPTEN) on the proliferation of malignant glioma cells were assessed under indirect co-culture conditions. The luciferase gene transfected U251 cells were plated at a density of 5,000 cells/well in a Falcon 96-well plate (BD Biosciences, Franklin Lakes, NJ, USA) in 100 µl of minimal essential medium (MEM) on day 0. The cells were cultured using CM that was collected from the cultures of native MSCs (CMcontrol) and MSCPTEN (CMPTEN) on day 1. The culture volume was 100 µl with five different ratios of MSCPTEN to CM from MSCcontrol, as follows: 0, 25, 50, 75 and 100%. On days 0, 3 and 6, 1 µl (0.15 mg/ml) D-luciferin (Caliper Life Sciences, Waltham, MA, USA) was added into each well and the cells were incubated in the same culture conditions for an additional 10 min. Luminescence was measured using the IVIS Spectrum System (Caliper Life Sciences). The media were replaced every 36 h and the measurements of every group were repeated three times.
Migration assay of MSCs with Transwell co-culture system
A cell migration assay was performed, as previously described (32). U251 cells were placed on the plastic surface of 24-well plates (1×105 cells/well). On the following day, the wild-type MSCs and MSCPTEN in serum-free media were seeded onto the 8 µm microporous membranes of the Transwell insert (Corning Incorporated, Corning, NY, USA). MSCs were incubated for 48 h at 37°C and in a 5% CO2 atmosphere. The inserts were washed with phosphate buffered saline (PBS) and the upper surface of the membrane was scraped gently using a cotton swab to remove non-migrated cells. Then, the membranes were fixed with 95% ethanol (Sangon Biotech Co., Ltd., Shanghai, China) and stained using 0.1% crystal violet (Sangon Biotech Co., Ltd.). The average number of migrated cells was assessed by counting five randomly selected microscopic fields at x100 magnification. All experiments were repeated at least three times. To detect the migration status of the MSCs with normal cells, U251 glioma cells were replaced with native MSCs on the plastic surface of the 24-well plates.
Immunoblotting. Immunoblotting was used to detect the cellular expression of MSCs
Briefly, the MSCs were washed with PBS three times and collected using cell lysis buffer (Beyotime Institute of Biotechnology, Haimen, China). Cell lysates were incubated on ice for 30 min. The protein concentration was determined using bicinchoninic acid protein assay reagents (Beyotime Institute of Biotechnology), according to the manufacturer's protocol. Equal amounts of protein (50 µg/sample) were loaded on each lane and separated by electrophoresis in 12% sodium dodecyl sulfate polyacrylamide gel and electrotransferred to nitrocellulose membranes (Beyotime Institute of Biotechnology). The membrane was incubated in blocking buffer for 1 h at RT and then incubated overnight at 4°C with monoclonal rabbit anti-mouse PTEN primary antibodies (cat. no. 217702; dilution, 1:1,000; R&D Systems, Inc., Minneapolis, MN, USA). The blots were rinsed with Tris-buffered saline and Tween-20 (EMD Millipore, Billerica, MA, USA) three times, incubated with horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (cat. no. A0216; dilution, 1:1,000; Beyotime Institute of Biotechnology) for 60 min and detected by chemiluminescence using ECL Hyperfilm (EMD Millipore). The CM samples were filtered through a 0.22 µm membrane (EMD Millipore) and equally concentrated using a 10,000 molecular weight cut-off (catalog no., 42406; EMD Millipore). Then, PTEN was detected in the CM.
Fluorescence microscopy
The cell viability was also detected using an Invitrogen LIVE/DEAD Viability/Cytotoxicity assay kit (Thermo Fisher Scientific), according to the manufacturer's protocol, but with a slight modification (10). Briefly, a total of 1×105 U251 cells were plated onto 24-well plates in 500 µl of MEM on day 0. The media were replaced with 50 or 100% CM on day 1. On day 4, the cultures were washed twice with PBS. Freshly prepared working solution containing 1µM calcein acetoxymethyl and 2 µM ethdium homodimer-1 (Thermo Fisher Scientific) was then added directly to the cultures, with 250 µl solution added per well on 24-well plates, and the wells were incubated at RT for 10 min in the dark. The images were captured using a fluorescence microscope (IX71; Olympus Corporation, Tokyo, Japan) and the associated analysis was performed using ImageJ 1.45 k software (National Institute of Health of USA, Bethesda, MD, USA).
Statistical analysis
Numerical data were expressed as the mean ± standard error. Statistical differences between the means for the various groups were evaluated using Prism 4.0 software (GraphPad software, San Diego, CA, USA) using the Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
PTEN-bearing mRNA synthesis and expression in MSCs
Anticancer gene-bearing mRNA was constructed as illustrated in Fig. 1A. In order to make the PTEN product secretory (stPTEN), a leading sequence that corresponded to an 18 amino acid segment (sequence, MKFPSQLLLLLLFGIPGM) was introduced into the DNA template. A transacting activator of transcription (TAT; sequence, YGRKKRRQRRR) was inserted into the PTEN template, to act at the transmembrane (Fig. 1B). The stPTEN DNA template was cloned from previously constructed pDsRed1-TAT-PTEN vectors (Fig. 2Aa) (33) and the PCR product of the stPTEN sequence was then inserted into the pcDNA 3.3-TOPO TA vector. The inserted DNA sequence was confirmed by an analysis of the colony using PCR (Fig. 2Ab), restriction enzyme digestion (Fig. 2Ac) and sequencing (data not shown). The correct plasmid insert was excised by restriction enzyme digestion (using BAmHI and XhoI; Takara Biotechnology Co., Ltd., Dalian, China) and used to template tail PCR (Fig. 2Ad). As shown in Fig. 2B, the expression of PTEN was clearly enhanced in transfected MSCs. The endogenous PTEN was observed in control MSCs with a slightly varied size, the maximum expression of transfected PTEN mRNA was observed at 24 h post transfection, and subsequently decreased at 36 h. The stPTEN was also detectable in the corresponding CMs.
Figure 2.

In vitro synthesis of PTEN mRNA and transfection into MSCs. (A) Analysis of synthesized PTEN mRNA by agarose gel electrophorsis. (a) Analysis of the PTEN sequence PCR product. The PTEN sequence was inserted into the pcDNA 3.3-TOPO TA vector by (b) single colony PCR and (c) restriction enzyme digestion. (d) PTEN tail PCR product. Lanes 1–4 contain different clones. (B) Immunoblotting analysis of PTEN expression in (a) cell lysates and (b) conditioned media. H2O acted as the PCR template. DL100, 100-bp DNA ladder maker; PTEN, phosphatase and tensin homolog; mRNA, messenger RNA; MSCs, mesenchymal stem cells; PCR, polymerase chain reaction; Con, control.
Effect of mRNA transfection on MSC migration
MSC migration was analyzed using the Transwell system. Subsequent to 48 h of co-culture, a considerable number of cells migrated across the microporous membrane towards the U251 cells (Fig. 3A). However, few cells migrated towards the normal MSC control cells (Fig. 3B). Notably, the transfection of PTEN-mRNAs significantly enhanced the migration of MSCs towards the U251 glioma cells compared with control MSCs (Fig. 3C).
Figure 3.

In vitro migratory ability of MSCs. (A) The migratory ability of wild-type MSCs and MSCPTEN toward U251 glioma cells using a Transwell co-culture system. Representative photomicrographs of the stained membrane indicates migrated cells (magnification, x100). (B) The migration of MSCs was tested using wild-type MSCs as control cells (magnification, x100). (C) Summary of three independent experiments was presented as mean ± standard error of the mean. The cell number of each filter membrane was obtained from the average of five randomly selected microscopic fields. *P<0.05 vs. wild-type MSCs. MSCs, mesenchymal stem cells; PTEN, phosphatase and tensin homolog; MSCPTEN, PTEN-engineered MSCs.
Effect of PTEN-engineered MSCs on the viability of U251 glioma cells
The indirect co-culture was used to determine the effects of PTEN-engineered MSCs on the viability of U251 glioma cells by luminescence. As shown in Fig. 4A, the intensity of luminescence in U251 cells decreased with the increased incubation time and CM ratio. All test results are summarized in Fig. 4B. Starting at a low CM ratio of 25%, the cells incubated with CMPTEN revealed significant cell death at day 6 (47.7%; P<0.05). However, significant cell death (P<0.05) started to appear at day 3, at a CM ratio of 100%. The ratio of CMPTEN-induced cell death at day 3 was not statistically different from day 0.
Figure 4.

Assessment of U251 cell viability using bioluminescence determination. (A) Representative measurement of luminescence intensity. Cell culture medium is indicated on the left side of the graph. CM ratios and time points are labeled at the top and bottom of the images respectively. The luminescence intensity of each well was determined by IVIS Spectrum System, 10 min subsequent to adding D-luciferin. Luminescence scale: Color scale: Min = 6.31×106; Max = 1.19×108. (B) Summary of U251 cell viability. U251 cells were co-cultured with (a) CMcontrol and (b) CMPTEN. The relative cell viability is represented as luciferase activity. Data are presented as mean ± standard error of the mean. *P<0.05 vs. control (0%) at the same time point. CM, conditioned media; PTEN, phosphatase and tensin homolog; CMCONTROL, CM collected from the cultures of native MSCs; CMPTEN, CM collected fom the cultures of PTEN-engineered MSCs.
MSCPTEN-mediated U251 cell death in indirect co-cultures
CM-induced U251 cell death was also examined on day 4 by fluorescence microscopy (IX71; Olympus Corporation) subsequent to LIVE/DEAD staining. Two CM ratios, 50 and 100%, were used in this component of the study. As shown in Fig. 5, the cell death was proportionally associated with CMPTEN. The dose-dependent cell death indicates that MSCPTEN-derived PTEN is an important mediator responsible for U251 cell death during indirect co-culture. Marked cell death was not detected in the CM obtained from the native MSCs in the current experimental conditions.
Figure 5.

U251 cell viability in indirect co-cultures. (A) Cells were incubated in various CMs (indicated on the left side of the graph) at various ratios (indicated at the top). LIVE/DEAD staining was performed on day 4 following the initiation of the indirect co-culture (orginal magnification, x400). (B) Summary of cell viability of indirect co-cultures. Data are expressed as the mean ± standard error of the mean for three independent experiments. *P<0.05 vs. control. Brightfield, whole population of cells attached to the culture surface; Live, live cells stained with calcein in green; Dead, dead cells stained with ethdium homodimer-1 in red; Merged, merged images; CM, conditioned media, PTEN, phophatase and tensin homolog; CMCONTROL, CM collected from the cultures of native MSCs; CMPTEN, CM collected fom the cultures of PTEN-engineered MSCs.
Discussion
Malignant glioma is one of the most challenging cancers to treat successfully, mainly due to the particularity of its location and biological characteristics, including the infiltrative nature, resistance to apoptosis, propensity for recurrence and resistance to conventional therapies (3). Since the identification of the tumor-oriented migration capacity of MSCs, the application of specific anticancer gene-engineered MSCs has demonstrated potential for the development of targeted therapy of malignant gliomas (34–36). While MSCs may carry anticancer genes with various viral or plasmid vectors, these methods may not be used in clinical applications (37).
Synthetic mRNA in vitro is a novel technique that presents a safe, efficient strategy for gene therapy (25). The use of synthetic mRNA completely removes the risk of modifying the host genome (38–39), and the safety of synthetic mRNA has been demonstrated in clinical studies (40). Therefore, the synthetic mRNA-based strategy is a simple and non-integrating approach for the application of specific anticancer gene-engineered MSCs.
The loss of PTEN expression in a wide range of cancer cells, including gliomas, reflects the importance of PTEN in the maintenance of cancer cell survival, with >50% of human glioblastoma cases exhibiting loss of expression (41). PTEN function restoration may inhibit cancer cell growth and may induce cell death under certain circumstances, such as loss of PTEN expression (28–30). In consideration of the potential clinical applications, stPTEN mRNA was synthesized in vitro and used in the present MSC-mediated anticancer study. As demonstrated in Fig. 2B, the synthesized PTEN mRNA was expressed in MSCs with the peak activity at 24 h following transfection. The expression is consistent with a previous study, which used a DNA-based vector in the same cell model (33).
In the present study, the tumor cell-oriented migration of MSCs, which were engineered with DNA-based anticancer gene expression vectors, was investigated using the xCELLigence system in pancreatic cancer cells and glioma DBTRG cells (33,42). In order to make the strategy clinically practical, the effects of synthesized mRNA transfection on the migration ability of MSCs was detected. As shown in Fig. 3, the migration of MSCs was not obstructed by PTEN mRNA transfection. Notably, the migration of MSCs towards the U251 cells was significantly increased by transfected PTEN mRNA. The underlying mechanisms require additional studies in the future. The results of the present study suggest that synthesized mRNA may be applied to the MSC-mediated anticancer strategy.
To the best of our knowledge, the present study is the first demonstration that synthetic modified PTEN mRNA engineered MSC-mediated cytotoxic effects on glioma cells. In the present study, the cytotoxic effects of CM from MSCPTEN on U251 cells were examined using a luminescence technique and fluorescence microscopy. Under the current indirect co-culture conditions, U251 cells were sensitive to CMPTEN (Fig. 4). The significant cytotoxicity was observed on day 6 and at a CM ratio of 50%. The LIVE/DEAD assay possessed the advantage of being straightforward, and reflected the intact status of the detected cells at any time point. As shown in Fig. 5, dose-responsible death of U251 glioma cells revealed the anticancer effect of MSCs that were transfected with PTEN-mRNAs. However, the drawback of the LIVE/DEAD assay is that it only applies to the cells that remain on the culture surface during staining. The detached cells, the majority of which are dead cells, are not included in the assessment. Multiple assessments and in vivo experiments are required in additional studies.
In the present study, a safe and efficient strategy of mRNA-based gene transfer was successfully applied in the MSCs-mediated cytotoxicity of tumor cells. The approach of MSC-mediated gene therapy caused a dual-targeted killing effect to the tumor, mainly due to the tropism characteristics of MCSs and engineered anticancer agents. mRNA-based gene transfer may also kill tumor cells locally and consistently. The methodology used in the present study also holds the potential to use the MSCs of the patients and to switch the tumor attackers corresponding to the individual condition. In conclusion, the present study provides an essential foundation for additional in vivo and potentially clinical anticancer studies using synthesized mRNAs.
Acknowledgments
The present study was supported by the Ministry of Science and Technology (grant no. 2013ZX10001004-002-005), the Environmental Protection Department of Hubei Province (grant no. 2013HB13), the Science and Technology Department of Hubei Province (grant no. 2014CFA068) and the Public Health and Family Planning Department of Hubei Province (grant no. WJ2015MB223).
References
- 1.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westphal M, Lamszus K. The neurobiology of gliomas: From cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12:495–508. doi: 10.1038/nrn3060. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson SD. Malignant gliomas: Diagnosis and treatment. Dis Mon. 2011;57:558–569. doi: 10.1016/j.disamonth.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Sun XY, Nong J, Qin K, Warnock GL, Dai LJ. Mesenchymal stem cell-mediated cancer therapy: A dual-targeted strategy of personalized medicine. World J Stem Cells. 2011;3:96–103. doi: 10.4252/wjsc.v3.i11.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabatabai G, Wick W, Weller M. Stem cell-mediated gene therapies for malignant gliomas: A promising targeted therapeutic approach? Discov Med. 2011;11:529–536. [PubMed] [Google Scholar]
- 6.Loebinger MR, Janes SM. Stem cells as vectors for antitumour therapy. Thorax. 2010;65:362–369. doi: 10.1136/thx.2009.128025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat J, Warnock GL. Potential implications of mesenchymal stem cells in cancer therapy. Cancer Lett. 2011;305:8–20. doi: 10.1016/j.canlet.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Zhang L, Xu W, Qian H, Ye S, Zhu W, Cao H, Yan Y, Li W, Wang M, et al. Experimental therapy for lung cancer: Umbilical cord-derived mesenchymal stem cell-mediated interleukin-24 delivery. Curr Cancer Drug Targets. 2013;13:92–102. doi: 10.2174/156800913804486665. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Zhang L, Xu W, Qian H, Ye S, Zhu W, Cao H, Yan Y, Li W, Wang M, et al. Experimental therapy for lung cancer: umbilical cord-derived mesenchymal stem cell-mediated interleukin-24 delivery. Curr Cancer Drug Targets. 2013;13:92–102. doi: 10.2174/156800913804486665. [DOI] [PubMed] [Google Scholar]
- 10.Sun XY, Nong J, Qin K, Lu H, Moniri MR, Dai LJ, Warnock GL. MSC(TRAIL)-mediated HepG2 cell death in direct and indirect co-cultures. Anticancer Res. 2011;31:3705–3712. [PubMed] [Google Scholar]
- 11.Rodríguez R, García-Castro J, Trigueros C, Arranz García M, Menéndez P. Multipotent mesenchymal stromal cells: clinical applications and cancer modeling. Adv Exp Med Biol. 2012;741:187–205. doi: 10.1007/978-1-4614-2098-9_13. [DOI] [PubMed] [Google Scholar]
- 12.Fritz V, Jorgensen C. Mesenchymal stem cells: An emerging tool for cancer targeting and therapy. Curr Stem Cell Res Ther. 2008;3:32–42. doi: 10.2174/157488808783489462. [DOI] [PubMed] [Google Scholar]
- 13.Yagi H, Soto-Gutierrez A, Parekkadan B, Kitagawa Y, Tompkins RG, Kobayashi N, Yarmush ML. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–679. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amara I, Touati W, Beaune P, de Waziers I. Mesenchymal stem cells as cellular vehicles for prodrug gene therapy against tumors. Biochimie. 2014;105:4–11. doi: 10.1016/j.biochi.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Ren C, Kumar S, Chanda D, Chen J, Mountz JD, Ponnazhagan S. Therapeutic potential of mesenchymal stem cells producing interferon-alpha in a mouse melanoma lung metastasis model. Stem Cells. 2008;26:2332–2338. doi: 10.1634/stemcells.2008-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–3608. [PubMed] [Google Scholar]
- 17.Li X, Lu Y, Huang W, Xu H, Chen X, Geng Q, Fan H, Tan Y, Xue G, Jiang X. In vitro effect of adenovirus-mediated human Gamma Interferon gene transfer into human mesenchymal stem cells for chronic myelogenous leukemia. Hematol Oncol. 2006;24:151–158. doi: 10.1002/hon.779. [DOI] [PubMed] [Google Scholar]
- 18.Tang XJ, Lu JT, Tu HJ, Huang KM, Fu R, Cao G, Huang M, Cheng LH, Dai LJ, Zhang L. TRAIL-engineered bone marrow-derived mesenchymal stem cells: TRAIL expression and cytotoxic effects on C6 glioma cells. Anticancer Res. 2014;34:729–734. [PubMed] [Google Scholar]
- 19.Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G, Figueiredo JL, Martuza RL, Weissleder R, Shah K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci USA. 2009;106:4822–4827. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenecker J, Huth S, Rudolph C. Gene therapy for cystic fibrosis lung disease: Current status and future perspectives. Curr Opin Mol Ther. 2006;8:439–445. [PubMed] [Google Scholar]
- 21.Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonhardt C, Schwake G, Stögbauer TR, Rappl S, Kuhr JT, Ligon TS, Rädler JO. Single-cell mRNA transfection studies: Delivery, kinetics and statistics by numbers. Nanomedicine (Lond) 2014;10:679–688. doi: 10.1016/j.nano.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Wang XL, Hu P, Guo XR, Yan D, Yuan Y, Yan SR, Li DS. Reprogramming human umbilical cord mesenchymal stromal cells to islet-like cells with the use of in vitro-synthesized pancreatic-duodenal homebox 1 messenger RNA. Cytotherapy. 2014;16:1519–1527. doi: 10.1016/j.jcyt.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 24.Guo XR, Wang XL, Li MC, Yuan YH, Chen Y, Zou DD, Bian LJ, Li DS. PDX-1 mRNA-induced reprogramming of mouse pancreas-derived mesenchymal stem cells into insulin-producing cells in vitro. Clin Exp Med. 2014;10:152–160. doi: 10.1007/s10238-014-0319-0. [DOI] [PubMed] [Google Scholar]
- 25.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, Später D, Xu H, Tabebordbar M, Gorbatov R, et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31:898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Zhou Y, Reske SN, Shen C. PTEN mutation: Many birds with one stone in tumorigenesis. Anticancer Res. 2008;28:3613–3619. [PubMed] [Google Scholar]
- 28.Ciuffreda L, Falcone I, Incani UC, Del Curatolo A, Conciatori F, Matteoni S, Vari S, Vaccaro V, Cognetti F, Milella M. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv Biol Regul. 2014;56:66–80. doi: 10.1016/j.jbior.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Muniyan S, Ingersoll MA, Batra SK, Lin MF. Cellular prostatic acid phosphatase, a PTEN-functional homologue in prostate epithelia, functions as a prostate-specific tumor suppressor. Biochim Biophys Acta. 2014;1846:88–98. doi: 10.1016/j.bbcan.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moniri MR, Sun XY, Rayat J, Dai D, Ao Z, He Z, Verchere CB, Dai LJ, Warnock GL. TRAIL-engineered pancreas-derived mesenchymal stem cells: Characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012;19:652–658. doi: 10.1038/cgt.2012.46. [DOI] [PubMed] [Google Scholar]
- 32.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 33.Yang ZS, Tang XJ, Guo XR, Zou DD, Sun XY, Feng JB, Luo J, Dai LJ, Warnock GL. Cancer cell-oriented migration of mesenchymal stem cells engineered with an anticancer gene (PTEN): An imaging demonstration. Onco Targets Ther. 2014;7:441–446. doi: 10.2147/OTT.S59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009;27:2320–2330. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- 35.Dwyer RM, Khan S, Barry FP, O'Brien T, Kerin MJ. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoop K, Kolokythas M, Klutz K, Willhauck MJ, Wunderlich N, Draganovici D, Zach C, Gildehaus FJ, Böning G, Göke B, et al. Image-guided, tumor stroma-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated NIS gene delivery. Mol Ther. 2011;19:1704–1713. doi: 10.1038/mt.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Quintanilla JI, Bhere D, Heidari P, He D, Mahmood U, Shah K. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells. 2013;31:1706–1714. doi: 10.1002/stem.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, Sancho-Martinez I, Belmonte Izpisua JC. Cell fate conversion by mRNA. Stem Cell Res Ther. 2011;2:5. doi: 10.1186/scrt46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 40.Van Nuffel AM, Corthals J, Neyns B, Heirman C, Thielemans K, Bonehill A. Immunotherapy of cancer with dendritic cells loaded with tumor antigens and activated through mRNA electroporation. Methods Mol Biol. 2010;629:405–452. doi: 10.1007/978-1-60761-657-3_27. [DOI] [PubMed] [Google Scholar]
- 41.Leslie NR, Downes CP. PTEN function: How normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moniri Roshan M, Young A, Reinheimer K, Rayat J, Dai LJ, Warnock GL. Dynamic assessment of cell viability, proliferation and migration using real time cell analyzer system (RTCA) Cytotechnology. 2015;67:379–386. doi: 10.1007/s10616-014-9692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]