

Abstract
IBD has been associated with differential abundance of numerous organisms when compared to healthy controls, however few studies have investigated variability in the microbiome across intestinal locations, and how this variability might be related to disease location and phenotype.
In this study we have analyzed the microbiome of a large cohort of individuals recruited at Mount Sinai Hospital in Toronto, Canada. Biopsies were taken from subjects with Crohn's disease (CD) ulcerative colitis (UC), as well as healthy controls (HC), and individuals having undergone ileal pouch anal anastomosis (IPAA) for treatment of UC or familial adenomatous polyposis (FAP). Microbial 16S rRNA was sequenced using the Illumina MiSeq platform.
We observed a great deal of variability in the microbiome characterizing different sampling locations. Samples from pouch and afferent limb were comparable in microbial composition. When comparing sigmoid and TI samples, more differences were observed. The greatest number of differentially abundant microbes was observed when comparing either pouch or afferent limb samples to sigmoid or TI. Despite these differences, we were able to observe modest microbial variability between IBD phenotypes and healthy controls, even when controlling for sampling location and additional experimental factors. The majority of detected associations were observed between healthy controls and CD, with decreases in specific genera in the families Ruminococcaceae and Lachnospiraceae characterizing tissue samples from individuals with CD. This study highlights important considerations when analyzing the composition of the microbiome, and also provides useful insight into differences in the microbiome characterizing these seemingly related phenotypes.
Introduction
There is currently significant research focus on investigating the role of the human-associated microbiome in health and disease. Several previous studies have suggested that ‘dysbioses’ are associated with the pathogenesis of complex diseases including diabetes, obesity and inflammatory bowel disease (IBD)(1-5), yet have struggled to demonstrate causal associations. Location-specific microbiome heterogeneity has also been described across the gastrointestinal tract in small, healthy cohorts(6, 7), with location-specific patterns generally believed to arise from physiological variability in the different gastrointestinal environments(7). In the case of IBD, intestinal microenvironment heterogeneity leading to local patterns of microbial variation may be important in modulating site-specific inflammation and phenotypic outcomes. Stool and tissue-based studies have done little to address questions of how temporal and location-specific microbial patterns may favor the onset of disease in one location over another and often do not account for the strong effect of inflammation on the structure of the microbiome. Furthermore, variable microbial communities across sampling sites could impact the results and interpretation of microbiome-focused studies which investigate disease phenotypes using heterogeneous sample collections.
In order to adequately determine how disease risk may be modulated by location-specific microbial factors, human models of IBD are especially useful. Inflammation of the pouch following ileal pouch-anal anastomosis (IPAA) in patients with ulcerative colitis (UC) is relatively common(8, 9), and remains poorly understood. Similar to IBD, pouch inflammation is thought to result from a combination of genetic and microbial factors which together contribute to a dysregulated immune response and subsequent inflammation. Indeed, our group has illustrated that IBD-associated genetic polymorphisms are associated with chronic pouch inflammatory complications, including the development of a Crohn's disease (CD)-like phenotype(10). As has been observed in IBD, alterations in the tissue-associated microbiome have been found in ileal-pouch inflammation(11, 12). Both diseases are characterized by an overall decrease in organism diversity, however, the specific organisms associated with each of these phenotypes are not identical1, 12-14. Furthermore, the phenotypic differences between pouch inflammatory complications and IBD, most notably the responsiveness to antibiotic therapy among a majority of individuals with pouchitis, suggest that there are differences between the microbiota in these groups, which may have a role in pathogenesis. Given that pouches are constructed from ileal tissue, among patients with a disease phenotype which is not typically characterized by ileal inflammation (UC), it is interesting to speculate that changes in the tissue-associated microbiome resulting from fecal stasis generated in the pouch, may be responsible for de novo ileal inflammation. We have made use of a large cohort of patients, including healthy individuals and those with evidence of either acute or long term chronic inflammatory phenotypes, all on minimal medical therapy, to address these hypotheses by comparing and contrasting the tissue-associated microbiome of colonic and ileal tissue with that of the ileal pouch.
Methods
Patient recruitment and sample collection
Patients were recruited during regularly scheduled endoscopic follow-up, at Mount Sinai Hospital (MSH) in Toronto, Canada, in accordance with approval granted by the hospital's Research Ethics Board. Patients with confirmed UC or familial adenomatous polyposis (FAP) who had undergone IPAA a minimum of one year prior to enrollment were included in the study. In addition, individuals with confirmed UC, CD, or healthy controls (HC), with no previous surgery and who were not being treated with immunomodulators, steroids or anti-TNF therapy at the time of endoscopy, were also recruited. In all patients medication use was recorded. Biopsies were taken using standard forceps, from within the pouch itself and 5-10 cm into the afferent limb (AL) in patients with pouches, or 20cm from the anal verge (sigmoid) and 5-10cm into the terminal ileum (one from each site). These samples were immediately placed into sterile, empty freezer vials and snap frozen in liquid nitrogen. Additional biopsies taken from the same locations were sent to the MSH pathology lab for histological scoring. During the endoscopy, physicians documented the appearance of the biopsy sites using previously described criteria for inflammation(12, 13). In samples obtained from patients with a pouch this included assessing for the presence of erythema, friability, ulceration and histological evidence of polymorphonuclear leukocyte infiltration, and ulceration or erosions(12, 14, 15). Samples in which a combined score of greater than three among these characteristics were considered inflamed(12, 13). Patients with CD were endoscopically assessed by the physician performing their procedure for the presence of inflammation at the biopsy site.
All subjects were classified into outcome groups based on a combination of long-term disease characteristics and inflammatory activity at the time of the study procedure. Those without a diagnosis of IBD were classified as HC and individuals with FAP were classified using this designation. Within the non-surgical group, individuals were classified as CD or UC based on medical chart review, with phenotypes described as per the Montreal classification(16). Within the pouch, phenotypic outcome groups were defined as previously described(12, 13), with individuals who had no evidence of inflammation classified as “no pouchitis” (NP); individuals with inflammation of the pouch classified as either “acute” (AP) or “chronic pouchitis” (CP) based on both the number and length of episodes, and response to treatment; and individuals with documented evidence of inflammation of the afferent limb or proximal small bowel, or who developed a stricture or fistula at least one year following ileostomy closure classified into the “CD-like” group (CDL).
Microbial DNA Extraction and Analysis
Biopsies were processed using identical protocols at two separate time points, as a subgroup of the pouch and AL samples were included in a previous study(12). Total DNA was extracted using the QIAGEN DNeasy Blood and Tissue kit (QIAGEN, California, USA) with an additional bead beating step performed using the FastPrep system (MP Biomedicals, Solon, USA). Samples underwent 16S sequencing of the V4 hypervariable region of microbial DNA using the Illumina MiSeq platform (Illumina, San Diego, USA). Sequences were processed using the pick-otu pipeline in Qiime, with the usearch clustering algorithm(17). This algorithm includes both de novo and reference-based chimera searching. OTUs were generated based on 97% sequence identity and assigned to taxonomic outcome groups using the Greengenes taxonomy (4feb2011). OTUs not corresponding to bacterial taxonomy were excluded. Samples with fewer than 3000 reads were resequenced (from 16S libraries), and if remaining below this threshold, excluded from further analysis.
Principal coordinates analysis was conducted on the raw OTU table, with beta-diversity estimated using both the Spearman rank and Bray-Curtis-Faith(18) distances. 2D plots were generated for the first three components, with samples labeled according to location. Rarefaction was performed with a minimum sampling depth of 3000 OTUs per sample, and alpha diversity estimated using the Shannon and Chao1diversity and richness indices at this level of sampling. (19, 20) Community coverage was calculated with Goods coverage(21). Intra-individual analyses examining the differences between sampling sites within the same individual (sigmoid vs TI, and Pouch vs AL) were compared using the Wilcoxon signed rank test and an exact McNemar's paired test for relative abundance and dichotomized presence-absence data respectively, in order to assess differences in microbiome composition within individuals at different sites.
To determine which organisms were significantly associated with outcome and sampling location, filtering and Trimmed Mean of M-values (TMM) normalization on raw OTU count data using EdgeR(22) was performed in R (v 3.1.0). EdgeR is a computational tool designed for de novo sequencing studies, which offers an alternative approach to both rarefaction and the use of relative abundance calculations, with the advantage of not requiring data reduction to a common level and subsequent data loss(23). Additionally, normalized values are not bounded by 0 or 1, as is the case in relative abundance calculations. OTUs with assigned taxonomy to the genus level were combined into common genera. We filtered genera from our analysis which were not present at an abundance of one count per million in at least 30 of our samples. Following TMM normalization, count data was analyzed in the pouch/PPI vs Sigmoid/TI comparisons using both exact methods and a negative binomial generalized linear model. In each case, comparisons were performed between the locations of interest in all samples, as well as in samples obtained from exclusively non inflamed sites, and from individuals who had not been on antibiotics in the previous month. Additional comparisons were made in the sub-cohorts of individuals who were “healthy” (FAP, HC, NP), and among those with “ulcerative colitis type” disease (UC, AP, CP) or “Crohn's type” disease (CD, CDL). In the negative binomial analysis, these described groups were also used, along with sequencing flowcell as covariates in the analysis. Extraction batch also showed evidence of an association with community diversity (data not shown), however it was not included as a separate covariate in the analysis in order to retain power. False discovery rate (FDR) corrected p-values below 0.05 were considered significant.
Comparisons between phenotypes within sites was also performed using the above method. Samples taken from the sigmoid of patients with UC and CD, with inactive disease or minimal inflammation were compared to HC samples. The majority of terminal ileum (TI) samples were from inflamed tissue, and the sample numbers were too small to perform analyses between the different phenotypes in this group of samples.
Verification of the results obtained from this analysis method was performed using a more traditional approach, comparing the relative abundance between outcome groups, using non-parametric and exact statistics. In brief, comparisons between pouch and sigmoid, pouch and TI, afferent limb and sigmoid, and afferent limb and TI were conducted using Fisher's exact test and the non-parametric Kruskal-Wallis tests of significance. LEfSe (v. 1.1.0)(24) was used to explore whether additional organisms at different phylogenetic levels were also associated with biopsy location. Additional attention was paid to genus-level organisms which had been previously associated with IBD or pouchitis through either our own work, or that of others(12, 25, 26). The composition of the microbiome was then compared at the genus-level between disease subgroups (CD and UC). Analyses were carried out separately in the sigmoid and TI, with the primary outcome measure of interest being CD and UC compared to HC. Further analyses were conducted in which medication use and inflammation were controlled for using linear modeling. Finally, in order to investigate the relationship between sampling location and microbiome content, we also assessed the correlation between length of time since ileostomy closure and genera relative abundance using Spearman correlation.
Results
Cohort demographic and phenotypic characteristics
627 samples from 384 individuals were collected as part of this study, with 609 samples meeting our criteria for inclusion in the final analysis. Demographic and clinical data are described in table 1, with the number of samples collected from each location and antibiotic use demonstrated in table 2. Figure 1 illustrates the percent of samples in each of these groups in which inflammation was present. Within the non-surgical group, disease extent was classified via the Montreal classification. The majority of patients in the CD group had been diagnosed with ileocolonic disease (L3), and the majority in the UC group had extensive disease extending beyond the splenic flexure (E3), although not all had active inflammation at the time of endoscopy.
Table 1.
Demographic and clinical characteristics of study cohort.
| Total Cohort (n=384) | No Pouch (n=135) | Pouch (n=249) | |
|---|---|---|---|
| Gender (% Female) | 193 (50.2) | 67 (49.6) | 126 (50.6) |
| Mean age (SD) | 45.7 (13.7) | 42.0 (15.1) | 47.8 (12.5) |
| Smoking* (%) | 42 (10.9) | 14 (10.4) | 28 (11.2) |
| Medication use at endoscopy | 123 (32.0) | 51 (37.8) | 70 (28.1) |
| Antibioticsα | 77 (20.2) | 9 (6.7) | 68 (27.3) |
| Mesalamineα | 53 (13.8) | 50 (37.0) | 3 (1.2) |
subject reports regular smoking (≥1 cigarette per day) for 1 month prior to sample collection
subject reports medication use within the month prior to sample collection and recruitment
Table 2.
Number of samples analyzed for each site and phenotype.
| HC | CD | UC | FAP | |
|---|---|---|---|---|
| Pouch | - | - | 184 (27.7) | 30 (10.0) |
| Afferent Limb | - | - | 172 (27.3) | 27 (7.4) |
| Ileum | 28 (0) | 17 (11.8) | 25 (16.0) | - |
| Sigmoid | 44 (0) | 40 (17.1) | 41 (9.7) | 1 (0) |
In brackets is the percent of patients in each group reporting antibiotic use in the month preceding enrollment.
Figure 1.
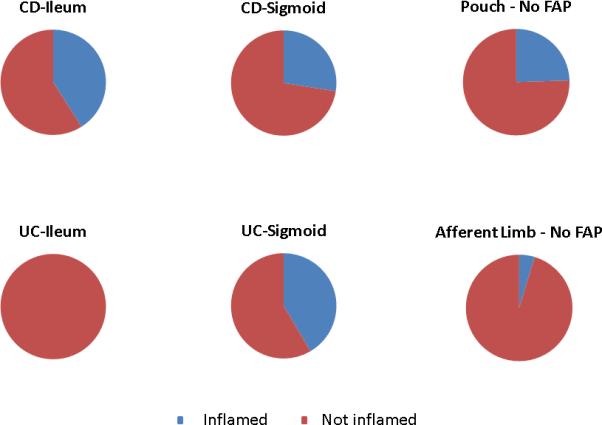
Pie charts illustrating the proportion of samples per site and inflammatory state which were classified as inflamed.
Measures of diversity in various sampling locations
We obtained a high degree of community coverage across our samples, with rarefaction curves illustrating consistency across samples (supplementary figure 1), and Goods coverage index above 0.95 in all, and above 0.97 in the vast majority of samples. Diversity varied across sampling locations, with samples taken from the pouch and AL demonstrating less diversity than those taken from either the TI or sigmoid (kruskal wallis p<0.05)(supplementary figure 2). In keeping with this, beta diversity plots appear to show separation between pouch and non-surgical samples (both sigmoid and TI), suggesting that there may be large scale community structure differences between microbial communities in surgical and non-surgical anatomic locations (supplementary figure 3). No significant differences in diversity were observed when comparing sites within the same individual (ie comparing sigmoid to TI, and pouch to afferent limb in each patient).
Site-specific structure of the microbiome
2963 OTUs were generated, which were classified to the genus level, belonging to 296 unique genera. The genera which were included in the analysis corresponded to four main phyla: the Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria (Figure 2). Bacteroidetes made up a significantly larger proportion of the microbiome (p<0.001) in sigmoid samples compared to pouch and AL, and in TI compared to pouch (p=0.02). In contrast, there was an increase in the Proteobacteria in both the pouch and AL compared to the sigmoid and TI (p<0.001) and a more modest increased abundance of Fusobacteria (p<0.001) and Firmicutes (p<0.01) at these sites compared to sigmoid samples, although no difference between the proportion of Firmicutes from the TI and pouch. Such changes are broad and difficult to characterize however, so the remainder of our analyses were performed at the genus level.
Figure 2.
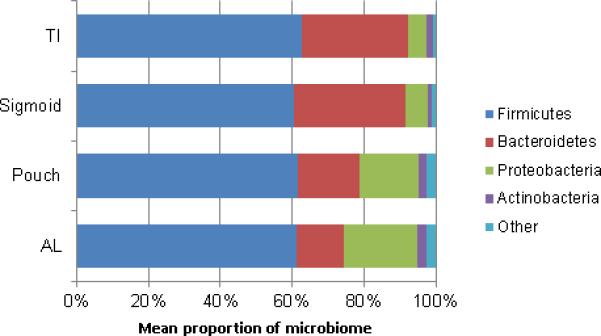
Dominant phyla and their relative abundance in specific sampling locations
We evaluated differences in microbial community structure between healthy sigmoid and TI, and pouch and AL within the same individuals. As we have previously reported(12), no significant differences were observed between microbial samples taken from the pouch and afferent limb at the genus level. In contrast with our observations of no significant differences between sigmoid and TI in terms of diversity, when we compare the relative abundance of individual genera, we were able to identify several significant differences in relative abundance between anatomical locations at different sites within the same individual. These associations occur even in the absence of inflammatory activity at either site (Figure 3).
Figure 3.
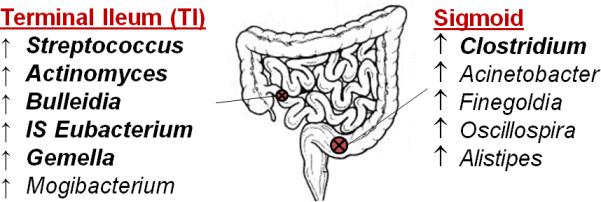
Organisms which were detected at differential abundance in the sigmoid compared to terminal ileum at a nominal significance threshold of p<0.01. Genera demonstrating an increased abundance at a specific sampling site (compared to the other) are listed. Organisms which are in bold remained significant after FDR correction for multiple testing. IS = incertae sedis
When we examined differences in the composition of the microbiome of samples taken from pouch, AL, sigmoid and TI, it was observed that the abundance of several organisms were significantly associated with sampling location, and depending on the genera in question, were detected at either higher or lower abundance in the pouch compared to either the sigmoid or TI. In healthy tissue (no evidence of inflammation at the time of endoscopy, and no antibiotic use in the previous month), 37 genera were differentially abundant in the pouch compared to the sigmoid, 46 when comparing the sigmoid and afferent limb, and 20 in the AL compared to TI (supplementary table 1). Described associations were observed in both the exact and negative binomial analyses with batch effect correction. Several of the genera which were found to be significantly associated with sampling location are described in figure 4 and 5.
Figure 4a-c.
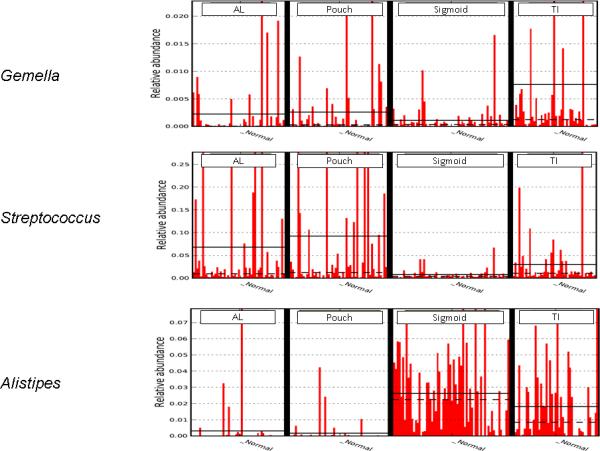
LEfSe image showing the abundance of Gemella, Streptococcus, and Alistipes across sampling sites. These organisms showed different abundance patterns across sites, some more closely resembling TI, others Sigmoid, some which were different from both. Solid line represents the mean, dashed line the median.
Figure 5.
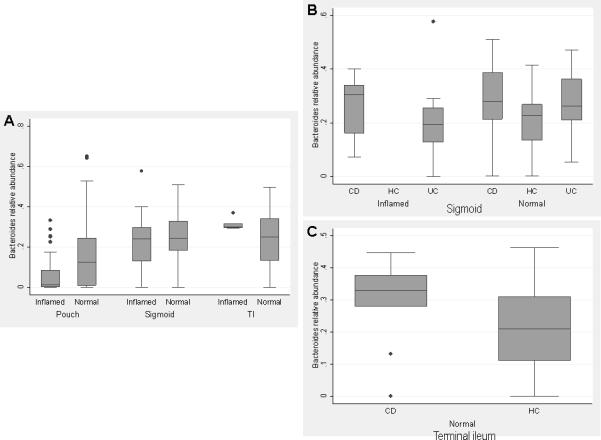
Comparison between relative abundance of Bacteroides in different inflammatory phenotypes detected at different sampling sites A) Comparison between surgical and non-surgical sites. AL samples not included as only four samples from this site were inflamed. B) Breakdown comparing IBD subtypes among inflamed and normal sigmoid samples. As would be expected, no healthy controls had inflammation at this site. C) Comparison between CD and HC in the terminal ileum. Significant differences are marked with a *.
Several patterns could be observed in the comparisons of organism abundance between sample sites (Figure 4). Several genera were detected in the pouch and afferent limb with relative abundance levels typical of either the sigmoid or TI, while others were detected at levels which were intermediate between these sites. Most genera measured, however, were detected at lower abundance in pouch and AL samples compared to the sigmoid (ie. Oscillospira, Alistipes). Somewhat surprisingly, for many organisms, this observation was also true when these sites were compared to the TI. On the other hand, Flavobacterium was detected at much higher abundance in the TI compared to both pouch and AL samples, yet while trending in a similar direction, was not significantly different in abundance compared to the sigmoid. Conversely, several organisms were detected at higher abundance in the pouch/AL compared to either of the non-surgical sites (ie. Escherichia, Streptococcus). Among organisms demonstrating association with biopsy location, several had been previously associated with IBD or pouch inflammatory outcomes (Table 3).
Table 3.
A selection of organisms previously associated with non-surgical IBD or pouchitis, and detected at significantly different relative abundance between sampling locations in our cohort.
| Genus | Direction of previous IBD association | Mean Relative Abundance | FDR Corrected p-value* | |||
|---|---|---|---|---|---|---|
| Sigmoid | TI | Pouch | AL | |||
| Faecalibacterium(25, 26) | ↓ CD | 0.10 | 0.12 | 0.03 | 0.02 | 0.04 |
| Ruminococcus (Ruminococcaceae)(25, 26) | ↓ CD | 0.004 | 0.003 | 0.002 | 0.002 | 0.09 |
| Ruminococcus (Lachnospiraceae)(25, 26) | ↑ CD | 0.06 | 0.06 | 0.02 | 0.02 | 0.002 |
| Streptococcus(42) | ↓ CD | 0.005 | 0.02 | 0.05 | 0.05 | <10−14 |
| Escherichia(25, 26) | ↑ CD/UC | 0.02 | 0.02 | 0.11 | 0.14 | <10−4 |
| Dorea(12) | ↓ pouchitis | 0.006 | 0.007 | 0.002 | 0.002 | <0.01 |
All organisms included in the table are those in which there was a highly significant difference in abundance in at least one of the pairwise comparisons, in the cohort without inflammation and on no antibiotics in the previous month.
Displayed p-values are those obtained from negative binomial EdgeR analysis.
When we explored whether changes in the composition of the microbiome in different locations was impacted by disease status, we evaluated location-specific microbiome differences in specific phenotypes, including CD (CD no-surgery vs CDL in the pouch), UC (UC no surgery vs pouchitis in the pouch), and overall presence of inflammation, regardless of specific IBD phenotype (inflammation of pouch/AL vs inflammation of sigmoid/TI). In each of these scenarios, we found that similar location-specific microbiome variability could be observed to that seen in the whole cohort, although due to the loss of power from including fewer samples in the analysis, several organisms did not remain significant at our stringent threshold (supplementary table 1).
Association between length of time since pouch surgery and bacterial relative abundance In general, in the cohort of pouch patients in whom no inflammation was detected at, or before study endoscopy, as well as for only the FAP pouch cohort, genera relative abundance correlated poorly with time since surgery (days). Spearman correlation coefficients (ρ) for each organism were between 0 and |0.2|. Organisms with ρ of greater magnitude than 0.15 are illustrated in supplementary figure 4. The majority of the organisms which were detected at highly divergent abundance in pouch and non-pouch samples, were among those illustrating poor correlation with time since ileostomy closure. Several, Clostridium and Escherichia for example, which were detected at lower abundance in the sigmoid and TI compared to the pouch, trended towards decreased abundance over time in ileal pouches, suggesting that high levels of these organisms in a subset of patients might be a ‘new pouch specific’ phenomenon or a result of the surgical procedure.
Association between microbial relative abundance and disease phenotype
Our previous work has documented the importance of the microbiota in post-IPAA inflammatory disease in the majority of the pouch cohort included in this analysis(12). As such we focused our phenotypic analysis on the non-surgical CD and UC cohort. Associations between disease phenotype and the microbiome could be observed when sampling location was controlled for (samples from a single location analyzed together) although these were fewer than differences between surgical and non-surgical sample sites. In sigmoid samples among individuals with no inflammation or antibiotic use, when compared to HC, five genera were associated with CD, and two with UC (Figure 6). Several additional organisms were associated with IBD in the whole cohort. The majority of organisms associated with CD demonstrated a decreased abundance in disease. Fewer organisms, such as Veillonella appeared increased among individuals with this phenotype. No organisms were capable of distinguishing between CD and UC.
Figure 6.
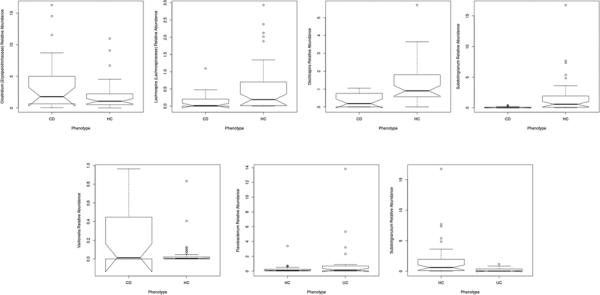
Box plots illustrating relative abundance of genera significantly associated with IBD phenotype in sigmoid samples (pFDR < 0.05)
Few organisms were significantly associated with disease phenotypes in both IPAA and non-surgical inflammatory phenotypes. For example, Dorea, which was previously documented at significantly decreased levels in pouch inflammation(12), was also decreased in CD compared to HC. However, following multivariate analysis in which samples from individuals on antibiotics and with inflammation were excluded, this association did not remain significant. Faecalibacterium abundance was not significantly associated with CD or UC, although trended towards decreased abundance in CD compared to HC.
Discussion
The large sample size of our cohort made it possible to observe common alterations in microbial profile which transcended individual-specific differences at a single time point, and ensured that we had sufficient power to observe microbiome differences in sampling locations and disease phenotypes despite the use of stringent analytical controls. Furthermore, the depth of phenotypic information available on our cohort, allowed us to conduct a variety of subgroup analyses to more fully elucidate the effect of phenotype on the microbiome in the absence of confounding features. These findings demonstrate that despite considerable differences in the composition of the tissue-associated microbiota at different sampling locations, and based on variable degrees of inflammation and surgical history, phenotype-specific changes in the microbiome can be observed. Such observations add strength to hypotheses implicating microbes in disease pathogenesis.
One of the strongest associations observed in our analysis, was between microbial genera and specific sample locations, with large differences detected between samples taken from individuals following surgery and those having not undergone pouch construction. Several associations at different taxonomic ranks were observed from the phylum to the genus level. Notably, Bacteroidetes comprised a significantly lower proportion of the tissue associated microbiome in pouches, while Proteobacteria and Fusobacteria were more abundant. At the genus level, a large number of taxa within these phyla were detected at differential abundance in pouches compared to ileal and colonic tissue. The majority of these associations demonstrated a decreased abundance of typically ubiquitous organisms in the pouch and AL compared to ileal and sigmoid tissue. Escherichia and Streptococcus were exceptions to this pattern and were detected at higher abundance in pouch tissue. Previous work has demonstrated that Escherichia and other Enterobacteriaceae thrive in conditions of inflammation, due in part to host-derived byproducts of nitric oxide synthase(27). This might suggest that despite a relatively healthy appearing pouch among individuals with no history of pouchitis or FAP, sub-clinical inflammation of the submucosa may be present to a greater degree than that seen in ileal or colonic tissue, exerting a profound effect on the structure of the microbiome. Alternatively, it is possible that past antibiotic use either following pouch construction or at the time of surgery, may influence the composition of the pouch microbiome with long term repercussions. The moderate levels of intrinsic antibiotic resistance characterizing both Escherichia and Streptococcus thus lead to the dominance of these groups in pouch tissue(28-30). Despite similarities in disease phenotype between UC and inflammatory complications of the pouch, our observations of different microbial profiles in these locations may provide important information regarding disease pathogenesis.
Although the number of significantly different organisms was highest when comparing samples from pouches versus both ileum and sigmoid, there were also alterations in the composition of the microbiome between sigmoid and ileal tissue. These observations in conjunction with previous work demonstrating an increase in microbial diversity from the stomach through to the anus(7), highlight the importance of selection of appropriate anatomical locations for studies examining the microbiome in the context of mucosal biopsies.
Perhaps surprisingly, we observed that organisms which have been previously implicated in pouchitis or non-surgical IBD(12, 26, 31-33) were detected at differential levels between pouch and non-surgical samples. Several organisms previously suggested to be protective against IBD, including Faecalibacterium, were also decreased in the ileal pouch(31). Our previous results documenting reduced diversity and reduced abundance of specific organisms with anti-inflammatory potential in the inflamed ileal pouch (reduced further from that seen in healthy pouches), suggests that incremental decreases in these organisms' abundance may not influence pouch inflammatory processes, rather severe reductions of these organisms accompany pouch inflammation. It is therefore possible that the high incidence of pouch inflammatory complications following IPAA, are a result of a reduction in the already inherently low abundance of beneficial organisms which characterize the pouch microbiome in general.
Several studies have demonstrated that a period of adaptation occurs within the pouch as it undergoes colonic metaplasia(34-37). In order to ensure that results of decreased prevalence of organisms in pouch tissue were not due to this phenomenon, we explored whether alterations in the pouch-associated microbiome were related to the amount of time which had lapsed since surgery. We saw little evidence to support the hypothesis that microbial changes were associated with the duration of time since surgery. However, samples were not collected longitudinally, and were specifically restricted to individuals who had undergone pouch surgery at least one year prior to recruitment. It is possible that the majority of adaptations of the pouch microbiome occur during the first year, mirroring the period when the greatest degree of colonic metaplasia is occurring(34, 38). Furthermore, the question of whether colonic adaptation of ileal tissue leads to changes in the microbiome, or whether alterations in the tissue associated microbiome result from the pelvic pouch procedure lead to changes in ileal tissue, remains unanswered. To further explore this, additional prospective studies are required. However, it would seem that pouch duration is not a confounding factor in our analysis.
Given the large number of organisms detected at differential abundance in varied sample locations, it was reassuring to note that alterations in the microbiome characterizing disease phenotypes could be detected. These associations were observed in a minimally medicated cohort and were not predicated on the presence of active inflammation at the site of biopsy. The most dramatic associations between specific genera and phenotype, could be observed when comparing individuals with CD to those who were healthy, with fewer differences detected between UC and controls. Alterations in community structure in IBD have been reported previously by other groups(26, 31, 32, 39), however few have controlled for all the experimental conditions included in our analysis. We suggest that the organisms which are robustly associated with outcome in our analysis, including genera in the families Ruminococcaceae and Lachnospiraceae, would be valuable targets for future study. Additional organisms showing more modest associations, or which have been reported by other groups but failed to remain significant following multivariate analyses in our cohort, should be evaluated for significance in larger cohorts with application of similarly stringent criteria.
The question of whether an ileal pouch can be considered a model for de novo IBD has been discussed on numerous occasions, and our group has demonstrated that there are indeed similarities, specifically genetic and serological, between pouch inflammation and non-surgical IBD(40, 41). Yet, the question of whether the pouch is a useful model of de novo CD of the ileum, or recurrence of colonic UC has not been well elucidated. On the one hand, the occurrence of inflammation in tissue of ileal origin comprising the pouch would seem to suggest processes similar to CD are involved. Yet colonic metaplasia which is often undergone by ileal tissue following pouch construction(34, 38), could over time lead to recreation of more ‘colonic’ conditions necessary for the development of UC. Commensurate with these hypotheses, our observations demonstrate that the formation of the pouch results in drastic changes to the composition of the microbiome, with the final composition resembling neither ileal nor colonic tissue, but rather a novel hybrid of the two. Thus, the ileal origin of tissue prior to surgery may be influential in defining the composition of the pouch microbiome, with the unique physiology and gene expression characteristics of this region of bowel impacting the architecture of microbial communities occupying this niche. However, niche-specific growth parameters characterizing the pouch such as fecal stasis, lead to alterations in the structure of the microbial community which are more in keeping with its colonic functionality. This would seem to suggest that the pouch may be a useful model for either colonic or ileal inflammation, with the important caveat that the described microbiome differences must be considered when interpreting results.
Perhaps most importantly, this study highlights the importance of experimental controls and appropriate study design and statistical methodologies for analyzing microbiome data in complex diseases, especially those involving the intestinal tract. Our data illustrating microbiome differences between sampling sites, even among healthy individuals emphasizes the importance of obtaining tissue biopsies from a common location in order to minimize sampling effects. While many studies to date have explored the differences in the tissue microbiome between inflamed and uninflamed tissue, it is important to consider that some of the observed differences in these studies might be the result of the variable sampling locations used, especially when multiple individuals in case or control groups have samples taken from locations differing from the other group. Taken together, we have demonstrated that the composition of the intestinal microbiome is dependent on the sampling location and the disease phenotype of the individual under study. Further questions relating to how these differences may interact with the host to modify either gene expression or intestinal permeability remain to be answered.
Supplementary Material
{kind=link}
Acknowledgments
Sources of Funding
This study is funded by a grant from Crohn's and Colitis Canada and The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK-062423). MSS is supported by the Gale and Graham Wright Chair in Digestive Diseases at Mount Sinai Hospital and by grants from the Crohn's and Colitis Foundation of Canada (CCFC) and The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK-062423).
Footnotes
Conflicts of Interest
The authors have no conflicts (financial, professional, or personal) to disclose.
References
- 1.Manichanh C, Rigottier-Gois L, Bonnaud E, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu X, Ma C, Han L, et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol. 2010;61:69–78. doi: 10.1007/s00284-010-9582-9. [DOI] [PubMed] [Google Scholar]
- 3.Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stearns JC, Lynch MD, Senadheera DB, et al. Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170. doi: 10.1038/srep00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murrell ZA, Melmed GY, Ippoliti A, et al. A prospective evaluation of the long-term outcome of ileal pouch-anal anastomosis in patients with inflammatory bowel disease-unclassified and indeterminate colitis. Dis Colon Rectum. 2009;52:872–878. doi: 10.1007/DCR.0b013e31819f5d4c. [DOI] [PubMed] [Google Scholar]
- 9.Tyler AD, Milgrom R, Xu W, et al. Antimicrobial antibodies are associated with a Crohn's disease-like phenotype after ileal pouch-anal anastomosis. Clin gastro and hepatol. 2012;10:507–512. e501. doi: 10.1016/j.cgh.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Tyler AD, Milgrom R, Stempak JM, et al. The NOD2insC polymorphism is associated with worse outcome following ileal pouch-anal anastomosis for ulcerative colitis. Gut. 2012 doi: 10.1136/gutjnl-2011-301957. [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin SD, Walker AW, Churcher C, et al. The bacteriology of pouchitis: a molecular phylogenetic analysis using 16S rRNA gene cloning and sequencing. Ann Surg. 2011;252:90–98. doi: 10.1097/SLA.0b013e3181e3dc8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyler AD, Knox N, Kabakchiev B, et al. Characterization of the gut-associated microbiome in inflammatory pouch complications following ileal pouch-anal anastomosis. PLoS ONE. 2013;8:e66934. doi: 10.1371/journal.pone.0066934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabakchiev B, Tyler AD, Stempak JM, et al. Down regulation of expression of xenobiotic efflux genes is associated with pelvic pouch inflammation in ulcerative colitis. Inflamm Bowel Dis. 2014;20:1157–1164. doi: 10.1097/MIB.0000000000000078. [DOI] [PubMed] [Google Scholar]
- 14.Sandborn WJ, Tremaine WJ, Batts KP, et al. Pouchitis after ileal pouch-anal anastomosis: a Pouchitis Disease Activity Index. Mayo Clin Proc. 1994;69:409–415. doi: 10.1016/s0025-6196(12)61634-6. [DOI] [PubMed] [Google Scholar]
- 15.Heuschen UA, Allemeyer EH, Hinz U, et al. Diagnosing pouchitis: comparative validation of two scoring systems in routine follow-up. Dis Colon Rectum. 2002;45:776–786. doi: 10.1007/s10350-004-6297-7. discussion 786-778. [DOI] [PubMed] [Google Scholar]
- 16.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19(Suppl A):5–36. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 17.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faith DP. Conservation evaluation and phylogenetic diversity. Biological Conservation. 1992;61:1–10. [Google Scholar]
- 19.Shannon CE. The mathematical theory of communication. 1963. MD Comput. 1997;14:306–317. [PubMed] [Google Scholar]
- 20.Chao AS, T-J. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environmental and Ecological Statistics. 2003;10:429–443. [Google Scholar]
- 21.Esty W. The Efficiency of Good's Nonparametric Coverage Estimator. The Annals of Statistics. 1986;14:1257–1260. [Google Scholar]
- 22.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joossens M, Huys G, Cnockaert M, et al. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 26.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter SE, Winter MG, Xavier MN, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorlozano A, Jimenez-Pacheco A, de Dios Luna Del Castillo J, et al. Evolution of the resistance to antibiotics of bacteria involved in urinary tract infections: A 7-year surveillance study. American journal of infection control. 2014;42:1033–1038. doi: 10.1016/j.ajic.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Hodges TL, Zighelboim-Daum S, Eliopoulos GM, et al. Antimicrobial susceptibility changes in Enterococcus faecalis following various penicillin exposure regimens. Antimicrob Agents Chemother. 1992;36:121–125. doi: 10.1128/aac.36.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan SS, Schreckenberger PC, Zheng X, et al. An intrinsic pattern of reduced susceptibility to fluoroquinolones in pediatric isolates of Streptococcus pyogenes. Diagnostic microbiology and infectious disease. 2008;62:205–209. doi: 10.1016/j.diagmicrobio.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang S, Denman SE, Morrison M, et al. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 32.Lepage P, Hasler R, Spehlmann ME, et al. Twin Study Indicates Loss of Interaction Between Microbiota and Mucosa of Patients With Ulcerative Colitis. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 33.Willing B, Halfvarson J, Dicksved J, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 34.de Silva HJ, Millard PR, Kettlewell M, et al. Mucosal characteristics of pelvic ileal pouches. Gut. 1991;32:61–65. doi: 10.1136/gut.32.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donati OF, Weishaupt D, Weber A, et al. Colonic transformation of ileal pouch-anal anastomosis and of the distal ileum: MRI findings. The British journal of radiology. 2010;83:e185–187. doi: 10.1259/bjr/72125476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shepherd NA. The pelvic ileal reservoir: pathology and pouchitis. Neth J Med. 1990;37(Suppl 1):S57–64. [PubMed] [Google Scholar]
- 37.Shepherd NA, Healey CJ, Warren BF, et al. Distribution of mucosal pathology and an assessment of colonic phenotypic change in the pelvic ileal reservoir. Gut. 1993;34:101–105. doi: 10.1136/gut.34.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawaguchi AL, Dunn JC, Saing MS, et al. Functional and morphologic changes of the ileal mucosa after ileoanal pouch procedure. J Am Coll Surg. 2000;190:310–314. doi: 10.1016/s1072-7515(99)00276-8. [DOI] [PubMed] [Google Scholar]
- 39.Willing BP, Dicksved J, Halfvarson J, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. e1841. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 40.Tyler AD, Milgrom R, Stempak JM, et al. The NOD2insC polymorphism is associated with worse outcome following ileal pouch-anal anastomosis for ulcerative colitis. Gut. 2013;62:1433–1439. doi: 10.1136/gutjnl-2011-301957. [DOI] [PubMed] [Google Scholar]
- 41.Tyler AD, Milgrom R, Xu W, et al. Antimicrobial antibodies are associated with a Crohn's disease-like phenotype after ileal pouch-anal anastomosis. Clin Gastroenterol Hepatol. 2012;10:507–512. e501. doi: 10.1016/j.cgh.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Li Q, Wang C, Tang C, et al. Molecular-phylogenetic characterization of the microbiota in ulcerated and non-ulcerated regions in the patients with Crohn's disease. PLoS ONE. 2012;7:e34939. doi: 10.1371/journal.pone.0034939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.