

Abstract
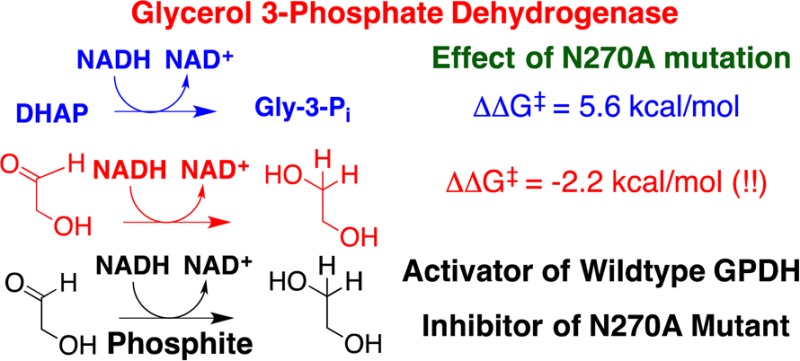
The side chains of R269 and N270 interact with the phosphodianion of dihydroxyacetone phosphate (DHAP) bound to glycerol 3-phosphate dehydrogenase (GPDH). The R269A, N270A, and R269A/N270A mutations of GPDH result in 9.1, 5.6, and 11.5 kcal/mol destabilization, respectively, of the transition state for GPDH-catalyzed reduction of DHAP by the reduced form of nicotinamide adenine dinucleotide. The N270A mutation results in a 7.7 kcal/mol decrease in the intrinsic phosphodianion binding energy, which is larger than the 5.6 kcal/mol effect of the mutation on the stability of the transition state for reduction of DHAP; a 2.2 kcal/mol stabilization of the transition state for unactivated hydride transfer to the truncated substrate glycolaldehyde (GA); and a change in the effect of phosphite dianion on GPDH-catalyzed reduction of GA, from strongly activating to inhibiting. The N270A mutation breaks the network of hydrogen bonding side chains, Asn270, Thr264, Asn205, Lys204, Asp260, and Lys120, which connect the dianion activation and catalytic sites of GPDH. We propose that this disruption dramatically alters the performance of GPDH at these sites.
Orotidine monophosphate decarboxylase (OMPDC),1 triosephosphate isomerase (TIM),2 and glycerol 3-phosphate dehydrogenase (GPDH)3 achieve a high specificity for expression of 6–8 kcal/mol of dianion binding energy at the transition states for the enzyme-catalyzed decarboxylation, proton transfer, and hydride transfer reactions, respectively (Figure 1).4−16 Previous studies of dianion activation of OMPDC and TIM provide support for the proposal that their active sites consist of functionally independent catalytic and dianion activation loci.4,5,7−9,12 Less is known about the mechanism for dianion activation of hydride transfer catalyzed by GPDH.17
Figure 1.
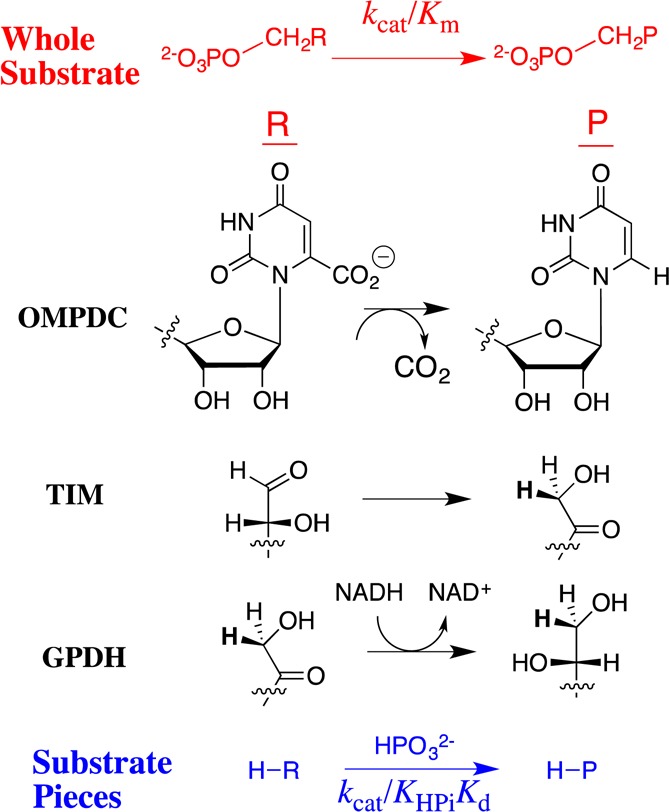
Reactions of whole substrates RCH2OPO32– (kcat/Km) and substrate pieces RH + HPO32– (kcat/KHPiKd) catalyzed by OMPDC, TIM, and GPDH. In each case, 6–8 kcal/mol of the dianion binding energy is used to activate the enzyme for catalysis of the respective reactions of whole and truncated substrates.
The hydrogen bond between the amide side chain of N270 and the phosphodianion of substrate dihydroxyacetone phosphate (DHAP) is shown in Figure 2.18 We predicted that the elimination of this hydrogen bond by the N270A mutation would result in small and similar decreases in the activation barriers (ΔG⧧) for GPDH-catalyzed reactions of whole substrate DHAP, and the substrate pieces glycolaldehyde (GA) and phosphite dianion, as observed in a study of the effect of a Q215A mutation of a similar dianion-gripper residue at OMPDC.12,19 We report, instead, that the N270A mutation causes large and complex changes in the activation barriers for the GPDH-catalyzed reaction of the whole substrate DHAP and the substrate pieces glycolaldehyde (GA) and phosphite dianion. The results suggest that this amide side chain is a linchpin in an assembly of side chains (Figure 2) that connect the catalytic and dianion activation sites and is essential to the optimal functioning of these sites.
Figure 2.
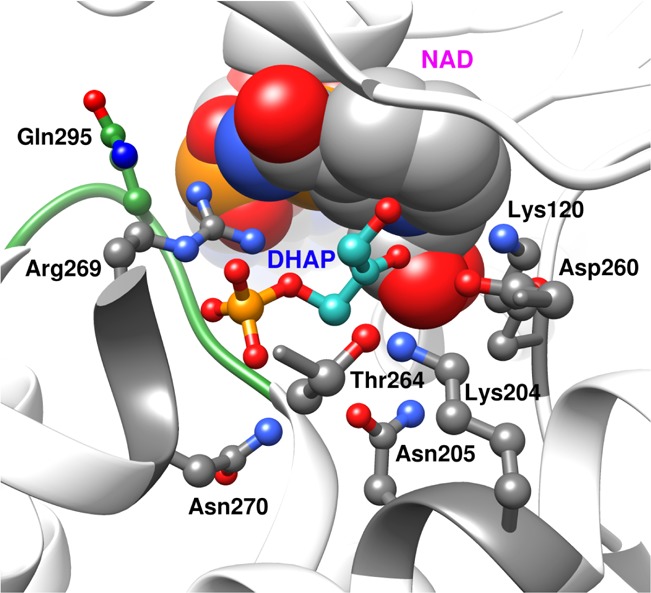
Representation of the X-ray crystal structure of the nonproductive ternary complex of hlGPDH, DHAP, and NAD+ (Protein Data Bank entry 1WPQ), which shows (a) loop residues 292–297 (green) that fold over DHAP and (b) Arg269 and Asn270 that interact with the substrate phosphodianion. (c) Network of hydrogen-bonded side chains (Asn270, Thr264, Asn205, Lys204, Asp260, and Lys120) that connect the catalytic and dianion activation sites.
The N270A and R269A/N270A mutants of GPDH from human liver (hlGPDH) were prepared by standard methods, described in the Supporting Information, and their activity was assayed at 25 °C and pH 7.5 (triethanolamine buffer) by monitoring the reduction of DHAP by the reduced form of nicotinamide adenine dinucleotide (NADH).15 The Michaelis–Menten plots of initial velocity data, v/[E], against [DHAP] for N270A (Figure S1A) and R269A/N270A (Figure S1B) mutant hlGPDH-catalyzed reactions, conducted at 100 or 200 μM NADH, show excellent fits to single sets of kinetic parameters kcat and Km (Table 1), so that Km ≪ 100 μM for NADH.15,17 There is no significant rescue of the activity of the N270A mutant by 60 mM formamide.20
Table 1. Kinetic Parameters for Wild-Type and Mutant hlGPDH-Catalyzed Reactions of Whole Substrate DHAP and Truncated Substrate Glycolaldehyde at pH 7.5 (triethanolamine buffer) and an Ionic Strength of 0.12 (NaCl)a.
| enzyme | kcat (s–1)b | Km (M)b | kcat/Km (M–1 s–1)b | (kcat/Km)E (M–1 s–1)c |
|---|---|---|---|---|
| WT,d 10.8 kcal/mole | 240 ± 10 | (5.2 ± 0.3) × 10–5 | (4.6 ± 0.3) × 106 | (5.0 ± 0.6) × 10–2 |
| R269A,d 9.1 kcal/molf | (5.9 ± 0.4) × 10–3 | (5.7 ± 0.5) × 10–3 | 1.0 ± 0.1 | ≤0.003g |
| N270A, 3.1 kcal/mole 5.6 kcal/molf | 9.0 ± 0.5 | (2.5 ± 0.2) × 10–2 | 360 ± 35 | 2.0 ± 0.2 |
| R269A/N270A, 11.5 kcal/molf | (2.8 ± 0.1) × 10–4 | (1.5 ± 0.1) × 10–2 | (1.7 ± 0.1) × 10–2 | ≤0.003g |
The uncertainty in the kinetic parameters is the standard error from least-squares fits of the kinetic data.
Kinetic parameter for hlGPDH-catalyzed reactions at saturating [NADH], calculated for the carbonyl form of DHAP.15
Calculated for the carbonyl form of GA at saturating [NADH].15
From ref (17).
The intrinsic dianion binding energy, calculated from the ratio of kcat/Km for hlGPDH-catalyzed reduction of DHAP and GA.
Effect of the mutation on the activation barrier ΔG⧧ for wild-type hlGPDH (Scheme 1), determined as described in the text.
Estimated upper limit for (kcat/Km)E, calculated as described in the Supporting Information.
A comparison of the values for kcat/Km (M–1 s–1) for the wild-type and mutant hlGPDH-catalyzed reactions shows that the N270A and R269A/N270A mutations result in 5.6 and 11.5 kcal/mol destabilization, respectively, of the transition states for hlGPDH-catalyzed reduction of DHAP by NADH. The latter is slightly larger than the 10.8 kcal/mol intrinsic phosphodianion binding energy for wild-type hlGPDH, calculated from the ratio of second-order rate constants kcat/Km for hlGPDH-catalyzed reduction of the whole and phosphodianion-truncated substrates.17
The effect of R269A and N270A mutations of hlGPDH on the stability of the transition state for enzyme-catalyzed reduction of DHAP depends upon their order (Scheme 1). The 5.6 kcal/mol effect of the N270A mutation of wild-type hlGPDH is 3.2 kcal/mol larger than the effect of the N270A mutation at the R269A mutant. The 9.1 kcal/mol effect of the R269A mutation is, likewise, larger than the 5.9 kcal/mol effect of this mutation at the N270A mutant. These results do not reflect the elimination of a 3.2 kcal/mol direct stabilizing interaction between the side chains of R269 and N270,21 because the side chains are well-separated (Figure 2). We propose that the N270A mutation of wild-type hlGPDH results in the loss of a 2.4 kcal/mol interaction between the amide side chain and the phosphodianion, plus an additional ∼3.2 kcal/mol weakening of the 9.1 kcal/mol interaction between the phosphodianion and the side chain of R269 (Scheme 1). The remaining 5.9 kcal/mol interaction of this side chain cation is then eliminated by the R269A mutation of the N270A mutant. By comparison, the N270A mutation at R269A hlGPDH results in the loss of the intrinsic ∼2 kcal/mol side chain–dianion interaction.
Scheme 1.
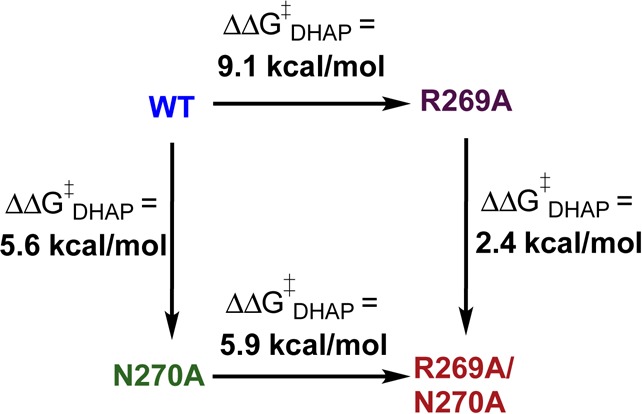
The following observations show that the N270A mutation of hlGPDH results in a dramatic structural reorganization at the active site of hlGPDH.
(1) The N270A mutation results in a 7.7 kcal/mol decrease in the 10.8 kcal/mol intrinsic phosphodianion binding energy for wild-type hlGPDG,5 to 3.1 kcal/mol (Table 1). This 7.7 kcal/mol effect is 2.1 kcal/mol larger than the 5.6 kcal/mol effect of the mutation on the stability of the transition state for reduction of DHAP. We conclude that the N270A mutation results in a reorganization of the active site side chains of hlGPDH, which alters the binding conformation for DHAP. The result is that the large 7.7 kcal/mol reduction in transition state stabilization from phosphodianion binding interactions is offset by a 2.1 kcal/mol transition state stabilization from new interactions with the catalyst.
(2) The N270A mutation cripples hlGPDH-catalyzed reduction of DHAP but results in a 40-fold increase in (kcat/Km)E for hlGPDH-catalyzed reduction of the truncated substrate GA by NADH (Table 1). This corresponds to a 2.2 kcal/mol stabilization of the transition state for hydride transfer to the truncated substrate, in contrast to the 5.6 kcal/mol destabilization of the transition state for reduction of DHAP (Scheme 1). We suggest that similar enzyme–ligand interactions account for this stabilization of the transition state for reduction of GA, and for reduction of DHAP (see above).
(3) The N270A mutation results in a change in the effect of phosphite dianion on (kcat/Km)obs for hlGPDH-catalyzed reduction of GA, from strongly activating15 to inhibiting (Figure 3). Figure 3 (inset) shows the nonlinear least-squares fit of data at 0–1.0 mM HPO32– to eq 1, derived for Scheme 2, obtained using a (kcat/Km)E of 2.0 M–1 s–1 (Table 1), a (kcat/Km)E·HPi of 1.3 M–1 s–1, and a Kd of 60 μM. By comparison, a (kcat/Km)E of 0.05 M–1 s–1 (Table 1), a (kcat/Km)E·HPi of 1100 M–1 s–1, and a Kd of 70 mM were reported for wild-type hlGPDH.5 The further decrease in (kcat/Km)obs as [HPO32–] is increased to 30 mM cannot be fit to a kinetic scheme that shows a second phosphite dianion binding site. This decrease may be partly or entirely due to a specific phosphite dianion salt effect on (kcat/Km)obs.
Figure 3.

Effect of increasing [HPO32–] on the observed second-order rate constant for N270A hlGPDH-catalyzed reduction of GA at 25 °C and pH 7.5 (10 mM triethanolamine buffer). The data from the inset show a good fit to eq 1 derived for Scheme 2.
Scheme 2.

(4) The linear 50% decrease and increase in the values of (kcat/Km)obs for wild-type hlGPDH-catalyzed reduction of 10 μM DHAP and the N270A mutant-catalyzed reduction of 2 mM DHAP by 0.2 mM NADH, respectively, was observed as [HPO32–] is increased from 0 to 30 mM (Figure S2). The former decrease is consistent with weak competitive inhibition of the wild-type hlGPDH-catalyzed reaction by phosphite dianion, but we are uncertain of the explanation for the apparent weak phosphite dianion activation of the reaction catalyzed by N270A mutant hlGPDH.
These results contrast sharply with observations from mutagenesis studies of dianion-gripper side chains at OMPDC and TIM, whose catalyzed reactions are also activated by phosphite dianion.12,14,19,22,23 The following earlier results are consistent with only small effects of these mutations on protein structure.
(1) The effect of individual amino acid substitutions, determined for consecutive Q215A, Y217F, and R235A mutations of gripper side chains of OMPDC, is nearly independent of the order of the mutations.12,23 By contrast, the effect of R269A and N270A mutations depends strongly upon their order (Scheme 1).
(2) The Q215A, Y217F, and R235A mutations of OMPDC do not cause significant changes in the second-order rate constant [(kcat/Km)E] for OMPDC-catalyzed reduction of the phosphodianion-truncated substrate.12,23 By contrast, the N270A mutation results in a 40-fold increase in (kcat/Km)EhlGPDH-catalyzed reduction of GA by NADH.
(3) The Q215A, Y217F, and R235A mutations each result in the same increase in activation barrier ΔG⧧ for the catalyzed reactions of whole substrate OMP, and the substrate pieces (Figure 1).12 This supports the conclusion that the entire effect of each mutation results from the elimination of single side chain interactions with enzyme-bound phosphite dianion or the substrate phosphodianion. By contrast, we propose that the N270A mutation results in a dramatic structural reorganization at the active site of hlGPDH that extensively alters ligand–side chain interactions.
(4) The side chain cations of K12, R235, and R269 from TIM,24 OMPDC,25 and hlGPDH,18 respectively, form ion pairs to the substrate phosphodianion. The K12G, R235A, and R269A mutations result in 7.8, 5.8, and 9.1 kcal/mol destabilization of the respective enzymatic transition states.17,22,26 The binding of alkyl ammonium cations (K12G)27 or guanidine cation (R235A and R269A)17,22 at the site of the excised side chain results in a large rescue of enzymatic activity. The previous results were cited as evidence of a similar catalytic architecture for TIM, OMPDC, and GPDH that optimizes stabilizing ion pairing interactions. The results reported in this paper show that this similarity in architecture for GPDH does not extend beyond the cationic side chain of R269.
To summarize, the restructuring of the active site of N270A mutant hlGPDH results in (a) a 7.7 kcal/mol reduction in the intrinsic phosphodianion binding energy, (b) a 2.2 kcal/mol stabilization of the transition state for reduction of GA, and (c) the elimination of the specificity for expression of dianion binding at the transition state for GPDH-catalyzed reduction of GA.5,15 These effects are consistent with the alteration of interactions between the site for catalysis of hydride transfer and for dianion activation. We note that the N270A mutation disrupts the following network of hydrogen-bonded side chains, Asn270, Thr264, Asn205, Lys204, Asp260, and Lys120 (Figure 2 and Figure S3), which forms a bridge between the Asn270 side chain, which interacts with the substrate phosphodianion, and Lys120, which interacts with the carbonyl of DHAP. The distances between the interacting side chains are listed in Table S1 of the Supporting Information. We propose that this bridge plays a key role in enabling the differential expression of dianion interactions at the Michaelis complex and the transition state for GPDH-catalyzed hydride transfer.
These results demonstrate a previously unrecognized plasticity in the active site of GPDH, which is reflected by the opposing effects of the N270A mutation on enzyme activity toward reduction of whole substrate DHAP and truncated substrate glycolaldehyde. We emphasize, on the one hand, that the nature of this plasticity is not understood while, on the other, such unexpected and dramatic results are often the driving force for new discoveries.
Finally, we note that wild-type GPDH shows a large intrinsic dianion binding energy, but a low activity for catalysis of reduction of GA,5,15 while the N270A mutation shows a dramatic reduction in the intrinsic dianion binding energy but an increase in the level of direct stabilization of the transition state for reduction of GA. Consequently, wild-type hlGPDH compared with N270A hlGPDH shows optimal transition state stabilization from dianion interactions, but this is obtained only at the expense of protein–ligand interactions, which stabilize the transition state for reduction of GA catalyzed by the N270A mutant. This emphasizes the imperative for effective catalysis, through specificity in the expression of intrinsic dianion interactions at the transition state for GPDH-catalyzed hydride transfer.16,28
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00116.
Procedures for the preparation of mutant hlGPDHs, kinetic protocols, Michaelis–Menten plots for N270A and R269A/N270A mutant enzyme-catalyzed reactions, the dependence of the velocity of wild-type and N270A mutant enzyme-catalyzed reactions on [HPO32–], a representation of the enzyme active site, and a table that lists the distances between interacting side chains (PDF)
We acknowledge the National Institutes of Health (Grants GM39754 and GM116921) for generous support.
The authors declare no competing financial interest.
Supplementary Material
References
- Radzicka A.; Wolfenden R. (1995) Science 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Richard J. P. (2012) Biochemistry 51, 2652–2661. 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J.; Guerra D.; Michels P. A. M.; Hol W. G. J. (2003) J. Mol. Biol. 329, 335–349. 10.1016/S0022-2836(03)00421-2. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. (2015) J. Am. Chem. Soc. 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. (2015) J. Am. Chem. Soc. 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Go M. K.; O'Donoghue A. C.; Amyes T. L.; Pegan S. D.; Wang Y.; Loria J. P.; Mesecar A. D.; Richard J. P. (2014) Biochemistry 53, 3486–3501. 10.1021/bi500458t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. (2014) J. Am. Chem. Soc. 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. (2014) Curr. Opin. Chem. Biol. 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Wierenga R. K.; Loria J. P.; Richard J. P. (2013) Biochemistry 52, 5928–5940. 10.1021/bi401019h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Nitsch-Velasquez L.; Amyes T. L.; Richard J. P. (2013) J. Am. Chem. Soc. 135, 5978–5981. 10.1021/ja401504w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Koudelka A. P.; Amyes T. L.; Richard J. P. (2012) J. Am. Chem. Soc. 134, 10286–10298. 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. (2014) J. Am. Chem. Soc. 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. (2013) Biochemistry 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2013) Biochemistry 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. (2008) Biochemistry 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2013) Biochemistry 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. (2015) J. Am. Chem. Soc. 137, 5312–5315. 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X.; Ji C.; Han X.; Zhao X.; Li X.; Mao Y.; Wong L.-L.; Bartlam M.; Rao Z. (2006) J. Mol. Biol. 357, 858–869. 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. (2008) Biochemistry 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney M. D.; Kirsch J. F. (1989) Science 243, 1485–1488. 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- Horovitz A.; Fersht A. R. (1990) J. Mol. Biol. 214, 613–617. 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; McKay Wood B.; Gerlt J. A.; Richard J. P. (2010) Biochemistry 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. (2012) Biochemistry 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph-McCarthy D.; Lolis E.; Komives E. A.; Petsko G. A. (1994) Biochemistry 33, 2815–2823. 10.1021/bi00176a010. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Koudelka A.; Amyes T. L.; Richard J. P. (2010) Biochemistry 49, 5377–5389. 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. (2010) J. Am. Chem. Soc. 132, 13525–13532. 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (2006) Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.