

Abstract
Uterine fibroids are benign hormone-sensitive tumors of uterine smooth muscle cells leading to heavy menstrual bleeding and pelvic pain. Ulipristal acetate (UPA) is an emerging medical treatment of fibroids with the potential to be used for long-term treatment. In this context, the present article summarizes UPA’s main clinical pharmacology and pharmacokinetic (PK) properties. Ulipristal acetate has good oral bioavailability and a half-life allowing one single oral administration per day for the management of fibroids. As a steroid, UPA is a substrate for cytochrome P450 (CYP) 3A4 but does not act as an inducer or inhibitor of the CYP system or transporter proteins. With the exception of drugs modulating CYP3A4 activity, risks of drug–drug interactions with UPA are unlikely. In conclusion, besides its pharmacodynamic characteristics, UPA shows favorable PK properties that contribute to a good efficacy–safety ratio for the long-term management of uterine fibroids in clinical practice.
Keywords: clinical pharmacology, pharmacokinetic, ulipristal acetate, uterine fibroid
Introduction
Leiomyomas or fibroids are benign hormone-sensitive tumors of uterine smooth muscle cells often resulting in heavy menstrual bleeding, pelvic pressure, and pain.1 Surgical interventions, especially hysterectomy, are the current mainstay of treatment.2 Options for medical therapy are limited to preoperative reduction in symptoms related to uterine bleeding and fibroid size since there is no effective long-term medical treatment. The levonorgestrel-releasing intrauterine device, while not approved for this indication, has been found to reduce menstrual blood loss in women with uterine fibroids, but its efficacy is reduced in patients with distorted uterus,3 and gonadotropin-releasing hormone agonists can only be used for short-term therapy due to safety concerns (loss of bone mass) and adverse reactions (hot flashes).4,5 A third treatment option is the selective progesterone receptor modulator ulipristal acetate (UPA), which was recently shown to be safe and efficacious during more than 1 year of intermittent treatment.6 A European marketing authorization has been granted in February 2012, for Esmya 5 mg UPA tablets, for the preoperative treatment of moderate to severe symptoms of uterine fibroids in adult women of reproductive age with a treatment duration limited to 3 months.7 The initial authorized treatment duration of 3 months has been extended to 2 courses of 3-month treatment in early 2014.7 This new oral agent may raise interest for the medical management of uterine fibroids as a novel and much-needed therapy not only for use presurgically but also for long-term chronic use in uterine fibroids in the near future. Therefore, the present work aims at providing a summary of the main clinical pharmacology and pharmacokinetic (PK) properties, that is, absorption, distribution, metabolism and elimination of UPA, the potential of UPA to interact with food and other drugs, and its use in special populations. To identify relevant studies, a literature search of Google Scholar and Pubmed was performed from January 2003 to March 2014, with the key word “ulipristal acetate” combined with “pharmacokinetics.” No language restrictions were imposed. Reports of clinical trials describing the PKs or metabolism of UPA were selected. In addition, previous reviews and, where available, original study data were examined. Finally, selected information from companies developing UPA was also consulted.
Materials and Methods
Ulipristal Acetate: Drug Substance, Main Metabolite, and Drug Product
Ulipristal acetate has a steroidal structure (Figure 1). Its N-monodemethylated and N-didemethylated metabolites were denominated PGL4002 and PGL4004 (Figure 1). All studies presented in this article used UPA tablets with dosage strengths of 5, 10, or 30 mg with the exception of the mass balance study that used a water/ethanol drug substance solution. The tablets were produced using manufacturing processes similar to marketed UPA formulations and in compliance with Good Manufacturing Practice.
Figure 1.

Structure of ulipristal acetate and its oxidative metabolites PGL4002 and PGL4004.
Clinical Studies
The protocols of all clinical studies presented in this article were approved by the appropriate regulatory agencies and Independent Ethics Committees before the start of the study. All studies were conducted according to the “Declaration of Helsinki” and International Conference on Harmonisation-Good Clinical Practice (ICH GCP) consolidated guidelines.
Measurement of UPA and Its Main Metabolite PGL4002 in Plasma
Concentrations of UPA and PGL4002 in plasma were determined by SGS Cephac (Saint-Benoit, France) using a validated liquid chromatography followed by tandem mass spectrometry (LC-MS/MS) assay. Over the calibration range 0.100 to 40.0 ng/mL, the overall precision and accuracy of the assay were within 15% (20% at the lowest level of quantification) for both UPA and PGL4002.
Clinical Pharmacology
Healthy Participants
Mass balance
The metabolism and excretion of 14C-UPA were investigated in 5 healthy premenopausal women after a single oral dose of 20 mg/59 μCi with a radiochemical purity of at least 97% in an ethanol/water solution.8 After dosing, blood, plasma, urine, and fecal samples were collected for up to 11 days and analyzed for concentrations of radioactivity. Ulipristal acetate metabolite profiles in plasma were determined by high-performance liquid chromatography with radioactivity flow detection; metabolite structures were confirmed by LC-MS/MS.
The primary route of excretion of radioactivity was via the liver, with a mean value of 73% of the administered dose recovered in feces. Mean renal excretion was 6% of the administered dose. Ulipristal acetate underwent extensive metabolism, and only 6% of plasma area under the plasma concentration time curve (AUC) of total radioactivity was accounted for by UPA. Radiochromatograms of plasma revealed that oxidative demethylation was the major metabolic pathway, most likely via cytochrome P450 (CYP) isoenzyme 3A4 as described subsequently. At peak plasma radioactivity, the majority of circulating radioactivity was constituted by UPA (58.0%), N-monodemethylated UPA (PGL4002 [20.5%]), and N-didemethylated UPA (PGL4004) eluting together with PGL4002+2H (8.3%). Metabolite profiles in feces contained some of the plasma metabolites as well as acetylated and glucuronidated UPA; however, unchanged UPA was not present in the feces.
Ulipristal acetate at a dose of 20 mg was well tolerated in this study.
Drug interaction
Cytochrome enzymes
CYP3A4 is the major CYP isoenzyme responsible for the oxidative metabolism of UPA, with some minor contribution from CYP1A2 and CYP2D6.9 Thus, concomitant administration of UPA with potent CYP3A4 inhibitors such as ketoconazole or telithromycin may inhibit the metabolism of UPA and cause increased plasma levels. In addition, concomitant administration with potent CYP3A4 inducers like rifampicin or carbamazepine may reduce plasma concentrations of UPA and result in decreased efficacy. Therefore, in vivo drug interaction studies with a strong CYP3A4 inducer (rifampicin) and with strong and moderate CYP3A4 inhibitors (ketoconazole and erythromycin, respectively) were performed.
Induction of CYP3A4 was investigated in 22 healthy women of reproductive age during an open-label, single sequence, 2-period crossover study. Administration of the potent CYP3A4 inducer rifampicin (300 mg twice daily for 9 days) markedly decreased Maximum plasma concentration (Cmax) and AUC of UPA and PGL4002 by 90% or more and decreased UPA half-life by 2.2-fold corresponding to an approximately 10-fold decrease in UPA exposure.10
A clinical CYP3A4 inhibitor study investigated the effects of multiple doses of ketoconazole (200 mg, twice daily during 7 days) on the PKs of a single 10mg dose of UPA in 18 healthy female individuals of reproductive age.11 This study was a randomized, 2-period, crossover, open-label design with a 14-day washout period between UPA doses. Ulipristal acetate was either administered alone or concomitantly with the day 4 morning dose of ketoconazole. The oral coadministration of UPA and ketoconazole reduced UPA metabolism and resulted in an approximate 2-fold increase in the average maximum plasma UPA levels and an approximate average 5.8-fold increase in bioavailability with a reduced elimination rate (kel) from 0.018 h−1 to 0.009 h−1 and clearance and an approximate doubling of the median elimination half-life (t1/2) from about 41 to 78 hours. A concomitant decrease in plasma PGL4002 levels subsequently occurred as a result of this ketoconazole-induced metabolic inhibition of UPA with a reduced kel (from 0.028 h−1 to 0.010 h−1) and an increased t1/2 (from about 25 to 69 hours) leading to a somewhat increased extent of PGL4002 exposure.
Moderate CYP3A4 inhibitors are more likely to be prescribed to women of reproductive age with uterine myoma, and thus, a drug interaction study with the moderate CYP3A4 inhibitor erythromycin was performed.12 Similar to the induction trial, this study used an open-label, single sequence, 2-period crossover design. Eighteen healthy women of reproductive age received oral UPA (20 mg) once daily on days 1 and 13 and twice-daily erythromycin propionate administrations (500 mg) from days 9 through 17. Based on the PK results, CYP3A4 inhibition induced by erythromycin led to a very limited increase in Cmax (1.2-fold) and a just under 3-fold increase in the AUC for UPA. In line with the observations during the ketoconazole study, erythromycin inhibition of CYP3A4 induced a significant decrease in PGL4002 Cmax (approximately 2-fold) and a 1.5-fold increase in AUC of PGL4002 which is related to the prolongation of the t1/2 (from about 24 to 48 hours).
The results of in vivo erythromycin and ketoconazole drug interaction studies, in vitro data as well as the PK characteristics of UPA described in the multiple dose study13 allowed the development of a physiologically based PK model of UPA using the Simcyp Population-based Simulator (version 11.1).
The combination of the UPA fraction metabolized by CYP3A4 of 94% and intrinsic metabolic clearance inputs to produce an oral clearance of 57.5 L/h gave the best fit to recover the in vivo data.
This model was then used to describe the PKs of drug interactions of UPA with a mild CYP3A4 inhibitor in virtual populations.14 Ten virtual trials of 18 healthy women aged 19 to 42 years receiving 9 days of fluconazole (50, 100, and 200 mg daily) and a single dose of UPA (10 mg) on day 6 were generated to assess variability across groups in the cohort. A range of doses of fluconazole was used to assess the effects of both low (50/100 mg) and a more frequently used higher dose (200 mg). The predicted median UPA Cmax ratio values were 1.3, 1.4, and 1.6 for the 50-, 100-, and 200mg fluconazole dose, respectively. Corresponding median AUC ratios were 1.5, 1.9, and 2.5, respectively.
Membranes transporters
During UPA development, particular attention has been paid to its interaction with membrane transporters expressed in epithelia of the intestine, liver, and kidney because of their key role in drug disposition, therapeutic efficacy, and adverse drug reactions. Ulipristal acetate is not a substrate of clinically relevant drug transporters such as the permeability glycoprotein 1 (P-gp) and organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1 and -B3). Furthermore, it is not an inhibitor of the bile salt export pump, breast cancer resistance protein, organic cation transporter 1 and 2, OATP1B1 and OATP1B3, and organic anion transporter 1 and 3 (OAT1 and -3).15 However, in vitro studies suggested that UPA is a potent P-gp inhibitor with half maximal inhibitory concentration of 348 ng/mL (0.732 µmol/L).16 To assess the clinical relevance of this finding, 18 healthy female volunteers of reproductive age were randomized to receive either the sensitive P-gp substrate fexofenadine (60 mg) alone or concomitantly with UPA at 10 mg (1.5 hours after administration of UPA) in a 2-period, crossover, open-label study.17
Based on the median Time of occurrence of maximum plasma concentration (Tmax) of UPA of approximately 0.75 hours with a t1/2 of approximately 36 hours, the administration of UPA 1.5 hours prior to the fexofenadine administration produced a high UPA concentration in the intestinal tract during the major absorption process of fexofenadine.
Following the coadministration of UPA and fexofenadine (60 mg), mean fexofenadine AUC and Cmax were all minimally decreased in the presence of UPA, with no effect on the time to maximum fexofenadine concentration. The AUC and Cmax geometric mean ratios (P-gp alone and UPA/P-gp) were 0.97 (90% confidence intervals [CIs]: 0.86-1.11) and 0.91 (0.77-1.06), respectively. The CIs for AUC were contained within the bounds of (0.80-1.25) consistent with bioequivalence and just slightly below the lower 0.80 limit and within the 0.80 to 1.25 interval for the upper limit of bioequivalence for Cmax.
Gastric pH enhancers
Ulipristal acetateis a lipophilic compound with an octanol/water partition coefficient exceeding 4, and a weak base (pKa = 5.49), and therefore is nearly insoluble in water at neutral pH. Its solubility is seen to increase as pH decreased. Hence, concomitant administration of drugs resulting in a transient increase in gastric pH, such as proton pump inhibitors, antacids, and H2-receptor antagonists are likely to reduce the solubility of UPA and thus to alter plasma exposure. An in vivo drug interaction study with esomeprazole, a proton pump inhibitor, was conducted to determine the effects of an increased gastric pH on the PKs of UPA. In this single sequence, open-label study, 18 healthy women of reproductive age received oral UPA tablets (10 mg) once on days 1 and 13 and daily esomeprazole administrations (20 mg) from days 9 through 14. Coadministration of esomeprazole decreased geometric mean Cmax of UPA by 65% (geometric mean ratio point estimate [90% CI]: 0.35 [0.28-0.42]) and delayed median Tmax from 0.75 to 1.00 hours but had only a minor effect on AUC (geometric mean ratio [90% CI]: 1.11 [0.98-1.27]). Ratios of PGL4002 PK parameters were similar to what was previously observed for UPA; geometric mean Cmax was decreased by 64% (geometric mean ratio point estimate [90% CI]: 0.36 [0.30-0.43]) and Tmax was delayed from 0.75 to 1.75 hours. The AUC of PGL4002 remained within the limits of bioequivalence (geometric mean ratio [90% CI]: 0.98 [0.89-1.08]).
Iron deficiency/anemia
Heavy uterine bleeding is a frequent symptom of uterine fibroids which may lead to iron deficiency anemia secondary to chronic blood loss.18 Iron salts are commonly used to treat iron deficiency anemia.19 However, iron-containing preparations are known to decrease the absorption and clinical effectiveness of several commonly used drugs when coadministered.20 As the concurrent use of oral iron preparations and UPA is likely to occur, the effect of oral iron administration on the bioavailability and PKs of UPA was determined in a single dose, randomized, 2-way crossover study in 22 healthy women of reproductive age.21 Each participant received 2 doses of 10 mg UPA, once by itself and once concurrently with one 200-mg ferrous sulfate tablet once, equivalent to 65 mg of elemental iron (Fe2+). The concomitant administration of UPA and oral iron resulted in nonbioequivalence with a 32% (geometric mean ratio point estimate [90% CI]: 0.68 [0.58-0.79]) and 33% (geometric mean ratio point estimate [90% CI]: 0.67 [0.59-0.76]) reduction in UPA and PGL4002 Cmax, respectively. The AUC, however, was bioequivalent between treatments with CIs between 80% and 125%, as reflected by a minimally reduced AUC of 10% (geometric mean ratio point estimate [90% CI]: 0.90 [0.82-0.98]) for UPA and 11% (geometric mean ratio point estimate [90% CI]: 0.89 [0.82-0.96]) for PGL4002 compared to UPA administration without iron. Median UPA t1/2 was somewhat shorter (40 vs 43 hours) with a slightly faster median kel (0.017 vs 0.016 h−1) in the absence of iron while PGL4002 t1/2 was prolonged (35 vs 29 hours) with a slower kel (0.020 vs 0.024 h−1) in the absence of iron. Thus, the coadministration of UPA and oral iron reduced peak plasma UPA and subsequently PGL4002 concentrations with only minimal effect on bioavailability. This effect of oral iron on UPA PKs, however, is not felt to be of any clinical relevance.
Administration of UPA did not lead to discontinuation of treatment or serious or severe adverse events in any of the interaction studies. There were no clinically significant changes in clinical laboratory evaluations, vital signs, physical findings, or any other observations related to safety.
Single and repeated dose PK
The reported PK of single oral doses of UPA (10-50 mg) was assessed in healthy female volunteers.11–13,21–23 After administration, UPA rapidly reached peak concentrations within 1 hour, with the apparent terminal t1/2 ranging from 35 to 43 hours. The AUC and Cmax for UPA increased in an approximately dose-proportional manner (Table 1). No clinically meaningful differences in Tmax or apparent t1/2 were noted across the dose range. Ulipristal acetate was quickly metabolized to PGL4002. Its PK parameters are presented in Table 2.
Table 1.
Between-Study Comparison of Ulipristal Acetate (UPA) Pharmacokinetic Parameters Following UPA Single Administration of 10, 20, and 50 mg.a
| Study | UPA Dose | N | Tmax, (h, Median (Min-Max) | Cmax, ng/mL, Mean (SD) | AUC, ng·h/mL, Mean (SD) |
|---|---|---|---|---|---|
| pH drug interaction study 23 | 10 | 18 | 0.75 (0.5-1.5) | 61.0 (27.2) | 189.1 (102.0) |
| Iron drug interaction study21 | 10 | 22 | 0.8 (0.5-1.0) | 81.9 (27.0) | 209.1 (93.3) |
| Food interaction study22 | 10 | 18 | 0.8 (0.5-2.0) | 83.6 (32.5) | 257.0 (126.3) |
| CYP3A4 strong inhibitor drug interaction study11 | 10 | 18 | 0.7 (0.5-1.0) | 73.2 (22.1) | 205.4 (88.9) |
| Multiple dose study13 | 10 | 8 | 0.88 (0.5-1.5) | 42.2 (23.6) | 216.0 (152.2) |
| CYP3A4 moderate inhibitor drug interaction study12 | 20 | 18 | 0.75 (0.5-3.0) | 106.6 (56.8) | 275.0 (153.9) |
| Multiple dose study13 | 20 | 8 | 0.75 (0.5-2.0) | 130.9 (69.3) | 583.0 (359.1) |
| Multiple dose study13 | 50 | 8 | 0.89 (0.75-1.0) | 354.8 (187.6) | 1538.3 (586.9) |
Abbreviations: CYP, cytochrome P450; SD, standard deviation.
aTmax: median value (min-max), Mean: arithmetic mean, AUC: AUC0–∞.
Table 2.
Between-study Comparison of PGL4002 Pharmacokinetic Parameters Following Ulipristal Acetate (UPA) Single Administration of 10, 20, and 50 mg.a
| Study | UPA Dose | N | Tmax, h, Median (Min-Max) | Cmax, ng/mL, Mean (SD) | AUC, ng·h/mL, Mean (SD) |
|---|---|---|---|---|---|
| pH drug interaction study23 | 10 | 18 | 0.75 (0.50-1.50) | 16.3 (5.6) | 54.8 (18.9) |
| Iron drug interaction study21 | 10 | 22 | 0.8 (0.5-2.0) | 26.1 (5.7) | 82.7 (24.2) |
| Food interaction study22 | 10 | 18 | 0.8 (0.5-2.0) | 26.4 (7.4) | 87.4 (36.1) |
| CYP3A4 strong inhibitor drug interaction study11 | 10 | 18 | 0.8 (0.5-1.5) | 21.0 (5.4) | 65.1 (23.3) |
| Multiple dose study13 | 10 | 8 | 1.0 (0.5-5.0) | 15.6 (7.8) | 80.6 (31.5) |
| CYP3A4 moderate inhibitor drug interaction study12 | 20 | 18 | 0.75 (0.5-3.0) | 40.2 (56.8) | 119.5 (46.5) |
| Multiple dose study13 | 20 | 8 | 0.75 (0.75-2.0) | 37.9 (15.6) | 202.1 (77.2) |
| Multiple dose study13 | 50 | 8 | 1.0 (0.75-1.5) | 89.9 (44.0) | 471.5 (158.3) |
Abbreviations: CYP, cytochrome P450; SD, standard deviation; AUC, area under the plasma concentration time curve.
aTmax: median value (min-max), Mean: arithmetic mean, AUC: AUC0–∞.
To determine the effect of a high-fat meal on UPA PK, 18 healthy participants were randomized to the following open-label treatments: a single oral 10 mg tablet of UPA administered following a high-fat meal, and a single oral 10 mg UPA administered fasted. Administration of UPA after a high-fat meal resulted in a slower rate of UPA absorption as indicated by a 27% decrease in Cmax (geometric means ratio [fed/fasted] 0.73; 90% CIs: 0.60-0.89) but an increase in the extent of absorption (AUC) by 26% (geometric means ratio [fed/fasted] 1.26; 90% CIs: 1.15-1.38). Similar results were obtained for PGL4002 (geometric mean ratios and 90% CIs for Cmax: 0.74, 90% CIs: 0.63-0.88, and for AUC 1.26; 90% CIs: 1.17-1.37). In addition, the high-fat meal delayed median Tmax for both UPA and PGL4002 from 0.8 to 1.5 hours but with no significant effect on apparent terminal t1/2.22 Similar results were obtained in another fed-fasted study in 19 healthy females utilizing a single oral 30-mg UPA dose. In this study, administration of UPA after a high-fat meal resulted in a slower rate of UPA absorption as indicated by a 45% decrease in Cmax and a delay of about 2.25 hours in median Tmax. However, the extent of absorption was increased in the presence of food as evidenced by an increase in AUC of 26% compared to that observed after UPA administration in the fasted state. Comparable results were obtained for PGL4002.10 Because of these modest degrees of changes, UPA may be taken with or without food.
An incremental oral dose study assessed the PK properties of multiple once-daily doses of UPA.13 Thirty-two normal weight women of reproductive age were randomly assigned to one of 4 treatment groups: UPA 10, 20, or 50 mg or placebo, once daily for 10 days. The plasma concentration–time profiles and principal PK parameters (Tmax, Cmax, and AUC) were similar at days 1 (single dose) and 10 (steady state; Table 3). In the groups receiving UPA once-daily doses, accumulation of UPA was modest with AUC accumulation ratios (day 10/day 1) ranging between 1.1 and 1.2 and a Cmax ratios from 1.3 to 2.0. A supraproportional increase with dose in Cmax and AUC was observed from 10 to 50 mg for UPA plasma bioavailability in this study. It is, however, worth noting that when normalized to dose, a supraproportional increase in UPA exposure was found from 10 to 20 mg, whereas from 20 to 50 mg, AUC/dose and Cmax/dose remained within the same order of magnitude. The apparent terminal elimination t1/2 was 38.0 to 48.6 hours. Steady state plasma concentrations of UPA were achieved within 4 or 5 days of starting treatment at 10 and 50 mg, respectively. For PGL4002, Tmax and t1/2 values were similar to those of UPA. Exposure to PGL4002 was approximately one-third of that of UPA. Single and multiple administrations of UPA were safe and well tolerated under the conditions of the reported studies.
Table 3.
Summary Statistics of Ulipristal Acetate (UPA) and PGL4002 Pharmacokinetic Parameters following UPA Single and Repeated Administration of 10, 20 and 50 mg (N = 8 per group).a
| UPA Dose | Day | UPA | PGL4002 | ||||
|---|---|---|---|---|---|---|---|
| Tmax, h, Median (Min-Max) | Cmax, ng/mL, Mean (SD) | AUC ng·h/mL, Mean (SD) | Tmax, h, Median (Min-Max) | Cmax, ng/mL, Mean (SD) | AUC, ng·h/mL, Mean (SD) | ||
| 10 | 1 | 0.88 (0.5-1.5) | 42.2 (23.6) | 216.0 (152.2) | 1.0 (0.5-5.0) | 15.6 (7.8) | 80.6 (31.5) |
| 10 | 0.75 (0.5-1.0) | 63.7 (26.1) | 216.6 (103.9) | 0.75 (0.5-1.5) | 21.6 (8.3) | 84.7 (31.5) | |
| 20 | 1 | 0.75 (0.5-2.0) | 130.9 (69.3) | 583.0 (359.1) | 0.75 (0.75-2.0) | 37.9 (15.6) | 202.1 (77.2) |
| 10 | 0.75 (0.5-3.0) | 169.8 (103.6) | 602.8 (311.8) | 0.88 (0.75-3.0) | 46.9 (21.0) | 203.6 (72.1) | |
| 50 | 1 | 0.89 (0.75-1.0) | 354.8 (187.6) | 1538.3 (586.9) | 1.0 (0.75-1.5) | 89.9 (44.0) | 471.5 (158.3) |
| 10 | 0.75 (0.5-1.5) | 454.9 (186.3) | 1655.7 (577.7) | 0.91 (0.75-1.5) | 104.7 (38.1) | 452.1 (151.7) | |
Abbreviations: SD, standard deviation; AUC, area under the plasma concentration time curve.
aTmax: median value (min–max), Mean: arithmetic mean, AUC: AUC0–∞ on day 1; AUCSS on day 10.
Patients with uterine fibroids
Trough concentrations of UPA have been assessed in 2 patient studies in which UPA at 5 and 10 mg/d was administered to women with symptomatic uterine fibroids for 12 weeks.24,25 Predose plasma samples collected at baseline and during the weeks 5, 9, and 13 study visits were analyzed. Results from these studies indicate that trough concentrations increase in a dose-dependent manner following once-daily dosing of 5 and 10 mg of UPA and were roughly similar to the results obtained from the Phase I studies. Mean plasma levels from the patient studies were slightly higher than the trough levels observed in the multiple dose Phase I study, which probably reflects the fact that the time of plasma sampling in relation to previous dose could not strictly be controlled in a patient setting. Furthermore, the patient studies recruited a less standardized group of participants in terms of age, body weight, habits, and other confounding factors.
At UPA trough concentrations of 3.7 and 3.0 ng/mL, uterine bleeding was controlled in 90% and 91% of patients receiving 5 mg, respectively. Treatment at 10 mg UPA resulted in uterine bleeding control in 98% and 92% of patients; associated Minimal concentrations (Cmins) were 8.8 and 7.8 ng/mL, respectively (Figure 2).24,25
Figure 2.
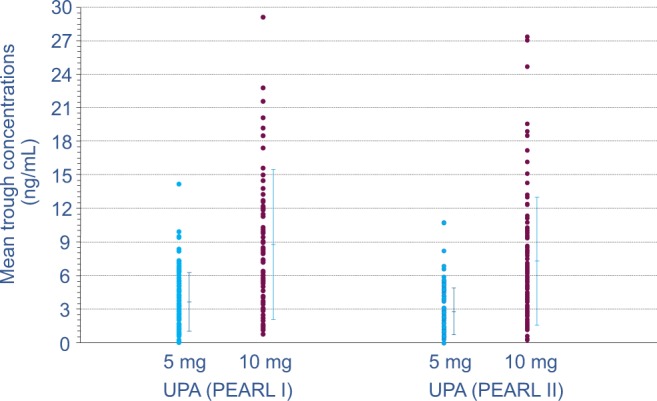
Trough UPA plasma concentrations assessed during patient studies PEARL I24 and PEARL II25. UPA was administered daily at 5 or 10 mg for 3 months to patients with uterine fibroids. Through UPA, plasma concentrations were assessed during weeks 5, 9, and 13 of treatment. Individual (•) and mean values (with standard deviation) are plotted. PEARL indicates PGL4001’s Efficacy Assessment in Reduction of symptoms due to uterine Leiomyomata. UPA indicates ulipristal acetate.
Numerous UPA trough exposure values at 5 and 10 mg/d were overlapping, thus supporting the observed comparable efficacy data for the 5-mg and 10-mg doses.
Ulipristal acetate was safe and well tolerated in patient studies. Headache and breast tenderness were the most common adverse events associated with UPA but did not occur significantly more frequently than with placebo.
Special Populations
Patients with hepatic impairment
One open-label study evaluated the PK of single doses of UPA of 10 mg in 8 women with a moderate degree of hepatic impairment (HI; defined as a Child-Pugh score of 7-9) but otherwise healthy.26 These women were pair matched with control participants on age, weight, and tobacco use. Following an overnight fast, UPA was administered, serial blood samples for PK evaluation were collected, and safety variables assessed over the next 120 hours. Decreases in bound UPA AUC and Cmax were 1.6-fold and 2.3-fold lower for patients with moderate HI, respectively, when compared to levels obtained in paired participants with normal hepatic function. Tmax was slightly increased in women with HI, and the terminal t1/2 increased with impaired hepatic function. Compared to values in women with normal hepatic function (Tmax: 0.9 hours, t1/2: 53 hours), the Tmax and t1/2 values of UPA in those with HI were 1.6 and 72 hours, respectively. Plasma protein and albumin concentrations were not altered in patients with HI. Ulipristal acetate was well tolerated and no adverse events or study discontinuations were recorded in this trial.
Discussion and Conclusion
The PK characteristics of the new selective progesterone receptor modulator UPA developed to treat uterine fibroids have been evaluated in clinical trials performed in healthy women of reproductive age, patients with fibroids, and volunteers with HI.
All studies selected for the present article used the same analytical laboratory and analysis method for UPA, thus ensuring consistency of results.
Ulipristal acetate has a good oral bioavailability. It is mainly excreted via the feces and extensively metabolized by the liver. Similar to the metabolism of other progesterone receptor modulators,27–29 CYP3A4-mediated N-demethylation of UPA resulted in the main metabolites N-monodemethylated (PGL4002) and N-didemethylated UPA (PGL4004), the former of which has some pharmacological activity,9,30 and downstream derivatives of UPA were the remaining identified metabolites.
A clinical CYP3A4 induction study with rifampicin showed that healthy volunteers had significantly reduced UPA plasma level and thus concomitant use of UPA and potent CYP3A4 inducers is not recommended. Clinical drug–drug interaction studies with strong (ketoconazole) and moderate (erythromycin) CYP3A4 inhibitors proofed UPA to be a sensitive substrate. Although the safety and tolerability of UPA was not negatively impacted by either ketoconazole or erythromycin-induced increases in UPA and PGL4002 exposure, a recommendation to avoid concomitant therapy with moderate or strong CYP3A4 inhibitors is appropriate.10,31
Although UPA proved to be a potent P-gP inhibitor in vitro which indicated a potential interaction with P-gP substrates, the coadministration of UPA is not expected to result in a clinically relevant effect on the PKs of P-gp substrates.10,31 However, the labeling recommends to separate the administration of UPA and P-gp substrates such as dabigatran etexilate, digoxin, or fexofenadine by at least 1.5 hours, mimicking the studied exposure scenario.10
Concomitant use of UPA with proton pump inhibitor (gastric pH enhancer) esomeprazole led to a modified absorption rate, while exposure in terms of AUC remained close to bioequivalence limits. In the context of chronic administration of UPA, no clinically significant effects are expected from coadministration with drugs increasing gastric pH.
Similarly, coadministered oral iron sulfate decreased peak plasma concentrations, but overall exposure (AUC) was only slightly decreased and no effect on Tmax was observed compared to UPA administration without iron. This small effect on UPA PKs with the coadministration of oral iron is therefore not expected to be clinically relevant with long-term multidose administration.
Exposures after single-dose administrations of 10 to 50 mg UPA were similar across the reported studies and comparable to those reported previously by Melis et al after a single administration of 30 mg UPA.32 Other authors reported UPA serum concentrations after single dose administrations between 1 and 200 mg using a radioimmunoassay that detected UPA and cross-reactive metabolites.33,34 Given the difference in analysis methodology, these exposures are generally comparable to our results.
The multiple dosing of UPA exhibited a PK profile consistent with that of a once-daily regimen. Ulipristal acetate PK parameters were similar to those obtained from single-dose studies in healthy female participants.
Concomitant administration with food resulted in no dramatic effect on the exposure of UPA and PGL4002 and is not expected to be of clinical relevance for daily administration.9,10,31 The observed modifications of a reduced and delayed Cmax and a slightly increased AUC are not considered clinically relevant, especially in the context of chronic administration of UPA.
Trough UPA concentrations were generally comparable in healthy participants and in patients with fibroids. Beneficial pharmacodynamic effects were observed in a large majority of patients at mean trough levels of 3 to 4 ng/mL. This is comparable with observations in an UPA contraception trial using a radioimmunoassay for UPA and cross-reactive metabolites. In this study, oral doses inducing amenorrhea through ovulation suppression and a direct effect on the endometrium had serum levels above 10 ng/mL.35
In volunteers with moderate HI, exposure to UPA was somewhat reduced and well tolerated. According to the labeling, no dosage adjustment of UPA is required for patients with mild HI, but UPA is not recommended in patients with moderate or severe HI unless the patient is closely monitored.10
In conclusion, UPA has a good oral bioavailability and can be administered with or without food. The biological half-life allows one oral administration per day. The PK characteristics of UPA should probably rule out clinically relevant drug–drug interactions, with the exception of those related to CYP3A4 in most patients. Main UPA clinical pharmacology characteristics are summarized in a graphical abstract (Figure 3). The PK advantages offered by this new long-term fibroid treatment should be considered positively besides the already well-described PD benefits, that is, the rapid induction of amenorrhea in a majority of patients with a concomitant improvement in fibroid-related anemia, a reduction in fibroid volume, a reduction in fibroid-related pain, and an improvement in the women’s fibroid-related impairment of quality of life.
Figure 3.

Graphical abstract of ulipristal acetate clinical pharmacology. Examples of relevant CYP3A4 inducers include rifampicin, rifabutin, carbamazepine, oxcarbazepine, phenytoin, fosphenytoin, phenobarbital, primidone, St John wort, efavirenz, nevirapine, and long term use of ritonavir. Relevant moderate and strong CYP3A4 inhibitors comprise moderate: erythromycin, grapefruit juice, verapamil; strong: ketoconazole, ritonavir, nefazodone, itraconazole, telithromycin, clarithromycin. Clinically relevant effects of UPA on P-gp substrates are not expected, but coadministration should be separated by at least 1.5 hours. Examples of P-gp substrates include dabigatran etexilate, digoxin, and fexofenadine. UPA indicates ulipristal acetate; CYP, cytochrome P450; P-gp, permeability glycoprotein 1.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: This work was funded by PregLem SA, a member of the Gedeon Richter Group, and by Watson Laboratories, Inc. a subsidiary of Actavis, Inc. OP is a salaried employee of ObsEva SA (formerly employee of PregLem SA, a subsidiary of Gedeon Richter plc). RHZ is a salaried employee of Watson Laboratories, Inc. A subsidiary of Actavis, Inc. and JPG is a salaried employee of PregLem SA, a subsidiary of Gedeon Richter plc.
References
- 1. Stewart EA. Uterine fibroids. Lancet. 2001;357 (9252):293–298. [DOI] [PubMed] [Google Scholar]
- 2. Stewart EA. Uterine fibroids and evidence-based medicine--not an oxymoron. N Engl J Med. 2012;366 (5):471–473. [DOI] [PubMed] [Google Scholar]
- 3. Zapata LB, Whiteman MK, Tepper NK, Jamieson DJ, Marchbanks PA, Curtis KM. Intrauterine device use among women with uterine fibroids: a systematic review. Contraception. 2010;82 (1):41–55. [DOI] [PubMed] [Google Scholar]
- 4. Donnez J, Schrurs B, Gillerot S, Sandow J, Clerckx F. Treatment of uterine fibroids with implants of gonadotropin-releasing hormone agonist: assessment by hysterography. Fertil Steril. 1989;51 (6):947–950. [DOI] [PubMed] [Google Scholar]
- 5. Lethaby A, Vollenhoven B. Fibroids (uterine myomatosis, leiomyomas). Clin Evid (Online). 2011;pii:0814. [Google Scholar]
- 6. Donnez J, Vazquez F, Tomaszewski J, et al. Long-term treatment of uterine fibroids with ulipristal acetate. Fertil Steril. 2014;pii: S0015–0282(14)00146–0. [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency. Esmya-ulipristal. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002041/human_med_001542.jsp&mid=WC0b01ac058001d124. Published March 15, 2012. Updated May 25, 2014.
- 8. Pohl O, Kendrick J, Gotteland JP. Metabolic disposition of [14C] ulipristal acetate in healthy premenopausal women. J Bioequiv Availab. 2013;(5):177–184. [Google Scholar]
- 9. European Medicines Agency. CHMP assessment report for Ellaone. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001027/WC500023673.pdf. Published November 13, 2009. Updated September 23, 2013.
- 10. European Medicines Agency. Esmya - Summary of Product Characteristics. Web site http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002041/WC500124085.pdf. Published March 15, 2012. Updated May 25, 2014.
- 11. Watson Laboratories Inc. Watson Laboratories study UL1102: An Open-Label, Crossover Study to Investigate the Effects of Multiple Doses of Ketoconazole on the Pharmacokinetics of a Single Dose of Ulipristal Acetate in Healthy Female Subjects. Study Report 2011. [Google Scholar]
- 12. Pohl O, Osterloh I, Gotteland JP. Effects of erythromycin at steady-state concentrations on the pharmacokinetics of ulipristal acetate. J Clin Pharm Ther. 2013;38 (6):512–517. [DOI] [PubMed] [Google Scholar]
- 13. Pohl O, Osterloh I, Gotteland JP. Ulipristal acetate - safety and pharmacokinetics following multiple doses of 10-50 mg per day. J Clin Pharm Ther. 2013;38 (4):314–320. [DOI] [PubMed] [Google Scholar]
- 14. PregLem SA. PregLem study PGL12-011: Quantitative Predicition of Systemic Exposure to PGL4001 (Ulipristal Acetate) and the Effects of Concomitant CYP3A4 Inhibitors/Inducers Using Prior in Vitro and in Vivo Data. Study Report 2012.
- 15. PregLem S.A. PregLem study PGL12-010: Assessment of Potential Substrate and Inhibitor of PGL4001 of Efflux (BCRP and BSEP) and Uptake (OCT1, OCT2, OAT1, OAT3, OATP1B1 and OATP1B3) Transporter. Study Report 2012.
- 16. PregLem S.A. PregLem study PGL09-007: P-gp Interaction Studies of Ulipristal Acetate and PGL4002 in Caco-2 Cell Monolayer System. Study Report 2009.
- 17. Watson Laboratories Inc. Watson Laboratories study UL1105: An Open-Label, Crossover Study to Assess the Effects of 10 mg Ulipristal Acetate on the Pharmacokinetics of Fexofenadine (Allegra) in Healthy Female Subjects. Study Report 2012.
- 18. Wallach EE, Vlahos NF. Uterine myomas: an overview of development, clinical features, and management. Obstet Gynecol. 2004;104 (2):393–406. [DOI] [PubMed] [Google Scholar]
- 19. Harju E. Clinical pharmacokinetics of iron preparations. Clin Pharmacokinet. 1989;17 (2):69–89. [DOI] [PubMed] [Google Scholar]
- 20. Campbell NR, Hasinoff BB. Iron supplements: a common cause of drug interactions. Br J Clin Pharmacol. 1991;31 (3):251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watson Laboratories Inc. Watson Laboratories study UL1103: An Open-Label, Crossover Study to Investigate the Effects of Oral Iron Administration on the Pharmacokinetics of a Single 10 mg Dose of Ulipristal Acetate in Healthy Female Subjects. Study Report 2012.
- 22. Watson Laboratories Inc. Watson Laboratories study UL1101: An Open-Label, Crossover, Comparative Bioavailability Study of 10 mg Ulipristal Acetate After a High-Fat Meal or Under Fasting Conditions. Study Report 2011.
- 23. Pohl O, Osterloh I, Lecomte V, Gotteland JP. Changes in gastric pH and in pharmacokinetics of ulipristal acetate - a drug-drug interaction study using the proton pump inhibitor esomeprazole. Int J Clin Pharmacol Ther. 2013;51(1):26–33. [DOI] [PubMed] [Google Scholar]
- 24. Donnez J, Tatarchuk TF, Bouchard P, et al. Ulipristal acetate versus placebo for fibroid treatment before surgery. N Engl J Med. 2012;366 (5):409–420. [DOI] [PubMed] [Google Scholar]
- 25. Donnez J, Tomaszewski J, Vazquez F, et al. Ulipristal acetate versus leuprolide acetate for uterine fibroids. N Engl J Med. 2012;366 (5):421–432. [DOI] [PubMed] [Google Scholar]
- 26. Watson Laboratories Inc. Watson Laboratories study UL1106: An Open-Label, Prospective Study to Evaluate the Pharmacokinetics and Safety of a Single Oral 10 mg Dose of Ulipristal Acetate in Female Subjects with Moderately Impaired Hepatic Function Compared to Healthy Matched Subjects with Normal Hepatic Function. Study Report 2012.
- 27. Jang GR, Benet LZ. Cytochrome P4503A4-mediated N-demethylation of the antiprogestins lilopristone and onapristone. Drug Metab Dispos. 1997;25 (10):1119–1122. [PubMed] [Google Scholar]
- 28. Khan KK, He YQ, Correia MA, Halpert JR. Differential oxidation of mifepristone by cytochromes P450 3A4 and 3A5: selective inactivation of P450 3A4. Drug Metab Dispos. 2002;30 (9):985–990. [DOI] [PubMed] [Google Scholar]
- 29. Zurth C, Kagels F. Determination of onapristone and its N-desmethyl metabolite in human plasma or serum by high-performance liquid chromatography. J Chromatogr. 1990;532 (1):115–123. [DOI] [PubMed] [Google Scholar]
- 30. Attardi BJ, Burgenson J, Hild SA, Reel JR. In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone. J Steroid Biochem Mol Biol. 2004;88(3):277–288. [DOI] [PubMed] [Google Scholar]
- 31. Watson Laboratories Inc. FIBRISTAL(TM) PRODUCT MONOGRAPH. http://www.fibristal.ca/docs/Fibristal_Product_Monograph_E.pdf. published on June 2013.
- 32. Melis GB, Piras B, Marotto MF, et al. Pharmacokinetic evaluation of ulipristal acetate for uterine leiomyoma treatment. Expert Opin Drug Metab Toxicol. 2012;8 (7):901–908. [DOI] [PubMed] [Google Scholar]
- 33. Blithe DL, Nieman LK, Blye RP, Stratton P, Passaro M. Development of the selective progesterone receptor modulator CDB-2914 for clinical indications. Steroids. 2003;68 (10-13):1013–1017. [DOI] [PubMed] [Google Scholar]
- 34. Passaro MD, Piquion J, Mullen N, et al. Luteal phase dose-response relationships of the antiprogestin CDB-2914 in normally cycling women. Hum Reprod. 2003;18 (9):1820–1827. [DOI] [PubMed] [Google Scholar]
- 35. Chabbert-Buffet N, Pintiaux-Kairis A, Bouchard P. Effects of the progesterone receptor modulator VA2914 in a continuous low dose on the hypothalamic-pituitary-ovarian axis and endometrium in normal women: a prospective, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2007;92 (9):3582–3589. [DOI] [PubMed] [Google Scholar]