

Abstract
Sodium potassium chloride co-transporter (NKCC) belongs to cation-dependent chloride co-transporter family, whose activation allows the entry of Na+, K+ and 2Cl- inside the cell. It acts in concert with K+ Cl- co-transporter (KCC), which extrudes K+ and Cl- ions from cell. NKCC1 is widely distributed throughout the body, while NKCC2 is exclusively present in kidney. Protein kinase A, protein kinase C, Ste20-related proline-alanine-rich kinase, oxidative stress responsive kinases, With No K=lysine kinase and protein phosphatase type 1 control the phosphorylation/dephosphorylation of key threonine residues of in regulatory domain of NKCC1. The selective inhibitors of NKCC1 including bumetanide and furosemide are conventionally employed as diuretics. However, recent studies have indicated that NKCC1 may be involved in the pathophysiology of anxiety, cerebral ischemia, epilepsy, neuropathic pain, fragile X syndrome, autism and schizophrenia. The inhibitors of NKCC1 are shown to produce anxiolytic effects; attenuate cerebral ischemia-induced neuronal injury; produce antiepileptic effects and attenuate neuropathic pain. In the early developing brain, GABAA activation primarily produces excitatory actions due to high NKCC1/KCC2 ratio. However, as the development progresses, the ratio of NKCC1/KCC2 ratio reverses and there is switch in the polarity of GABAA actions and latter acquires the inhibitory actions. The recapitulation of developmental-like state during pathological state may be associated with increase in the expression and functioning of NKCC1, which decreases the strength of inhibitory GABAergic neurotransmission. The present review describes the expanding role and mechanism of NKCC1 in the pathophysiology of different diseases.
Keywords: Anxiety, cerebral ischemia, neuropathic pain, sodium potassium chloride co-transporter.
INTRODUCTION
Na+ K+ 2Cl- co-transporter(NKCC) belongs to cation-dependent chloride co-transporter family and mediates transport of Na+, K+ and Cl- ions into the cell [1, 2]. Activation of NKCC allows the entry of Na+, K+ and 2Cl- ions inside the cell. Other types of cation-dependent chloride co-transporters include K+ Cl- co-transporter (KCC)and Na+ Cl- co-transporter [3-5]. NKCC1 and NKCC2 are two members of NKCC family and NKCC2 is exclusively present on the thick ascending limb of Henle in the kidneys [6, 7]. However, NKCC1 is widely distributed in various types of tissues including stomach, heart, skeletal muscle, lungs, brain and also in kidney [8]. NKCC1 usually acts in concert with other cation-dependent chloride co-transporter such as KCC, which extrudes K+ and Cl- ions from the cell [9, 10].
The activation or inactivation of NKCC1 is primarily controlled by phosphorylation or dephosphorylation of key threonine residues of its structure. In shark rectal gland, three phosphoacceptor sites at Thr184, Thr189 and Thr202 constitute the regulatory domain in the N-terminal of NKCC1. These phosphoacceptor sites are highly conserved between NKCC1 and NKCC2, thus, both NKCC1 and NKCC2 are activated by phosphorylation in a similar manner. The phosphorylation of Thr189 is essential for the NKCC1 activation, on the other hand, Thr184 and Thr202 has modulatory action [11]. The shark Thr175/Thr179/Thr184/Thr189 residues are homologous to human Thr203/Thr207/Thr212/Thr217residues and phosphorylation of these cause activation of human NKCC1 [12, 13]. On the other hand, dephosphorylation of threonineresidues of KCC causes its activation [14]. The role of different enzymes controlling phosphorylation and dephosphorylation of key residues in regulatory domain of NKCC1 including protein kinase A, protein kinase C, Ste20-related proline-alanine-rich kinase (SPAK), oxidative stress responsive kinases (OSR1), With No K=lysine kinase (WNK) and protein phosphatase type 1 has been documented in various studies [15-19]. The neuronal actions of NKCC1 activation are primarily related to reversal of inhibitory actions of GABA neurotransmitter in the brain regions. Although, activation of GABAA primarily produces inhibitory actions due to inward movement of Cl- ions (hyperpolarization), yet, increase in NKCC1/KCC2 ratio on neurons may lead to intracellular accumulation of chloride ions to alter the Cl- gradient. It is followed by net outward movement of Cl- ions (depolarization) due to opening of GABAA-regulated Cl- channels and there is weakening of inhibitory signals of GABA [20, 21, 22, 23]. The shift in polarity of GABAA in pathological states may be responsible for deleterious effects in number of CNS diseases (Fig. 1).
Fig. (1).
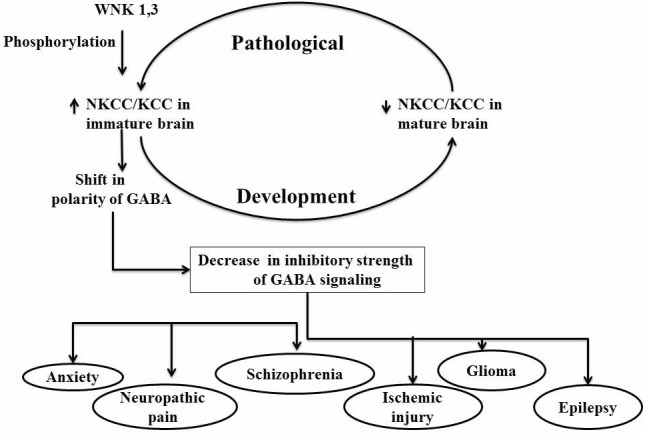
The reversal of NKCC/KCC ratio decreases the inhibitory strength of GABAA signaling, which in turn leads to different pathological states.
The pathophysiological role of NKCC1 has been illustrated in different studies using selective inhibitors of NKCC1. Furthermore, the use of NKCC1 null rodents has also contributed immensely in identifying the role of NKCC1 in normal physiology and pathophysiology of different diseases. Initially, Flagella et al. developed homozygous mutant NKCC1-deficient mouse which exhibited growth retardation, difficulties in maintaining balance, reduced blood pressure and collapsed membranous labyrinth leading to deafness [24]. Yang et al. developed SPAK-null mice with an impaired NKCC1 functioning and these mice exhibited hypotension due to impaired vasoconstriction and recapitulated Gitelman syndrome with hypokalemia, hypomagnesemia, and hypocalciuria [25]. These studies in NKCC1 null mice suggest the important contribution of NKCC1 in maintaining the blood pressure. The selective inhibitors of NKCC1 including bumetanide and furosemide are conventionally employed as diuretics. However, more recent studies have indicated that NKCC1 also plays an important role in various other pathological states including anxiety, cerebral ischemia, epilepsy, neuropathic pain, schizophrenia, fragile X syndrome and glioma and the inhibitors of NKCC1 are shown to produce beneficial effects in different disease models (Table 1). During cerebral ischemia, there is an up-regulation of NKCC1 expression in different brain regions, which is secondary to elevation of glutamate and activation of p38 and JNK MAP kinases [26, 27]. An increased expression of NKCC1 causes intracellular accumulation of chloride ion to shift GABA-mediated hyperpolarisation to depolarization, which may be responsible for inducing seizures [28]. GABAA-mediated depolarization leads to the up-regulation of p75NTR, which may be involved in neuronal cell death and application of NKCC inhibitor prevents p75NTR up-regulation and neuronal death in the injured areas [29]. Nerve injury may up-regulate the expression of NKCC1 in the dorsal horn due to activation of Ca2+/CaM kinases through stimulation of transient receptor potential vanilloid type 1 to induce allodynia [30]. The present review describes the role of NKCC1 in the pathophysiology of these different diseases.
Table 1.
Pharmacological actions of NKCC1 inhibitors, bumetanide and furosemide along with their doses in different disease models.
| Sr. No. | Intervention | Effect | Reference |
|---|---|---|---|
| Anxiety | |||
| 1. |
Furosemide (100 mg/kg)/ Bumetanide (70 mg/kg) |
|
Krystal et al., 2012 |
| Cerebral ischemia | |||
| 2. | Bumetanide (100 µmol/L) 10 µM 7.6 -30.4 mg/kg SB-239063 SP-600125 |
|
Yan et al., 2001 Yan et al., 2003 Pond et al., 2006 Beck et al., 2003 O'Donnell et al., 2004 Wallance et al., 2012 |
| Epilepsy | |||
| 3. | Furosemide (50 mg/kg) Bumetanide (10 µM) Bumetanide Or genetic knockdown of NKCC1 |
|
Hochman, 2012 Ostby et al., 2009 Nardou et al., 2009 Dzhala et al., 2010 Haas and Sontheimer 2012 |
| Neuropathic pain | |||
| 4. | Furosemide (32.0±6.9 µg) Bumetanide (105.6±99.1 µg/paw) (10-500µM) (30 mg/kg) Antisense oligodeoxynucleotides Tyrosine receptor kinase B blocker (K252a 2µg) |
|
Granados-Soto et al., 2005; Prescott et al., 2006 Lin et al., 1999 Ita et al., 2006; Pitcher and Cervero, 2010 Cramer et al., 2008 Zhang et al., 2008 Yong et al., 2010 |
TYPES, STRUCTURE AND DISTRIBUTION
NKCC is a unique family of transporters, and its two members NKCC1 and NKCC2 have been identified so far [4]. NKCC1 is widely distributed throughout the body and is present on variety of secretory epithelial and non-epithelial cells, while NKCC2 is specifically located on kidney [1]. Structurally, these co-transporters have ~1,200 amino acids that are arranged into 12 α-helical transmembrane-spanning domains [31]. It is reported that the central ~500-amino acid residue region is arranged into 12 transmembrane domains with connecting loops and has N-linked glycosylation between 7 and 8 transmembrane segments [8]. The second trans-membrane region is highly conserved for NKCC and is an important site that determines the binding of cations (Na+ and K+) as well as antagonist (including bumetanide). Furthermore, bumetanide binding determinants are also present on transmembrane 7, -11, and -12. On the other hand, the chloride binding determinants are present on transmembrane 4 and transmembrane 7. Studies have shown that the binding of different ions and bumetanide to NKCC, and ion transport depends on the central ~500 amino acids residue. NKCC1 has mainly three regions, hydrophobic region, the amino-terminal-region with at least one phosphorylation site and the carboxy-terminal region with several phosphorylation sites [7] (Fig. 2).
Fig. (2).

Structure of NKCC1 with important binding sites for chloride ions and bumetanide along with important phosphorylation sites at the N terminal end of channel. NKCC1 is secondarily active transporter and is activated depending on sodium gradient created by Na+ K+ ATPase. The decrease in intracellular sodium activates NKCC1 which mediates transport of Na+, K+ and Cl- ions into the cell.
The trans-membrane regions of both members are highly conserved, with 75–90% similarity between the NKCC1 and NKCC2 [8]. Both have N-linked glycosylation sites on their extracellular loop [32]. These two members are encoded by two different set of genes. Genes encoding mouse NKCC1 are localized on chromosome 18 [3], whereas the genes encoding NKCC2 are located on chromosome 2 [33]. The genes encoding human NKCC1 are localized on chromosome 5 and genes for NKCC1 are localized on chromosome 15 [34]. In the kidneys, NKCC1 is present on the luminal surface of the macula densa, apical membrane of thick ascending limbs, juxtaglomerular granular cells, in the outer and inner medullary collecting duct, loop of Henle, afferent arteriole and extraglomerular mesangium [35]. The localization of NKCC1 in the central nervous system has also been demonstrated by northern blotting and in situ hybridization studies [36, 37]. Immunocytochemical studies have shown the NKCC1 protein expression on the plasma membranes of the neurons throughout the brain, especially on pyramidal cells of the cerebral cortex, striatum, hippocampus [38] and also on the dorsal root ganglia of the spinal cord [39]. Furthermore, the localization of NKCC1 has been reported on the glial cells [40]. Within the cardiovascular system, extensive distribution of NKCC1 in cardiomyocytes, fibroblasts, vascular endothelial cells, smooth muscle cells and red blood cells has been described [1, 41]. NKCC1 is also localized throughout the outer plexiform layer of the distal retina, a synaptic lamina that is comprised of the axon terminals of photoreceptors and the dendrites of horizontal and bipolar cells [42]. The NKCC2 isoform has been identified only in the medullary regions of the kidney, the loop of Henle and the juxtaglomerular apparatus [5, 43].
ACTIVATION AND INHIBITION OF NKCC1
The wide varieties of protein-mediated biological activities are regulated by protein phosphorylation and dephosphorylation, and it is shown that NKCC1 protein must be phosphorylated to mediate cotransport of ions. It is reported that NKCC1 protein present in the shark rectal gland is regulated by direct and reversible phosphorylation at serine and threonine residues in response to cAMP and calyculin-A [44]. Studies have demonstrated a direct relationship between degree of ATP phosphorylation and NKCC1 activation. ATP is required to generate cAMP in response to the agonist for cAMP-mediated response and an increase in cellular levels of cAMP stimulate NKCC1 transporter [45]. ATP also provides the necessary phosphate groups for cAMP-activated protein kinases, such as protein kinase A and protein kinase C, which mediate protein phosphorylation of NKCC1 and NKCC2 [3, 18]. It is also proposed that cAMP may also stimulate NKCC1 indirectly by increasing the permeability of the chloride channels present on apical membrane leading to Cl- secretory efflux and reducing intracellular Cl- ion concentration [46].
However, cAMP-mediated NKCC1 activation is primarily mediated through activation of protein kinase A and protein kinase C. Khurihara et al. demonstrated that protein kinase A is involved in NKCC1 phosphorylation in rat acinar basolateral membranes by activation of endogenous membrane-associated kinases or inhibition of phosphatases present in acinar basolateral membranes [15]. Protein phosphatase type 1 is an important regulatory enzyme which binds to the NKCC1 via RVXFXD sequence present in N-terminus sites of regulatory subunit to promote dephosphorylation and inactivation of NKCC1 [47]. Protein kinase C may activate Ste20-related proline-alanine-rich kinase (SPAK), which subsequently binds to the N-terminal of NKCC1 and causes its phosphorylation during hyperosmotic stress in human airway epithelial cells [18, 48]. Various studies also have investigated the involvement of With No K=lysine kinases (WNK) in phosphorylation and activation of Ste20-related proline/alanine-rich kinase/Oxidative stress responsive kinases (SPAK/OSR1) that subsequently regulate phosphorylation and activation of NKCC1 [19, 49]. Hypertonic stress (cell shrinkage), and decreased intracellular Cl- ion levels trigger the phosphorylation and activation of WNKs to promote NKCC activation and KCC inhibition via phosphorylation of critical amino acids. Furthermore, silencing of WNK kinase activity promotes NKCC inhibition and KCC activation via dephosphorylation [50,51]. In fact, the three isoforms of WNK including WNK1, WNK2 and WNK3 (but not WNK4) are activated by auto-phosphorylation of their Thr243, Ser382, Ser383, Ser1261 residues in response to stress. In turn, activated SPAK/OSR1 regulates the phosphorylation of Thr203, Thr207 and Thr212 residues of NKCC [52]. However, it is proposed that WNK4 in association with calcium binding protein 39 (cbp39) may also activate NKCC1 in a SPAK/OSR1-independent manner (Table 2) [17, 53]. Among the different isoforms, WNK1 and WNK3 are highly expressed in the brain and their modulation may control the activation and deactivation of cation cotransporters to produce the beneficial effects in diseases involving altered ratio and functioning of NKCC1 and KCC2 [50].
Table 2.
Different enzymes involved in phosphorylation and dephosphorylation of NKCC1
| Sr. No. | Kinases/ Phosphatase Enzyme |
Phosphorylation/ Dephosphorylation Site |
Comment | Reference |
|---|---|---|---|---|
| 1. | Ca2+/Camodulin-kinase-II | NKCC1 phosphorylation |
|
Galena and Cervero, 2005 |
| 2. | Protein kinase A | NKCC1 phosphorylation |
|
Khurihara et al., 2002 |
| 3. | Protein phosphatase type 1 | NKCC1 dephosphorylation |
|
Darman et al.,2001; Matskevich et al., 2005 |
| 4. | Protein kinase C | NKCC1 phosphorylation |
|
Dowd and Forbush, 2003; Smith et al., 2008 |
| 5. | With No K=lysine kinases (WNK) WNK1, WNK2 and WNK3 WNK4 |
NKCC1 phosphorylation NKCC1 phosphorylation NKCC1 phosphorylation |
|
Susa et al., 2012; Zeniya et al., 2013 Thastrup et al., 2012 Gagnon et al., 2011; Ponce-Coria et al., 2014 |
Darman and Forbush demonstrated that the activation of NKCC isolated from shark rectal gland is directly controlled by phosphorylation of three phosphoacceptor sites at Thr184, Thr189 and Thr202 which constitute the regulatory domain present in the N-terminal of NKCC1. These phosphoacceptor sites are highly conserved between NKCC1 and NKCC2, thus both NKCC1 and NKCC2 are activated by phosphorylation in a similar manner. The phosphorylation of Thr189 is essential for the NKCC1 activation, on the other hand, Thr184 and Thr202 has modulatory action [11]. Similarly, it was reported that shark Thr175/Thr179/Thr184/Thr189 residues are homologous to human Thr203/Thr207/Thr212/Thr217residues and phosphorylation of these cause activation of human NKCC [12, 13]. Gimenez and Forbush investigated the activation of NKCC2 expressed in Xenopus laevis oocytes through the phosphorylation of three N-terminal threonines such as Thr99, Thr104 and Thr117 [54].
Besides ATP and cAMP, increased levels of excitatory neurotransmitters such as glutamate, N methyl-D-aspartate and glutamate receptor agonist trans-ACPD (1-amino-1,3 dicarboxycyclopentane) may also contribute significantly in activating the cotransporter activity in the neurons [55]. The activation of NKCC1 is also regulated by sodium potassium ATPase pump (Na+ K+ ATPase), which tends to decrease the intracellular sodium levels. Indeed, NKCC1 is secondarily active transporter and is activated depending on sodium gradient created by Na+ K+ ATPase. The decrease in intracellular sodium activates NKCC1 which mediates transport of Na+, K+ and Cl- ions into the cell. Bumetanide is a member of sulfamoylbenzoic acid loop diuretic family and exerts its diuretic action by blocking the NKCC in the thick ascending limb of loop of Henle [1]. It blocks both NKCC1 and KCC2 cation-chloride co-transporters with about 500-fold greater affinity for NKCC1 than for KCC2 [56]. Studies have shown that bumetanide is heavily bound to plasma proteins (~98%) and is highly ionized at physiological pH, so that it poorly penetrates into the brain [57]. Furthermore, it is rapidly eliminated in rodents because of extensive oxidation of its N-butyl side chain. The elimination half-life of bumetanide in mice is 47 min and in rats is 13 min. In humans, the half-life is between 1 and 1½ hours. Accordingly, scientists have employed piperonyl butoxide inhibitors to inhibit the oxidation and increase the activity of bumetanide in rodents [58]. Furthermore, scientists have developed the lipophilic and uncharged prodrugs of bumetanide that penetrate the blood-brain barrier more easily than the parent drug [57]. Furosemide is a loop diuretic and is involved in blockade of both NKCC1 and the KCC2, but it produces more potent action against the KCC2 [8, 50]. Torasemide 1-isopropyl-3-{4-(3-methyl-phenylamino) pyridine-3-sulfonyl} urea is a loop diuretic of the class pyridine sulfonylurea, which blocks the action of NKCC [59]. Piretanide is also an inhibitor of NKCC [60].
NKCC1 AND GABAA RECEPTOR-MEDIATED NEUROTRANSMISSION
GABA is the primary inhibitory neurotransmitter present throughout the mammalian central nervous system. However, activation of GABAA receptors may also produce excitatory actions depending upon the Cl- gradient [61, 62]. Generally, low intracellular Cl- ions concentration is maintained in the neurons and activation of GABAA receptors causes hyperpolarization due to inward flow of Cl- ions. However, maintenance of high intracellular Cl- ion levels (due to activation of NKCC1) imparts excitatory actions to GABAA due to outward movement of Cl- ions [22, 63]. Indeed in the early developing brain, activation of GABAA primarily produces excitatory actions. However, there is developmental switch in GABA polarity in which the excitatory actions of GABAA are switched to inhibitory actions with the progression of development [64]. During an early development (early after birth), the excitatory GABA acts a trophic factor in the cortex [65] and it activates a number of signaling cascades to promote the dendritic development, and synaptogenesis [66]. The early excitatory depolarizing actions of GABAA activation are due to high neuronal expression of NKCC1, which tends to increase the intracellular Cl- ions levels. However, during the later stages of development, there is switch in polarity of GABA i.e., there is change from excitatory to inhibitory functions of GABAA with the progression of development. The shift in polarity of GABAA responses occurs in rodent hippocampus during the first 2 postnatal weeks [67]. This developmental switch of polarity is attributed to the increase in KCC2 (which extrudes Cl- from cells) and decrease in NKCC1 expression [68]. Accordingly, the relative ratio of these transporters determines the intracellular Cl- concentration and polarity of GABAA actions [64, 68]. In pathological conditions, there may be the recapitulation of developmental-like state, i.e., increase in the ratio of NKCC1/KCC2 leading to shift in the polarity of GABA actions and decreasing the strength of inhibitory GABAergic neurotransmission (Fig. 3).
Fig. (3).

In the early developing brain, GABA primarily produces excitatory actions due to high NKCC1/KCC2 ratio. As a consequence, there is an increased intracellular Cl-ions, which leads to depolarizing actions of GABA due to outward movement of Cl- ions. However, as the development progresses, the ratio of NKCC1/KCC2 ratio reverses and there is switch in the polarity of GABA actions due to decreased intracellular Cl- ions and GABA acquires inhibitory actions due to inward movement of Cl- ions. The recapitulation of developmental-like state during pathological state may be associated with increase in the expression and functioning of NKCC1, which decreases the strength of inhibitory GABAergic neurotransmission.
NKCC1 AND GLUTAMATE
There have been studies suggesting that activation of NKCC1 may participate in glutamate-mediated excitotoxicity. A study by Schomberg et al. described that the activation of NMDA receptors stimulates the NKCC1 activity in neurons [69]. Beck et al. described that pretreatment of cortical neurons with bumetanide prevents glutamate-induced neuronal cell death. Furthermore, bumetanide significantly attenuated glutamate-induced rise in intracellular Na and Cl ions in a significant manner suggesting that activation of NMDA receptors is followed by activation of NKCC1, which contributes in increasing Na and Cl- ions inside the cells. However, inhibition of NKCC1 after glutamate treatment had no effect on neuronal death suggesting that NKCC1 plays an important role during the initial stages of glutamate-mediated neurotoxicity [26].
KCC2 AS NEUROPROTECTIVE AND ITS COMPLEX REGULATION BY BRAIN DERIVED NEURO-TROPHIC FACTOR (BDNF)
KCC2 gene generates two isoforms, KCC2a and KCC2b and during the development, there is a significant increase in the expression of KCC2 in the brain. Among its two isoforms, the expression levels of KCC2a mRNA in hippocampus are relatively constant; whereas KCC2b mRNA increases significantly suggesting that the developmental increase in the expression of KCC2 is mainly due to the KCC2b isoform [70, 71]. The role of KCC2 is implicated in the regulation of neuronal migration, dendritic outgrowth and formation of the excitatory and inhibitory synaptic connections. Its function or expression is suppressed under several pathological conditions including neuronal trauma, epilepsies, axotomy of motoneurons, neuronal inflammations and cerebral ischemia. Pellegrino et al. demonstrated the neuroprotective role of KCC2 as knock out of the KCC2 gene in rat hippocampal neurons was shown to increase the intracellular chloride concentration and compromise neuronal survival. The rescue of the KCC2 activity through its over-expression effectively restored chloride concentration and enhanced neuronal resistance to oxidative stress and glutamate-induced excitotoxicity suggesting the novel neuroprotective role of endogenous KCC2 [72]. Khalilov et al. demonstrated the enhanced synaptic activity and epileptiform events in the embryonic KCC2 deficient hippocampus [73]. Lee et al. reported that the enhanced glutamatergic activity may possibly down-regulate the KCC2 to enhance the depolarizing and excitotoxicity actions of GABAA receptors [74] suggesting that glutamate-mediated neuronal toxicity may be partially attributed to decreased expression of neuroprotective KCC2.
The expression of KCC2 is regulated by BDNF through a complex signaling cascade during normal development and under pathological conditions. It has been shown that the levels of KCC2 mRNA are increased in BDNF overexpressing embryos [75]. Furthermore, the expression of KCC2 is decreased in TrkB (tyrosine receptor kinase B) receptor knock-out mice [76]. Ludwig et al. demonstrated that BDNF robustly increases the expression of dependent KCC2 gene via the ERK1/2 pathway in immature neurons [71]. In contrast to immature condition, into more mature conditions the expression of BDNF decreases the KCC2 expression. In contrast to physiological role of BDNF in increasing the expression of KCC2, an increase in BDNF during neuronal injury tends to decrease the expression of KCC2 [29, 77] suggesting the dual role of BDNF in regulating the KCC2 expression in normal and pathological states.
ROLE OF NKCC1 IN DIFFERENT DISEASES:
NKCC1 in Anxiety
Krystal et al. studied the anti-anxiety role of the NKCC1 blockers in rat model of anxiety. Administration of furosemide (100 mg/kg I.V.) and bumetanide (70 mg/kg I.V.) was shown to exert significant anxiolytic effects in the conditioned models of anxiety (contextual fear-conditioning and fear-potentiated startle), but not in the unconditioned models of anxiety (elevated plus maze and open- field test). The exact reason for their selective anxiety attenuating actions in conditioned models of anxiety (but not in unconditioned models) was not explained in the study. Since in that study only a single dose of each drug was employed, it may not be completely ruled out that these drugs may produce anxiolytic effects in the unconditioned models of anxiety at higher doses [40]. Studies have demonstrated that furosemide and bumetanide antagonize the NKCC1 (present on both neurons and glial cells), and KCC2 cotransporter (present on neurons) [63, 78, 79, 80]. However, as compared to furosemide, bumetanide is more specific inhibitor of NKCC1 than KCC2 [60]. Based on this, it was proposed that antagonism of NKCC1 rather than KCC2 is responsible for the documented anxiolytic effects of furosemide and bumetanide. Had the KCC2 antagonism be responsible for antianxiety effects, then bumetanide would have been ineffective in anxiety models. The authors linked the anxiety relieving actions of NKCC1 antagonists to enhanced GABAA signaling in the central nervous system. As a consequence of inhibition of Cl- cotransporter, these medications increase the transmembrane chloride gradients in the neurons and thus, increase GABA-induced hyperpolarizing inhibitory postsynaptic potentials [67, 78]. The possible changes in the GABAergic neurotransmission may be associated with the changes in connectivity triggered by dendritic spines loss and sprouting. Furthermore, due to close inter-relationship between GABA and glutamate, the changes in glutamatergic action may also be altered. However, experimental studies are needed to explore these changes associated with anti-anxiety effects of bumetanide and furosemide.
NKCC1 in Cerebral Ischemia
Loss of blood supply to the brain (cerebral ischemia) initiates a cascade of cellular events, which significantly disturb the neuronal ionic homeostasis and produce neuronal death in the hippocampus and other brain regions. However, the molecular events and the consequences are not same in all ischemic conditions and depend upon the severity of ischemic insult. Studies have documented that intracellular Cl- concentrations rises in the rat hippocampal neurons in response to oxygen glucose deprivation, an in vitro model of ischemia [81, 82] and the maintenance of extracellular Cl- at 10 mM during reoxygenation and glucose supplementation has been shown to reduce the neuronal damage [38]. During ischemia, increased NKCC1 activity may be responsible for ischemia-induced intracellular Cl- and Na+ ions accumulation, which in turn causes neuronal injury due to hyperexcitability of neurons [83]. Yan et al. demonstrated the increased expression of NKCC1 in the brain cortex in cerebral ischemia model of 2 h left middle cerebral artery occlusion (focal cerebral ischemia) and 24 h reperfusion in male spontaneously hypertensive rats. Microdialysis of 100 µmol/L of bumetanide transiently inhibited the NKCC1 activity in the brain and inhibition of co-transporter with bumetanide decreased ischemic infarct volume and brain edema. It was proposed that bumetanide inhibits the co-transporter mediated Cl- influx in neurons and consequently, reduces ischemic neuronal damage [84]. Pond and co-workers demonstrated that bumetanide reduces intracellular Cl- accumulation and neuronal damage in the hippocampal neurons of adult transgenic mice subjected to 8 min of oxygen-glucose deprivation and reoxygenation in the presence of glucose [38].
Beck et al. reported the neuroprotective role of bumetanide in two in vitro models of ischemic cell death i.e., glutamate (100 µM)-induced excitotoxicity (in incubated cortical neurons for 24 hr) and oxygen glucose deprivation (3 hrs)-reoxygenation (21 hrs) (an in vitro model for ischemia and reperfusion). It was demonstrated that inhibition of NKCC1 with 10 µM bumetanide abolishes glutamate-mediated neurotoxicity as well as oxygen glucose deprivation-induced neuronal death. However, neuroprotective effects of bumetanide due to NKCC1 inhibition were observed only with its 30 min pretreatment before glutamate and oxygen glucose deprivation suggesting the involvement of NKCC1 in initial stages of cell damage during excitotoxicity [26]. Yan et al. demonstrated the role of NKCC1 in cerebral edema and neuronal damage during cerebral ischemia in male spontaneously hypertensive rats. In cerebral cortex and striatum of rat brain, the expression of NKCC1 was increased during 2 h of cerebral ischemia and was persistent during 24 h of reperfusion. Furthermore inhibition of NKCC1 by bumetanide 100 µM was shown to reduce cerebral edema and neuronal damage during cerebral ischemia [84].
O'Donnell and co-workers demonstrated that intravenous administration of bumetanide (7.6 mg/kg) inhibits ischemia-induced cerebral edema in rats subjected to middle cerebral artery occlusion [85]. Brillault et al. demonstrated that hypoxia-induced increase in cells swelling is associated with late increase in Na+ content of the cells (after 3-5 h of hypoxia, but not after 1 h). Furthermore, administration of bumetanide was shown to reduce hypoxia-induced cell swelling along with rise in Na+ content. Based on this, it was hypothesized that during early stages of cerebral ischemia, various ischemic factors cause stimulation of NKCC localized on luminal side of blood brain barrier to transport Na+ and water from the blood to the brain to produce cerebral edema [86]. Liu et al. demonstrated the role of NKCC in edema formation in young (10-12 weeks) and aged (15-16 months) mice in middle cerebral artery occlusion model of ischemic stroke. Using western blot analysis, it was demonstrated that NKCC1 expression is increased in ischemic penumbra of young mice as compared to aged mice. Intravenous administration of bumetanide (30.4 mg/kg) was shown to decrease ischemia-induced edema formation in young mice more effectively as compared to aged mice. It was concluded that young mice had more edema formation probably due to increased NKCC1 expression during an ischemic stroke than aging animals, and accordingly bumetanide was more effective in blocking edema in young ones [87].
Yang et al. linked the neuroprotective effects of biphalin (a non-selective opioid receptor agonist) in in vitro and in vivo models of stroke to decreased expression of NKCC1 in the brain [88]. During cerebral ischemia, various mechanisms may be responsible for increased expression or activity of NKCC1 in the brain region. Lee et al. described the increase in NKCC1 mRNA and protein expression in the cortex region in middle cerebral artery model of neuronal injury in rats. This study suggested that during cerebral ischemia the epigenetic mechanisms such as methylation and demethylaton regulate the expression of NKCC1. The promoter sites of NKCC are formed by cytosine guanine dineucleotide in the form of cytosine-phosphodiester-guanine and the process of methylation of the promoter sites of NKCC decreases the expression of NKCC. During methylation of the promoter site of NKCC, methyl group is transferred to cytosine in cytosine-phosphodiester-guanine dineucleotide (catalysed by DNA methyltransferases) and expression of NKCC1 is decreased. During cerebral ischemia, down-regulation of DNA methyltransferases leads to demethylation of promoter region and subsequently, increases the expression of NKCC1 [89]. Recently, Wallace and co-workers demonstrated that activation of p38 and JNK MAP kinases may be involved in NKCC activation in cerebral microvascular endothelial cells during ischemia. This study reported that selective inhibition of these kinases by SB-239063 and SP-600125 decreases the ischemic stimulation of NKCC and reduces the brain damage [27].
Furthermore, an increase in NKCC1 expression or activation during ischemia may be ascribed indirectly to ischemia-induced rise in glutamate and calcium levels [2, 55, 69]. Schomberg and co-workers described that administration of glutamate receptor agonists i.e. trans-(6)-1-aminocyclopentanetrans-1,3-dicarboxylic acid and 3,5-dihydroxyphenylglycine causes stimulation of NKCC1 in the cortical neurons. It was suggested that activation of glutamate receptors by trans-(6)-1-aminocyclopentanetrans-1,3-dicarboxylic acid and 3,5-dihydroxyphenylglycine increases the intracellular Ca2+ levelsand activation of Ca2+/CaM kinases stimulates NKCC1 [69]. Similarly, Sun and Murali described that glutamate ionotropic receptor (NMDA) and the metabotropic receptor agonists (trans-(6)-1-aminocyclopentanetrans-1,3-dicarboxylic acid) stimulate NKCC by increasing intracellular Ca2+ ion concentration in the human neuroblastoma (SH-SY5Y) cells [55]. Su and co-workers demonstrated an upregulation in NKCC1 protein expression in the cortical astrocytes cultures of rats due to high extracellular K+ ion and stimulation of NKCC1 in astrocytes in a Ca2+-dependent manner [2]. However, an increase in NKCC1 expression may not be solely responsible for the deleterious effects observed during cerebral ischemia. It has been shown that the activation of NMDA receptors is also associated with the down-regulation of KCC2 [74], therefore, it may be possible that glutamate-induced neuronal toxicity may be secondary to increased expression of NKCC1 and decreased expression of neuroprotective KCC2. Furthermore, it is shown that bumetanide prevents the up-regulation of pan-neurotrophin receptor (p75(NTR)) and reduces the levels of endogenous BDNF in the injured areas to rescue injured neurons [29]. Along with it is also shown that BDNF reduces the expression of KCC2 in injured neurons [77]. Therefore, it is also possible to suggest that NKCC1 blockers may additionally produce the beneficial effect by decreasing the dependency of injured neurons on BDNF and increasing the expression of neuroprotective KCC2 (Fig. 4).
Fig. (4).

Ischemicinjury causes stimulation of glutamate receptors which increases the release of Ca2+/CaM kinases and cAMP that enhances the expression of NKCC. Similarly p38 and MAP kinases activated due to ischemia that causes upregulation of NKCC. Ischemic injury also leads to down-regulation of DNA methyltransferases which causes increased expression of NKCC.
NKCC1 in Epilepsy
Epilepsy is one of the common neurological disorders characterized by seizures resulting from abnormal, excessive or hyper-synchronous neuronal activity in the brain [90]. Research evidences suggest that neuronal hyper-excitability and hyper-synchronization is the result of disruption of delicate balance between the excitatory and inhibitory synaptic activity. Dzhala et al. described that the NKCC1 blocker (bumetanide) decreases the Cl- accumulation in the immature neurons, suppresses epileptiform activity in the hippocampal slices and attenuates electrographic seizures in neonatal rats. However, bumetanide had no effect in the presence of the bicuculline (GABAA receptor antagonist) and in brain slices from NKCC1-knockout mice suggesting that NKCC1 facilitates seizures in the developing brain [91, 92]. The same group described the role of NKCC1 in neonatal seizures in intact hippocampal slices prepared from neonatal rats and transgenic mice expressing clomeleon, a fusion protein consist of the Cl--sensitive yellow fluorescent protein and the Cl--insensitive cyan fluorescent protein. By using clomeleon imaging, the study suggested that NKCC1 gradually increases the intracellular chloride concentration of neurons, which shifts GABA-mediated hyperpolarization to depolarization to facilitate the seizures. It was shown that bumetanide inhibits the chloride accumulation in neurons and consequently reduces recurrent seizures [93]. Sen et al. demonstrated the increased expression of NKCC1 in pharmacotherapy resistant refractory human epilepsy including hippocampal sclerosis and focal cortical dysplasia. An increased expression was found in surgically resected adult human hippocampal sclerosis and in dysplastic neurons, whereas the immunoreactivity for NKCC1 was uniform in normal nonepileptic cortex. The hippocampus with granule cells dispersion had higher concentrations of GABAA on their dendrites along with high expression of NKCC1 within the granule cell layer as compared to cells without granule cell dispersion suggesting that in former cases GABA is rendered less capable of hyperpolarisation due to abundant NKCC1. It was proposed that in refractory human epilepsy (hippocampal sclerosis and focal cortical dysplasia), the over-expression of NKCC1 may lead to higher intracellular chloride concentrations within the neurons. Therefore, it becomes difficult for GABA to generate inhibitory hyperpolarizing potentials or even causes GABA to become excitatory leading to development of seizures [94].
Conti and co-workers demonstrated that alterations in the balance of NKCC1 and KCC2 activity may decrease the hyperpolarizing effects of GABA and therefore, contribute to epileptogenesis in human brain tumors. By injecting surgically resected peritumoral cortical tissues of epileptic patients afflicted by gliomas and cortical tissues of non-epileptic patients into Xenopus oocytes, an increased immunoreactivity of NKCC1 in oocytes leading to GABA-evoked depolarizing potential was demonstrated as compared to those from nonepileptic healthy cortex. The blockade of NKCC1 using bumetanide or activation of KCC2 using TPEN N,N,N¢,N¢-tetrakis-(2-pyridylmethyl)-ethylenediamine (Zn2+ chelator), reverted the depolarization shift in oocytes injected with epileptic peritumoral tissue similar to that observed in oocytes injected with nonepileptic cortex. It was proposed that increased expression of these co-transporters might cause a decrease of GABAergic inhibition leading to increased excitotoxicity [95]. Oriaifo and co-workers described the antiepileptic effects of furosemide (50 mg/kg) in leptazol-induced seizures in mice [96]. Studies have suggested that the non-synaptic mechanisms are also very important in producing antiepileptic effects of these NKCC blockers. It was described that the non-synaptic mechanisms change the ionic concentrations by transporting extracellular potassium and chloride ions into glial cells through NKCC1. It leads to generation of osmotic gradients between extracellular and intracellular compartments, which cause the movement of water from extracellular to intracellular compartments and thereafter, to glial cells. These changes are associated with reduction in extracellular volume fraction (defined as proportion of a volume of brain tissue that is composed of extracellular space) [97, 98]. The treatment with bumetanide and furosemide blocks the changes in osmolarity of extracellular spaces to produce antiepileptic effect. The normalization of extracellular volume fraction (non-synaptic mechanism) in the presence of NKCC1 blocker to produce antiepileptic effects was demonstrated with the help of optical imaging of hippocampal slices [99].
Nardou and collaborators demonstrated that inhibition of NKCC1 by bumetanide blocks the expression of seizures, but does not efficiently prevent the long-lasting episodes of recurrent seizures. Using preparation of two intact interconnected hippocampal formations of neonatal Wistar rats, this study described that administration of bumetanide (10 µM) does not prevent generation of epileptogenic focus, but blocks the spontaneous focus seizures. It was proposed that during recurrent seizures, GABA mediates the massive influx of Cl- to produce permanent accumulation of intracellular chloride ions that overcome the inhibitory effect of bumetanide on NKCC1 [22]. Using immunocytochemistry, Munoz et al. demonstrated the changes in the relative expression of NKCC1 and KCC2 in the neurons of the subicular region adjacent to sclerotic areas of the hippocampus. In the transition between the subiculum with sclerotic regions of CA1, the double immunolabeling of NKCC1 and KCC2 revealed that 20% of the NKCC1-immunoreactive neurons do not express KCC2 [100]. Earlier Huberfeld et al. showed that nearly all dentate granule cells express both mRNA and proteins of KCC2. However, KCC2 was absent from approximately 30% of CaMKIIalpha (calcium/calmodulin-dependent protein kinase IIalpha)-positive subicular pyramidal cells. Furthermore, it was reported that all cells that were hyperpolarized during interictal events were immunopositive for KCC2, whereas the majority of depolarized cells were KCC2 immunonegative [101]. A preclinical study by de Guzman et al. reported that the excitability of subiculum network is increased in the rodent model of temporal lobe epilepsy, which was attributed to reduction in mRNA of KCC2 in the subiculum of pilocarpine-treated rats [102] (Fig. 5).
Fig. (5).

Epileptogenic stimuli enhance the expression of NKCC that causes intracellular accumulation of chloride ions which shift GABA mediated hyperpolarization to depolarization. Thus, depolarization lead to development of seizures. Also epileptogenic stimuli decreases the expression of KCC that causes intracellular accumulation of chloride ions lead to generation of osmotic gradient between intracellular and extracellular components. This gradient causes movement of fluid to intracellular components that decreases extracellular fluid fraction lead to seizure generation.
Rangroo Thrane et al. demonstrated that ammonia induces seizures by compromising the potassium buffering capacity of astrocytes and over-activating NKCC1 in neurons. Furthermore, genetic deletion of NKCC1 or pharmacological inhibition with bumetanide potently suppressed ammonia-induced neurological dysfunction [103]. Töpfer et al. investigated the effects of inhibition of bumetanide metabolism on the anticonvulsant effects in chronic model of epilepsy. The authors reported that inhibition of bumetanide metabolism by inhibiting the piperonyl butoxide (PBO) increases the brain levels of bumetanide. In the rat kindling model, bumetanide (with or without PBO) did not exert anticonvulsant effects on fully kindled seizures, but dose-dependently altered the kindling development [58]. Töllner et al. demonstrated that the administration of 2 ester prodrugs of bumetanide, the pivaloyloxymethyl and N,N-dimethylaminoethylester results in significantly higher brain levels of bumetanide. Furthermore, these prodrugs were more effective than bumetanide in potentiating the anticonvulsant effect of phenobarbital in the kindling model in rats [57].
Gliomas are frequently associated with the development of seizures, termed as peritumoral epilepsy. Campbell et al. demonstrated the reduced numbers of peritumoral parvalbumin-positive GABAergic inhibitory interneurons and the elevated intracellular Cl- concentration in the remaining peritumoral neurons in mouse glioma model. The increase in intracellular Cl concentration in neurons was attributed to decreased plasmalemmal expression of KCC2, which subsequently induced the depolarizing actions of GABAA. The depolarizing actions of due to activation of GABAA receptors render the peritumoral neuronal networks hyper-excitable and susceptible to seizures triggered by excitatory stimuli [104]. Pallud et al. demonstrated that important contribution of cortical GABAergic excitation to epileptic activities in the neocortical slices from the peritumoral surgical margin resected around human brain gliomas. In glioma cells, chloride homeostasis is perturbed due to reduction in KCC2 and increase in NKCC1 expression in neurons surrounding the glioma. The increase in Cl- ions is in turn associated with depolarizing GABAergic signaling leading to epileptic discharges in glioma patients [105].
In contrast to well documented preclinical as well as clinical reports suggesting the clear role of NKCC1 and its blockers in epilepsy, Brandt et al. did not report the beneficial effects of bumetanide in epilepsy model. The authors investigated the effect of GABA-mimetic antiepileptic drug, phenobarbital (25 mg/kg) alone or in combination with bumetanide i.v infusion (0.8 mg/kg/h) in the pilocarpine model of temporal lobe of epilepsy in adult female rats. Bumetanide infusion did not produce adequate antiepileptic effect. Moreover, antiepileptic effect of phenobarbital was not potentiated during co-administration of bumetanide for 5 days after development of status epilepticus. It was suggested that NKCC1 expression is up-regulated shortly after status epilepticus in rats, but this alteration in expression is not adequate for two weeks after pilocarpine-induced status epilepticus. Therefore, neither inhibition of NKCC1 with bumetanide prevented spontaneous recurrent seizure during status epilepticus nor the effect of phenobarbital was potentiated by concomitant administration of bumetanide [28].
NKCC1 in Neuropathic Pain
The complex pathophysiology of neuropathic pain involves neuronal hyperexcitability in damaged areas of the peripheral or central nervous system characterized by allodynia (pain response to non-noxious stimuli), hyperalgesia (exaggerated pain sensation) and dysthesia [106, 107]. KCC2 strongly modulates the excitability of spinal cord networks and these spinal neuronal networks are more excitable in KCC2 mutant mice [108, 109]. Boulenguez et al. described the role of KCC2 in hyper-excitability of spinal reflexes, decreased synaptic inhibition and spasticity in spinal cord injury (SCI) model. The authors described the down-regulation of KCC2 in the motor-neuron membranes after SCI in rats. The spinal excitability was also increased in KCC2-deficient mice and in intact rats after intrathecal injection of BDNF, which down-regulates KCC2. Furthermore, the sequestration of BDNF at the time of SCI prevented the decrease in KCC2 after SCI and restored the spinal excitability [110]. Morales-Aza and collaborators reported the alteration in mRNA and protein expression of NKCC1 and KCC2 in the dorsal root ganglia and spinal sensory neurons in Mycobacterium tuberculosis-induced arthritis model of neuropathic pain. After 4 days of injection of Mycobacterium tuberculosis suspension (acute arthritis), an increased mRNA and proteins expression of NKCC1 and KCC2 in the superficial layers, but not in deep dorsal horn, was documented. During chronic arthritis (at 10th day), the mRNA expression of NKCC1 remained elevated, but expression of KCC returned to the basal levels. This study suggested that alterations in expression of cation chloride co-transporter (NKCC1 and KCC2) result in GABA-mediated depolarization, which causes increase in neuronal excitability and produce inflammatory pain [111]. Granados-Soto and co-workers demonstrated the role of NKCC1 located on intraspinal and peripheral sites of sensory neurons in formalin- induced neuropathic pain in female rats. This study reported that intrathecal injection of furosemide (32.0±6.9 µg) inhibits both phases, but more potently inhibits phase 2 of formalin-induced pain than bumetanide (194.6±97.9 µg) and piretanide (254.4±104.9 µg). Peripheral (intradermal) injection of bumetanide (105.6±99.1 µg/paw) more potently inhibited phase 1 suggesting that inhibition of intraspinal and peripheral NKCC1 may prevent formalin-induced neuropathic pain [112]. Galena and Cervero demonstrated that mechanical hyperalgesia due to the intracolonic administration of capsaicin is associated with transient induction of NKCC1 phosphorylation due to stimulation of Ca2+/Camodulin-kinase-II in the mouse spinal cord. There was no change observed in the mRNA or protein expression of NKCC1, however, NKCC1 translocation was shown to be increased by 50% in plasma membrane than cytosol. It was proposed that in the lumbosacral portion of the spinal cord, rapid phosphorylation and recruitment of NKCC1 may play a role in development and maintenance of hyperalgesia in response to painful visceral stimulus [113]. Intrathecal administration of bumetanide (10-100µM) reduces dorsal root reflexes, vasodilation, plasma extravasation, allodynia and hyperalgesia in a dose-dependent manner, in response to capsaicin injection in the plantar region of hind paw. It was suggested that bumetanide decreases capsaicin-induced dorsal root reflexes and neurogenic inflammation due to inhibition of NKCC1 in the spinal afferent neurons. NKCC1 activation may enhance primary depolarizations due to the opening of GABAA receptors to generate dorsal root reflexes in the spinal cord [114, 115].
Cramer and collaborators also described the role of NKCC1 and KCC2 in development of neuropathic pain after spinal cord injury in adult male rats using MASCIS impactor. In this study, an increased expression of NKCC1 and reduced expression of KCC2 was reported in the spinal cord tissue on day 14 after spinal cord injury. Furthermore, administration of bumetanide (30 mg/kg i.p.) resulted in reduction of pain behavior in animals. It was suggested that disruption of balance between NKCC1 and KCC2 causes elevation of intracellular chloride, which stimulates interneuronal GABA release and enhances primary afferent depolarizations to produce neuropathic pain [116]. Research study has reported that reduction in expression of KCC2 in rat dorsal horn is involved in development of peripheral inflammatory hyperalgesia in response to complete Freund’s adjuvant injection. Intrathecal injection of antisense oligodeoxynucleotides against KCC2 into naive rats and pharmacological inhibition of KCC2 with furosemide (in dose 3.3µg) was associated with development of hyperalgesia. It was proposed that decreased expression of KCC2 leading to reduction in efflux of chloride ions shifts GABA mediated hyperpolarisation to depolarization, which is responsible for development of hyperalgesia [23, 117]. Earlier research study has also described the reduction in KCC2 expression in phase 1 of flinching behavior in formalin induced pain model [118].
The role of NKCC1 and KCC2 in trigeminal caudalis of mice in lipopolysaccharide-induced dental pain due to inflammation of tooth pulp has also been described. Real-time reverse transcriptase-polymerase chain reaction revealed the decrease in mRNA expression of KCC2 and increase in expression of NKCC1 in dental tissue. Furthermore, blockade of brain-derived neurotrophic factor-tyrosine receptor kinase B pathway by intracisternal injection of tyrosine receptor kinase B blocker (K252a 2µg) produced anti-nociceptive behavior in lipopolysaccharide treated mice [119]. It is proposed that lipopolysaccharide increases the release of endogenous BDNF which may activate tyrosine receptor kinase B to down-regulate the expression of KCC2. The latter changes are responsible for hyperexcitability and pain due to loss of GABA-mediated inhibition in lipopolysaccharide treated mice [23, 77, 119, 120]. Various other studies have reported that increased release of endogenous brain-derived neurotrophic factor leads to reduction of KCC2 expression via activation of tyrosine receptor kinase B in the spinal cord neurons [77, 118, 121]. Intrathecal administration of bumetanide has been shown to reduce rapid eye movement sleep deprivation (48hr)-induced mechanical pain hypersensitivity in adult male rats [122]. Pitcher and Cervero demonstrated that local application of bumetanide (500 µM) reduces capsaicin-induced sensitization of wide dynamic range and nociceptor specific neurons in dorsal root ganglia in male rats. It suggests that enhanced activity of NKCC1 may increase primary afferent depolarization leading to touch-evoked pain (allodynia) and hyperalgesia [123].
Austin and Delpire employed intrathecal injection of selective inhibitor of KCC2 (D4), an inactive structural variant (D4.14) and bumetanide in mice to demonstrate the role of NKCC1 and KCC in hypersensitivity and neuropathic pain model. Bumetanide was used in concentration of 20 µM, as it selectively inhibits NKCC1 without any significant effect on KCC2 at this particular dose. An inactive structural variant (D4.14) produced no effect, but inhibition of NKCC1 by bumetanide produced anti-nociceptive effect in terms of increase withdrawal latency. In contrast, inhibition of KCC2 was shown to decrease the latency period in heat stimulation test suggesting the development of hypersensitization [124]. Pitcher and co-workers described the key role of spinal transient receptor potential vanilloid type 1 and NKCC1 activation in intracolonic capsaicin injection induced visceral allodynia in mice [30]. Intrathecal injection of bumetanide, either before or 4 hrs after establishment of referred allodynia, was shown to attenuate allodynia suggesting that capsaicin-induced allodynia was secondary to NKCC1 activation and was transient receptor potential vanilloid type 1 dependent. The endogenous transient receptor potential vanilloid type 1 agonists released in response to painful stimulus stimulates transient receptor potential vanilloid type 1 receptors present on the dorsal horn to activate extracellular regulated kinase 1 and 2 [125] and Ca2+/Calmodulin-dependent kinase II α [126]. Activation of these kinases increases the phosphorylation of NKCC1, which in turn increases the levels of intracellular chloride ions to induce depolarizing action of GABAA. These changes persistently stimulate Aβ fibers to produce allodynia in neuropathic pain conditions [30, 115, 127] (Fig. 6).
Fig. (6).

Nerve injury or inflammation causes activation of TRPV1 which causes activation of extracellular receptor kinases and Ca2+/CaM kinasesthat lead to the phosphorylation and activation of NKCC. On the other hand nerve injury or inflammation also causes stimulation of BDNF that activate tyrosine receptor B which decreases the expression of KCC. The increased expression of NKCC and reduced expression of KCC increases intracellular accumulation of chloride ions produces depolarization effect of GABA. Thus, GABA mediated depolarization causes stimulation of fiber Aβ that lead to generation of allodynia.
NKCC1 and Schizophrenia
There have been a large number of studies showing the important role of NKCC1 in the pathogenesis of schizophrenia [128]. In a meta-analysis, the NKCC1 locus has been linked to schizophrenia [129] and NKCC1 has been described as a potential susceptibility gene for schizophrenia [130]. Aron and Lewis described the potential involvement of NKCC1 and KCC2 in the pathogenesis of schizophrenia. Although the authors reported no alteration in the mRNA expression of chloride cotransporters, yet the mRNA levels of their regulators such as oxidative stress response kinases (OxSRL) and WNK3 were increased in the dorsolateral prefrontal cortex of schizophrenia patients [131]. WNK 3 is a potent activator of NKCC1 and inhibitor of KCC2 [132, 133]. It also possesses an OXSR1 binding motif [132]. Accordingly, the authors proposed that an increase in WNK3 expression in schizophrenia patients may lead to increased NKCC1 and decreased KCC2 activity through OXSR1 [131]. The resulting alterations in chloride cotransporters in turn alter the GABA signaling and there have been abundant studies showing the alteration in GABA functioning in schizophrenia [134, 135]. There have been other studies also showing the decreased levels of KCC2 mRNA in the hippocampus region of schizophrenia [136,137]. It is described that the ratio of NKCC1/KCC2 determines the maturity of brain development and nature of actions of GABA. The change in NKCC1/KCC2 expression ratio parallels the change of GABA from an excitatory to an inhibitory neurotransmitter. The transition of GABA from excitatory to inhibitory is important for brain development [78, 138]. Hyde et al. described the increased ratio of NKCC1/KCC2 and decreased KCC2 levels in the hippocampus of schizophrenic patients suggesting the immature state of the GABA system. The authors described the abnormalities in GABA signaling due to altered ratio of NKCC1/KCC2 are critical in the development of schizophrenia [139]. A study of Kim et al. describes the critical role of depolarizing GABA, involving NKCC1 and KCC2, in DISC1 (Disrupted-in-Schizophrenia) dependent regulation of neuronal development [140].
NKCC1, Fragile X Syndrome and Autism
Fragile X syndrome is the most common heritable form of mental retardation and is caused by silencing of the Fmr1 gene (fragile X mental retardation 1) and absence of fragile X mental retardation protein. Adusei et al. described the early developmental alterations in GABAergic protein expression in fragile X knockout mice without any significant effect on the levels of NKCC1 and KCC2 [141]. Other studies have also reported the down-regulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome [142, 143]. The decreased GABAergic functioning in fragile X syndrome suggests the possible involvement of NKCC in this mental retardation. He et al. described that the timing of the switch from depolarizing to hyperpolarizing actions of GABA is delayed in the fragile X mice, which may possibly due to concurrent alteration in the expression of the neuronal chloride co-transporter NKCC1 that promotes the accumulation of intracellular chloride [64].
A clinical study has documented that daily administration of bumetanide (1 mg) during a 3-month period decreases the autistic behavior without any side effects suggesting the potential usefulness of NKCC inhibitors in infantile autistic syndrome. It was proposed that reduction in intracellular Cl- ion levels in the presence of butamanide may have prevented the excitatory actions of GABA in the neurological disorder [144]. The same group of workers demonstrated the efficacy of chloride-importer antagonist bumetanide (1 mg daily) in autism in 60 children in double-blind clinical trial for 3 months [145]. Very recently, Tyzio et al. described the elevated intracellular chloride levels and increased excitatory GABA in fragile X rodent models of autism. However, maternal pretreatment with bumetanide was shown to produce long lasting effects and restore the electro-physiological and behavioral alteration in adult offsprings of these models suggesting that over-activation of NKCC may be critical in elevating intracellular chloride levels in autism [146, 147].
NKCC1 in Glioma
Gliomas are the primary brain tumors derived from glial cells with their unusual propensity to diffusely invade the surrounding brain areas. Studies have shown that Cl− ions accumulate intracellularly in immature neurons and glioma. The intracellular Cl− may be maintained at ∼100 mmol/L in glioma cells and electrochemical gradient for Cl− provides the energetic driving force for cell shrinkage during cell invasion. Haas and Sontheimer proposed that the high intracellular Cl- in gliomas may be probably achieved through the action of the NKCC1. It was described that the invading glioma cells preferentially localize NKCC1 at the leading edge and Cl- transport induces local cell volume changes without affecting overall cell volume. By employing pharmacological inhibitor of NKCC1 (bumetanide) or genetic knockdown of NKCC1, using short hairpin RNA (shRNA) constructs, the authors documented the reduced glioma cell migration and described the potential usefulness of NKCC1 inhibitors in treating glioma [148].
DISCUSSION AND FUTURE DIRECTIONS
Summarized role of NKCC1 in Different Diseases
NKCC1 and KCC2 are cation-dependent chloride co-transporters and their activation or inactivation is primarily controlled by phosphorylation or dephosphorylation via enzymes including WNK 1 and 3 [50, 51]. The neuronal actions of NKCC1 activation are primarily related to reversal of inhibitory actions of GABA neurotransmitter. The recapitulation of developmental (immature brain)-like state during pathological state is associated with increase in the ratio of NKCC1/KCC2, which decreases the strength of inhibitory GABAergic neurotransmission [64]. Furthermore, activation of NMDA receptors stimulates NKCC1 [69] and down-regulates KCC2 [74]. Along with it, bumetanide prevents glutamate-induced neuronal cell death [26] suggesting that glutamate-mediated excitotoxicity may be secondary to increased expression of NKCC1 and decreased expression of neuroprotective KCC2. Studies have reported the beneficial effects of NKCC1 blockers in different diseases including anxiety, cerebral ischemia, epilepsy, schizophrenia, glioma etc. Furosemide and bumetanide selectively produce anxiolytic effects in the conditioned models of anxiety without any significant effect in unconditioned models of anxiety [40]. The expression of NKCC1 in the brain regions is increased during cerebral ischemia and NKCC1 blockers attenuate cerebral ischemic injury in vivo models [84; 86] and in in vitro models of ischemia [26]. NKCC1 blocker (bumetanide) suppresses epileptiform activity in the hippocampal slices and attenuates electrographic seizures in neonatal rats [91] and in vivo models of epilepsy [96]. The expression of NKCC1 is increased in pharmacotherapy resistant refractory human epilepsy including hippocampal sclerosis and focal cortical dysplasia [94]. Indeed, the relative increase in expression of NKCC1 with respect to KCC2 in the neurons of the subicular region adjacent to sclerotic areas of the hippocampus may contribute to higher excitability in temporal lobe epilepsy [100, 102]. The down-regulation of KCC2 and increased expression of NKCC1 in response to nerve injury may contribute increase the excitability of spinal cord networks and produce spasticity [108, 109, 116]. The down-regulation of KCC2 during nerve injury and cerebral ischemia has been attributed to increase in BDNF, which tends to decrease KCC2 through activation of TRkB pathway [29, 77]. This is in contrast to physiological signaling during which increase in BDNF tends to increase the expression of KCC2 [75, 76]. In the similar lines, the relative changes in cation chloride cotransporters may contribute in the pathogenesis of schizophrenia, fragile X syndrome, Autism and glioma.
NKCC1 and Cell Volume Regulation
Cation chloride cotransporters help to regulate the cell volume as cell shrinkage activates the kinases, including WNK 1 and 3, which phosphorylate the NKCCs and KCCs to promote their activation and inactivation, respectively. On the contrary, cell swelling or increase in intracellular chloride inhibits kinase activity to promote net dephosphorylation of both cotransporters, thereby resulting in the inhibition of NKCC and activation of KCC. The maintenance of cell volume is critical for proper cell function and survival and alterations of cell volume can jeopardize the structural integrity and intracellular milieu of cells. In pathological conditions including cerebral ischemia, the changes in cellular volume in the form of cellular edema is an important factor in inducing neuronal injury. Accordingly, scientists have shown the effectiveness of NKCC1 blockers including bumetanide in reducing cerebral edema and neuronal damage during cerebral ischemia [84, 86, 87]. Furthermore, treatment with bumetanide and furosemide is shown to block the changes in osmolarity of extracellular spaces and the normalization of extracellular volume fraction is assumed to contribute in producing antiepileptic effect [99].
Future Directions
In anxiety, the possible changes in the GABAergic neurotransmission may be associated with the changes in connectivity triggered by loss and sprouting of dendritic spines. However, experimental studies are needed to explore these changes associated with anti-anxiety effects of bumetanide and furosemide. Furthermore, the changes in dendritic spines as well as changes in dendritic axonal arborization may change the strength of neurotransmission in ischemia and temporal lobe epilepsy. However, the role of NKCC1 in such changes has not been ben explored yet.
Although there have been some studies suggesting the inter-relationship between glutamate and NKCC1, yet most of the studies have implicated the role of shift in polarity of GABA as the possible mechanism in the pathophysiology of diseases in association with NKCC1 and KCC2. Nevertheless, experimental studies are required to elucidate the role of glutamate in NKCC1-induced deleterious effects in different diseases.
The role of cell volume regulation is mainly explored in cerebral ischemia and to some extent in epilepsy. However, due to important role of NKCC1 in the pathophysiology of other diseases also, the important role of cell volume regulation cannot be ruled out in that group of diseases. Accordingly, studies are required to understand the contribution of cell volume regulation in producing beneficial effects of NKCC1 blockers in anxiety, schizophrenia, neuropathic pain etc.
CONCLUSION
The development of immature brain like state during pathological conditions with relative increase in the ratio of NKCC1/KCC2 on neurons may shift the polarity of GABA and there is decrease in the strength of inhibitory GABAergic neurotransmission. The depolarizing actions of GABA produce neuronal excitability to produce deleterious effects in number of CNS diseases. Accordingly, apart from the diuretic action, NKCC1 blockers have been shown to produce beneficial in attenuating the development of diseases including anxiety, cerebral ischemia, epilepsy, neuropathic pain, schizophrenia, fragile X syndrome, autism and glioma. The development of new pharmacological agents with more selective NKCC1 inhibitors may expand their therapeutic spectrum.
ACKNOWLEDGEMENTS
The authors are grateful to Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, India for supporting this study and providing technical facilities for the work.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Haas M., Forbush B., III The Na-K-Cl cotransporter of secretory epithelia. Annu. Rev. Physiol. 2000;62:515–534. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 2.Su G., Haworth R.A., Dempsey R.J., Sun D. Regulation of Na(+)-K(+)-Cl(-) cotransporter in primary astrocytes by dibutyryl cAMP and high [K(+)](o). Am. J. Physiol. Cell Physiol. 2000;279(6):C1710–C1721. doi: 10.1007/s002329900016. [DOI] [PubMed] [Google Scholar]
- 3.Delpire E., Rauchman M.I., Beier D.R., Hebert S.C., Gullans S.R. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J. Biol. Chem. 1994;269(41):25677–25683. [PubMed] [Google Scholar]
- 4.Park J.H., Saier M.H., Jr Phylogenetic, structural and functional characteristics of the Na-K-Cl cotransporter family. J. Membr. Biol. 1996;149(3):161–168. doi: 10.1007/s002329900016. [DOI] [PubMed] [Google Scholar]
- 5.Payne J.A., Forbush B., III Molecular characterization of the epithelial Na-K-Cl cotransporter isoforms. Curr. Opin. Cell Biol. 1995;7(4):493–503. doi: 10.1016/0955-0674(95)80005-0. [DOI] [PubMed] [Google Scholar]
- 6.Geck P., Pietrzyk C., Burckhardt B.C., Pfeiffer B., Heinz E. Electrically silent cotransport on Na+, K+ and Cl- in Ehrlich cells. Biochim. Biophys. Acta. 1980;600(2):432–447. doi: 10.1016/0005-2736(80)90446-0. [DOI] [PubMed] [Google Scholar]
- 7.Isenring P., Forbush B. Characterization of the renal absorptive NKCC2., comparative studies including hNKCC1, sNKCC1 and The HEK-293 NKCC1. J. Biol. Chem. 1998;273:11295–11301. doi: 10.1074/jbc.273.18.11295. [DOI] [PubMed] [Google Scholar]
- 8.Russell J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000;80(1):211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 9.Adragna N.C., Di Fulvio M., Lauf P.K. Regulation of K-Cl cotransport: from function to genes. J. Membr. Biol. 2004;201(3):109–137. doi: 10.1007/s00232-004-0695-6. [DOI] [PubMed] [Google Scholar]
- 10.Payne J.A., Rivera C., Voipio J., Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26(4):199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 11.Darman R.B., Forbush B. regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J. Biol. Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- 12.Flemmer A.W., Gimenez I., Dowd B.F., Darman R.B., Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem. 2002;277(40):37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- 13.Vitari A.C., Thastrup J., Rafiqi F.H., Deak M., Morrice N.A., Karlsson H.K., Alessi D.R. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 2006;397(1):223–231. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jennings M.L., Schulz R.K. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J. Gen. Physiol. 1991;97(4):799–817. doi: 10.1085/jgp.97.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurihara K., Nakanishi N., Moore-Hoon M.L., Turner R.J. Phosphorylation of the salivary Na(+)-K(+)-2Cl(-) cotransporter. Am. J. Physiol. Cell Physiol. 2002;282(4):C817–C823. doi: 10.1152/ajpcell.00352.2001. [DOI] [PubMed] [Google Scholar]
- 16.Matskevich I., Hegney K.L., Flatman P.W. Regulation of erythrocyte Na–K–2Cl cotransport by threonine phosphorylation. Biochimica. Biophysica. Acta. 2005;1714:25–34. doi: 10.1016/j.bbamem.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Ponce-Coria J., Markadieu N., Austin T., Flammang L., Rios K., Welling P.A., Delpire E. A novel SPAK-independent pathway involving Cab39 and WNK4 in the activation of Na-K-Cl cotransporters. J. Biol. Chem. 2014;289:17680–17688. doi: 10.1074/jbc.M113.540518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith L., Smallwood N., Altman A., Liedtke C.M. PKCdelta acts upstream of SPAK in the activation of NKCC1 by hyperosmotic stress in human airway epithelial cells. J. Biol. Chem. 2008;283:22147–22156. doi: 10.1074/jbc.M801752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Susa K., Kita S., Iwamoto T., Yang S.S., Lin S.H., Ohta A., Sohara E., Rai T., Sasaki S., Alessi D.R., Uchida S. Effect of heterozygous deletion of WNK1 on the WNK-OSR1/ SPAK-NCC/NKCC1/NKCC2 signal cascade in the kidney and blood vessels. Clin. Exp. Nephrol. 2012;16(4):530–538. doi: 10.1007/s10157-012-0590-x. [DOI] [PubMed] [Google Scholar]
- 20.Brumback A.C., Staley K.J. Thermodynamic regulation of NKCC1-mediated Cl- cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J. Neurosci. 2008;28(6):1301–1312. doi: 10.1523/JNEUROSCI.3378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen G., Trombley P.Q., van den Pol A.N. Excitatory actions of GABA in developing rat hypothalamic neurones. J. Physiol. 1996;494(Pt 2):451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nardou R., Ben-Ari Y., Khalilov I. Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J. Neurophysiol. 2009;101(6):2878–2888. doi: 10.1152/jn.90761.2008. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W., Liu L.Y., Xu T.L. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience. 2008;152(2):502–510. doi: 10.1016/j.neuroscience.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 24.Flagella M., Clarke L.L., Miller M.L., Erway L.C., Giannella R.A., Andringa A., Gawenis L.R., Kramer J., Duffy J.J., Doetschman T., Lorenz J.N., Yamoah E.N., Cardell E.L., Shull G.E. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 1999;274(38):26946–26955. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 25.Yang S.S., Lo Y.F., Wu C.C., Lin S.W., Yeh C.J., Chu P., Sytwu H.K., Uchida S., Sasaki S., Lin S.H. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010;21(11):1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck J., Lenart B., Kintner D.B., Sun D. Na-K-Cl cotransporter contributes to glutamate-mediated excitotoxicity. J. Neurosci. 2003;23(12):5061–5068. doi: 10.1523/JNEUROSCI.23-12-05061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace B.K., Jelks K.A., O’Donnell M.E. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am. J. Physiol. Cell Physiol. 2012;302(3):C505–C517. doi: 10.1152/ajpcell.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandt C., Nozadze M., Heuchert N., Rattka M., Löscher W. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J. Neurosci. 2010;30(25):8602–8612. doi: 10.1523/JNEUROSCI.0633-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shulga A., Magalhães A.C., Autio H., Plantman S., di Lieto A., Nykjær A., Carlstedt T., Risling M., Arumäe U., Castrén E., Rivera C. The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J. Neurosci. 2012;32(5):1757–1770. doi: 10.1523/JNEUROSCI.3282-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitcher G.M., Ritchie J., Henry J.L. Peripheral neuropathy induces cutaneous hypersensitivity in chronically spinalized rats. Pain Med. 2013;14(7):1057–1071. doi: 10.1111/pme.12123. [DOI] [PubMed] [Google Scholar]
- 31.Xu J.C., Lytle C., Zhu T.T., Payne J.A., Benz E., Jr, Forbush B., III Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. USA. 1994;91(6):2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reshkin S.J., Lee S.I., George J.N., Turner R.J. Identification, characterization and purification of a 160 kD bumetanide-binding glycoprotein from the rabbit parotid. J. Membr. Biol. 1993;136(2):243–251. doi: 10.1007/BF02505766. [DOI] [PubMed] [Google Scholar]
- 33.Quaggin S.E., Payne J.A., Forbush B., III, Igarashi P. Localization of the renal Na-K-Cl cotransporter gene (Slc12a1) on mouse chromosome 2. Mamm. Genome. 1995;6(8):557–558. doi: 10.1007/BF00356178. [DOI] [PubMed] [Google Scholar]
- 34.Payne J.A., Forbush B., III Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc. Natl. Acad. Sci. USA. 1994;91(10):4544–4548. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan M.R., Plotkin M.D., Brown D., Hebert S.C., Delpire E. Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal inner medullary collecting duct, the glomerular and extraglomerular mesangium, and the glomerular afferent arteriole. J. Clin. Invest. 1996;98(3):723–730. doi: 10.1172/JCI118844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clayton G.H., Owens G.C., Wolff J.S., Smith R.L. Ontogeny of cation-Cl- cotrasporter expression in rat neocortex. Dev. Brain Res. 1998;109:281–292.. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- 37.Plotkin M.D., Kaplan M.R., Peterson L.N., Gullans S.R., Hebert S.C., Delpire E. Expression of the Na(+)-K(+)-2Cl- cotransporter BSC2 in the nervous system. Am. J. Physiol. 1997;272(1 Pt 1):C173–C183. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- 38.Pond B.B., Berglund K., Kuner T., Feng G., Augustine G.J., Schwartz-Bloom R.D. The chloride transporter Na(+)-K(+)-Cl- cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J. Neurosci. 2006;26(5):1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitcher M.H., Price T.J., Entrena J.M., Cervero F. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol. Pain. 2007;3:17. doi: 10.1186/1744-8069-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krystal A.D., Sutherland J., Hochman D.W. Loop Diuretics Have Anxiolytic Effects in Rat Models of Conditioned Anxiety. Plos One. 2012;7:e35417. doi: 10.1371/journal.pone.0035417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orlov S.N., Koltsova S.V., Tremblay J., Baskakov M.B. Pavel, Hamet. NKCC1 and hypertension: Role in the regulation of vascular smooth muscle contractions and myogenic tone. Ann. Med. 2012;44:111–118. doi: 10.3109/07853890.2011.653395. [DOI] [PubMed] [Google Scholar]
- 42.Shen W., Purpura L.A., Li B., Nan C., Chang I.J., Ripps H. Regulation of synaptic transmission at the photoreceptor terminal: a novel role for the cation–chloride co-transporter NKCC1. J. Physiol. 2013;591:133, 147. doi: 10.1113/jphysiol.2012.241042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gamba G., Miyanoshita A., Lombardi M., Lytton J., Lee W.S., Hediger M.A., Hebert S.C. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 1994;269(26):17713–17722. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- 44.Lytle C. Common Sites Calyculin-A Involves Phosphorylation at Shrinkage, cAMP, Fluoride, and Na-K-Cl Cotransport Protein by Cell Activation of the Avian Erythrocyte. J. Biol. Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- 45.Ueberschär S., Bakker-Grunwald T. Effects of ATP and cyclic AMP on the (Na+ + K+ + 2Cl-)-cotransport system in turkey erythrocytes. Biochim. Biophys. Acta. 1985;818(2):260–266. doi: 10.1016/0005-2736(85)90566-8. [DOI] [PubMed] [Google Scholar]
- 46.Greger R., Schlatter E., Wang F., Forrest J.N., Jr Mechanism of NaCl secretion in rectal gland tubules of spiny dogfish (Squalus acanthias). III. Effects of stimulation of secretion by cyclic AMP. Pflugers Arch. 1984;402(4):376–384. doi: 10.1007/BF00583938. [DOI] [PubMed] [Google Scholar]
- 47.Darman R.B., Flemmer A., Forbush B. Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J. Biol. Chem. 2001;276(37):34359–34362. doi: 10.1074/jbc.C100368200. [DOI] [PubMed] [Google Scholar]
- 48.Dowd B.F., Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 2003;278(30):27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- 49.Zeniya M., Sohara E., Kita S., Iwamoto T., Susa K., Mori T., Oi K., Chiga M., Takahashi D., Yang S.S., Lin S.H., Rai T., Sasaki S., Uchida S. Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension. 2013;62(5):872–878. doi: 10.1161/HYPERTENSIONAHA.113.01543. [DOI] [PubMed] [Google Scholar]
- 50.Kahle K.T., Rinehart J., Lifton R.P. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kahle K.T., Ring A.M., Lifton R.P. Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 2008;70:329–355. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 52.Thastrup J.O., Rafiqi F.H., Vitari A.C., Pozo-Guisado E., Deak M., Mehellou Y., Alessi D.R. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 2012;441(1):325–337. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gagnon K.B., Rios K., Delpire E. Functional insights into the activation mechanism of Ste20-related kinases. Cell. Physiol. Biochem. 2011;28(6):1219–1230. doi: 10.1159/000335854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giménez I., Forbush B. Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am. J. Physiol. Renal Physiol. 2005;289(6):F1341–F1345. doi: 10.1152/ajprenal.00214.2005. [DOI] [PubMed] [Google Scholar]
- 55.Sun D., Murali S.G. Na+-K+-2Cl- cotransporter in immature cortical neurons: A role in intracellular Cl- regulation. J. Neurophysiol. 1999;81(4):1939–1948. doi: 10.1152/jn.1999.81.4.1939. [DOI] [PubMed] [Google Scholar]
- 56.Bassett P., Meehan S., Lonjaret J.G., Sheridan K., Hempleman S. Dual response to bumetanide in avian intrapulmonary chemoreceptors (1092.1). FASEB J. 2014;28:1092–1. [Google Scholar]
- 57.Töllner K., Brandt C., Töpfer M., Brunhofer G., Erker T., Gabriel M., Feit P.W., Lindfors J., Kaila K., Löscher W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 2014;75(4):550–562. doi: 10.1002/ana.24124. [DOI] [PubMed] [Google Scholar]
- 58.Töpfer M., Töllner K., Brandt C., Twele F., Bröer S., Löscher W. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur. J. Neurosci. 2014;39(4):673–687. doi: 10.1111/ejn.12424. [DOI] [PubMed] [Google Scholar]
- 59.Knauf H., Mutschler E. Clinical pharmacokinetics and pharmacodynamics of torasemide. Clin. Pharmacokinet. 1998;34(1):1–24. doi: 10.2165/00003088-199834010-00001. [DOI] [PubMed] [Google Scholar]
- 60.Hannaert P., Alvarez-Guerra M., Pirot D., Nazaret C., Garay R.P. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedebergs Arch. Pharmacol. 2002;365(3):193–199. doi: 10.1007/s00210-001-0521-y. [DOI] [PubMed] [Google Scholar]
- 61.Ben-Ari Y., Cherubini E., Corradetti R., Gaiarsa J.L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Misgeld U., Deisz R.A., Dodt H.U., Lux H.D. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232(4756):1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- 63.Chen H., Sun D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol. Res. 2005;27(3):280–286. doi: 10.1179/016164105X25243. [DOI] [PubMed] [Google Scholar]
- 64.He Q., Nomura T., Xu J., Contractor A. The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 2014;34(2):446–450. doi: 10.1523/JNEUROSCI.4447-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Represa A., Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28(6):278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 66.Cellot G., Cherubini E. Functional role of ambient GABA in refining neuronal circuits early in postnatal development. Front. Neural Circuits. 2013;7:136. doi: 10.3389/fncir.2013.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farrant M., Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog. Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- 68.Rivera C., Voipio J., Payne J.A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397(6716):251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 69.Schomberg S.L., Su G., Haworth R.A., Sun D. Stimulation of Na-K-2Cl cotransporter in neurons by activation of Non-NMDA ionotropic receptor and group-I mGluRs. J. Neurophysiol. 2001;85(6):2563–2575. doi: 10.1152/jn.2001.85.6.2563. [DOI] [PubMed] [Google Scholar]
- 70.Uvarov P., Ludwig A., Markkanen M., Soni S., Hu¨bner C.A., Rivera C., Airaksinen M.S. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J. Biol. Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ludwig A., Uvarov P., Soni S., Thomas-Crusells J., Airaksinen M.S., Rivera C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 2011;31(2):644–649. doi: 10.1523/JNEUROSCI.2006-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pellegrino C., Gubkina O., Schaefer M., Becq H., Ludwig A., Mukhtarov M., Chudotvorova I., Corby S., Salyha Y., Salozhin S., Bregestovski P., Medina I. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J. Physiol. 2011;589(Pt 10):2475–2496. doi: 10.1113/jphysiol.2010.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khalilov I., Chazal G., Chudotvorova I., Pellegrino C., Corby S., Ferrand N., Gubkina O., Nardou R., Tyzio R., Yamamoto S., Jentsch T.J., Hübner C.A., Gaiarsa J.L., Ben-Ari Y., Medina I. Enhanced Synaptic Activity and Epileptiform Events in the Embryonic KCC2 Deficient Hippocampus. Front. Cell. Neurosci. 2011;5:23. doi: 10.3389/fncel.2011.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee H.H., Deeb T.Z., Walker J.A., Davies P.A., Moss S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011;14(6):736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aguado F., Carmona M.A., Pozas E., Aguiló A., Martínez-Guijarro F.J., Alcantara S., Borrell V., Yuste R., Ibañez C.F., Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development. 2003;130(7):1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- 76.Carmona M.A., Pozas E., Martínez A., Espinosa-Parrilla J.F., Soriano E., Aguado F. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb. Cortex. 2006;16(1):47–63. doi: 10.1093/cercor/bhi083. [DOI] [PubMed] [Google Scholar]
- 77.Rivera C., Li H., Thomas-Crusells J., Lahtinen H., Viitanen T., Nanobashvili A., Kokaia Z., Airaksinen M.S., Voipio J., Kaila K., Saarma M. BDNF-induced TrkB activation down-regulates the K+–Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blaesse P., Airaksinen M.S., Rivera C., Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61(6):820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 79.Jayakumar A.R., Norenberg M.D. The Na-K-Cl Co-transporter in astrocyte swelling. Metab. Brain Dis. 2010;25(1):31–38. doi: 10.1007/s11011-010-9180-3. [DOI] [PubMed] [Google Scholar]
- 80.MacVicar B.A., Feighan D., Brown A., Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37(2):114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- 81.Galeffi F., Sah R., Pond B.B., George A., Schwartz-Bloom R.D. Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: effect of diazepam. J. Neurosci. 2004;24(18):4478–4488. doi: 10.1523/JNEUROSCI.0755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Inglefield J.R., Schwartz-Bloom R.D. Optical imaging of hippocampal neurons with a chloride-sensitive dye: early effects of in vitro ischemia. J. Neurochem. 1998;70(6):2500–2509. doi: 10.1046/j.1471-4159.1998.70062500.x. [DOI] [PubMed] [Google Scholar]
- 83.Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79(4):1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 84.Yan Y., Dempsey R.J., Sun D. Na+-K+-Cl- cotransporter in rat focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001;21(6):711–721. doi: 10.1097/00004647-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 85.O'Donnell M.E., Tran L., Lam T.I., Liu X.B., Anderson S.E. Bumetanide inhibition of the blood–brain barrier Na–K–Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J. Cereb. Blood Flow Metab. 2004;24:1046–1056.. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- 86.Brillault J., Lam T.I., Rutkowsky J.M., Foroutan S., O’Donnell M.E. Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 2008;294(1):C88–C96. doi: 10.1152/ajpcell.00148.2007. [DOI] [PubMed] [Google Scholar]
- 87.Liu F., Akella P., Benashski S.E., Xu Y., McCullough L.D. Expression of Na-K-Cl cotransporter and edema formation are age dependent after ischemic stroke. Exp. Neurol. 2010;224(2):356–361. doi: 10.1016/j.expneurol.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang L., Shah K., Wang H., Karamyan V.T., Abbruscato T.J. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J. Pharmacol. Exp. Ther. 2011;339(2):499–508. doi: 10.1124/jpet.111.184127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee H.A., Hong S.H., Kim J.W., Jang I.S. Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. J. Neurochem. 2010;114(2):520–529. doi: 10.1111/j.1471-4159.2010.06772.x. [DOI] [PubMed] [Google Scholar]
- 90.Chang B.S., Lowenstein D.H. Epilepsy. N. Engl. J. Med. 2003;349(13):1257–1266. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- 91.Dzhala V.I., Talos D.M., Sdrulla D.A., Brumback A.C., Mathews G.C., Benke T.A., Delpire E., Jensen F.E., Staley K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005;11(11):1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 92.Nardou R., Yamamoto S., Chazal G., Bhar A., Ferrand N., Dulac O., Ben-Ari Y., Khalilov I. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134(Pt 4):987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- 93.Dzhala V.I., Kuchibhotla K.V., Glykys J.C., Kahle K.T., Swiercz W.B., Feng G., Kuner T., Augustine G.J., Bacskai B.J., Staley K.J. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J. Neurosci. 2010;30(35):11745–11761. doi: 10.1523/JNEUROSCI.1769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sen A., Martinian L., Nikolic M., Walker M.C., Thom M., Sisodiya S.M. Increased NKCC1 expression in refractory human epilepsy. Epilepsy Res. 2007;74(2-3):220–227. doi: 10.1016/j.eplepsyres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 95.Conti L., Palma E., Roseti C., Lauro C., Cipriani R., de Groot M., Aronica E., Limatola C. Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia. 2011;52(9):1635–1644. doi: 10.1111/j.1528-1167.2011.03111.x. [DOI] [PubMed] [Google Scholar]
- 96.Oriaifo S.E., Otokiti I., Omogbai E.K. The effect of furosemide on experimentally-induced seizures in mice. J.C.M.R. 2012;7:89–93. [Google Scholar]
- 97.Østby I., Øyehaug L., Einevoll G.T., Nagelhus E.A., Plahte E., Zeuthen T., Lloyd C.M., Ottersen O.P., Omholt S.W. Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space. PLOS Comput. Biol. 2009;5(1):e1000272. doi: 10.1371/journal.pcbi.1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simard M., Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129(4):877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 99.Hochman D.W. The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia. 2012;53(Suppl. 1):18–25. doi: 10.1111/j.1528-1167.2012.03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Muñoz A., Méndez P., DeFelipe J., Alvarez-Leefmans F.J. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48(4):663–673. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 101.Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007;27(37):9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Guzman P., Inaba Y., Biagini G., Baldelli E., Mollinari C., Merlo D., Avoli M. Subiculum network excitability is increased in a rodent model of temporal lobe epilepsy. Hippocampus. 2006;16(10):843–860. doi: 10.1002/hipo.20215. [DOI] [PubMed] [Google Scholar]
- 103.Rangroo Thrane V., Thrane A.S., Wang F., Cotrina M.L., Smith N.A., Chen M., Xu Q., Kang N., Fujita T., Nagelhus E.A., Nedergaard M. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 2013;19(12):1643–1648. doi: 10.1038/nm.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Campbell S.L., Robel S., Cuddapah V.A., Robert S., Buckingham S.C., Kahle K.T., Sontheimer H. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia. 2015;63(1):23–36. doi: 10.1002/glia.22730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pallud J., Le Van Quyen M., Bielle F., Pellegrino C., Varlet P., Labussiere M., Cresto N., Dieme M.J., Baulac M., Duyckaerts C., Kourdougli N., Chazal G., Devaux B., Rivera C., Miles R., Capelle L., Huberfeld G. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014;6:244ra89. doi: 10.1126/scitranslmed.3008065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jaggi A.S., Singh N. Intrathecal delivery of farnesyl thiosalicylic acid and GW 5074 attenuates hyperalgesia and allodynia in chronic constriction injury-induced neuropathic pain in rats. Neurol. Sci. 2013;34(3):297–304. doi: 10.1007/s10072-012-0991-3. [DOI] [PubMed] [Google Scholar]
- 107.Kaur G., Jaggi A.S., Singh N. Exploring the potential effect of Ocimum sanctum in vincristine-induced neuropathic pain in rats. J. Brachial Plex. Peripher. Nerve Inj. 2010;5:3. doi: 10.1186/1749-7221-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vinay L., Jean-Xavier C. Plasticity of spinal cord locomotor networks and contribution of cation-chloride cotransporters. Brain Res. Rev. 2008;57:103–10. doi: 10.1016/j.brainresrev.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Stil A., Jean-Xavier C., Liabeuf S., Brocard C., Delpire E., Vinay L., Viemari J.C. Contribution of the potassium-chloride co-transporter KCC2 to the modulation of lumbar spinal networks in mice. Eur. J. Neurosci. 2011;33:1212–22. doi: 10.1111/j.1460-9568.2010.07592.x. [DOI] [PubMed] [Google Scholar]
- 110.Boulenguez P., Liabeuf S., Bos R., Bras H., Jean-Xavier C., Brocard C., Stil A., Darbon P., Cattaert D., Delpire E., Marsala M., Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 2010;16(3):302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- 111.Morales-Aza B.M., Chillingworth N.L., Payne J.A., Donaldson L.F. Inflammation alters cation chloride cotransporter expression in sensory neurons. Neurobiol. Dis. 2004;17(1):62–69. doi: 10.1016/j.nbd.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 112.Granados-Soto V., Arguellesb C.F., Francisco J.A. lvarez- Leefmans. Peripheral and central antinociceptive action of Na–K– 2Cl cotransporter blockers on formalin-induced nociception in rats. Pain. 2005;114:231–238. doi: 10.1016/j.pain.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 113.Galan A., Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience. 2005;133(1):245–252. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 114.Valencia-de Ita S., Lawand N.B., Lin Q., Castañeda-Hernandez G., Willis W.D. Role of the Na+-K+-2Cl- cotransporter in the development of capsaicin-induced neurogenic inflammation. J. Neurophysiol. 2006;95(6):3553–3561. doi: 10.1152/jn.01091.2005. [DOI] [PubMed] [Google Scholar]
- 115.Willis W.D., Jr Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp. Brain Res. 1999;124(4):395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- 116.Cramer S.W., Baggott C., Cain J., Tilghman J., Allcock B., Miranpuri G., Rajpal S., Sun D., Resnick D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol. Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prescott S.A., Sejnowski T.J., De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol. Pain. 2006;2:32. doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nomura H., Sakai A., Nagano M., Umino M., Suzuki H. Expression changes of cation chloride cotransporters in the rat spinal cord following intraplantar formalin. Neurosci. Res. 2006;56(4):435–440. doi: 10.1016/j.neures.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 119.Li Y.Q., Li H., Wei J., Qu L., Wu L.A. Expression changes of K+-Cl- co-transporter 2 and Na+-K+-Cl- co-transporter1 in mouse trigeminal subnucleus caudalis following pulpal inflammation. Brain Res. Bull. 2010;81(6):561–564. doi: 10.1016/j.brainresbull.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 120.Miletic G., Miletic V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain. 2008;137(3):532–539. doi: 10.1016/j.pain.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rivera C., Voipio J., Thomas-Crusells J., Li H., Emri Z., Sipilä S., Payne J.A., Minichiello L., Saarma M., Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 2004;24(19):4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wei H., Hao B., Huang J.L., Ma A.N., Li X.Y., Wang Y.X., Pertovaara A. Intrathecal administration of a gap junction decoupler, an inhibitor of Na(+)-K(+)-2Cl(-) cotransporter 1, or a GABA(A) receptor agonist attenuates mechanical pain hypersensitivity induced by REM sleep deprivation in the rat. Pharmacol. Biochem. Behav. 2010;97(2):377–383. doi: 10.1016/j.pbb.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 123.Pitcher M.H., Cervero F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151(3):756–762. doi: 10.1016/j.pain.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 124.Austin T.M., Delpire E. Inhibition of KCC2 in Mouse Spinal Cord Neurons Leads to Hypersensitivity to Thermal Stimulation. Anesth. Analg. 2011;113:1509–1515. doi: 10.1213/ANE.0b013e31822e0a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhuang Z.Y., Xu H., Clapham D.E., Ji R.R. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J. Neurosci. 2004;24(38):8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Price T.J., Jeske N.A., Flores C.M., Hargreaves K.M. Pharmacological interactions between calcium/calmodulin-dependent kinase II alpha and TRPV1 receptors in rat trigeminal sensory neurons. Neurosci. Lett. 2005;389(2):94–98. doi: 10.1016/j.neulet.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rudomin P., Schmidt R.F. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp. Brain Res. 1999;129(1):1–37. doi: 10.1007/s002210050933. [DOI] [PubMed] [Google Scholar]
- 128.Kalkman H.O. Alterations in the expression of neuronal chloride transporters may contribute to schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011;35(2):410–414. doi: 10.1016/j.pnpbp.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Lewis C.M., Levinson D.F., Wise L.H., DeLisi L.E., Straub R.E., Hovatta I., Williams N.M., Schwab S.G., Pulver A.E., Faraone S.V., Brzustowicz L.M., Kaufmann C.A., Garver D.L., Gurling H.M., Lindholm E., Coon H., Moises H.W., Byerley W., Shaw S.H., Mesen A., Sherrington R., O’Neill F.A., Walsh D., Kendler K.S., Ekelund J., Paunio T., Lönnqvist J., Peltonen L., O’Donovan M.C., Owen M.J., Wildenauer D.B., Maier W., Nestadt G., Blouin J.L., Antonarakis S.E., Mowry B.J., Silverman J.M., Crowe R.R., Cloninger C.R., Tsuang M.T., Malaspina D., Harkavy-Friedman J.M., Svrakic D.M., Bassett A.S., Holcomb J., Kalsi G., McQuillin A., Brynjolfson J., Sigmundsson T., Petursson H., Jazin E., Zoëga T., Helgason T. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am. J. Hum. Genet. 2003;73(1):34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Potkin S.G., Turner J.A., Guffanti G., Lakatos A., Fallon J.H., Nguyen D.D., Mathalon D., Ford J., Lauriello J., Macciardi F., FBIRN A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr. Bull. 2009;35(1):96–108. doi: 10.1093/schbul/sbn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arion D., Lewis D.A. Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry. 2011;68(1):21–31. doi: 10.1001/archgenpsychiatry.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kahle K.T., Rinehart J., de Los Heros P., Louvi A., Meade P., Vazquez N., Hebert S.C., Gamba G., Gimenez I., Lifton R.P. WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. USA. 2005;102(46):16783–16788. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.de Los Heros P., Kahle K.T., Rinehart J., Bobadilla N.A., Vázquez N., San Cristobal P., Mount D.B., Lifton R.P., Hebert S.C., Gamba G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. USA. 2006;103(6):1976–1981. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu S., Gullapalli R.P., Frost D.O. Olanzapine antipsychotic treatment of adolescent rats causes long term changes in glutamate and GABA levels in the nucleus accumbens. Schizophr. Res. 2015;161(2-3):452–457. doi: 10.1016/j.schres.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Taylor S.F., Tso I.F. GABA abnormalities in schizophrenia: A methodological review of in vivo studies. Schizophr. Res. 2014. [DOI] [PMC free article] [PubMed]
- 136.Hyde T.M. Cation chloride co-transporters in human brain development and schizophrenia: NKCC1 and KCC2 [abstract]. Biol. Psychiatry. 2008;63:(symposium 7)–47S. [Google Scholar]
- 137.Hyde T.M. The cation chloride co-transporters KCC2 and NKCC1 and schizophrenia [abstract]. Biol. Psychiatry. 2009;64:(symposium 8)–170S. [Google Scholar]
- 138.Owens D.F., Kriegstein A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002;3(9):715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 139.Hyde T.M., Lipska B.K., Ali T., Mathew S.V., Law A.J., Metitiri O.E., Straub R.E., Ye T., Colantuoni C., Herman M.M., Bigelow L.B., Weinberger D.R., Kleinman J.E. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 2011;31(30):11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim J.Y., Liu C.Y., Zhang F., Duan X., Wen Z., Song J., Feighery E., Lu B., Rujescu D., St Clair D., Christian K., Callicott J.H., Weinberger D.R., Song H., Ming G.L. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell. 2012;148(5):1051–1064. doi: 10.1016/j.cell.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Adusei D.C., Pacey L.K., Chen D., Hampson D.R. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology. 2010;59(3):167–171. doi: 10.1016/j.neuropharm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 142.Curia G., Papouin T., Séguéla P., Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex. 2009;19(7):1515–1520. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.D’Hulst C., De Geest N., Reeve S.P., Van Dam D., De Deyn P.P., Hassan B.A., Kooy R.F. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 144.Lemonnier E., Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 2010;99(12):1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x. [DOI] [PubMed] [Google Scholar]
- 145.Lemonnier E., Degrez C., Phelep M., Tyzio R., Josse F., Grandgeorge M., Hadjikhani N., Ben-Ari Y. A randomised controlled trial of bumetanide in the treatment of autism in children. 2012. [DOI] [PMC free article] [PubMed]
- 146.Eftekhari S., Shahrokhi A., Tsintsadze V., Nardou R., Brouchoud C., Conesa M., Burnashev N., Ferrari D.C., Ben-Ari Y. Response to Comment on “Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring”. Science. 2014;346(6206):176. doi: 10.1126/science.1256009. [DOI] [PubMed] [Google Scholar]
- 147.Tyzio R., Nardou R., Ferrari D.C., Tsintsadze T., Shahrokhi A., Eftekhari S., Khalilov I., Tsintsadze V., Brouchoud C., Chazal G., Lemonnier E., Lozovaya N., Burnashev N., Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343(6171):675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 148.Haas B.R., Sontheimer H. Inhibition of the Sodium-Potassium-Chloride Cotransporter Isoform-1 reduces glioma invasion. Cancer Res. 2010;70(13):5597–5606. doi: 10.1158/0008-5472.CAN-09-4666. [DOI] [PMC free article] [PubMed] [Google Scholar]