

Abstract
Although it is generally accepted that the abuse-related effects of amphetamines and cocaine result from the activation of the brain dopaminergic (DA) system, the psychostimulants also alter other neurotransmitter systems. In particular, they increase extracellular levels of norepinephrine (NE) and serotonin by inhibiting respective plasma membrane transporters and/or inducing release. The present review will discuss the preclinical findings on the effects of the NE system modulation (lesions, pharmacological and genetic approaches) on behaviors (locomotor hyperactivity, behavioral sensitization, modification of intracranial self-stimulation, conditioned place preference, drug self-administration, extinction/reinstatement of drug seeking behavior) related to the psychostimulant addiction.
Keywords: Amphetamine, behavioral sensitization, cocaine, conditioned place preference, drug self-administration, intracranial self-stimulation, locomotor activity, norepinephrine.
INTRODUCTION
The psychostimulants amphetamine, its derivatives and cocaine are commonly abused drugs. It is well known that the brain dopamine (DA) system plays a critical role in the mechanisms responsible for their behavioral effects, including reinforcing and rewarding properties [1-3]. However, it should be remembered that besides DA, cocaine and amphetamines increase also the extracellular levels of norepinephrine (NE) and serotonin by inhibiting respective plasma membrane transporters and/or stimulating monoamine release [4-7]. The literature contains a lot of reports examining the involvement of NE in the effects of addictive psychostimulants.
The locus coeruleus (LC) is the origin of an NE projection that as the dorsal noradrenergic bundle (DNB) innervates the cerebellum, hippocampus and forebrain. The second ascending noradrenergic system, named the ventral noradrenergic bundle (VNB), originates in the lateral tegmental nuclei (LTN) and projects to the hypothalamus, midbrain and amygdala [8].
NE produces its effects via adrenoceptors (ARs) that are G-protein coupled receptors. The AR family comprises 9 receptor subtypes encoded by separate genes, including three α1 ARs (α1a, α1b and α1d), three α2 ARs (α2a, α2b and α2c) and three β ARs (β1, β2 and β3). The α1 ARs are Gαq-coupled receptors that activate protein kinase C. The α2 ARs are coupled to Gαi-protein and are located on NE neurons, functioning as inhibitory autoreceptors. In addition, pre- and postsynaptic α2 heteroreceptors are located on other neurons in the brain. The β ARs are Gαs-coupled receptors that stimulate adenylyl cyclase activity and activate protein kinase A [9].
The present review aims to discuss the engagement of NE in several effects of the psychostimulants, including locomotor hyperactivity, behavioral sensitization, modification of intracranial self-stimulation, conditioned place preference (CPP) and drug self-administration.
LOCOMOTOR HYPERACTIVITY (TABLE 1)
Locomotor stimulation is one of the most characteristic effects of psychostimulants. It is usually monitored in mice and rats as horizontal activity in photoresistor actometers.
It has been reported that the inhibition of NE synthesis affects locomotor hyperactivity evoked by psychostimulants. Specifically, α-methyl-p-tyrosine (AMPT), an inhibitor of tyrosine hydroxylase [10] has been shown to attenuate locomotor activity increased by a single dose of amphetamine or cocaine in mice and rats [11-15], having been without effect on the basal locomotor activity of animals [11]. However, since AMPT inhibits not only NE but also DA synthesis, the above effects do not allow to conclude which of the monoamines plays a role in the behavioral response to the psychostimulants.
The results obtained in experiments with inhibitors of DA-β-hydroxylase (DBH), an enzyme that converts DA to NE in the final step of NE synthesis [16, 17], are also inconclusive. For instance, disulfiram has been shown to reduce hyperlocomotion evoked by amphetamine or cocaine in mice and rats, though in the case of the latter psychostimulant such an effect was observed only after subacute but not acute administration of the DBH inhibitor [18-20], which – at the same time – exhibited its own inhibitory effect on the basal locomotor activity, at least in mice [21]. Attenuation or no influence on amphetamine-induced locomotor hyperactivity in mice or rats has been described after other DBH inhibitors: FLA-63 [12-14, 22] or U-14,624 [14, 15], respectively. On the other hand, Gaval-Cruz et al. [23] have recently reported that subacute administration of disulfiram, but not nepicastat, another DBH inhibitor, increased the cocaine-evoked hyperlocomotion in mice. Interestingly, the latter observation is in line with the results obtained in DBH knockout mice, in which cocaine and a low dose of amphetamine produced more distinct locomotor hyperactivity than in wild type animals [23-25]. However, the augmentation of the amphetamine effect was diminished by SCH 23390 (DA D1 receptor antagonist) but not by prazosin (α1 AR blocker) suggesting the contribution of DA signaling in this behavioral response to the psychostimulant [24].
The effects of chemical or electrolytic lesions of the NE pathways also cast doubt on their importance for psychostimulant-induced hyperlocomotion. In particular, Harro et al. [26], Alttoa et al. [27] and Kõiv et al. [28] have found that DSP-4-induced LC NE neuron lesions attenuated locomotor response to amphetamine or cocaine in some experimental conditions. In contrast, Archer et al. [29] demonstrated that severe denervation of LC projections did not affect amphetamine-induced locomotor hyperactivity in rats. In addition, whereas Creese and Iversen [30] and Roberts et al. [31] reported that 6-hydroxydopamine (6-OHDA)-induced lesions of DNB or VNB did not affect locomotor stimulation after amphetamine, other authors did observe attenuation of this response following the neurotoxin injections into the LC or DNB [29, 32]. Reduction of the locomotor response to amphetamine was also found in rats with electrolytic lesions of the LC or VNB [33].
A number of studies were performed to examine the effects of α1 AR antagonists on the hyperlocomotion induced by psychostimulants. Actually, systemically administered prazosin was found to attenuate the response to amphetamine [24, 34-40] and cocaine [34, 36, 37, 41] injected systemically in mice and rats or amphetamine infused into the nucleus accumbens (NAc) in rats [42], though its lack of effect on cocaine-induced locomotor hyperactivity in rats has also been reported [38, 43, 44]. Interestingly, local administration of prazosin or terazosin into the prefrontal cortex (PFC) or the NAc shell, respectively, abolished the locomotor hyperactivity induced by intraaccumbally injected amphetamine or systemically administered cocaine in rats [35, 42, 45]. Results of further studies using animals with genetically inactivated subtypes of α1 ARs have indicated that α1b ARs play a crucial role in the locomotor hyperactivity induced by amphetamine [37, 39, 46] or cocaine [37], though no locomotor response to amphetamine was also observed in α1d AR knockout mice [47].
Antagonists of α2 ARs (e.g., yohimbine, dexefaroxan or idazoxan), which facilitate NE transmission [48-50] presumably by blocking the autoreceptor function [51], increased locomotor hyperactivity induced by amphetamine [52, 53] or cocaine [44] in rats. Correspondingly, genetic inactivation of either α2a or α2c AR subtype significantly enhanced the locomotor response to amphetamine, indicating that both subtypes are involved in the effect of the psychostimulant [54, 55]. It should be underlined, however, that enhancement of the locomotor activating effects of amphetamine observed in these two knockout models might depend on distinct mechanisms, since an increase in NE turnover was observed in α2a AR knockout mice [56], but not in α2c AR knockout mice [57].
Pharmacological stimulation of α2 ARs by clonidine or dexmedetomidine, known to reduce NE release [48, 49, 58, 59] attenuated amphetamine- or cocaine-induced hyperlocomotion [38, 44, 54]. Importantly, the inhibitory effect of dexmedetomidine on the response to amphetamine was much weaker in α2c AR knockout mice, while in mice with overexpressed α2c ARs, locomotor hyperactivity induced by the psychostimulant was significantly attenuated by the agonist [54]. However, the involvement of NE signaling in the latter effect seems doubtful, since in mice with overexpressed α2c ARs no changes in the brain NE were observed [57].
In contrast to α1 and α2 ARs, there are only a few studies that investigated the role of β ARs in the psychostimulant-induced locomotor hyperactivity and their results are not clear-cut. In fact, no effect and/or an increase in the behavioral response to cocaine was demonstrated after propranolol in mice and rats, while this β AR antagonist, depending on the dose, increased or reduced the behavioral effect of amphetamine in rats or mice, respectively [34, 38, 60, 61]. No effect on amphetamine-induced locomotor hyperactivity was found after timolol or nadolol in rats [40, 62].
BEHAVIORAL SENSITIZATION (TABLE 1)
Table 1.
The effects of NE system modulation on amphetamines- and cocaine-induced locomotor hyperactivity and sensitization.
| NE System Modulation | AMPHETAMINES | COCAINE | ||||
|---|---|---|---|---|---|---|
| Locomotor Hyperactivity | Sensitization | Locomotor Hyperactivity | Sensitization | |||
| Development | Expression | Development | Expression | |||
| Neurotoxic lesion | ↓ a[26,27], b[29,32] Ø a[29], b[30,31] |
↓ a[27] | ↓ a[28] |
|||
| DBH knockout | ↑ [24] | ↓ [24] | ↑ [23,25] | |||
| DBH inhibitors | ↓ c[18], d[12,13] Ø d[14,22], f[14,15] |
↓ c[19] Ø c[20], e[23] ↑ c[23] |
↑ c[20,23], e[23] | ↑ c[20,23], e[23] | ||
| α1 AR knockout | ↓ g[37,39,46] ↓ h[47] |
↓ g[37,39] | ↓ g[37] Ø h[47] |
↓ g[37] |
||
| α1 AR antagonists | ↓ i[24,34-40,42] | ↓ i[24,36] Ø i[38] |
↓ i[39] Ø i[24] |
↓ i[34,36,37,41], j[45] Ø i[38,43,44] |
↓ i[36,44] Ø i[38,43] |
↓ i[44] Ø i[43] |
| α2 AR overexpression | ↓ [54] | |||||
| α2 AR agonists | ↓ k[38], l[54] | ↓ k[59] Ø k[38] |
↓ k[44] Ø k[38] |
Ø k[38,44] |
↓ k[44] | |
| α2 AR knockout | ↑ m[55], n[54] | ↓ m[55] | ||||
| α2 AR antagonists | ↑ o[53], p[52] | ↑ q[59] ↓ p,r[65] |
↓ r[55,65] | ↑ s[44] Ø p[44] |
Ø p,s[44] | Ø p,s[44] |
| β AR antagonists | ↑ t[38] ↓ t[34] Ø u[62], v[40,62] |
Ø t[38], u[62] ↓ v[62] |
Ø u,v[62] | Ø t[38,61] ↑ t[60] |
Ø t[38] | |
↓ – inhibitory effect; ↑ – facilitating effect; Ø – no effect; AR – adrenoceptor; DBH – dopamine-β-hydroxylase; NE – norepinephrine; a – DSP-4; b – 6-OHDA; c – disulfiram; d – FLA-63; e – nepicastat; f – U-14,624; g – α1b AR; h – α1d AR; i – prazosin; j – terazosin; k – clonidine; l – dexmedetomidine; m – α2A AR; n – α2C AR; o – dexefaroxan; p – idazoxan; q – efaroxan; r – atipamezole; s – yohimbine; t – propranolol; u – nadolol; v – timolol.
Repeated, intermittent exposure to psychostimulants is known to induce sensitization characterized by an increase in the number of behavioral events, including locomotor hyperactivity, when a challenge dose of the psychostimulant is readministered after the repeated treatment regimen was discontinued. It has been suggested that the sensitization paradigm models drug craving [63], though recently some objections to such conclusion have been raised [64].
NE synthesis inhibition by DBH activity blockade was reported to have discrepant effects on behavioral sensitization. Weinshenker et al. [24] showed attenuation of the locomotor response to a challenge dose of amphetamine administered 2 days after withdrawal from repeated treatment with the psychostimulant in DBH knockout mice as compared with wild type animals, though they observed an increase in stereotypical behaviors at the expense of horizontal locomotion. In contrast, Gaval-Cruz et al. [23] and Haile et al. [20] found that the DBH inhibitors disulfiram or nepicastat facilitated the development and expression of sensitization to the locomotor effect of cocaine in mice and rats.
On the other hand, partial denervation of LC projections with a low dose of the selective neurotoxin DSP-4 abolished the development of amphetamine sensitization in rats, suggesting that this behavioral phenomenon requires intact NE projections ascending from the LC [27].
Blockade of α1 ARs by prazosin attenuated both development and expression of sensitization to the locomotor effects of amphetamine or cocaine paired with environmental context in mice or rats, but not in the unpaired paradigm [24, 36, 38, 39, 43, 44]. Further studies with genetically inactivated subtypes of α1 ARs demonstrated that α1b AR subtype contributes to the sensitizing effects of the psychostimulants [37, 39].
The role of α2 ARs in the behavioral sensitization to psychostimulants is not clear. Thus, in line with the facilitating effect of α2 AR antagonism on NE transmission, Doucet et al. [59] showed that the α2 AR blocker efaroxan administered in combination with amphetamine for four consecutive days potentiated the sensitization development in mice as measured on the challenge day three weeks later. In contrast, Juhila et al. [55, 65] found that the α2 AR antagonists atipamezole or idazoxan attenuated both the development and expression of sensitization to the locomotor effect of amphetamine in mice. Moreover, Juhila et al. [55] also showed that amphetamine sensitization did not develop in α2a AR deficient mice. In contrast, Jimenéz-Rivera et al. [44] reported no effect of yohimbine or idazoxan on the development and expression of cocaine sensitization in rats.
Studies examining the effects of pharmacological stimulation of α2 ARs demonstrated that clonidine attenuated the development of sensitization to the locomotor effect of amphetamine in mice [59], but not in rats [38] and expression, but not development, of cocaine sensitization in rats [38, 44].
Only a few studies examined the effect of β AR antagonists on behavioral sensitization to psychostimulants. Whereas Vanderschuren et al. [38] observed no effect of propranolol on the development of amphetamine or cocaine sensitization, Colussi-Mas et al. [62] reported an attenuating effect of timolol, injected systemically or locally into the bed nucleus of stria terminalis (BNST), but no influence of nadolol (peripherally acting β AR blocker), administered systemically, on the development of sensitization to amphetamine in rats.
INTRACRANIAL SELF-STIMULATION (ICSS)
Intracranial self-stimulation (ICSS) is a method used to study the neural mechanisms of reinforcement. In this model, animals are trained to deliver electrical stimulation (eliciting positive reinforcement) to certain regions of their brain by performing an operant response (e.g. pressing a lever). ICSS can be measured as self-stimulation rate (responses/min) or current threshold. Many studies reported that administration of cocaine or amphetamines, facilitated the self-stimulation responding with electrodes placed in several brain structures, including the substantia nigra, NAc, medial forebrain bundle (mFB) of the lateral hypothalamus, LC or DNB [66-68].
A number of studies have examined the effects of lesions of NE system on self-stimulation behavior affected by amphetamine. Indeed, it has been shown that neonatal administration of 6-OHDA into the LC did not alter the facilitating effect of the psychostimulant on self-stimulation elicited from the same region in adult rats, but this observation was made on a relatively small number of animals [69]. Reduction of brain NE induced by intracisternal injections of 6-OHDA in adult rats also did not affect the amphetamine effect on self-stimulation in the LC, lateral hypothalamus or substantia nigra [70-72]. In addition, administration of the DBH inhibitor U-14,624 in 6-OHDA-treated rats did not alter the amphetamine facilitating effect on hypothalamic self-stimulation behavior [70]. Other studies have demonstrated that 6-OHDA-induced lesions of DNB did not change the amphetamine effect on hippocampal self-stimulation responding [73]. Overall, these findings indicate that the NE system does not mediate the action of amphetamine on brain self-stimulation.
The effect of the pharmacological α1 ARs blockade on psychostimulant-potentiated self-stimulation has been studied to a limited extent. For example, it was shown that the α1 AR antagonist azaperone, but not phenoxybenzamine or phentolamine, attenuated the effect of cocaine on self-stimulation responding elicited from the posterior hypothalamus, but also reduced the baseline self-stimulation behavior [68, 74]. As for the amphetamine-induced facilitation of ICSS, it was demonstrated that central administration of phenoxybenzamine or phentolamine, but not dibenamine, attenuated the effect of the psychostimulant administered intracerebroventricularly on hypothalamic self-stimulation behavior; however, phentolamine also reduced baseline self-stimulation response [74]. On the contrary, when amphetamine was administered systemically, neither intraperitoneally nor centrally injected phenoxybenzamine affected the hypothalamic self-stimulation responding potentiated by the psychostimulant [74]. On the basis of these observations, the role of α1 ARs in the psychostimulant-facilitated brain stimulation seems doubtful.
As far as pharmacological activation of α2 AR subtype is concerned, only one research group tested the effects of clonidine and reported that it attenuated the facilitating effect of amphetamine on self-stimulation behavior in the mFB but also reduced the baseline self-stimulation responding [75], indicating a non-specific effect.
Limited data have shown that the β AR blocker propranolol administered intracerebroventricularly or systemically did not affect the facilitating effect of either cocaine or amphetamine on self-stimulation of the posterior hypothalamus when the psychostimulants were given intraperitoneally. When amphetamine was injected centrally, propranolol reduced the ICSS response to the psychostimulant but also reduced the baseline self-stimulation responding [74]. Thus, it can be concluded that amphetamine- or cocaine-facilitated ICSS elicited from hypothalamus is not mediated by β ARs.
CONDITIONED PLACE PREFERENCE (CPP)
Conditioned place preference (CPP) is a model for testing the rewarding properties of drugs of abuse. In the first phase of CPP (pre-test), animals are allowed a 15-25-min access to a two-compartment apparatus. In the second phase (conditioning) animals are given repeated injections of the drug in one chamber and vehicle in the other. Two methods can be used, the biased method where the drug is administered in the less preferred and vehicle in the preferred compartment or unbiased method in which the experimenter chooses the compartment to be conditioned. In a post-conditioning phase, animals are tested for the expression of CPP by allowing them a full access to both compartments in the absence of the drug; the so-called preference post-test is designed to retrieve the memory of the drug-cue association (retrieval trial). Following the post-test, animals can be introduced to the extinction sessions during which they are given saline injections and are placed in the apparatus with access to both compartments. After reaching “no-preference criterion” [76], reinstatement tests (induced by drug or stress) can be conducted.
Acquisition
Here we describe the expression of psychostimulant-induced CPP, when conditioning was performed in animals with altered NE system (lesions, genetic manipulations, AR ligands). Actually, Kõiv et al. [28] showed that partial LC lesion induced by a low dose of DSP-4 reduced whereas the widespread LC denervation with a high dose of DSP-4 completely disrupted the expression of cocaine-induced CPP in rats. Other authors reported that selective depletion of NE tissue level in the mPFC in mice following local 6-OHDA administration, blocked cocaine- and amphetamine-induced CPP and in the case of amphetamine, it evoked a slight preference for the vehicle-paired compartment, implying the place aversion to the psychostimulant [77, 78]. However, in these experimental conditions Ventura et al. [77, 78] demonstrated a decrease in cocaine- and amphetamine-induced release of not only NE in the PFC but also DA in the NAc. In contrast to the above findings, earlier studies of Spyraki et al. [79] revealed that systemic administration of 6-OHDA in neonatal or adult rats, leading to destruction of central and/or peripheral NE system, did not alter the CPP induced by cocaine in adults, however, the biased method was used there, opposite to the unbiased method used by Ventura et al. [77, 78]. Studies employing genetic approach to examination of the role of NE deficiency in cocaine-induced CPP, reported that cocaine, depending on the dose, evoked preference (5 mg/kg), no preference (10 mg/kg) or aversion (20 mg/kg) in DBH knockout mice [25, 80], in contrast to control animals in which that psychostimulant in a wide dose range (10-60 mg/kg) evoked CPP, but no aversion [25]. Noteworthy is the fact that restoration of NE levels in the brain by administration of a synthetic amino acid DOPS and carbidopa reinstated cocaine CPP in NE-depleted mice [80]. On the basis of the data obtained using DBH knockout mice, it can be concluded that NE depletion altered the balance between psychostimulant reward and aversion.
Little is known about the contribution of α ARs to the psychostimulant-induced CPP. Sershen et al. [81] reported that repeated administration of the preferential α1 AR antagonist phentolamine during conditioning phase did not alter the cocaine-induced CPP in mice. Further, Davis et al. [82] and Juhila et al. [55] demonstrated that α2a AR knockout mice exhibited similar to wild type animals preference to amphetamine or cocaine.
Analysis of β AR signaling revealed that administration of the β AR blocker timolol before each conditioning session, attenuated the expression of 3,4-methylenedio-xymethamphetamine (MDMA)-induced CPP in mice [83]. On the other hand, in the case of cocaine-induced CPP, neither systemic administration of propranolol in mice nor infusions of a mixture of betaxolol (β1 AR antagonist) and ICI-118,551 (β2 AR antagonist) into the central nucleus of the amygdala (CeA) or BNST in rats before each conditioning session altered the expression of the psychostimulant-induced CPP [61, 84]. Consistent with these findings, CPP developed to the same extent in mice deficient in both β1 and β2 ARs as in wild type animals [85].
Retrieval/Extinction
There are some reports investigating the contribution of β ARs to retrieval of cocaine-associated memories. Briefly, data show that the β AR antagonists propranolol administered systemically or nadolol injected locally into the mPFC or dorsal hippocampus, but not into the basolateral amygdala, attenuated the retrieval of cocaine CPP memory [86-88].
As far as extinction phase of cocaine CPP is concerned, the impairing effect of the α2 AR antagonist yohimbine was demonstrated in mice [82, 89]. However, this effect did not seem to be related to α2 ARs since atipamezole, a more selective antagonist of these receptors was ineffective and also because that the impairing effect of yohimbine was exacerbated in α2a AR knockout animals [82]. Extinction of cocaine CPP was also insensitive to the α1 AR antagonist prazosin [90]. In addition, extinction of the psychostimulant CPP has been shown to be facilitated in mice deficient in β1 and β2 ARs [85].
Reinstatement
It has been shown that the α2 AR antagonists yohimbine and BRL-44408, possibly via enhancement of NE neurotransmission, produced reinstatement of the extinguished cocaine-induced CPP in mice [85, 91]. Importantly, the effect of yohimbine was blocked by the β AR blocker propranolol, but not by α1 AR antagonist prazosin or α2 AR agonist clonidine [91], while the reinstatement produced by BRL-44408 was blocked by the β2 (ICI-118551), but not β1 (betaxolol), AR antagonist and was not observed in mice lacking β1 and β2 ARs [85]. Interestingly, the reinstatement of the extinguished cocaine-induced CPP was also evoked by the nonselective β AR agonist isoproterenol or the selective β2 AR agonist clenbuterol [85, 92]. Noteworthy is the fact that isoproterenol-induced reinstatement was blocked by betaxolol or ICI-118,551, while reinstatement evoked by clenbuterol was not present in mice with targeted deletion of β1 and β2 ARs, but was still present after pretreatment with β1 AR antagonist betaxolol [85, 92]. Reinstatement of cocaine CPP in mice was also induced by a high dose of the α2 AR agonist clonidine [91] and involvement of postsynaptic α2 ARs in this effect cannot be excluded.
While the α1 AR antagonist prazosin failed to alter reinstatement of CPP induced by either cocaine or stress, the α2 AR agonist clonidine at a low dose blocked stress-, but not cocaine-primed reinstatement of the drug-induced CPP in mice [91].
Regarding the role of β ARs, neither propranolol administered just before priming nor permanent inactivation of β1 and β2 ARs affected cocaine-primed reinstatement of CPP in mice [61, 85, 91]. On the other hand, acute intrahippocampal infusion of nadolol or repeated systemic administration of propranolol during retrieval or extinction sessions prevented subsequent cocaine-primed reinstatement of CPP in rats [86, 88].
Importantly, the administration of propranolol prior to the reinstatement session blocked stress-evoked reinstatement of cocaine CPP in mice [91] and these findings are in agreement with genetic studies in mice deficient in β1 and β2 ARs in which stress failed to induce reinstatement [85]. When receptor subtype-selective antagonists were used in order to establish which receptor subtype mediated stress-induced reinstatement, it was shown that both ICI-118,551 and betaxolol blocked reinstatement of cocaine CPP in response to stress [85, 91, 92], although both antagonists reduced animals’ locomotor activity [85].
DRUG SELF-ADMINISTRATION (TABLE (TABLE 2))
Table 2.
The effects of the NE system modulation on self-administration of amphetamines and cocaine.
| NE System Modulation |
AMPHETAMINES | COCAINE | ||||
|---|---|---|---|---|---|---|
| Maintenance | Extinction | Reinstatement | Maintenance | Extinction | Reinstatement | |
| Neurotoxic lesion | Ø a*[3] | |||||
| DBH inhibitors | ↓ b●▲[114] | Ø cd*[96] ↓ d#[97] |
↓ cd*●[96], d#▲■[97] Ø cd●[125] |
|||
| α1 AR antagonists | ↓ e*[1,2] Ø e*[98], f *[2,99] |
Ø e*[94,100], f *[94,100,101], g*[102], g[103], h*[104] ↓ g*#[105], g#[106] |
↓ g*●[122] Ø g*▲[112] Ø g●[124] |
|||
| α2 AR agonists | Ø i*[107] | Ø j#[106] | ↓ i*[112] | ↓ ijk*▲[112], ilm*■[121] Ø ilm*●[121] ↓ i●[124] |
||
| α2 AR antagonists | ↑ n*[111] | ↑ n*▲[118, 119] Ø o*▲[112] |
||||
| β AR antagonists | ↓ p*[1,2] | ↓ p*[60,110], s*[60] Ø r#[106] |
↑ q*[113] | ↓ q+r*■[123] Ø q+r*●[123] Ø p●[124] |
||
| β AR agonists | ↓ t*[113] | |||||
↓ – inhibitory effect; ↑ – facilitating effect; Ø – no effect; AR – adrenoceptor; DBH – dopamine-β-hydroxylase; NE – norepinephrine; a – 6-OHDA; b – U-14,624; c – disulfiram; d – nepicastat; e– phentolamine; f – phenoxybenzamine; g– prazosin; h– terazosin; i – clonidine; j – UK 14,304; k – guanfacine; l – lofexidine; m – guanabenz; n – yohimbine; o – RS-79948; p – propranolol; q – ICI-118,551; r – betaxolol; s – atenolol; t – clenbuterol. Reinstatement induced by: ● – drug; ▲ – cue; ■ – stress. Schedule of reinforcement: * – fixed-ratio (FR); #– progressive-ratio (PR).
The drug self-administration model is designed to test the rewarding properties of drugs of abuse. In this technique, an animal is introduced into the chamber to self-administer a drug using operant responding (a lever press or nose poke). There are different operant schedules of drug self-administration, including ratio (i.e., drug infusion depends on a specified number of responses) and interval (i.e., drug is infused after a specific amount of elapsed time) schedule [93]. Among those most often used are: fixed-ratio (FR, completion of the FR requirement, usually ranging from 1 to 5, results in drug infusion) or progressive-ratio (PR, drug infusion is contingent on exponential increase in response requirements) schedules, with the latter method used to measure the motivating effect of drug reinforcement.
The drug self-administration paradigm consists of several experimental phases: acquisition, maintenance, extinction and reinstatement. During extinction training, saline is infused instead of the addictive drug and the number of presses on the active lever gradually decreases until extinction criteria are met. Then the reinstatement of drug seeking behavior, which models some aspects of relapse and is manifested as an increase in the number of active lever presses, can be precipitated by administration of the priming dose of the addictive drug (drug-induced reinstatement), presentation of drug-associated cues (cue-induced reinstatement) or stress, including footshock or forced swim (stress-induced reinstatement).
Maintenance
Studies investigating the effects of depletion of the functional pool of NE with the catecholamine synthesis-blocking agent AMPT demonstrated an increase in cocaine or amphetamine self-administration behavior in rhesus monkeys or rats under the FR schedule of reinforcement [94, 95], suggesting that catecholamine depletion attenuates the reinforcing effects of the psychostimulants. However, since disulfiram and nepicastat, DBH inhibitors administered systemically [96] as well as lesions of DNB and VNB induced by 6-OHDA [3] did not affect the maintenance phase of cocaine self-administration in rats, the importance of NE for rewarding activity of the psychostimulants or at least cocaine seems doubtful. Nevertheless, when cocaine was self-administered under a PR reinforcement schedule in rats, it has been found that DBH inhibition by nepicastat reduced the breakpoint for the psychostimulant, but not for food or sucrose, responding [97]. The latter finding may indicate that NE signaling plays an important role in the motivating effects of cocaine.
Studies examining the effects of α1 AR ligands on the maintenance of psychostimulant self-administration rather exclude the role of these receptors in drug reinforcement. In combination tests, it has been shown that phentolamine reduced the rate of responding for amphetamine in rats [1, 2], but not in pigeons [98], while another α1 AR antagonist phenoxybenzamine had no effect either in rats or dogs self-administering amphetamine [2, 99]. As suggested by Yokel and Wise [2], the inhibitory effect of phentolamine on amphetamine responding could be due to its action on metabolism of the psychostimulant. In the case of cocaine reinforcement, neither phenoxybenzamine nor phentolamine changed cocaine self-administration in rhesus monkeys, dogs or rats [94, 100, 101]. Moreover, the more selective α1 AR blockers prazosin and terazosin were also shown to be ineffective in the maintenance phase of psychostimulant self-administration. In fact, prazosin did not alter cocaine self-administration in squirrel or rhesus monkeys [102, 103], while terazosin administered locally into the mPFC or ventral tegmental area (VTA) did not affect cocaine self-administration in rats [104]. Finally, another evidence against the involvement of α1 AR signaling was presented by Risner and Jones [99], who found in substitution studies that the α1 AR agonist methoxamine failed to maintain responding for amphetamine in dogs. In addition, it has been shown that rats with extended access to cocaine (6-h sessions of self-administration or passive administration of the psychostmulant for a few days) exhibited the higher breakpoint for the drug using PR schedule and this increased breakpoint was reduced or blocked by prazosin. Such an effect of prazosin was not observed in cocaine self-administration under PR schedule in non-cocaine-pretreated rats [105, 106]. Moreover, the latter authors [106] showed that the number of neurons with α1 AR-like immuno-reactivity was significantly lower in the BNST of rats with extended access to cocaine. The data suggest that activation of α1 ARs may be associated with increased motivation for cocaine administration and that the extended amygdala, such as the BNST, may be regarded as neuroanatomical target of this phenomenon.
As far as α2 ARs are concerned, the results of studies in which α2 AR agonists clonidine or UK 14,304 were used indicated that the activation of these receptors was engaged neither in reinforcing properties of amphetamine nor increased motivation for cocaine in rats [106, 107]. However, Weerts and Griffiths [108] and Woolverton et al. [109] showed that clonidine was successfully substituted for cocaine in rhesus monkeys and baboons self-administering the psychostimulant.
A number of reports investigated the role of β AR system in psychostimulant reinforcement. It was shown that pretreatment with the nonselective β AR blocker propranolol attenuated cocaine and amphetamine self-administration in squirrel monkeys and rats, but, especially at higher doses, also slightly decreased the responding for food in rats [1, 2, 60, 110]. Furthermore, Harris et al. [60] found that atenolol, a peripherally restricted β1 AR blocker also reduced cocaine self-administration in rats, but to a much lesser extent than propranolol. On the other hand, when the selective and centrally acting β1 AR antagonist betaxolol was tested in cocaine self-administration paradigm performed under the PR schedule, it failed to alter the breakpoint for cocaine in rats [106], which suggested that central β1 ARs were not involved in motivation for the psychostimulant.
Extinction/Reinstatement
The involvement of NE signaling in the extinction of psychostimulant self-administration has been studied in rats to a limited extent. Whereas the α2 AR antagonist yohimbine, increasing NE release, augmented the number of responses on the active lever during consecutive extinction sessions [111], the α2 AR agonist clonidine, inhibiting NE release, attenuated the responding on the active lever, but it also reduced the number of the inactive lever presses [112]. In some opposition to the above observations are the results of the experiments, in which β2 AR ligands were used. In fact, while the activation of these receptors in the infralimbic cortex facilitated extinction of cocaine self-administration, their blockade in this region hampered it [113].
On the other hand, the results demonstrating the involvement of NE in the reinstatement of psychostimulant drug seeking behavior in rats are much more consistent. Actually, Schroeder et al. [96, 97] showed that the DBH inhibitors nepicastat and disulfiram reduced cue- and completely blocked cocaine- or stress-induced seeking behavior, and did not alter the responses on the inactive lever. Similar results were reported by Davis et al. [114], who found inhibitory effect of another DBH inhibitor U-14,624 on amphetamine seeking behavior. In line with the above observations, intracerebroventricular infusion of NE evoked reinstatement of cocaine seeking behavior [115, 116] while systemic administration of the α2 AR antagonist yohimbine (but not RS-79948) in doses increasing the NE release induced reinstatement of cocaine or methamphetamine seeking behavior [97, 111, 112, 115, 117-120]. Importantly, the effects of yohimbine were attenuated by DBH inhibitors and by the α2 AR agonist guanfacine, but not by clonidine [97, 115, 120]. It was also found that yohimbine (but not RS-79948) potentiated cue-primed cocaine seeking behavior [112, 118, 119]. In addition, the agonists of α2 ARs clonidine, guanfacine or UK 14,304 attenuated cue-induced cocaine seeking behavior, though their effects may be non-specific as they also attenuated inactive lever presses, inhibited locomotor activity or decreased food self-administration [112]. Nevertheless, the inhibitory effect of clonidine on the cue-induced reinstatement of cocaine seeking behavior was blocked by RS-79948, indicating that it was mediated by α2 ARs [112]. Clonidine and other α2 AR agonists lofexidine and guanabenz were also shown to block or reduce stress-, but not cocaine-, primed reinstatement of drug seeking behavior, though lofexidine was also found to attenuate the number of inactive lever presses and sucrose reinforcement [121]. The antagonist of α1 ARs prazosin administered in doses that did not affect inactive lever responding or operant responding for food, reduced active lever responding during cocaine- [122], but not cue-induced reinstatement of cocaine seeking behavior [112]. A mixture of the β1 and β2 AR antagonists betaxolol and ICI-118,551, respectively, infused into the BNST or CeA attenuated or blocked, respectively, stress-, but not cocaine-induced reinstatement of cocaine seeking behavior, and were without effect on inactive lever responding or responding for sucrose [123]. In other words, in rats NE, via distinct ARs, seems to facilitate reinstatement of psychostimulant seeking behavior depending on the kind of priming.
However, in contrast to rats, the results obtained in nonhuman primates do not support the above conclusion. In fact, although cocaine-induced reinstatement of cocaine seeking behavior was attenuated by clonidine, it was not affected by prazosin or propranolol in squirrel monkeys [124]. Moreover, in the same species DBH inhibitors (disulfiram, nepicastat) not only were ineffective in the above paradigm, but, unexpectedly, nepicastat given alone induced a modest reinstatement effect [125]. Contradictory results were also reported after α2 AR antagonists [96, 126].
CONCLUSIONS
The data presented above indicate that the brain NE system is involved in different effects of psychostimulants though the obtained results depend on experimental tools used to modify its activity and on behavioral paradigm employed in the study. Thus, regarding psychostimulant-induced locomotor hyperactivity, the results obtained with NE synthesis inhibitors or neurotoxins lesioning NE pathways are inconclusive, however, the data from pharmacological or genetic manipulations in α1 or α2, but not β ARs, indicate that NE neurotransmission has a facilitating effect. Similar conclusion may be drawn from the results on development and expression of sensitization to the locomotor effects induced by psychostimulants with additional contribution of β ARs, particularly those located in the BNST, to the development of amphetamine sensitization.
The results obtained in more specific models of drug rewarding activity indicate that the brain NE system does not seem to be specifically involved in psychostimulant-induced facilitation of ICSS, whereas it plays a role in some aspects of CPP and drug self-administration behavior. In particular, the NE system (especially β ARs) is engaged in stress-induced reinstatement of extinguished psychostimulant-induced CPP, but results concerning its importance for reinstatement evoked by psychostimulants themselves or other aspects of the behavior (acquisition, retrieval, extinction) are not conclusive. In drug self-administration model, it has been demonstrated that NE signaling (via different ARs) plays an important role in the motivating effects of psychostimulants and in reinstatement of drug seeking behavior (in rodents, but not in primates). At the same time, the NE system does not seem to be involved in either extinction of drug self-administration or rewarding activity of psychostimulants.
Several observations indicate that facilitating effects of NE signaling on addictive responses to amphetamines or cocaine may depend on the interaction between NE and DA systems, the latter one being a key neurotransmitter system involved in the psychostimulant-induced primary rewarding/reinforcing activity. In fact, it is well established that two DA systems, i.e. mesolimbic (consisting of the VTA containing DA cell bodies and the NAc where DA terminals are located) and mesocortical (the VTA projecting DA fibres to the PFC) systems, receive NE input. Actually, NE neurons originating from the LC and LTN innervate the VTA, while the NAc and PFC are innervated directly by NE projections from the LC and the LTN, respectively (Fig. 1; [127]). Besides the anatomical connections, there is a functional interaction between NE and DA systems, as evidenced by the induction of burst firing of the VTA neurons after electrical stimulation of the LC [128] and by the decreased neuronal activity of the VTA neurons after systemic administration of α2 AR agonists, inhibiting NE release, or α1 AR antagonists [129,130]. At the same time, it has been reported that lesions of the LC or intraaccumbal infusion of the α1 AR antagonist prazosin attenuated DA release in the NAc [131-133].
Fig. (1).
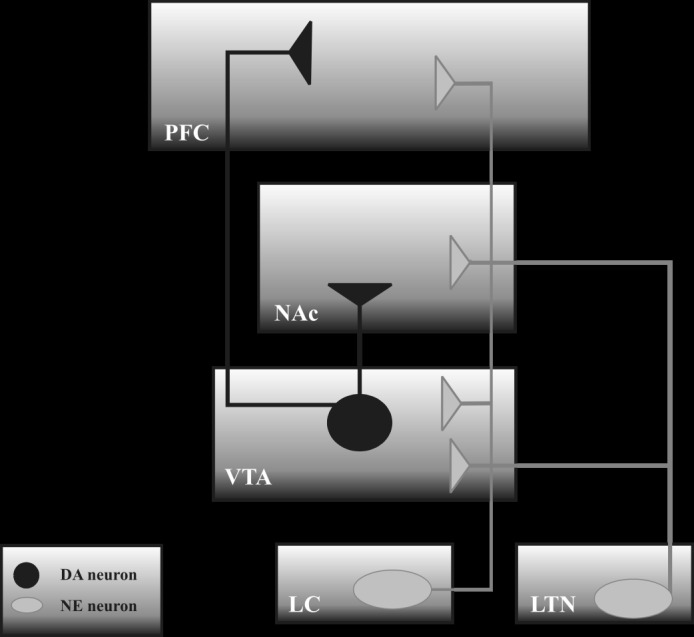
Schematic illustration of the norepinephrine (NE) innervation of the dopamine (DA) structures. LC – locus coeruleus; LTN – lateral tegmental nuclei; NAc – nucleus accumbens; PFC – prefrontal cortex; VTA – ventral tegmental area.
Importantly, modulation of NE signaling has also been shown to affect psychostimulant-induced DA release. For example, inhibition of the amphetamine-induced increase in the extracellular level of DA in the NAc was demonstrated in α1b AR or DBH knockout mice and in rats systemically pretreated with prazosin [25, 42, 46]. Similarly, DBH inhibitors and intraaccumbally administered α1 AR antagonist terazosin attenuated cocaine-induced DA overflow in the NAc in squirrel monkeys and rats, respectively [45,125].
Somehow in contrast to the above findings, animals with chronic deficiency of NE system (DBH knockout animals, chronic treatment with DBH inhibitors) that showed up-regulation of high-affinity state postsynaptic DA receptors and DA release from NE neurons, sometimes displayed behavioral hypersensitivity to psychostimulants [23, 25, 127].
Further studies are necessary to obtain more data on molecular mechanisms and neuroanatomical substrates responsible for the involvement of the NE system in psychostimulant addiction so that it could become a target for antiaddictive therapy.
ACKNOWLEDGEMENTS
This paper was supported by the statutory funds from the Department of Pharmacology (Laboratory of Drug Addiction Pharmacology), Institute of Pharmacology Polish Academy of Sciences (Kraków, Poland).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Yokel R.A., Wise R.A. Increased lever pressing for amphetamine after pimozide in rats: implications for a dopamine theory of reward. Science. 1975;187(4176):547–549. doi: 10.1126/science.1114313. [DOI] [PubMed] [Google Scholar]
- 2.Yokel R.A., Wise R.A. Attenuation of intravenous amphetamine reinforcement by central dopamine blockade in rats. Psychopharmacology. 1976;48(3):311–318. doi: 10.1007/BF00496868. [DOI] [PubMed] [Google Scholar]
- 3.Roberts D.C., Corcoran M.E., Fibiger H.C. On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol. Biochem. Behav. 1977;6(6):615–620. doi: 10.1016/0091-3057(77)90084-3. [DOI] [PubMed] [Google Scholar]
- 4.Koe B.K. Molecular geometry of inhibitors of the uptake of catecholamines and serotonin in synaptosomal preparations of rat brain. J. Pharmacol. Exp. Ther. 1976;199(3):649–661. [PubMed] [Google Scholar]
- 5.Kuczenski R., Segal D.S., Cho A.K., Melega W. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J. Neurosci. 1995;15(2):1308–1317. doi: 10.1523/JNEUROSCI.15-02-01308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reith M.E., Li M.Y., Yan Q.S. Extracellular dopamine, norepinephrine, and serotonin in the ventral tegmental area and nucleus accumbens of freely moving rats during intracerebral dialysis following systemic administration of cocaine and other uptake blockers. Psychopharmacology (Berl.) 1997;134(3):309–317. doi: 10.1007/s002130050454. [DOI] [PubMed] [Google Scholar]
- 7.Rothman R.B., Baumann M.H., Dersch C.M., Romero D.V., Rice K.C., Carroll F.I., Partilla J.S. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39(1):32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Kostowski W. Two noradrenergic systems in the brain and their interactions with other monoaminergic neurons. Pol. J. Pharmacol. Pharm. 1979;31(4):425–436. [PubMed] [Google Scholar]
- 9.Bylund D.B., Eikenberg D.C., Hieble J.P., Langer S.Z., Lefkowitz R.J., Minneman K.P., Molinoff P.B., Ruffolo R.R., Jr, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994;46(2):121–136. [PubMed] [Google Scholar]
- 10.Spector S., Sjoerdsma A., Udenfriend S. Blockade of endogenous norepinephrine synthesis by alpha-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. J. Pharmacol. Exp. Ther. 1965;147:86–95. [PubMed] [Google Scholar]
- 11.Menon M.K., Dandiya P.C., Bapna J.S. Modification of the effect of some central stimulants in mice pretreated with alpha-methyl-1-tyrosine. Psychopharmacology (Berl.) 1967;10(5):437–444. doi: 10.1007/BF00403985. [DOI] [PubMed] [Google Scholar]
- 12.Svensson T.H. The effect of inhibition of catecholamine synthesis on dexamphetamine induced central stimulation. Eur. J. Pharmacol. 1970;12(2):161–166. doi: 10.1016/0014-2999(70)90061-0. [DOI] [PubMed] [Google Scholar]
- 13.Rolinski Z., Scheel-Krüger J. The effect of dopamine and noradrenaline antagonists on amphetamine induced locomotor activity in mice and rats. Acta Pharmacol. Toxicol. (Copenh.) 1973;33(5):385–399. doi: 10.1111/j.1600-0773.1973.tb01540.x. [DOI] [PubMed] [Google Scholar]
- 14.Thornburg J.E., Moore K.E. The relative importance of dopaminergic and noradrenergic neuronal systems for the stimulation of locomotor activity induced by amphetamine and other drugs. Neuropharmacology. 1973;12(9):853–866. doi: 10.1016/0028-3908(73)90038-5. [DOI] [PubMed] [Google Scholar]
- 15.Hollister A.S., Breese G.R., Cooper B.R. Comparison of tyrosine hydroxylase and dopamine-beta-hydroxylase inhibition with the effects of various 6-hydroxydopamine treatments on d-amphetamine induced motor activity. Psychopharmacology (Berl.) 1974;36(1):1–16. doi: 10.1007/BF00441377. [DOI] [PubMed] [Google Scholar]
- 16.Musacchio J.M., Goldstein M., Anagnoste B., Poch G., Kopin I.J. Inhibition of dopamine-beta-hydroxylase by disulfiram in vivo. J. Pharmacol. Exp. Ther. 1966;152(1):56–61. [PubMed] [Google Scholar]
- 17.Thomas S.A., Matsumoto A.M., Palmiter R.D. Noradrenaline is essential for mouse fetal development. Nature. 1995;374(6523):643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 18.Maj J., Przegaliński E. Disulfiram and some effects of amphetamine in mice and rats. J. Pharm. Pharmacol. 1967;19(5):341–342. doi: 10.1111/j.2042-7158.1967.tb08101.x. [DOI] [PubMed] [Google Scholar]
- 19.Maj J., Przegaliński E., Wielosz M. Disulfiram and the drug-induced effects on motility. J. Pharm. Pharmacol. 1968;20(3):247–248. doi: 10.1111/j.2042-7158.1968.tb09735.x. [DOI] [PubMed] [Google Scholar]
- 20.Haile C.N., During M.J., Jatlow P.I., Kosten T.R., Kosten T.A. Disulfiram facilitates the development and expression of locomotor sensitization to cocaine in rats. Biol. Psychiatry. 2003;54(9):915–921. doi: 10.1016/S0006-3223(03)00241-5. [DOI] [PubMed] [Google Scholar]
- 21.Maj J., Przegaliński E. Some central effects of disulfiram. Dissertationes Pharmaceuticae et Pharmacologicae. XIX. 1967;5:505–513. doi: 10.1016/0006-8993(70)90185-X. [DOI] [Google Scholar]
- 22.Corrodi H., Fuxe K., Ljungdahl A., Ogren S.O. Studies on the action of some psychoactive drugs on central noradrenaline neurons after inhibition of dopamine-beta-hydroxylase. Brain Res. 1970;24(3):451–470. doi: 10.1016/0006-8993(70)90185-x. [DOI] [PubMed] [Google Scholar]
- 23.Gaval-Cruz M., Liles L.C., Iuvone P.M., Weinshenker D. Chronic inhibition of dopamine β-hydroxylase facilitates behavioral responses to cocaine in mice. PLoS One. 2012;7(11):e50583. doi: 10.1371/journal.pone.0050583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinshenker D., Miller N.S., Blizinsky K., Laughlin M.L., Palmiter R.D. Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. Proc. Natl. Acad. Sci. USA. 2002;99(21):13873–13877. doi: 10.1073/pnas.212519999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schank J.R., Ventura R., Puglisi-Allegra S., Alcaro A., Cole C.D., Liles L.C., Seeman P., Weinshenker D. Dopamine beta-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacology. 2006;31(10):2221–2230. doi: 10.1038/sj.npp.1301000. [DOI] [PubMed] [Google Scholar]
- 26.Harro J., Meriküla A., Lepiku M., Modiri A.R., Rinken A., Oreland L. Lesioning of locus coeruleus projections by DSP-4 neurotoxin treatment: effect on amphetamine-induced hyperlocomotion and dopamine D2 receptor binding in rats. Pharmacol. Toxicol. 2000;86(5):197–202. doi: 10.1034/j.1600-0773.2000.d01-35.x. [DOI] [PubMed] [Google Scholar]
- 27.Alttoa A., Eller M., Herm L., Rinken A., Harro J. Amphetamine-induced locomotion, behavioral sensitization to amphetamine, and striatal D2 receptor function in rats with high or low spontaneous exploratory activity: differences in the role of locus coeruleus. Brain Res. 2007;1131(1):138–148. doi: 10.1016/j.brainres.2006.10.075. [DOI] [PubMed] [Google Scholar]
- 28.Kõiv K., Zobel R., Raudkivi K., Kivastik T., Harro J. The effect of denervation of the locus coeruleus projections with N-(2- chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) on cocaineinduced locomotion and place preference in rats. Behav. Brain Res. 2011;216(1):172–179. doi: 10.1016/j.bbr.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 29.Archer T., Fredriksson A., Jonsson G., Lewander T., Mohammed A.K., Ross S.B., Söderberg U. Central noradrenaline depletion antagonizes aspects of d-amphetamine-induced hyperactivity in the rat. Psychopharmacology (Berl.) 1986;88(2):141–146. doi: 10.1007/BF00652230. [DOI] [PubMed] [Google Scholar]
- 30.Creese I., Iversen S.D. The pharmacological and anatomical substrates of the amphetamine response in the rat. Brain Res. 1975;83(3):419–436. doi: 10.1016/0006-8993(75)90834-3. [DOI] [PubMed] [Google Scholar]
- 31.Roberts D.C., Zis A.P., Fibiger H.C. Ascending catecholamine pathways and amphetamine-induced locomotor activity: importance of dopamine and apparent non-involvement of norepinephrine. Brain Res. 1975;93(3):441–454. doi: 10.1016/0006-8993(75)90182-1. [DOI] [PubMed] [Google Scholar]
- 32.Mohammed A.K., Danysz W., Ogren S.O., Archer T. Central noradrenaline depletion attenuates amphetamine-induced locomotor behavior. Neurosci. Lett. 1986;64(2):139–144. doi: 10.1016/0304-3940(86)90089-3. [DOI] [PubMed] [Google Scholar]
- 33.Kostowski W., Płaźnik A., Puciłowski O., Malatyńska E. Effect of lesions of the brain noradrenergic systems on amphetamine-induced hyperthermia and locomotor stimulation. Acta Physiol. Pol. 1982;33(4):383–387. [PubMed] [Google Scholar]
- 34.Snoddy A.M., Tessel R.E. Prazosin: effect on psychomotor-stimulant cues and locomotor activity in mice. Eur. J. Pharmacol. 1985;116(3):221–228. doi: 10.1016/0014-2999(85)90156-6. [DOI] [PubMed] [Google Scholar]
- 35.Blanc G., Trovero F., Vezina P., Hervé D., Godeheu A.M., Glowinski J., Tassin J.P. Blockade of prefronto-cortical alpha 1-adrenergic receptors prevents locomotor hyperactivity induced by subcortical D-amphetamine injection. Eur. J. Neurosci. 1994;6(3):293–298. doi: 10.1111/j.1460-9568.1994.tb00272.x. [DOI] [PubMed] [Google Scholar]
- 36.Drouin C., Blanc G., Villégier A.S., Glowinski J., Tassin J.P. Critical role of alpha1-adrenergic receptors in acute and sensitized locomotor effects of D-amphetamine, cocaine, and GBR 12783: influence of preexposure conditions and pharmacological characteristics. Synapse. 2002;43(1):51–61. doi: 10.1002/syn.10023. [DOI] [PubMed] [Google Scholar]
- 37.Drouin C., Darracq L., Trovero F., Blanc G., Glowinski J., Cotecchia S., Tassin J.P. Alpha1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J. Neurosci. 2002;22(7):2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanderschuren L.J., Beemster P., Schoffelmeer A.N. On the role of noradrenaline in psychostimulant-induced psychomotor activity and sensitization. Psychopharmacology (Berl.) 2003;169(2):176–185. doi: 10.1007/s00213-003-1509-8. [DOI] [PubMed] [Google Scholar]
- 39.Auclair A., Drouin C., Cotecchia S., Glowinski J., Tassin J.P. 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur. J. Neurosci. 2004;20(11):3073–3084. doi: 10.1111/j.1460-9568.2004.03805.x. [DOI] [PubMed] [Google Scholar]
- 40.Alsene K.M., Fallace K., Bakshi V.P. Ventral striatal noradrenergic mechanisms contribute to sensorimotor gating deficits induced by amphetamine. Neuropsychopharmacology. 2010;35(12):2346–2356. doi: 10.1038/npp.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wellman P., Ho D., Cepeda-Benito A., Bellinger L., Nation J. Cocaine-induced hypophagia and hyperlocomotion in rats are attenuated by prazosin. Eur. J. Pharmacol. 2002;455(2-3):117–126. doi: 10.1016/S0014-2999(02)02616-X. [DOI] [PubMed] [Google Scholar]
- 42.Darracq L., Blanc G., Glowinski J., Tassin J.P. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J. Neurosci. 1998;18(7):2729–2739. doi: 10.1523/JNEUROSCI.18-07-02729.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filip M., Nowak E., Papla I. On the role of serotonin2A/2C receptors in the sensitization to cocaine. J. Physiol. Pharmacol. 2001;52(3):471–481. [PubMed] [Google Scholar]
- 44.Jiménez-Rivera C.A., Feliu-Mojer M., Vázquez-Torres R. Alpha-noradrenergic receptors modulate the development and expression of cocaine sensitization. Ann. N. Y. Acad. Sci. 2006;1074:390–402. doi: 10.1196/annals.1369.039. [DOI] [PubMed] [Google Scholar]
- 45.Mitrano D.A., Schroeder J.P., Smith Y., Cortright J.J., Bubula N., Vezina P., Weinshenker D. α-1 Adrenergic receptors are localized on presynaptic elements in the nucleus accumbens and regulate mesolimbic dopamine transmission. Neuropsychopharmacology. 2012;37(9):2161–2172. doi: 10.1038/npp.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auclair A., Cotecchia S., Glowinski J., Tassin J.P. D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J. Neurosci. 2002;22(21):9150–9154. doi: 10.1038/sj.mp.4001351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sadalge A., Coughlin L., Fu H., Wang B., Valladares O., Valentino R., Blendy J.A. alpha 1d Adrenoceptor signaling is required for stimulus induced locomotor activity. Mol. Psychiatry. 2003;8(7):664–672. doi: 10.1038/sj.mp.4001351. [DOI] [PubMed] [Google Scholar]
- 48.Tanda G., Bassareo V., Di Chiara G. Mianserin markedly and selectively increases extracellular dopamine in the prefrontal cortex as compared to the nucleus accumbens of the rat. Psychopharmacology (Berl.) 1996;123(2):127–130. doi: 10.1007/BF02246169. [DOI] [PubMed] [Google Scholar]
- 49.Devoto P., Flore G., Pani L., Gessa G.L. Evidence for co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex. Mol. Psychiatry. 2001;6(6):657–664. doi: 10.1038/sj.mp.4000904. [DOI] [PubMed] [Google Scholar]
- 50.Rizk P., Salazar J., Raisman-Vozari R., Marien M., Ruberg M., Colpaert F., Debeir T. The alpha2-adrenoceptor antagonist dexefaroxan enhances hippocampal neurogenesis by increasing the survival and differentiation of new granule cells. Neuropsychopharmacology. 2006;31(6):1146–1157. doi: 10.1038/sj.npp.1300954. [DOI] [PubMed] [Google Scholar]
- 51.Curet O., Dennis T., Scatton B. Evidence for the involvement of presynaptic alpha-2 adrenoceptors in the regulation of norepinephrine metabolism in the rat brain. J. Pharmacol. Exp. Ther. 1987;240(1):327–336. [PubMed] [Google Scholar]
- 52.Dickinson S.L., Gadie B., Tulloch I.F. Alpha 1- and alpha 2-adrenoreceptor antagonists differentially influence locomotor and stereotyped behaviour induced by d-amphetamine and apomorphine in the rat. Psychopharmacology (Berl.) 1988;96(4):521–527. doi: 10.1007/BF02180034. [DOI] [PubMed] [Google Scholar]
- 53.Villégier A.S., Drouin C., Bizot J.C., Marien M., Glowinski J., Colpaërt F., Tassin J.P. Stimulation of postsynaptic alpha1b- and alpha2-adrenergic receptors amplifies dopamine-mediated locomotor activity in both rats and mice. Synapse. 2003;50(4):277–284. doi: 10.1002/syn.10267. [DOI] [PubMed] [Google Scholar]
- 54.Sallinen J., Haapalinna A., Viitamaa T., Kobilka B.K., Scheinin M. D-amphetamine and L-5-hydroxytryptophan-induced behaviours in mice with genetically-altered expression of the alpha2C-adrenergic receptor subtype. Neuroscience. 1998;86(3):959–965. doi: 10.1016/S0306-4522(98)00100-6. [DOI] [PubMed] [Google Scholar]
- 55.Juhila J., Honkanen A., Sallinen J., Haapalinna A., Korpi E.R., Scheinin M. alpha(2A)-Adrenoceptors regulate d-amphetamineinduced hyperactivity and behavioural sensitization in mice. Eur. J. Pharmacol. 2005;517(1-2):74–83. doi: 10.1016/j.ejphar.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 56.Lähdesmäki J., Sallinen J., MacDonald E., Kobilka B.K., Fagerholm V., Scheinin M. Behavioral and neurochemical characterization of alpha(2A)-adrenergic receptor knockout mice. Neuroscience. 2002;113(2):289–299. doi: 10.1016/S0306-4522(02)00185-9. [DOI] [PubMed] [Google Scholar]
- 57.Sallinen J., Link R.E., Haapalinna A., Viitamaa T., Kulatunga M., Sjöholm B., Macdonald E., Pelto-Huikko M., Leino T., Barsh G.S., Kobilka B.K., Scheinin M. Genetic alteration of alpha 2C-adrenoceptor expression in mice: influence on locomotor, hypothermic, and neurochemical effects of dexmedetomidine, a subtype-nonselective alpha 2-adrenoceptor agonist. Mol. Pharmacol. 1997;51(1):36–46. doi: 10.1124/mol.51.1.36. [DOI] [PubMed] [Google Scholar]
- 58.Ihalainen J.A., Tanila H. In vivo regulation of dopamine and noradrenaline release by alpha2A-adrenoceptors in the mouse prefrontal cortex. Eur. J. Neurosci. 2002;15(11):1789–1794. doi: 10.1046/j.1460-9568.2002.02014.x. [DOI] [PubMed] [Google Scholar]
- 59.Doucet E.L., Bobadilla A.C., Houades V., Lanteri C., Godeheu G., Lanfumey L., Sara S.J., Tassin J.P. Sustained impairment of α2A-adrenergic autoreceptor signaling mediates neurochemical and behavioral sensitization to amphetamine. Biol. Psychiatry. 2013;74(2):90–98. doi: 10.1016/j.biopsych.2012.11.029. [DOI] [PubMed] [Google Scholar]
- 60.Harris G.C., Hedaya M.A., Pan W.J., Kalivas P. beta-adrenergic antagonism alters the behavioral and neurochemical responses to cocaine. Neuropsychopharmacology. 1996;14(3):195–204. doi: 10.1016/0893-133X(95)00089-V. [DOI] [PubMed] [Google Scholar]
- 61.Al-Hasani R., McCall J.G., Foshage A.M., Bruchas M.R. Locus coeruleus kappa-opioid receptors modulate reinstatement of cocaine place preference through a noradrenergic mechanism. Neuropsychopharmacology. 2013;38(12):2484–2497. doi: 10.1038/npp.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colussi-Mas J., Panayi F., Scarna H., Renaud B., Bérod A., Lambás-Señas L. Blockade of beta-adrenergic receptors prevents amphetamine-induced behavioural sensitization in rats: a putative role of the bed nucleus of the stria terminalis. Int. J. Neuropsychopharmacol. 2005;8(4):569–581. doi: 10.1017/S1461145705005298. [DOI] [PubMed] [Google Scholar]
- 63.Robinson T.E., Berridge K.C. Incentive-sensitization and addiction. Addiction. 2001;96(1):103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- 64.Steketee J.D., Kalivas P.W. Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol. Rev. 2011;63(2):348–365. doi: 10.1124/pr.109.001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Juhila J., Haapalinna A., Sirviö J., Sallinen J., Honkanen A., Korpi E.R., Scheinin M. The alpha2-adrenoceptor antagonist atipamezole reduces the development and expression of d-amphetamine-induced behavioural sensitization. Naunyn Schmiedebergs Arch. Pharmacol. 2003;367(3):274–280. doi: 10.1007/s00210-003-0695-6. [DOI] [PubMed] [Google Scholar]
- 66.Fibiger H.C., Phillips A.G. Role of dopamine and norepinephrine in the chemistry of reward. J. Psychiatr. Res. 1974;11:135–143. doi: 10.1016/0022-3956(74)90084-3. [DOI] [PubMed] [Google Scholar]
- 67.Wise R.A. Catecholamine theories of reward: a critical review. Brain Res. 1978;152(2):215–247. doi: 10.1016/0006-8993(78)90253-6. [DOI] [PubMed] [Google Scholar]
- 68.Aulakh C.S., Ghosh B., Pradhan S.N. Actions and interactions of cocaine on self-stimulation behavior in rats. Psychopharmacology (Berl.) 1979;63(1):75–79. doi: 10.1007/BF00426925. [DOI] [PubMed] [Google Scholar]
- 69.Umemoto M., Olds M.E. Presynaptic alpha-adrenergic mediation of self-stimulation in locus coeruleus in rats treated neonatally with 6-hydroxydopamine. Brain Res. 1981;219(1):107–119. doi: 10.1016/0006-8993(81)90271-7. [DOI] [PubMed] [Google Scholar]
- 70.Cooper B.R., Cott J.M., Breese G.R. Effects of catecholamine-depleting drugs and amphetamine on self-stimulation of brain following various 6-hydroxydopamine treatments. Psychopharmacology (Berl.) 1974;37(3):235–248. doi: 10.1007/BF00421537. [DOI] [PubMed] [Google Scholar]
- 71.Cooper B.R., Konkol R.J., Breese G.R. Effects of catecholamine depleting drugs and d-amphetamine on self-stimulation of the substantia nigra and locus coeruleus. J. Pharmacol. Exp. Ther. 1978;204(3):592–605. [PubMed] [Google Scholar]
- 72.Cooper B.R., Breese G.R. A role for dopamine in the psychopharmacology of electrical self-stimulation. Natl. Inst. Drug Abuse Res. Monogr. Ser. 1975;(3):63–70. doi: 10.1037/e469652004-001. [DOI] [PubMed] [Google Scholar]
- 73.Phillips A.G., Van Der Kooy D., Fibiger H.C. Maintenance of intracranial self-stimulation in hippocampus and olfactory bulb following regional depletion of noradrenaline. Neurosci. Lett. 1977;4(2):77–84. doi: 10.1016/0304-3940(77)90148-3. [DOI] [PubMed] [Google Scholar]
- 74.Bailey P.T., Pradhan S.N. Interactions of adrenergic stimulants and blockers on self-stimulation behavior in rats. Res. Commun. Chem. Pathol. Pharmacol. 1975;11(4):543–552. [PubMed] [Google Scholar]
- 75.Vetulani J., Leith N.J., Stawarz R.J., Sulser F. Effect of clonidine on the noradrenergic cyclic AMP generating system in the limbic forebrain and on medial forebrain bundle self-stimulation behavior. Experientia. 1977;33(11):1490–1491. doi: 10.1007/BF01918827. [DOI] [PubMed] [Google Scholar]
- 76.Fricks-Gleason A.N., Marshall J.F. Post-retrieval beta-adrenergic receptor blockade: effects on extinction and reconsolidation of cocaine-cue memories. Learn. Mem. 2008;15(9):643–648. doi: 10.1101/lm.1054608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ventura R., Cabib S., Alcaro A., Orsini C., Puglisi-Allegra S. Norepinephrine in the prefrontal cortex is critical for amphetamine-induced reward and mesoaccumbens dopamine release. J. Neurosci. 2003;23(5):1879–1885. doi: 10.1523/JNEUROSCI.23-05-01879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ventura R., Morrone C., Puglisi-Allegra S. Prefrontal/accumbal catecholamine system determines motivational salience attribution to both reward- and aversion-related stimuli. Proc. Natl. Acad. Sci. USA. 2007;104(12):5181–5186. doi: 10.1073/pnas.0610178104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spyraki C., Fibiger H.C., Phillips A.G. Cocaine-induced place preference conditioning: lack of effects of neuroleptics and 6-hydroxydopamine lesions. Brain Res. 1982;253(1-2):195–203. doi: 10.1016/0006-8993(82)90686-2. [DOI] [PubMed] [Google Scholar]
- 80.Jasmin L., Narasaiah M., Tien D. Noradrenaline is necessary for the hedonic properties of addictive drugs. Vascul. Pharmacol. 2006;45(4):243–250. doi: 10.1016/j.vph.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 81.Sershen H., Hashim A., Lajtha A. Differences between nicotine and cocaine-induced conditioned place preferences. Brain Res. Bull. 2010;81(1):120–124. doi: 10.1016/j.brainresbull.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 82.Davis A.R., Shields A.D., Brigman J.L., Norcross M., McElligott Z.A., Holmes A., Winder D.G. Yohimbine impairs extinction of cocaine-conditioned place preference in an alpha2-adrenergic receptor independent process. Learn. Mem. 2008;15(9):667–676. doi: 10.1101/lm.1079308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robledo P., Balerio G., Berrendero F., Maldonado R. Study of the behavioural responses related to the potential addictive properties of MDMA in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2004;369(3):338–349. doi: 10.1007/s00210-003-0862-9. [DOI] [PubMed] [Google Scholar]
- 84.Wenzel J.M., Cotten S.W., Dominguez H.M., Lane J.E., Shelton K., Su Z.I., Ettenberg A. Noradrenergic β-receptor antagonism within the central nucleus of the amygdala or bed nucleus of the stria terminalis attenuates the negative/anxiogenic effects of cocaine. J. Neurosci. 2014;34(10):3467–3474. doi: 10.1523/JNEUROSCI.3861-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vranjkovic O., Hang S., Baker D.A., Mantsch J.R. β-adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for β1 and β2 adrenergic receptors. J. Pharmacol. Exp. Ther. 2012;342(2):541–551. doi: 10.1124/jpet.112.193615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Otis J.M., Mueller D. Inhibition of β-adrenergic receptors induces a persistent deficit in retrieval of a cocaine-associated memory providing protection against reinstatement. Neuropsychopharmacology. 2011;36(9):1912–1920. doi: 10.1038/npp.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Otis J.M., Dashew K.B., Mueller D. Neurobiological dissociation of retrieval and reconsolidation of cocaine-associated memory. J. Neurosci. 2013;33(3):1271–81a. doi: 10.1523/JNEUROSCI.3463-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Otis J.M., Fitzgerald M.K., Mueller D. Inhibition of hippocampal β-adrenergic receptors impairs retrieval but not reconsolidation of cocaine-associated memory and prevents subsequent reinstatement. Neuropsychopharmacology. 2014;39(2):303–310. doi: 10.1038/npp.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Conrad K.L., Davis A.R., Silberman Y., Sheffler D.J., Shields A.D., Saleh S.A., Sen N., Matthies H.J., Javitch J.A., Lindsley C.W., Winder D.G. Yohimbine depresses excitatory transmission in BNST and impairs extinction of cocaine place preference through orexin-dependent, norepinephrine-independent processes. Neuropsychopharmacology. 2012;37(10):2253–2266. doi: 10.1038/npp.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bernardi R.E., Lattal K.M. A role for alpha-adrenergic receptors in extinction of conditioned fear and cocaine conditioned place preference. Behav. Neurosci. 2010;124(2):204–210. doi: 10.1037/a0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mantsch J.R., Weyer A., Vranjkovic O., Beyer C.E., Baker D.A., Caretta H. Involvement of noradrenergic neurotransmission in the stress- but not cocaine-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: role for β-2 adrenergic receptors. Neuropsychopharmacology. 2010;35(11):2165–2178. doi: 10.1038/npp.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McReynolds J.R., Vranjkovic O., Thao M., Baker D.A., Makky K., Lim Y., Mantsch J.R. Beta-2 adrenergic receptors mediate stress-evoked reinstatement of cocaine-induced conditioned place preference and increases in CRF mRNA in the bed nucleus of the stria terminalis in mice. Psychopharmacology (Berl.) 2014;231(20):3953–3963. doi: 10.1007/s00213-014-3535-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson T., Pickens R. Stimulant self-administration by animals: some comparisons with opiate self-administration. Fed. Proc. 1970;29(1):6–12. [PubMed] [Google Scholar]
- 94.Wilson M.C., Schuster C.R. Aminergic influences on intravenous cocaine self-administration by Rhesus monkeys. Pharmacol. Biochem. Behav. 1974;2(5):563–571. doi: 10.1016/0091-3057(74)90021-5. [DOI] [PubMed] [Google Scholar]
- 95.Baxter B.L., Gluckman M.I., Scerni R.A. Apomorphine self-injection is not affected by alpha-methylparatyrosine treatment: support for dopaminergic reward. Pharmacol. Biochem. Behav. 1976;4(5):611–612. doi: 10.1016/0091-3057(76)90205-7. [DOI] [PubMed] [Google Scholar]
- 96.Schroeder J.P., Cooper D.A., Schank J.R., Lyle M.A., Gaval-Cruz M., Ogbonmwan Y.E., Pozdeyev N., Freeman K.G., Iuvone P.M., Edwards G.L., Holmes P.V., Weinshenker D. Disulfiram attenuates drug-primed reinstatement of cocaine seeking via inhibition of dopamine β-hydroxylase. Neuropsychopharmacology. 2010;35(12):2440–2449. doi: 10.1038/npp.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schroeder J.P., Epps S.A., Grice T.W., Weinshenker D. The selective dopamine β-hydroxylase inhibitor nepicastat attenuates multiple aspects of cocaine-seeking behavior. Neuropsychopharmacology. 2013;38(6):1032–1038. doi: 10.1038/npp.2012.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tessel R.E., Barrett J.E. Antagonism of the behavioral effects of cocaine and d-amphetamine by prazosin. Psychopharmacology (Berl.) 1986;90(4):436–440. doi: 10.1007/BF00174057. [DOI] [PubMed] [Google Scholar]
- 99.Risner M., Jones B.E. Role of noradrenergic and dopaminergic processes in amphetamine self-administration. Pharmacol. Biochem. Behav. 1976;5(4):477–482. doi: 10.1016/0091-3057(76)90113-1. [DOI] [PubMed] [Google Scholar]
- 100.De Wit H., Wise R.A. Blockade of cocaine reinforcement in rats with the dopamine receptor blocker pimozide, but not with the noradrenergic blockers phentolamine or phenoxybenzamine. Can. J. Psychol. 1977;31(4):195–203. doi: 10.1037/h0081662. [DOI] [PubMed] [Google Scholar]
- 101.Risner M.E., Jones B.E. Intravenous self-administration of cocaine and norcocaine by dogs. Psychopharmacology (Berl.) 1980;71(1):83–89. doi: 10.1007/BF00433258. [DOI] [PubMed] [Google Scholar]
- 102.Woolverton W.L. Evaluation of the role of norepinephrine in the reinforcing effects of psychomotor stimulants in rhesus monkeys. Pharmacol. Biochem. Behav. 1987;26(4):835–839. doi: 10.1016/0091-3057(87)90618-6. [DOI] [PubMed] [Google Scholar]
- 103.Howell L.L., Byrd L.D. Characterization of the effects of cocaine and GBR 12909, a dopamine uptake inhibitor, on behavior in the squirrel monkey. J. Pharmacol. Exp. Ther. 1991;258(1):178–185. [PubMed] [Google Scholar]
- 104.Ecke L.E., Elmer G.I., Suto N. Cocaine self-administration is not dependent upon mesocortical α1 noradrenergic signaling. Neuroreport. 2012;23(5):325–330. doi: 10.1097/WNR.0b013e3283517628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang X.Y., Kosten T.A. Previous exposure to cocaine enhances cocaine self-administration in an alpha 1-adrenergic receptor dependent manner. Neuropsychopharmacology. 2007;32(3):638–645. doi: 10.1038/sj.npp.1301120. [DOI] [PubMed] [Google Scholar]
- 106.Wee S., Mandyam C.D., Lekic D.M., Koob G.F. Alpha 1-noradrenergic system role in increased motivation for cocaine intake in rats with prolonged access. Eur. Neuropsychopharmacol. 2008;18(4):303–311. doi: 10.1016/j.euroneuro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yokel R.A., Wise R.A. Amphetamine- type reinforcement by dopaminergic agonists in the rat. Psychopharmacology (Berl.) 1978;58(3):289–296. doi: 10.1007/BF00427393. [DOI] [PubMed] [Google Scholar]
- 108.Weerts E.M., Griffiths R.R. Evaluation of the intravenous reinforcing effects of clonidine in baboons. Drug Alcohol Depend. 1999;53(3):207–214. doi: 10.1016/S0376-8716(98)00130-6. [DOI] [PubMed] [Google Scholar]
- 109.Woolverton W.L., Wessinger W.D., Balster R.L. Reinforcing properties of clonidine in rhesus monkeys. Psychopharmacology (Berl.) 1982;77(1):17–23. doi: 10.1007/BF00436094. [DOI] [PubMed] [Google Scholar]
- 110.Goldberg S.R., Gonzalez F.A. Effects of propranolol on behavior maintained under fixed-ratio schedules of cocaine injection or food presentation in squirrel monkeys. J. Pharmacol. Exp. Ther. 1976;198(3):626–634. [PubMed] [Google Scholar]
- 111.Kupferschmidt D.A., Tribe E., Erb S. Effects of repeated yohimbine on the extinction and reinstatement of cocaine seeking. Pharmacol. Biochem. Behav. 2009;91(3):473–480. doi: 10.1016/j.pbb.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 112.Smith R.J., Aston-Jones G. α(2) Adrenergic and imidazoline receptor agonists prevent cue-induced cocaine seeking. Biol. Psychiatry. 2011;70(8):712–719. doi: 10.1016/j.biopsych.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.LaLumiere R.T., Niehoff K.E., Kalivas P.W. The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn. Mem. 2010;17(4):168–175. doi: 10.1101/lm.1576810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Davis W.M., Smith S.G., Khalsa J.H. Noradrenergic role in the self-administration of morphine or amphetamine. Pharmacol. Biochem. Behav. 1975;3(3):477–484. doi: 10.1016/0091-3057(75)90059-3. [DOI] [PubMed] [Google Scholar]
- 115.Brown Z.J., Tribe E., D’souza N.A., Erb S. Interaction between noradrenaline and corticotrophin-releasing factor in the reinstatement of cocaine seeking in the rat. Psychopharmacology (Berl.) 2009;203(1):121–130. doi: 10.1007/s00213-008-1376-4. [DOI] [PubMed] [Google Scholar]
- 116.Brown Z.J., Nobrega J.N., Erb S. Central injections of noradrenaline induce reinstatement of cocaine seeking and increase c-fos mRNA expression in the extended amygdala. Behav. Brain Res. 2011;217(2):472–476. doi: 10.1016/j.bbr.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 117.Shepard J.D., Bossert J.M., Liu S.Y., Shaham Y. The anxiogenic drug yohimbine reinstates methamphetamine seeking in a rat model of drug relapse. Biol. Psychiatry. 2004;55(11):1082–1089. doi: 10.1016/j.biopsych.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 118.Feltenstein M.W., See R.E. Potentiation of cue-induced reinstatement of cocaine-seeking in rats by the anxiogenic drug yohimbine. Behav. Brain Res. 2006;174(1):1–8. doi: 10.1016/j.bbr.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 119.Buffalari D.M., See R.E. Inactivation of the bed nucleus of the stria terminalis in an animal model of relapse: effects on conditioned cue-induced reinstatement and its enhancement by yohimbine. Psychopharmacology (Berl.) 2011;213(1):19–27. doi: 10.1007/s00213-010-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Buffalari D.M., Baldwin C.K., See R.E. Treatment of cocaine withdrawal anxiety with guanfacine: relationships to cocaine intake and reinstatement of cocaine seeking in rats. Psychopharmacology (Berl.) 2012;223(2):179–190. doi: 10.1007/s00213-012-2705-1. [DOI] [PubMed] [Google Scholar]
- 121.Erb S., Hitchcott P.K., Rajabi H., Mueller D., Shaham Y., Stewart J. Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology. 2000;23(2):138–150. doi: 10.1016/S0893-133X(99)00158-X. [DOI] [PubMed] [Google Scholar]
- 122.Zhang X.Y., Kosten T.A. Prazosin, an alpha-1 adrenergic antagonist, reduces cocaine-induced reinstatement of drug-seeking. Biol. Psychiatry. 2005;57(10):1202–1204. doi: 10.1016/j.biopsych.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 123.Leri F., Flores J., Rodaros D., Stewart J. Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J. Neurosci. 2002;22(13):5713–5718. doi: 10.1523/JNEUROSCI.22-13-05713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Platt D.M., Rowlett J.K., Spealman R.D. Noradrenergic mechanisms in cocaine-induced reinstatement of drug seeking in squirrel monkeys. J. Pharmacol. Exp. Ther. 2007;322(2):894–902. doi: 10.1124/jpet.107.121806. [DOI] [PubMed] [Google Scholar]
- 125.Cooper D.A., Kimmel H.L., Manvich D.F., Schmidt K.T., Weinshenker D., Howell L.L. Effects of pharmacologic dopamine β-hydroxylase inhibition on cocaine-induced reinstatement and dopamine neurochemistry in squirrel monkeys. J. Pharmacol. Exp. Ther. 2014;350(1):144–152. doi: 10.1124/jpet.113.212357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee B., Tiefenbacher S., Platt D.M., Spealman R.D. Pharmacological blockade of alpha2-adrenoceptors induces reinstatement of cocaine-seeking behavior in squirrel monkeys. Neuropsychopharmacology. 2004;29(4):686–693. doi: 10.1038/sj.npp.1300391. [DOI] [PubMed] [Google Scholar]
- 127.Weinshenker D., Schroeder J.P. There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology. 2007;32(7):1433–1451. doi: 10.1038/sj.npp.1301263. [DOI] [PubMed] [Google Scholar]
- 128.Grenhoff J., Nisell M., Ferré S., Aston-Jones G., Svensson T.H. Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J. Neural Transm. 1993;93(1):11–25. doi: 10.1007/BF01244934. [DOI] [PubMed] [Google Scholar]
- 129.Grenhoff J., Svensson T.H. Clonidine modulates dopamine cell firing in rat ventral tegmental area. Eur. J. Pharmacol. 1989;165(1):11–18. doi: 10.1016/0014-2999(89)90765-6. [DOI] [PubMed] [Google Scholar]
- 130.Grenhoff J., Svensson T.H. Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur. J. Pharmacol. 1993;233(1):79–84. doi: 10.1016/0014-2999(93)90351-H. [DOI] [PubMed] [Google Scholar]
- 131.Lategan A.J., Marien M.R., Colpaert F.C. Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res. 1990;523(1):134–138. doi: 10.1016/0006-8993(90)91646-X. [DOI] [PubMed] [Google Scholar]
- 132.Lategan A.J., Marien M.R., Colpaert F.C. Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci. 1992;50(14):995–999. doi: 10.1016/0024-3205(92)90093-5. [DOI] [PubMed] [Google Scholar]
- 133.Sommermeyer H., Frielingsdorf J., Knorr A. Effects of prazosin on the dopaminergic neurotransmission in rat brain. Eur. J. Pharmacol. 1995;276(3):267–270. doi: 10.1016/0014-2999(95)00062-P. [DOI] [PubMed] [Google Scholar]