

ABSTRACT
The cellular machinery responsible for Cu+-stimulated delivery of the Wilson-disease-associated protein ATP7B to the apical domain of hepatocytes is poorly understood. We demonstrate that myosin Vb regulates the Cu+-stimulated delivery of ATP7B to the apical domain of polarized hepatic cells, and that disruption of the ATP7B-myosin Vb interaction reduces the apical surface expression of ATP7B. Overexpression of the myosin Vb tail, which competes for binding of subapical cargos to myosin Vb bound to subapical actin, disrupted the surface expression of ATP7B, leading to reduced cellular Cu+ export. The myosin-Vb-dependent targeting step occurred in parallel with hepatocyte-like polarity. If the myosin Vb tail was expressed acutely in cells just prior to the establishment of polarity, it appeared as part of an intracellular apical compartment, centered on γ-tubulin. ATP7B became selectively arrested in this compartment at high [Cu+] in the presence of myosin Vb tail, suggesting that these compartments are precursors of donor–acceptor transfer stations for apically targeted cargos of myosin Vb. Our data suggest that reduced hepatic Cu+ clearance in idiopathic non-Wilsonian types of disease might be associated with the loss of function of myosin Vb.
KEY WORDS: ATP7B, Trafficking, Hepatocyte, Apical, Cu+, Myosin, Endosome
Summary: Myosin Vb regulates Cu+-induced recycling of ATP7B at the apical membrane in polarized hepatocytes.
INTRODUCTION
The Cu+-transporting ATPases 1 and 2 (ATP7A and ATP7B, respectively), transport cellular Cu+ and regulate cellular Cu+ homeostasis in chordates (Lutsenko et al., 2007). Disruption of ATP7B function in humans leads to Wilson disease, which affects liver and brain (Scheinberg and Sternlieb, 1959, 1960). ATP7B is expressed most abundantly in hepatocytes, where it transfers Cu+ into the lumen of the trans-Golgi network (TGN), a step required for the biosynthesis of cuproenzymes. When Cu+ is in excess, ATP7B sequesters Cu+ within vesicles that traffic to the canalicular (apical) membrane (Braiterman et al., 2009; Forbes and Cox, 2000; Nyasae et al., 2014), where ATP7B facilitates the extrusion of excess Cu+ into the bile (Fanni et al., 2005). Although a few proteins that affect trafficking of ATP7B have been identified, the molecular mechanisms responsible for polarized apical delivery of ATP7B remain poorly understood (Lim et al., 2006a,b; Materia et al., 2012).
Here, we have focused on myosin Vb (MYO5B, hereafter referred to as MyoVb) and its role in delivery of ATP7B to the luminal surface. MyoVb is the physiologically occurring isoform of myosin V in hepatocytes, whereas myosin Va (MYO5A) and myosin Vc (MYO5C; hereafter referred to as MyoVa and MyoVc, respectively) are not highly expressed. Mutations in the human MYO5B gene lead to microvillus inclusion disease (MVID), in which the filamentous actin (F-actin)-rich apical microvilli of enterocytes are absent, with concomitantly disrupted localization of apical membrane proteins that include P-type ATPases (Knowles et al., 2014; Müller et al., 2008). The apical microvilli of hepatocytes are also disrupted in MVID, and clinically MVID is associated with diarrhea and cholestasis (Girard et al., 2014; Knowles et al., 2014; Thoeni et al., 2014). The cholestasis may be explained as a complication secondary to hyperalimentation therapy. However, recent studies of MVID patients have shown disorganization of the canalicular pole of hepatocytes, and altered expression of MyoVb and RAB11A, suggesting that cholestasis in MVID sometimes is a direct effect of the loss of MyoVb function in hepatocytes (Girard et al., 2014; Thompson and Knisely, 2014).
Previous studies have shown that a tripartite targeting complex consisting of MyoVb, Rab11a and Rab11-FIP2 mediates the surface expression of many apical proteins (Chu et al., 2009; Ducharme et al., 2011; Hales et al., 2002; Lindsay and McCaffrey, 2002; Nedvetsky et al., 2007). Loss of MyoVb function causes mislocalization of ABC transporters involved in the maintenance of apical polarity in hepatocytes to Rab11a-rich subapical endosomes (Wakabayashi et al., 2005). These and other studies indicate that MyoVb is likely to participate physiologically in the apical delivery of ATP7B in hepatocytes. We tested this idea by using WIF-B cells as a model for polarized hepatocytes, in conjunction with overexpression of the dominant-negative mutant of MyoVb – the cargo-binding tail (MyoVbT) – and manipulation of cellular Cu+ levels.
RESULTS
Myosin Vb is the main myosin V isoform in WIF-B cells
It has been reported in the Human Protein Atlas, that MyoVb is the physiologically occurring isoform of MyoV in hepatocytes, wheareas MyoVa and MyoVc are not highly expressed. To determine the levels of the three isoforms of MyoV in WIFB cells, we measured their transcripts in WIF-B cells; their respective mRNA expression levels in fibroblasts were used as control by using real-time PCR. We found that expression levels of MyoVb are exponentially higher compared with those of MyoVc in WIF-B cells. MyoVa was not expressed in these cells (Fig. 1A). Our results in WIF-B cells corroborated the findings in mammalian hepatocytes. Hence, in our investigation we focused on the role and the mechanism of MyoVb in regulation of Cu+-induced ATP7B trafficking in WIF-B cells.
Fig. 1.

Expression and localization of endogenous MyoVb in WIF-B cells. (A) Determination of relative mRNA abundance of MyoVa, MyoVb, MyoVc by using real-time PCR in WIF-B cells compared with control cells (fibroblasts). mRNA abundance was calculated in relation to the β-actin mRNA in the same sample. mRNA abundance in WIF-B cells relative to fibroblasts measured shows that MyoVb is expressed in a much higher level in comparison to the other isoforms MyoVa and Vc. (B,C) Cells were grown on coverslips in low [Cu+], then some were switched to high Cu+ (10 µM) for 1.5 h, followed by fixation and dual staining for endogenous ATP7B (green) and MyoVb (red). (B) In low [Cu+] (top rows) ATP7B localizes to the TGN, and it does not overlap with MyoVb located near the apical membrane of the bile canaliculus (BC). Following Cu+ treatment (bottom rows), an increase in ATP7B and MyoVb overlap occurs near the apical membrane (white arrow). Scale bars: 5 μm. The zoomed image depicts detail in the apical region (BC), with areas of overlap (yellow) indicated by the arrow. (C) Histograms showing Pearson's coefficient of colocalized volume (PC value) of ATP7B and MyoVb within the bile canaliculus region under the two Cu+ conditions. Error bars in A and C are the mean±s.e.m.
Myosin Vb colocalizes with ATP7B at the bile canaliculus in the presence of high [Cu+]
Cu+-directed trafficking of endogenous ATP7B can be studied in polarized WIF-B cells, a cell culture model of hepatocytes (Braiterman et al., 2009, 2015; Guo et al., 2005; Nyasae et al., 2014). Following an overnight incubation in low [Cu+] (basal medium), cells are exposed to Cu+ (10–15 µM), causing ATP7B to exit the TGN as vesicles that traffic – via basolateral endosomes – to the apical domain within 1 h (Nyasae et al., 2014). Retrograde trafficking, initiated when Cu+-treated cells were transferred into medium supplemented with the Cu+ chelator bathocuproinedisulfonic acid (BCS), leads to re-accumulation of ATP7B in the TGN within ∼3 h (Nyasae et al., 2014). WIF-B cells were exposed to low or high [Cu+], then fixed and co-stained with antibodies against ATP7B and MyoVb. At low [Cu+], ATP7B localized to the TGN, whereas MyoVb localized to the bile canaliculus, which is juxtaposed at the apical junction of two or more polarized WIF-B cells (Fig. 1B). At low [Cu+], colocalization of ATP7B and endogenous MyoVb within a region of interest demarking the apical membrane was absent. At high [Cu+], the canalicular localization of MyoVb did not change appreciably. By contrast, ATP7B relocalized to sites of or very near to MyoVb on the apical membrane (Fig. 1B, bottom row). Colocalization analysis confirmed that ATP7B and MyoVb appear together near the apical membrane at high [Cu+] only (Fig. 1C). Thus, the two proteins selectively colocalized to the apical region under conditions that promote ATP7B-dependent Cu+ extrusion at the apical membrane.
ATP7B colocalizes with the MyoVbT–rich compartments at high [Cu+]
Cu+-induced trafficking of ATP7B involves a series of discrete trafficking steps, including (a) enhanced vesicular exit from the TGN, (b) Cu+-dependent retrieval from basolateral endosomes and delivery as minus-end directed cargos destined for the subapical region and (c) apical transfer and/or recycling of ATP7B between endosomes with associated Cu+ export (Nyasae et al., 2014). On the basis of its localization and previously reported functions (Knowles et al., 2014; Lapierre et al., 2001; Lapierre and Goldenring, 2005; Lise et al., 2006), we hypothesized that MyoVb is a strong candidate to be a motor that regulates the final step of ATP7B apical delivery. To test this, we employed adenovirus-mediated overexpression of the tail domain of MyoVbT, which functions as a dominant-negative by displacing Rab11a-positive vesicular cargos from endogenous MyoVb bound to subapical actin, thereby disrupting the cargo's myosin-dependent capture from endosomes (Lapierre et al., 2001; Lapierre and Goldenring, 2005).
It has previously been shown that long-term overexpression of the MyoVbT (≥72 h) negatively affects polarization in WIF-B9 cells (Wakabayashi et al., 2005). After 3 days in culture, 30% of WIF-B9 cells formed bile canaliculi; infection with adenoviral constructs of GDP-blocked Rab11a, and MyoVbT domain significantly reduced polarization (to ∼10% of control) when examined 4 days later (Wakabayashi et al., 2005). Under the growth condition that we used (plating densities 7×105 cells/ml), polarization in WIF-B cells begins about 9 days after plating, and reach full polarization at days 13–14, when almost all cells display a bile canaliculus. We infected day-9 cells with Tomato-tagged MyoVbT encoding adenovirus and recorded expression of the protein after 6, 8 and 16 h. Although MyoVbT expression was observed in all instances, fluorescence was strongest at 16 h, and we adopted this time point to study the acute effects of MyoVbT expression in our model. We compared the effects of short-term (16 h) vs long-term (96 h) expression of MyoVbT on cell polarization (Fig. 2A). In WIF-B cells infected on day 9 and imaged after 16 h and 96 h, the total number of bile canaliculi formed in control and MyoVbT cells was the same after 16 h of MyoVbT adenovirus infection compared with non-infected cells, whereas, as previously reported, at 96 h almost all bile canaliculi had disappeared. We conclude that a short-term (16 h) expression of MyoVbT does not reduce bile canaliculi formation. Therefore, in all the subsequent experiments in this study, MyoVbT was expressed for 16 h post infection and before Cu+ treatment or other experimental manipulations. The differences that we observed in our study as compared to the one by Wakabayashi et al., 2005 are probably attributed to the clonal difference between WIF-B and WIF-B9 cells.
Fig. 2.

Expression of exogenous MyoVbT in polarized WIF-B cells. (A) Histogram showing the number of bile canaliculi (NO of BC) in WIF-B cells infected with Ad-MyoVbT fused to tdTomato for 16 or 96 h (cells were then fixed and bile canaliculi counted). Short-term overexpression (16 h) of MyoVbT in WIF-B cells did not affect the number of bile canaliculi, whereas long-term expression greatly diminished them. (B) WIF-B cells expressing MyoVbT were grown in medium containing low [Cu+] (top row, -Cu), initially grown in medium containing low [Cu+] for 24 h and then transferred to medium containing high [Cu+] for 1.5 h (middle row, -Cu>+Cu), or were initially grown in low [Cu+] for 24 h, transferred to medium containing high [Cu+] for 1.5 h and then transferred to BCS-containing Cu+-free medium for 4 h (bottom row, -Cu>+Cu Cu>-Cu). Cells were triple-stained for ATP7B (green), MyoVbT (red) or TGN38 (blue). MyoVb exhibits an apical ring pattern that is unaltered by Cu+ (second column). ATP7B colocalizes with TGN38 but not with MyoVbT in low [Cu+] (top row). Following Cu+ treatment, ATP7B colocalizes with MyoVbT near the apical ring (middle row). On subsequent Cu+ chelation, ATP7B again overlaps with TGN38 (bottom row). Image detail is shown on the right in all three panels. Scale bars: 5 μm. (C) Spatial organization of ATP7B and MyoVbT near the apical actin ring. MyoVbT surrounds the cytosolic face of the apical F-actin ring labeled by phalloidin (blue, left), whereas ATP7B is distributed between F-actin (blue, right) and MyoVbT (red, middle; arrowheads indicate directionality from lumen to cytosol). Overlapping signals of ATP7B and MyoVbT (yellow) are indicated by arrows. The graph (right) shows the mean normalized pixel intensities for all three markers across the ten lines, with arrowheads shown in the triple-labeled image, plotted to show their spatial distribution relative to the luminal face of the membrane.
In WIF-B cells, MyoVbT exhibited a ring-like pattern around the bile canaliculus as found for endogenous MyoVb; this localization did not change with Cu+ concentration (Fig. 2B). When Cu+-dependent ATP7B trafficking was examined in cells acutely overexpressing MyoVbT, the overall itinerary of Cu+-directed trafficking of ATP7B appeared unaffected: that is, ATP7B exited the TGN in vesicles and accumulated in the apical region within 1 h (Fig. 2B, middle). Cu+ chelation triggered its return to the TGN (Fig. 2B, bottom panel). We wondered whether this apparent lack of MyoVbT effect on trafficking can be explained by the selective site of action of MyoVbT, i.e. near intracellular compartments that are located directly underneath the apical membrane. Thus, microscopic resolution might be insufficient to distinguish the pools of ATP7B on the apical membrane from subapical compartments closely associated with the non-luminal side of the apical membrane (Ihrke et al., 1998). Therefore, we employed deconvolution confocal microscopy to examine the microarchitechture around the F-actin-rich apical ring in greater detail (Fig. 2C).
MyoVbT concentrates under the cytosolic side of the apical F-actin, whereas ATP7B partially overlaps with both F-actin and MyoVbT structures. We identified MyoVbT-positive compartments in which ATP7B appeared to stall near the actin ring (Fig. 2C, middle arrowheads). However, colocalization analysis of apical ring markers revealed that the level of overlap between ATP7B and F-actin near the apical membrane was unchanged in the presence of MyoVbT. That is, regardless of the presence of MyoVbT, approximately half of the amount of ATP7B within the local apical region (within a 2-µm radius around the apical ring marked by phalloidin) colocalized with F-actin (47.7±2.1% in control cells versus 48.2±5.2% in cells overexpressing MyoVbT; mean±s.e.m., n=20 bile canaliculi per condition). Our microscopic analysis did not yield results of sufficient resolution in order to show changes of ATP7B microlocalization with respect to luminal versus cytosolic face of the apical ring; however, ATP7B did not appear to disassociate from F-actin in the presence of MyoVbT, suggesting that its attachment to the apical actin cytoskeleton is likely to involve F-actin-binding proteins in addition to MyoVb.
To explore whether ATP7B stays in MyoVbT compartments, we compared ATP7B colocalization with the Rho GTPase Rab11 in the absence and presence of MyoVbT. Cells infected with or without MyoVbT-expressing adenovirus were treated with Cu+ to induce trafficking of ATP7B to the apical region (Fig. 3). Colocalization of ATP7B and Rab11 increases threefold in the apical region following overexpression of MyoVbT, thus confirming that MyoVbT arrests ATP7B in Rab11 compartments.
Fig. 3.

ATP7B accumulates in Rab11-positive compartments. (Left) After treatment with 10 µM Cu+, polarized WIF-B cells were fixed and immunolabeled for ATP7B (green) and Rab11 (blue), in the absence or presence of MyoVbT (red). Arrowheads indicate regions of overlap between ATP7B and Rab11 near the apical membrane around the bile canaliculus, in the presence of MyoVbT. (Right) Pearsons's coefficient (PC) of colocalized ATP7B and Rab11 volumes within the bile canaliculus region under the two experimental conditions (histograms). Error bars are the mean±s.e.m.
Apical ATP7B shows a mixed partitioning between microtubules and actin
It has previously been reported that microtubule-depolymerizing drugs disrupt MyoVbT-labeled compartments (Lapierre et al., 2001). At high [Cu+], upon treatment of polarized cells that overexpress MyoVbT with Nocodazole, both MyoVbT and ATP7B dispersed into mostly separate punctae [Fig. 4, compare panel B with panel A (untreated control)], indicating ATP7B and MyoVbT differ in their ‘micro-location’ within the same larger structure. In addition, part of the ATP7B signal at the bile canaliculus persisted after Nocodazole treatment, indicating a pool of ATP7B resistant to microtubule disruption. We, thus, examined the distributions of ATP7B and MyoVbT in the presence of high [Cu+] and 10 µM of the microfilament disruptor cytochalasin D in polarized WIF-B cells. This higher concentration of cytochalasin D is required to disrupt the dense actin web surrounding the apical PM (Tuma et al., 2002). Similar to the results observed following Nocodazole treatment, cytochalasin D dispersed both MyoVbT and ATP7B (Fig. 4C). These experiments are consistent with a model in which, in Cu+-treated cells, ATP7B is distributed between the apical actin ring associated with the bile canaliculus (disrupted by cytochalasin D) and the subapical (vesicular, microtubule) compartment (disrupted by Nocodazole).
Fig. 4.
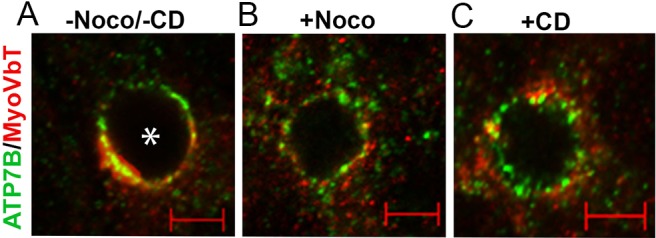
Microtubule and actin disruption completely disperses apical MyoVbT, but partly disperses apical ATP7B. (A) In cells not treated with nocadazole and/or cytocalasin D (−Noco/−CD) ATP7B (green) and MyoVbT (red) colocalize near the bile canaliculus. (B) Disruption of microtubules with Nocodazole (+Noco) disperses MyoVbT to numerous peripheral vesicles. ATP7B is also dispersed into vesicles following Nocodazole treatment, and a Nocodazole-insensitive pool remains associated on the apical membrane. (C) Disruption of actin polymerization with cyotchalasin D (+CD) also disperses ATP7B and MyoVbT into a discreet vesicular pool. This pool of vesicles is associated with the cortical actin adjacent to the bile canaliculus. Asterisk indicates a bile canaliculus. Scale bars: 2 µM.
MyoVbT overexpression prevents ATP7B localization at the apical membrane and disrupts Cu+ export
If MyoVbT arrests ATP7B intracellularly, apical surface expression of ATP7B should then be reduced. To test this idea we employed a surface-biotinylation assay to determine the amount of ATP7B at the apical membrane in Cu+-treated cells that do or do not overexpress MyoVbT. We first established that ATP7B can be selectively surface biotinylated at high [Cu+] when it is expressed at the apical surface. We compared extent of biotinylation between cells (day 13) treated with basal or Cu+-containing media. Biotinylation and immuno-isolation of detergent-solubilized membrane fraction by using streptavidin beads, and quantification of ATP7B in immunoblots revealed a large increase in biotinylated ATP7B within cells treated with Cu+ compared with untreated cells (Fig. 5A). To measure the effect of MyoVbT on localization of ATP7B to the apical surface, day-13 cells that expressed MyoVbT or not were treated with Cu+ and the extent of ATP7B biotinylation was determined. Immunofluorescence revealed partial colocalization of MyoVbT, ATP7B and biotin at the bile canaliculus (Fig. 5B). Quantification of ATP7B in immunoblots revealed an ∼50% reduction of biotinylated ATP7B in MyoVbT-overexpressing cells compared to non-infected ones (Fig. 5C).
Fig. 5.

Expression of MyoVbT reduces ATP7B at apical surface. (A) Cu+-dependent biotinylation of ATP7B. WIF-B cells grown in high-[Cu+] medium with [10 µM BCS (+)] or in low-[Cu+] medium with [25 µM BCS (−)] were biotinylated on ice, sonicated and cell membranes solubilized with detergent were incubated with streptavidin-coupled beads. Total membrane fractions or eluates from the beads were western blotted for ATP7B. Surface biotinylated ATP7B is detected at high-[Cu+] only. (B) Apical biotinylation in regions of overlap between ATP7B (blue), FITC-biotin (green) and MyoVbT (red) in the apical region of Cu+-treated cells. (C) Histogram shows immunoblot analysis of biotinylated ATP7B comparing cells overexpressing MyoVbT or not in high [Cu+]. Apical biotinylation is reduced in the presence of MyoVbT. The western blots (lower) show ATP7B in the total lysate (lanes 1, 2) or surface biotinylated in the presence (+; lanes 3,5) or absence (−; lanes 4, 6) of MyoVbT. TM denotes total membrane. E1 and E2 signify two serial eluates from the streptavidin agarose beads. (D) MyoVbT slows cellular Cu+ extrusion. Cu+ content of cells expressing MyoVbT (+; yellow bars) or not (−; black bars) was measured by atomic absorption spectroscopy at high [Cu+] or under conditions that had been changed from high to low [Cu+] (addition of BCS). Cu+ uptake appeared normal in MyoVbT-expressing cells (black bars). We observed a modest reduction in the rate of Cu+ extrusion following return to Cu+ chelation in BCS in the presence of myoVbT (yellow bars). Western blotting (bottom) showed similar levels of ATP7B protein under all conditions. Error bars in C and D are the mean±s.e.m.
If MyoVbT prevents access of ATP7B to the apical surface, one predicted effect would be a reduced Cu+ extrusion from the cell. To test this, we first measured the Cu+ content within WIF-B cells that had received and were cultured as follows: (1) cells cultured in basal growth medium, (2) cells cultured after treatment with 20 µM Cu+ for 2 h (high [Cu+]) or, (3) cells cultured after treatment with 20 µM Cu+ for 2 h followed by Cu+ removal with 50 µM BCS for 2 h. Cu+ treatment of cells resulted in a twofold increase of cellular [Cu+] compared with cells cultured in basal medium. Subsequent treatment with BCS reduced the Cu+ content by ∼75%, when cells extruded Cu+ they had taken up. Overexpression of MyoVbT did not affect basal Cu+ levels or change the ability of cells to accumulate Cu+ upon exposure to high [Cu+], indicating that the Cu+ uptake machinery remains unaffected. However, cells expressing MyoVbT showed reduced Cu+ efflux as judged by a higher level of intracellular [Cu+] (∼30%) compared to uninfected controls (Fig. 5D). To further substantiate our findings that disruption of microtubule machinery causes Cu+ accumulation, Cu+ loaded cell were treated with Nocodazole and Cu+ extrusion was measured. We recorded that Nocodazole treatment also reduced Cu+ export in cells by ∼37% of non-treated cell.
To ensure that Cu+ export from the cells primarily occurs at the apical surface and not at the basolateral membrane, we compared colocalization of ATP7B and MyoVbT between the apical membrane at the bile canaliculus (2 µM around the bile canaliculus) and rest of the cell. We did not see any substantial overlap of the two signals in any region of the cell as compared to high overlap at the bile canaliculus (Fig. S1).
Apical compartment precedes the apical ring during WIF-B polarization
In polarized WIF-B cells (∼day 10 to day 14), MyoVbT exhibited a ring-like pattern around the F-actin ring at the bile canaliculus, and our data from polarized cells provided several lines of evidence indicating that disruption of MyoVb interactions with ATP7B cargo abrogates a selective late apical targeting step in the Cu+-induced trafficking itinerary of ATP7B to the apical membrane. However, between day 12 and 13 of WIF-B cell polarization, ∼5–10% of the cells remain unpolarized and lack a bile canaliculus. Interestingly, in these pre-polarized cells, MyoVbT localizes to a tight juxtanuclear intracellular site that we have previously described as ‘apical compartment’ (Tuma et al., 2002). This apical compartment is an intracellular membranous structure clustered near the minus ends of microtubules that are labeled with γ-tubulin, where apically destined cargos accumulate selectively. Upon Cu+ treatment in the presence of MyoVbT, ATP7B loses TGN localization and moves to the apical compartment, which is strongly labeled with MyoVbT. In cells that do not express MyoVbT, ATP7B disperses into vesicular compartments in response to Cu+ treatment. Subsequent Cu+ chelation, whether MyoVbT is present or not, causes ATP7B relocate to the TGN (Fig. 6). Thus, the presence of MyoVbT did not affect the ability of ATP7B to modify its trafficking itinerary upon changes in Cu+ levels, but it did cause ATP7B to arrest and accumulate in the apical compartment in a Cu+-dependent manner.
Fig. 6.

MyoVbT arrests ATP7B in the apical compartment of unpolarized cells in high [Cu+]. WIF-B cells were transformed with virus on day 11 in culture and fixed 16 h later; ∼10% of the cells remained unpolarized. MyoVbT (red) localizes to the apical compartment under all Cu+ conditions (white arrows, second column). ATP7B (green) colocalizes with TGN38 (blue) in Cu+ depleted conditions and shows no overlap with MyoVbT in the apical compartment (top panel). Following Cu+ treatment, ATP7B traffics from the TGN and colocalizes with MyoVbT in the apical compartment (middle), and at the bile canaliculus in polarized cells (asterisks). In cell not expressing MyoVbT, ATP7B disperses to vesicles (arrowhead). On subsequent Cu+ chelation, ATP7B exhibits retrograde trafficking denoted by its overlap with TGN38 (bottom). Scale bars: 5 μm. Details of the boxed areas are magnified in the three right panels, showing the arrangement of ATP7B, MyoVbT, and TGN38 near the apical compartment.
Apical compartments become subapical endosomes in polarized cells
We next examined whether the apical compartment is a precursor of subapical endosomes. The Rab GTPase Rab11 has been reported to localize to the subapical region and regulate cargo recycling between the endosomes and the apical surface (Hoekstra et al., 2004). Our laboratory has previously demonstrated that exogenously expressed ATP7B partially localizes in Rab11 vesicles in the presence of high [Cu+] (Guo et al., 2005). To test for the presence of native trafficking components in the apical compartments of day-13 WIF-B cells, we overexpressed MyoVbT and stained for Rab11. We found a clear overlap between ATP7B and Rab11 around the bile canaliculus and at the apical compartment of day-13 polarized and unpolarized cells, respectively (Fig. 7A). Similar to MyoVbT itself, Rab11 was present in the apical compartment, regardless of the Cu+ concentration status. At high [Cu+], we observed an increase in the concomitant occurrence of ATP7B and of MyoVbT-infected cells, consistent with the idea that apical compartments are the precursors of the subapical recycling endosomes.
Fig. 7.

ATP7B colocalizes with Rab11 in the apical compartment and sub-apical endosomal compartments. (A) In Cu+-treated cells expressing myoVbT (red), ATP7B (green) accumulates with Rab11 (blue) both in polarized (top) and unpolarized (bottom) WIF-B cells. (B) Localization of MyoVbT (red) with respect to γ-tubulin (green) around the bile canaliculus (BC) in a polarized cell (top) and in proximity to the apical compartment in an unpolarized cell (bottom). MyoVbT compartments of polarized and unpolarized cells both surround the MTOC labeled by γ-tubulin (white arrows). Boxed areas indicate the enlarged image (last column). (C) Summary diagram showing pathways of Cu+-dependent ATP7B trafficking in polarized control WIF-B cells (left) and cells overexpressing MyoVbT (right). Our data suggest that ATP7B is transported to the apical luminal surface through a process that requires MyoVB. Overexpression of the MyoVbT (red) arrests ATP7B (green) in a subapical vesicular compartment (brown). BC, bile canaliculus; Nuc, nucleus.
If these two distinct MyoVbT-localizing subcellular entities (subapical endosomes in polarized cells and apical compartment in unpolarized cells) are analogous and serve as Cu+-sensitive pre-actin staging sites for ATP7B, then they should have a similar spatiotemporal relationship with the microtubule organizing center (MTOC), near minus ends of microtubules. In polarized hepatocytes, the minus ends of microtubules lie near the apical surface and the plus ends lie towards the basolateral membrane. We investigated whether MyoVbT compartments colocalize with γ-tubulin, an established marker of the MTOC. In polarized cells (Fig. 7B, top row), γ-tubulin also lies in a crescent-shaped location, underneath the MyoVbT ring at the bile canaliculus. In unpolarized cells, MyoVbT-positive apical compartments surrounded the MTOC (Fig. 7B, bottom row). These two minus-end-rich regions in the cell represent the terminal stop for microtubule-directed long-range transport – where cargos are captured at specialized subapical sites and transported through a mesh of F-actin, with the assistance of actin-based motors. The summary of Cu+-dependent trafficking of ATP7B regulated by MyoVb is depicted in Fig. 7C.
Inhibitory effect of MyoVbT on ATP7B trafficking increases with cell polarity
The MyoVbT-dependent arrest of ATP7B in the apical compartment in pre-polarized WIF-B cells (day 12) led us to test whether a similar or stronger effect would be observed in cells at earlier stages of polarization (<day 12). Infecting cells with MyoVbT at days 5, 8 and 11 revealed that the inhibitory effect of MyoVbT on Cu+-dependent trafficking of ATP7B increases with the polarization time (Fig. 8A). A vesicular pattern of ATP7B predominates in day-5 cells, with no indication of accumulation in the apical compartment, despite this compartment being present. As the cells mature during polarization, a typical clustered apical compartment pattern of ATP7B appears, which overlaps with MyoVbT. Pearson's correlation coefficient of colocalized volume between ATP7B and MyoVbT indicated minimal colocalization on day 5 (Pearson's correlation coefficient of 0.2896) as compared with that on day 11 (Pearson's correlation coefficient of 0.7294); day 8 values were in the middle range (Pearson's correlation coefficient of 0.4698) (Fig. 8B).
Fig. 8.

MyoVbT-dependent arrest of ATP7B in the apical compartment depends on the state of cell polarization. (A) Images of unpolarized cells stained for ATP7B (green) and MyoVbT (red) under low [Cu+] condition at day 11, compared to unpolarized cells on days 5, 8 and 11 under high [Cu+] condition. As cells become polarized, ATP7B appears increasingly arrested in the apical compartment in high Cu+, as shown by the yellow regions near the apical compartment in the merged images. Scale bars: 5 μm. (B) Pearson's correlation coefficient of colocalized volume was calculated for days 5, 8, and 11 of polarization (data represent mean±s.d. for at least 15 apical compartments per condition. Error bars are the mean±s.e.m. (C) Immunoblot shows no change in endogenous levels of MyoVb at days 5, 8 and 11 WIF-B cells compared to tubulin levels.
We tested whether the developmental selectivity of the MyoVbT effect on ATP7B arrest in the apical compartment can be explained by changes in MyoVb during the course of cell polarization. However, western blot analysis showed that expression levels for endogenous myosinVb on day-5, -8 and -11 cells were similar (Fig. 8C). Thus, the data suggest that one or more elements of molecular machinery to direct ATP7B to the apical compartment are lacking in unpolarized WIF-B cells; however, that lack of MyoVb is unlikely to account for our inability to observe ATP7B arrest within the apical compartment prior to the onset of cellular polarity. Further experiments will be required to determine the molecular basis of the developmental change that causes the alteration in MyoVb regulation of ATP7B trafficking near the apical membrane at high [Cu+].
DISCUSSION
The main finding of this study is that the Cu+-dependent delivery of the Wilson disease protein ATP7B to the apical membrane of hepatocyte-like cells requires MyoVb located near the F-actin-rich apical membrane. Myosin Vb participates in an apical-specific trafficking pathway that only operates in the presence of high [Cu+], and its function emerged later in development, when cells became polarized.
The Cu+-directed trafficking pathway of ATP7B can be subdivided into several steps that correspond to different intracellular locations. Our study shows that MyoVb participates in the local transfer of ATP7B, directing it from subapical intracellular membranes towards the apical lumen, where excess Cu+ is eliminated. Previous studies have shown that, when Cu+ levels rise ATP7B is released from the TGN because vesicles that move towards the plus ends of microtubules locate predominantly at the basolateral side of the cell (Nyasae et al., 2014). At high [Cu+], ATP7B is redirected from basolateral endosomes, followed by long-distance motor-directed transport towards the microtubule minus ends, located near the MTOC and the apical membrane. Our study indicates that MyoVb participates in ATP7B delivery specifically to this intracellular location, where microtubule-directed cargos are concentrated at γ-tubulin-rich sites and captured by subapical actin-dependent mechanisms prior to delivery to the apical lumen. At this location, cargo is, subsequently, transferred by MyoVb motor proteins that deliver the cargo to the apical membrane and also act as a tether for the subapical compartment at F-actin (Kapitein et al., 2013; Provance et al., 2008). Thus, MyoVb operates specifically near the penultimate molecular step prior to the ATP7B-dependent elimination of Cu+ in hepatocytes.
ATP7B is known to undergo multiple rounds of recycling at the apical membrane (Roelofsen et al., 2000). Biotinylation of the apical membrane revealed that a 50% reduction of ATP7B at the apical membrane occurs in cells overexpressing MyoVbT (this study). In our study, we were unable to easily dissect the relative contributions to Cu+ transport that are attributed to the initial delivery of nascent ATP7B molecules (arriving via microtubule motors) from the local recycling of resident apical ATP7B. The endosomal system that operates underneath the apical membrane is difficult to resolve because it is partly surrounded by a mesh of F-actin. Membrane internalized from the apical surface might remain associated with F-actin throughout its rounds of local recycling and, thus, rely on mechanisms that require the coordination of different myosin motors. Between 70 and 80% of polarized WIF-B cells were infected with adenovirus encoded MyoVbT, and expression levels per cell were heterogeneous, further complicating our estimate of surface expression. Differences in Cu+ content and surface biotinylation of ATP7B between control cells and MyoVbT-overexpressing WIF-B cells would have probably been higher, if 100% infectivity and homogenous MyoVbT expression in all cells could have been attained.
Our study revealed that regulation of the molecular interaction between ATP7B and MyoVb is not only Cu+ dependent but also emerges late in development, occurring in parallel to apical polarity. As they develop in culture, WIF-B cells progress through a pre-polarized stage, during which they express unique apical intracellular compartments that surround the MTOCs and are thought to be the precursors of the mature apical domain (Tuma et al., 2002). We observed that MyoVbT caused ATP7B to accumulate in apical compartments under conditions that rely both at high [Cu+] and on the developmental stage of the cultured cells. At early stages, when cells are unpolarized, MyoVbT was found in apical compartments, yet ATP7B did not accumulate there when Cu+ levels were raised. When cells approached their polarized state, the ability of Cu+ to direct ATP7B to apical compartments increased in a robust and obvious way, suggesting that development of polarity in culture leads to engagement of Cu+-bound ATP7B and specific donor sites for apically directed cargos. A similar phenomenon has been observed for apical protein p75 in polarized MDCK cells, in which the kinesin motor KIF5B selectively transports p75 in polarized cells but not in unipolar cells (Jaulin et al., 2007). This developmental shift could not be attributed to changes in levels of MyoVb itself. Changes in Rab11–FIP levels, phosphorylation (Overmeyer and Maltese, 2005), cellular localization or different motor proteins operating at different developmental stages, potentially, account for this. Although WIF-B cells polarize and are a useful model system to study polarized trafficking, further studies in primary hepatocytes and in situ are crucial to understand the physiological basis of the developmental onset in the role that MyoVb has in Cu+-induced trafficking of ATP7B during cell polarization.
Previously we have noticed that, in cells that do not express an apical membrane, Cu+ causes ATP7B to redistribute from the TGN to vesicles, but ATP7B does not fuse with the plasma membrane. In this study. we observed a similar situation for unpolarized WIF-B cells. In hepatocytes, apical proteins that span the membrane multiple times use a direct trafficking route – from the TGN to the apical membrane – without transient expression at the basolateral surface (Matter et al., 1990; Hubbard et al., 1989; Le Bivic et al., 1990). ATP7B, as a member of the ABC transporter class, seems to be similarly evolved. In situations where no apical destination exists, ATP7B arrests in intracellular vesicles that lack the ability to fuse with basolateral membranes. As apical polarity develops, WIF-B cells begin to express specific apical targeting machinery, such as the components of the MyoVb–Rab11–FIP2 complex.
MyoVb mutations cause MVID, which hinders nutrient uptake at the intestinal brush border, leading to intractable diarrhea. Current therapy for MVID involves parenteral feeding, which can lead to secondary cholestasis and damage to hepatocytes. A recent study by Girard et al. has suggested that some of the cholestasis in MVID is not secondary to therapy but, rather, a direct result of the lack of MyoVb at the bile canaliculus and the subsequent mislocalization of bile-salt export proteins (BSEP) (Girard et al., 2014). In support of such a mechanism, BSEP has been shown to localize to Rab11-positive compartments within the apical region of WIF-B9 cells, suggesting a direct role of MyoVb in its trafficking (Wakabayashi et al., 2004, 2005). It would, thus, be informative to measure hepatic Cu+ levels in individuals suffering from MVID, because our study suggests that ATP7B is mis-targeted, leading to accumulation of hepatic Cu+. Such measurements might be confounded by the secondary effects of hyperalimentation in these patients, which might affect the apical trafficking of ABC proteins in hepatocytes. Furthermore, screening the gene encoding MyoVb (MYO5B) and associated machinery for single-nucleotide polymorphisms and mutations might provide clues towards the understanding of idiopathic non-Wilsonian types of disease accompanied by Cu+ accumulation (Hayashi et al., 2012; Muller et al., 1998; Wijmenga et al., 1999).
MATERIALS AND METHODS
Cell culture and adenoviral infection
Cells of the rat hepatoma-derived hybrid cell line WIF-B were seeded in 10-cm tissue culture dishes containing six glass coverslips (22×22 mm); plating density was 7×105 cells/ml. Cells were cultured ∼9–12 days until maximal polarity had been reached (Braiterman et al., 2009). Tandem-tagged Tomato-tagged MyoVbT construct was a kind gift from Prof. Ora Weisz (University of Pittsburgh). The construct was packaged into adenoviruses and purified as described previously (Bastaki et al., 2002). To verify that viruses encoded the desired substitutions, adenoviral DNA was purified from infected HEK293A cells, PCR amplified and sequenced as previously described (Braiterman et al., 2009). WIF-B cells were infected as previously described (Braiterman et al., 2009), with the following modifications. Aggregates of virus were removed using 0.2 µm MILLEX GP filter units (Merck Millipore), infections were carried out for 30 min; infected cells were then cultured overnight with 10 μM bathocuproinedisulfonic acid (BCS; Sigma-Aldrich) or basal medium as required.
Trafficking assays
To study anterograde trafficking of ATP7B, WIF-B cells were grown in two separate culture dishes. Both sets were infected with MyoVbT-encoding adenovirus and they were cultured in 10 µM BCS for 16–24h. Both sets of cells were rinsed with 1× PBS, one set was fixed, and the other set was incubated for 1.5 h in 10–100 µM CuCl2 to induce anterograde trafficking of ATP7B. Cells were fixed, processed for indirect immunofluorescence and imaged.
To study retrograde trafficking of ATP7B, WIF-B cells were grown in two separate culture dishes. Both sets were infected with MyoVbT-encoding adenovirus. At 16–24 h after infection, both set of cells were incubated for 1.5 h in 10 µM CuCl2 to induce anterograde trafficking of ATP7B. They were then rinsed with 1× PBS, one set was fixed, and the other set was incubated in 50 µM BCS for 2 h to induce retrograde trafficking of ATP7B. Cells were fixed, processed for indirect immunofluorescence and imaged as described below.
Determination of relative mRNA abundance of MyoVa, MyoVb, MyoVc by real-time PCR
WIF-B and control cells (fibroblasts) were cultured on 6-cm tissue culture dishes. Cells were washed with PBS then lysed using a QIAshredder (Qiagen). RNA was extracted using a RNeasy minikit (Qiagen). cDNA was reverse transcribed by using the Transcriptor First Strand cDNA synthesis kit (Roche Applied Science). 1 μg of RNA was used for cDNA preparation in a final volume of 20 μl by using both anchored oligo(dT)18 and random hexamer primer following manufacturer's instructions.
Real-time PCR was performed by using 1 μl of the prepared cDNA in a 20-μl reaction volume with Power SYBR Green PCR Master Mix (Applied Biosystems). Transcript abundance of MyoVa, MyoVb and MyoVc was estimated with primers shown in Table S1; β-actin was used as control. The reaction conditions included initial denaturation at 95°C for 10 min, 40 cycles of PCR, which included 15 s of denaturing at 95°C and 1-min-long annealing and/or extension at 60°C. Fluorescence of each sample was measured at the end of each round of extension. mRNA abundance was calculated in relation to the β-actin mRNA in the same sample. mRNA abundance in WIF-B cells relative to fibroblasts was measured by using the 2−ΔΔCT method (Schmittgen and Livak, 2008).
Indirect immunofluorescence, imaging and quantification
Primary antibodies were obtained from the following sources: mouse anti-Tomato (Origene; 1:400), mouse anti-TGN38 (BD Biosciences, #610899; 1:400), rabbit anti-ATP7B (Abcam, #EPR6794; 1:300), mouse anti-γ-tubulin (Sigma, #T6557; 1:400), and mouse anti-Rab11 (BD Transduction, #610657; 1:200). Rabbit antibody (1:100) against endogenous MyoVb was a gift from Dr John Hammer (NHLBI). Secondary antibodies conjugated to Alexa-Fluor-488, Alexa-Fluor-568 or Alexa-Fluor-647 were from Invitrogen; those conjugated to Cy5 were from Jackson ImmunoResearch Laboratories. Immunolabeled WIF-B cells were analyzed by using a 100× PLAN-APO, 1.4 NA oil-immersion objective on LSM 510 META and LSM 700 confocal microscopes (Zeiss). For imaging, cells that expressed low levels of exogenous protein were selected. The distribution of the GFP-ATP7B variants was assessed relative to the organelle marker TGN38. Experiments were repeated twice or more and confocal images of 10–20 cells evaluated per experiment.
Imaris software was used to calculate the extent of colocalization. For quantification of colocalization around the bile canaliculus or at the apical spot, a tight volume (0.70 µM3) that included the compartment was cropped and the Pearson's correlation coefficient of colocalized volume was calculated between the two channels (red and green for MyoVbT and ATP7B, respectively, or blue and green for Rab11 and ATP7B, respectively). A low-intensity threshold of 5% was used for each channel. To measure colocalization of ATP7B and MyoVbT in unpolarized cells on days 5, 8 and 11, the cropped volume was larger to include the ATP7B vesicles. The volume was set at 0.80 µM3.
In the analysis depicted in Fig. 2C, control WIF-B cells or cells infected with MyoVbT-encoding adenovirus overnight were fixed in 4% PFA in PBS, permeabilized in TX-100 and stained for endogenous ATP7B. MyoVbT staining was enhanced using a rabbit anti-red fluorescent protein antibody (Rockland; 1:300). Apical rings were identified and confocal sections of 1 Airy unit were collected at 2× magnification. The stacks were deconvolved using the AutoQuant blind iterative method, as previously described (Nyasae et al., 2014). To determine the distribution of fluorescence markers near the bile canaliculus (Fig. 2C, left), ten lines were drawn through a bile canaliculus that had a fully formed lumen, by using the RGB profiler in ImageJ. The fluorescence of each line as normalized with respect to Z-axis (ImageJ-RGB profiler) and the average of the ten lines was plotted.
Western blots
WIF-B cells on coverslips were grown to confluency or until day 7, 9 or 11. Cells were trypsinized, washed with PBS and homogenized in 1.5 ml of lysis buffer [25 mM imidazole, 0.05 mM DTT, 2 mM AEBSF, 0.25 M sucrose, with protease inhibitor tablets (Roche)] with 20 strokes of loose-set pestle and 20 strokes of tight-set pestle in a dounce under ice. The homogenate was centrifuged at 600 g for 10 min at 4°C. The resulting supernatant was again centrifuged at 5000 g for 30 min and the pellet was resuspended in 100 μl of lysis buffer containing SDS. The sample was analyzed by 10% SDS PAGE, transferred to PVDF membrane (Millipore) using transfer buffer (10% methanol, 10% 100 mM CAPS pH 11 in water) and detected by using mouse anti-rat-ATP7B (1:5000), mouse anti rat-Myr-6 (rat myosin Vb, 1:2000) and α-tubulin antibody (equal loading control; Sigma; 1:5000). HRP-conjugated goat anti-mouse IgG was used as secondary antibody (1:10,000). The intensity of protein bands from three independent experiments was quantified using densitometry (Alpha Imager 2200) and normalized to the α-tubulin expression.
Surface biotinylation
Cells grown to confluency on coverslips (10–12 days) were washed three times with cold PBS and once with cold biotinylation buffer (10 mM borate, 137 mM NaCI, 3.8 mM KCI, 0.9 mM CaCl2, 0.52 mM MgCI2 and 0.16 mM MgSO4 pH 9). Cells were labeled with sNHS-LC-biotin (0.62 mg/ml in biotinylation buffer) twice for 15 min at 4°C. The reaction was quenched with 100 mM glycine in PBS. Cells were again washed (3×) with 1× PBS and sonicated using a probe sonicator (settings: 10 s at 10% amplitude). The slurry was centrifuged at 5000 g and the supernatant was collected and mixed with a 50% slurry of Neutravidin beads (Pierce, Cat# 29200) in 0.1% Triton X-100 in PBS (beads were pre-washed 5× in 0.1% Triton X-100 in PBS) for 2 h at 4°C
Bound protein was eluted twice at 95°C by using urea sample buffer (Ihrke, et al., 1993). Eluates were separated by SDS-electrophoresis, and the polypeptides transferred to nitrocellulose membrane. Twenty µl from 1 ml of the total membrane fraction was also analyzed, to estimate total ATP7B present in the starting extract. The blots were probed with a rat polyclonal antibody (1:5000) raised against the N-terminal of ATP7B. The relative amount of biotinylated ATP7B was determined by densitometry of the immunoblot bands, normalized to total ATP7B.
For immunofluorescence post-biotinylation, cells were fixed, blocked with 1% BSA in 1×PBS, then incubated with FITC-conjugated streptavidin (20 µg/ml) for 30 min at room temperature, washed with PBS and mounted with fluoromount for visualization.
[Cu+] measurement
WIF-B coverslips were rinsed twice with PBS. Cells were trypsinized and sonicated using a probe sonicator (10 s at 10% amplitude). Aliqouts of the cell homogenate (60 µl) were digested in 110 µl concentrated HNO3 at 65°C for 30 min. HPLC-grade water (230 µl) was added to reach a final volume of 400 µl. The Cu+ content of the cells was measured by using an atomic absorption spectrophotometer AA6650 (Shimadzu). Cu+ levels in the cell were normalized against ATP7B abundance determined by immunoblot. Since ATP7B traffics in WIF-B cells in response to treatment with Cu+ at concentrations of as low as 10–20 µM, it was necessary to ensure that such a minute intracellular Cu+ flux could be detected. Day-13 WIF-B cells were cultured in basal medium followed by treatment with CuCl2 (uptake) and subsequent removal of Cu+ with BCS (export) as mentioned above.
Acknowledgements
We acknowledge Cissy Li for help with the Atomic Absorption Spectroscopy.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
A.G. performed all experiments and analyzed data, A.B. performed some experiments and analyzed data. M.J.S. contributed data to their analyses. A.G., M.J.S., S.L. and A.L.H. wrote the manuscript.
Funding
This research was supported by Johns Hopkins University GI Core Center Pilot Project grant and Ramanujan Fellowship SB/S2/RJN-146/2014 (Department of Science and Technology, Govt. of India) to A.G.; and a National Institutes of Health (NIH) grant [grant numbers P01 DK-072084 and P01 GM-067166 to A.L.H.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.175307/-/DC1
References
- Bastaki M., Braiterman L. T., Johns D. C., Chen Y.-H. and Hubbard A. L. (2002). Absence of direct delivery for single transmembrane apical proteins or their “Secretory” forms in polarized hepatic cells. Mol. Biol. Cell 13, 225-237. 10.1091/mbc.01-07-0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiterman L., Nyasae L., Guo Y., Bustos R., Lutsenko S. and Hubbard A. (2009). Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am. J. Physiol. Gastrointest. Liver Physiol. 2, G433-G444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiterman L. T., Gupta A., Chaerkady R., Cole R. N. and Hubbard A. L. (2015). Communication between the N and C termini is required for copper-stimulated Ser/Thr phosphorylation of Cu(I)-ATPase (ATP7B). J. Biol. Chem. 290, 8803-8819. 10.1074/jbc.M114.627414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B.-B., Ge L., Xie C., Zhao Y., Miao H.-H., Wang J., Li B.-L. and Song B.-L. (2009). Requirement of myosin Vb.Rab11a.Rab11-FIP2 complex in cholesterol-regulated translocation of NPC1L1 to the cell surface. J. Biol. Chem. 284, 22481-22490. 10.1074/jbc.M109.034355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme N. A., Ham A.-J., Lapierre L. A. and Goldenring J. R. (2011). Rab11-FIP2 influences multiple components of the endosomal system in polarized MDCK cells. Cell. Logist. 1, 57-68. 10.4161/cl.1.2.15289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanni D., Pilloni L., Orru S., Coni P., Liguori C., Serra S., Lai M. L., Uccheddu A., Contu L., Van Eyken P. et al. (2005). Expression of ATP7B in normal human liver. Eur. J. Histochem. 4, 371-378. [DOI] [PubMed] [Google Scholar]
- Forbes J. R. and Cox D. W. (2000). Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum. Mol. Genet. 9, 1927-1935. 10.1093/hmg/9.13.1927 [DOI] [PubMed] [Google Scholar]
- Girard M., Lacaille F., Verkarre V., Mategot R., Feldmann G., Grodet A., Sauvat F., Irtan S., Davit-Spraul A., Jacquemin E. et al. (2014). MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 60, 301-310. 10.1002/hep.26974 [DOI] [PubMed] [Google Scholar]
- Guo Y., Nyasae L., Braiterman L. T. and Hubbard A. L. (2005). NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G904-G916. 10.1152/ajpgi.00262.2005 [DOI] [PubMed] [Google Scholar]
- Hales C. M., Vaerman J.-P. and Goldenring J. R. (2002). Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J. Biol. Chem. 277, 50415-50421. 10.1074/jbc.M209270200 [DOI] [PubMed] [Google Scholar]
- Hayashi H., Shinohara T., Goto K., Fujita Y., Murakami Y., Hattori A., Tatsumi Y., Shimizu A. and Ichiki T. (2012). Liver structures of a patient with idiopathic copper toxicosis. Med. Mol. Morphol. 45, 105-109. 10.1007/s00795-011-0556-1 [DOI] [PubMed] [Google Scholar]
- Hoekstra D., Tyteca D. and van IJzendoorn S. C. D. (2004). The subapical compartment: a traffic center in membrane polarity development. J. Cell Sci. 117, 2183-2192. 10.1242/jcs.01217 [DOI] [PubMed] [Google Scholar]
- Hubbard A. L., Stieger B. and Bariles J. R. (1989). Biogenesis of endogenous plasma membrane proteins in epithelial cells. Annu. Rev. Physiol. 51, 755-770. 10.1146/annurev.ph.51.030189.003543 [DOI] [PubMed] [Google Scholar]
- Ihrke G., Neufeld E. B., Meads T., Shanks M. R., Cassio D., Laurent M., Schroer T. A., Pagano R. E. and Hubbard A. L. (1993). WIF-B cells: an in vitro model for studies of hepatocyte polarity. J. Cell Biol. 123, 1761-1775. 10.1083/jcb.123.6.1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrke G., Martin G. V., Shanks M. R., Schrader M., Schroer T. A. and Hubbard A. L. (1998). Apical plasma membrane proteins and endolyn-78 travel through a subapical compartment in polarized WIF-B hepatocytes. J. Cell Biol. 141, 115-133. 10.1083/jcb.141.1.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaulin F., Xue X., Rodriguez-Boulan E. and Kreitzer G. (2007). Polarization-dependent selective transport to the apical membrane by KIF5B in MDCK cells. Dev. Cell 13, 511-522. 10.1016/j.devcel.2007.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein L. C., van Bergeijk P., Lipka J., Keijzer N., Wulf P. S., Katrukha E. A., Akhmanova A. and Hoogenraad C. C. (2013). Myosin-V opposes microtubule-based cargo transport and drives directional motility on cortical actin. Curr. Biol. 23, 828-834. 10.1016/j.cub.2013.03.068 [DOI] [PubMed] [Google Scholar]
- Knowles B. C., Roland J. T., Krishnan M., Tyska M. J., Lapierre L. A., Dickman P. S., Goldenring J. R. and Shub M. D. (2014). Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J. Clin. Invest. 124, 2947-2962. 10.1172/JCI71651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre L. A. and Goldenring J. R. (2005). Interactions of myosin vb with rab11 family members and cargoes traversing the plasma membrane recycling system. Methods Enzymol. 403, 715-723. 10.1016/S0076-6879(05)03062-4 [DOI] [PubMed] [Google Scholar]
- Lapierre L. A., Kumar R., Hales C. M., Navarre J., Bhartur S. G., Burnette J. O., Provance D. W. Jr, Mercer J. A., Bahler M. and Goldenring J. R. (2001). Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell 12, 1843-1857. 10.1091/mbc.12.6.1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bivic A., Quaroni A., Nichols B. and Rodriguez-Boulan E. (1990). Biogenetic pathways of plasma membrane proteins in Caco-2, a human intestinal epithelial cell line. J. Cell Biol. 111, 1351-1361. 10.1083/jcb.111.4.1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. M., Cater M. A., Mercer J. F. B. and La Fontaine S. (2006a). Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochem. Biophys. Res. Commun. 348, 428-436. 10.1016/j.bbrc.2006.07.067 [DOI] [PubMed] [Google Scholar]
- Lim C. M., Cater M. A., Mercer J. F. B. and La Fontaine S. (2006b). Copper-dependent interaction of dynactin subunit p62 with the N terminus of ATP7B but not ATP7A. J. Biol. Chem. 281, 14006-14014. 10.1074/jbc.M512745200 [DOI] [PubMed] [Google Scholar]
- Lindsay A. J. and McCaffrey M. W. (2002). Rab11-FIP2 functions in transferrin recycling and associates with endosomal membranes via its COOH-terminal domain. J. Biol. Chem. 277, 27193-27199. 10.1074/jbc.M200757200 [DOI] [PubMed] [Google Scholar]
- Lise M.-F., Wong T. P., Trinh A., Hines R. M., Liu L., Kang R., Hines D. J., Lu J., Goldenring J. R., Wang Y. T. et al. (2006). Involvement of myosin Vb in glutamate receptor trafficking. J. Biol. Chem. 281, 3669-3678. 10.1074/jbc.M511725200 [DOI] [PubMed] [Google Scholar]
- Lutsenko S., Barnes N. L., Bartee M. Y. and Dmitriev O. Y. (2007). Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011-1046. 10.1152/physrev.00004.2006 [DOI] [PubMed] [Google Scholar]
- Materia S., Cater M. A., Klomp L. W. J., Mercer J. F. B. and La Fontaine S. (2012). Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 287, 2485-2499. 10.1074/jbc.M111.302216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter K., Brauchbar M., Bucher K. and Hauri H.-P. (1990). Sorting of endogenous plasma membrane proteins occurs from two sites in cultured human intestinal epithelial cells (Caco-2). Cell 60, 429-437. 10.1016/0092-8674(90)90594-5 [DOI] [PubMed] [Google Scholar]
- Muller T., Muller W. and Feichtinger H. (1998). Idiopathic copper toxicosis. Am. J. Clin. Nutr. 5 Suppl., 1082S-1086S. [DOI] [PubMed] [Google Scholar]
- Müller T., Hess M. W., Schiefermeier N., Pfaller K., Ebner H. L., Heinz-Erian P., Ponstingl H., Partsch J., Rollinghoff B., Kohler H. et al. (2008). MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 40, 1163-1165. 10.1038/ng.225 [DOI] [PubMed] [Google Scholar]
- Nedvetsky P. I., Stefan E., Frische S., Santamaria K., Wiesner B., Valenti G., Hammer J. A. III, Nielsen S., Goldenring J. R., Rosenthal W. et al. (2007). A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic 8, 110-123. 10.1111/j.1600-0854.2006.00508.x [DOI] [PubMed] [Google Scholar]
- Nyasae L. K., Schell M. J. and Hubbard A. L. (2014). Copper directs ATP7B to the apical domain of hepatic cells via basolateral endosomes. Traffic 15, 1344-1365. 10.1111/tra.12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer J. H. and Maltese W. A. (2005). Tyrosine phosphorylation of Rab proteins. Methods Enzymol. 403, 194-202. 10.1016/S0076-6879(05)03016-8 [DOI] [PubMed] [Google Scholar]
- Provance D. W. Jr, Addison E. J., Wood P. R., Chen D. Z., Silan C. M. and Mercer J. A. (2008). Myosin-Vb functions as a dynamic tether for peripheral endocytic compartments during transferrin trafficking. BMC Cell Biol. 9, 44 10.1186/1471-2121-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofsen H., Wolters H., Van Luyn M. J. A., Miura N., Kuipers F. and Vonk R. J. (2000). Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119, 782-793. 10.1053/gast.2000.17834 [DOI] [PubMed] [Google Scholar]
- Scheinberg I. H. and Sternlieb I. (1959). The liver in Wilson's disease. Gastroenterology 37, 550-564. [PubMed] [Google Scholar]
- Scheinberg I. H. and Sternlieb I. (1960). Copper metabolism. Pharmacol. Rev. 13, 355-381. [PubMed] [Google Scholar]
- Schmittgen T. D. and Livak K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101-1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Thoeni C. E., Vogel G. F., Tancevski I., Geley S., Lechner S., Pfaller K., Hess M. W., Müller T., Janecke A. R., Avitzur Y. et al. (2014). Microvillus inclusion disease: loss of Myosin vb disrupts intracellular traffic and cell polarity. Traffic 15, 22-42. 10.1111/tra.12131 [DOI] [PubMed] [Google Scholar]
- Thompson R. J. and Knisely A. S. (2014). Microvilli as markers of disordered apical-membrane trafficking and assembly: bowel and liver. Hepatology 60, 34-36. 10.1002/hep.27148 [DOI] [PubMed] [Google Scholar]
- Tuma P. L., Nyasae L. K. and Hubbard A. L. (2002). Nonpolarized cells selectively sort apical proteins from cell surface to a novel compartment, but lack apical retention mechanisms. Mol. Biol. Cell 13, 3400-3415. 10.1091/mbc.02-04-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y., Lippincott-Schwartz J. and Arias I. M. (2004). Intracellular trafficking of bile salt export pump (ABCB11) in polarized hepatic cells: constitutive cycling between the canalicular membrane and rab11-positive endosomes. Mol. Biol. Cell 15, 3485-3496. 10.1091/mbc.E03-10-0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y., Dutt P., Lippincott-Schwartz J. and Arias I. M. (2005). Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc. Natl. Acad. Sci. USA 102, 15087-15092. 10.1073/pnas.0503702102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijmenga C., Müller T., Murli I. S., Brunt T., Feichtinger H., Schönitzer D., Houwen R. H., Müller W., Sandkuijl L. A. and Pearson P. L. (1999). Endemic Tyrolean infantile cirrhosis is not an allelic variant of Wilson's disease. Eur. J. Hum. Genet. 6, 624-628. 10.1038/sj.ejhg.5200235 [DOI] [PubMed] [Google Scholar]