

Abstract
A catalytic method for site- and enantioselective addition of commercially available di-B(pin)-methane to allylic phosphates is introduced (pin = pinacolato). Transformations may be facilitated by an NHC-Cu complex (NHC = N-heterocyclic carbene) and products obtained in 63–95% yield, 88:12 to >98:2 SN2′:SN2 selectivity and 85:15–99:1 enantiomeric ratio. The utility of the approach, entailing the involvement of different catalytic cross-coupling processes, is highlighted by application to formal synthesis of cytotoxic natural product rhopaloic acid A.
Keywords: allylic substitution, boron, copper, enantioselective synthesis, N-heterocyclic carbenes
Graphical abstract
Combination catalysis: The first examples of catalytic enantioselective allylic substitution reactions that introduce a CH2-B(pin) unit are introduced. In combination with various catalytic cross-coupling processes, a range of important organic molecules is now readily accessible.
Catalytic enantioselective allylic substitution (EAS) generates valuable products that contain a stereogenic center adjacent to a transposed alkene.[1] These processes may involve various organometallic reagents (e.g., Zn-, Mg-, or Al-based) and can be promoted by Cu complexes derived from chiral O-, N-, or P-based ligands or N-heterocyclic carbenes (NHCs).[1] Several applications in total synthesis have demonstrated utility.[2] Lately, organoboron species have been adopted in this area because of their robustness and tolerance toward some of the more common organic functional groups (e.g., carboxylic esters or ketones). The groundbreaking advance was achieved by Sawamura, who illustrated that trialkylboron molecules, formed in situ by hydroboration of α-olefins, can be made to participate in efficient, SN2′- and enantioselective phosphine-Cu-catalyzed EAS.[3] Allenyl-,[4] alkenyl-,[5] or propargyl-B(pin)[6] species (pin = pinacolato) have since been used in related NHC-Cu-catalyzed transformations. Another current development entails processes involving in situ-generated allylcopper species that furnish modifiable boron-containing products.[7]
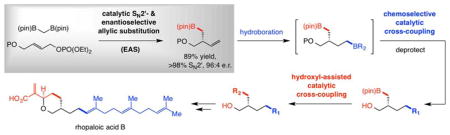
Nonetheless, key shortcomings remain, one of which revealed itself while we evaluated a possible route to cytotoxic natural product rhopaloic acid A[8],[9] (Scheme 1a). Preparation of enantiomerically enriched i called for an organoboron reagent that requires site-selective hydroboration of a somewhat sensitive dienyl-aldehyde (or ester derivative). The alternative pathway via diene ii, accessible by previously reported EAS protocols,[10] would demand differentiation of two terminal olefins, likely to generate difficult-to-separate isomeric mixtures. We thus envisioned the possibility of a catalytic EAS with commercially available di-B(pin)-methane 1,[11] a member of a class of compounds conceived by Matteson[12] and which has been the focus of several studies following the pioneering work of Endo and Shibata.[13] Such processes would not only involve an organoboron reagent, they would deliver products containing a versatile C-B(pin) bond as well. We imagined a subsequent sequence entailing hydroboration of the EAS product (iii), furnishing iv with differentiable C-B bonds that could then be converted chemoselectively to vi by a pair of catalytic cross-coupling reactions (via v); the first would involve the dialkylboron site chemoselectively and the second would benefit from the presence of the neighboring hydroxyl group[14] that wouild then be deployed to form the pyran ring. Herein, we describe the realization of these plans.
Scheme 1.
Problems associated with an application of catalytic enantioselective allylic substitution (EAS) to synthesis of rhopaloic acid B and a possible solution. Abbreviations: P, protecting group; LG, leaving group; pin, pinacolato.
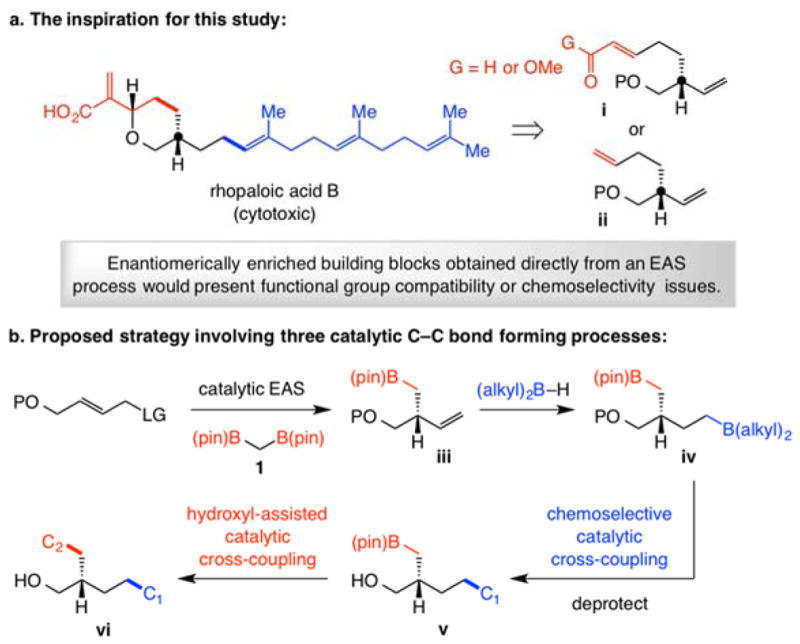
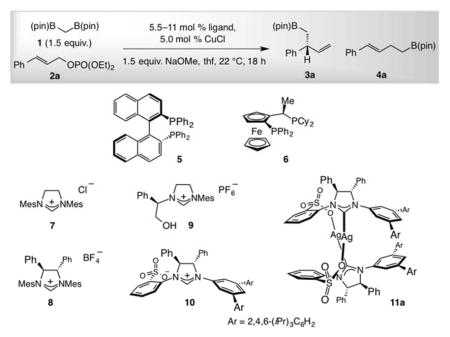
We favor allylic phosphates as substrates because the Lewis basic phosphate may bind to a chiral Cu complex, an association that is often pivotal to achieving high SN2′- and/or enantioselectivity.[4–6,7] We first found that EAS with only CuCl (no ligand) proceeds to 45% conversion, affording the linear isomer (4a) exclusively (Table 1, entry 1). A chiral ligand would have to bind efficiently to the transition metal and/or the derived Cu complex to promote formation considerably faster than a free Cu complex. We then determined that catalysts corresponding to bis-phosphines 5 and 6 generate the linear isomer (4a) selectively (entries 2–3). With imidazolinium salts 7-9[15] (entries 4–6), 4a was again favored. Matters improved with the NHC-Cu species derived from sulfonate-containing 10 (entry 7), which afforded 3a with 76% SN2′ selectivity and 97:3 enantiomeric ratio (e.r.). Based on the finding that 4a is produced by a ligand-free Cu catalyst (entry 1), we used the crystalline and air-stable dimeric Ag complex 11a[6], one of a set of complexes found to exchange ligands with exceptional efficiency;[16] the branched isomer 3a was thus formed in 95:5 SN2′:SN2 selectivity and 97:3 e.r. (entry 8, Table 1).
Table 1.
Examination of different types of Cu complexes.[a]
| ||||
|---|---|---|---|---|
| Entry | Ligand; Mol % | Conv. [%];[b] Yield [%][c] |
SN2′:SN2 (3a:4a)[b] | e.r.[d] |
| 1 | none[a] | 45; 21 | <2:>98 | NA |
| 2 | 5; 11 | 80; 76 | 2:98 | NA |
| 3 | 6; 11 | 65; 41 | 7:93 | ND |
| 4 | 7; 11 | >98; 73 | 5:95 | ND |
| 5 | 8; 11 | >98; 54 | 20:80 | ND |
| 6 | 9; 11 | >98; 69 | <2:>98 | NA |
| 7 | 10; 11 | >98; 77 | 76:24 | 97:3 |
| 8 | 11a; 5.5 | >98; 83 | 95:5 | 97:3 |
| Entry | Ligand; Mol % | Conv. [%];[b] Yield [%][c] |
SN2′:SN2 (3a:4a)[b] | e.r.[d] |
|---|---|---|---|---|
| 1 | None[a] | 45; 21 | <2:>98 | NA |
| 2 | 5; 11 | 80; 76 | 2:98 | NA |
| 3 | 6; 11 | 65; 41 | 7:93 | ND |
| 4 | 7; 11 | >98; 73 | 5:95 | ND |
| 5 | 8; 11 | >98; 54 | 20:80 | ND |
| 6 | 9; 11 | >98; 69 | <2:>98 | NA |
| 7 | 10; 11 | >98; 77 | 76:24 | 97:3 |
| 8 | 11; 5.5 | >98; 83 | 95:5 | 97:3 |
Performed under N2 atm; ≤5% linear methoxy addition products are observed in all cases.
Conv. and SN2′:SN2 ratios determined by analysis of 1H NMR spectra of unpurified mixtures; conv. (±2%) refers to disappearance of the 2a.
Yields of isolated and purified products but as a mixture of 3a and 4a (±5%).
E.r. values determined by HPLC (±1%). See the Supporting Information for details.
Abbreviations: Mes, 2,4,6-(Me)3-C6H2; NA, not applicable ND; not determined.
The protocol has considerable range (Chart 1). Various aryl-substituted substrates, including those with an electron donating or withdrawing substituent, whether it is at the ortho, meta or para position (12a-j) were converted to the corresponding primary alcohols in 61–95% yield (after oxidation), 88:12 to >98:2 SN2′:SN2 selectivity and 85:15–99:1 e.r. There were no complications due to the aryl ketone substituent in 12i, a functional unit not typically tolerated by the more nucleophilic organometallic species and, notably, in NHC-Cu-catalyzed transformations with a propargyl-B(pin) reagent.[6] The Cu catalyst remained operative in the presence of Lewis basic and potentially catalyst-deactivating N- and S-containing heterocyclic moieties: pyridyl- and thienyl-substituted 12k,l were obtained with similar efficiency and selectivity. Alkyl-substituted allylic phosphates (cf. 12m) were suitable. Reaction with a 1,3-dienyl substrate was somewhat less site- and enantioselective but none of the side product from the formation of a benzylic C-C bond could be detected (cf. 12n). Allylsilane 12o may be utilized in stereoselective synthesis.[17]
Chart 1.

Scope of the method. [a] Conv. refers to the consumption of allylic phosphate and was determined by analysis of 1H NMR spectra of unpurified reaction mixtures (the same for SN2′:SN2 ratios); ≤5% linear methoxy addition products are observed in all cases. For 12h-k the mixture was treated with NaIO4/NH4OAc at 22 °C for 1 h (1/1 acetone/H2O). [b] Yield refers to isolated and pure SN2′ isomers, except for 12k (5% linear product isomer), 12l (3% boryl addition byproduct) and 12n (3% product from Z isomer of the substrate). Enantioselectivities were determined by HPLC analysis. See the Supporting Information for details.
A distinct feature of the sulfonate-containing chiral NHC ligands (cf. 8 and 9) is that the Cu complexes promote highly SN2′-selective reactions (cf. Table 1 and Chart 1). This is congruent with the most recent mechanistic and computational studies,[6] revealing that the active species is likely a monodentate system wherein the sulfonate group, without the geometric constraints of chelation with the Cu center,[18] is oriented anti to the nearby phenyl substituent (I, Figure 1). Formation of an alkali metal bridge between the anionic tether of the chiral catalyst and the Lewis basic phosphate unit can engender a well-defined transition structure with the Cu-C bond disposed for SN2′ addition. There is indeed measurable dependence of branch selectivity on the identity of the base used (Figure 1). With the more Lewis acidic lithium salt there was >98% SN2′ selectivity, albeit at lower yield due to solubility issues. More of the achiral isomer was generated with the less Lewis acidic and larger potassium methoxide.
Figure 1.
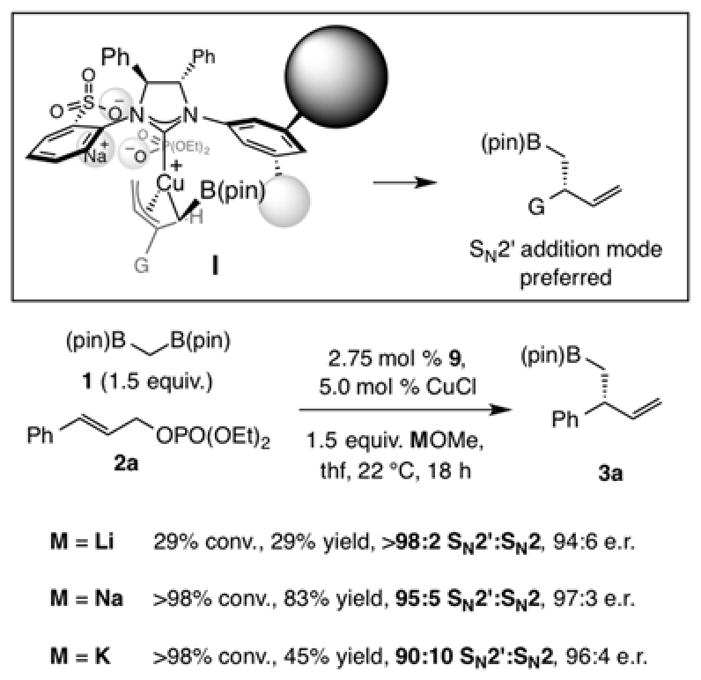
Mechanistic model to account for the origin of stereoselectivity and evidence pointing to the significance of counter-ion of the basic reagent used. See the Supporting Information for details.
We then probed the feasibility of application to synthesis of rhopaloic acid A[8,9] (Scheme 2). Organoboron compound 12p was purified by silica gel chromatography and isolated in 89% yield, >98:2 SN2′:SN2 selectivity and 96:4 e.r.; in this case, the Cu complex derived from 11b gave higher enantioselectivity (84:16 e.r. with 11a).[19] Hydroboration afforded diboron product 13, which was coupled in situ with E-alkenyl-iodide 14[20] by a phosphine-Pd-catalyzed process,[21] affording 15 (>98% chemoselectivity, 87% yield). Removal of the silyl ether and NHC-Cu-catalyzed cross-coupling involving the alkyl-B(pin) moiety and commercially available allylphosphate delivered tetraene 16 in 67% overall yield. As noted earlier, this latter process strongly benefits from the proximal hydroxyl group:[14] there was <2% C-C bond formation with silyl ether 15.[22] Cross-metathesis with Ru complex 17 and acrolein led to enal 18 (71% overall yield). Intramolecular conjugate addition[23] delivered pyran 19, which has previously been converted to rhopaloic acid A, in 85% yield and 89:11 diastereomeric ratio (d.r.).[9f]
Scheme 2.

Application to enantioselective synthesis of rhopaloic acid A; d.r. = diastereomeric ratio. See the Supporting Information for details.
To conclude, we introduce an EAS method involving commercially available and functional group tolerant di-B(pin)-methane as the reagent, furnishing products efficiently and with exceptional SN2′- and enantioselectivity. We demonstrate that in combination with chemoselective and hydroxyl-assisted catalytic cross-coupling transformations, the EAS protocol can provide access to an assortment of useful organic molecules efficiently and in high enantiomeric purity.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM-47480) and the NSF (CHE-1362763).
References & Footnotes
- 1.For reviews of catalytic enantioselective allylic substitution (EAS) reactions with “hard” organometallic reagents, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem Commun. 2004:1779–1785. doi: 10.1039/b401123f.Yorimitsu H, Oshima K. Angew Chem Int Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653.Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem Rev. 2008;108:2824–2852. doi: 10.1021/cr068424k.Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515.Baslé O, Denicourt-Nowicki A, Crévisy C, Mauduit M. In: Copper-Catalyzed Asymmetric Synthesis. Alexakis A, Krause N, Woodward S, editors. VCH-Wiley; 2014. pp. 85–125.
- 2.For a recent review regarding applications of catalytic EAS reactions to natural product synthesis, see: Calvo BC, Buter J, Minnaard AJ. In: Copper-Catalyzed Asymmetric Synthesis. Alexakis A, Krause N, Woodward S, editors. VCH-Wiley; 2014. pp. 373–447.
- 3.a) Ohmiya H, Yokobori U, Makida Y, Sawamura M. J Am Chem Soc. 2010;132:2895–2897. doi: 10.1021/ja9109105. [DOI] [PubMed] [Google Scholar]; b) Shido Y, Yoshida M, Tanabe M, Ohmiya H, Sawamura M. J Am Chem Soc. 2012;134:18573–18576. doi: 10.1021/ja3093955. [DOI] [PubMed] [Google Scholar]; c) Hajoh K, Shido Y, Ohmiya H, Sawamura M. Angew Chem Int Ed. 2014;53:5954–4958. doi: 10.1002/anie.201402386. [DOI] [PubMed] [Google Scholar]
- 4.Jung B, Hoveyda AH. J Am Chem Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Shintani R, Takatsu K, Takeda M, Hayashi T. Angew Chem Int Ed. 2011;50:8656–8659. doi: 10.1002/anie.201103581. [DOI] [PubMed] [Google Scholar]; b) Gao F, Carr JL, Hoveyda AH. Angew Chem Int Ed. 2012;51:6613–6617. doi: 10.1002/anie.201202856. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao F, Carr JL, Hoveyda AH. J Am Chem Soc. 2014;136:2149–2161. doi: 10.1021/ja4126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Jung B, Torker S, Hoveyda AH. J Am Chem Soc. 2015;137:8948–8964. doi: 10.1021/jacs.5b05805. [DOI] [PubMed] [Google Scholar]
- 7.Meng F, McGrath KP, Hoveyda AH. Nature. 2014;513:367–374. doi: 10.1038/nature13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Yanai M, Ohta S, Ohta E, Ikegami S. Tetrahedron. 1998;54:15607–15612. [Google Scholar]; b) Ohta S, Uno M, Yoshimura M, Hiraga Y, Ikegami S. Tetrahedron Lett. 1996;37:2265–2266. [Google Scholar]
- 9.For previous studies regarding synthesis of rhopaloic acids, see: Snider BB, He F. Tetrahedron Lett. 1997;31:5453–5454.Takagi R, Sasaoka A, Kojima S, Ohkata K. Chem Commun. 1997:1887–1888.Takagi R, Asasaoka, Nishitani H, Kojima S, Hiraga Y, Ohkata K. J Chem Soc, Perkin Trans. 1998;1:925–934.Nishitani H, Sasaoka A, Tokumasu M, Ohkata K. Heterocycles. 1999;50:35–38.Kadota K, Ogasawara K. Heterocycles. 2003;59:485–490.Brioche JCR, Goodenough KM, Whatrup DJ, Harrity JPA. Org Lett. 2007;9:3941–3943. doi: 10.1021/ol701553j.Brioche JCR, Goodenough KM, Whatrup DJ, Harrity JPA. J Org Chem. 2008;73:1946–1953. doi: 10.1021/jo702620e.
- 10.For example, see: Tissot-Croset K, Polet D, Alexakis A. Angew Chem Int Ed. 2004;43:2426–2428. doi: 10.1002/anie.200353744.Lopez F, van Zijl AW, Minnaard AJ, Feringa BL. Chem Commun. 2006:409–411. doi: 10.1039/b513887f.
- 11.While this manuscript was being prepared, a report appeared regarding related non-enantioselective processes with allylic chlorides promoted by achiral NHC-Cu complexes. See: Kim J, Park S, Park J, Cho SH. Angew Chem Int Ed. 2015 doi: 10.1002/anie.201509480.
- 12.Matteson DS, Moody RJ. Organometallics. 1982;1:20–28. [Google Scholar]
- 13.Endo K, Ohkubo T, Hirokami M, Shibata T. J Am Chem Soc. 2010;132:11033–11035. doi: 10.1021/ja105176v.Endo K, Ohkubo T, Shibata T. Org Lett. 2011;13:3368–3371. doi: 10.1021/ol201115k.Endo K, Ohkubo T, Ishioka T, Shibata T. J Org Chem. 2012;77:4826–4831. doi: 10.1021/jo3004293.Endo K, Ishioka T, Ohkubo T, Shibata T. J Org Chem. 2012;77:7223–7231. doi: 10.1021/jo3015165.For related investigations, see: Zhang ZQ, Yang CT, Liang LJ, Xiao B, Lu X, Liu JH, San YY, Marder TB, Fu Y. Org Lett. 2014;16:6342–6345. doi: 10.1021/ol503111h.Endo K, Ishioka T, Shibata T. Synlett. 2014;25:2184–2188.Sun C, Potter B, Morken JP. J Am Chem Soc. 2014;136:6534–6537. doi: 10.1021/ja500029w.Potter B, Szymaniak AA, Edelstein EK, Morken JP. J Am Chem Soc. 2014;136:17918–17921. doi: 10.1021/ja510266x.Joannou MV, Moyer BS, Meek SJ. J Am Chem Soc. 2015;137:6176–6179. doi: 10.1021/jacs.5b03477.Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ. Angew Chem Int Ed. 2015;54:14141–14145. doi: 10.1002/anie.201507171.
- 14.a) Meng F, Haeffner F, Hoveyda AH. J Am Chem Soc. 2014;136:11304–11307. doi: 10.1021/ja5071202. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blaisdell TP, Morken JP. J Am Chem Soc. 2015;137:8712–8715. doi: 10.1021/jacs.5b05477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clavier H, Coutable L, Toupet L, Guillemin JC, Mauduit M. J Organomet Chem. 2005;690:5237–5254. [Google Scholar]
- 16.May TL, Brown MK, Hoveyda AH. Angew Chem Int Ed. 2008;47:7358–7362. doi: 10.1002/anie.200802910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For synthesis of analogous homoallylic alcohols by other catalytic EAS strategies, see: Morken JP, Didiuk MT, Hoveyda AH. J Am Chem Soc. 1993;115:6997–6998.Didiuk MT, Johannes CW, Morken JP, Hoveyda AH. J Am Chem Soc. 1995;117:7097–7104.Yu B, Menard F, Isono N, Lautens M. Synthesis. 2009:853–859.
- 18.Lee Y, Li B, Hoveyda AH. J Am Chem Soc. 2009;131:11625–11633. doi: 10.1021/ja904654j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The reason for this variation in e.r. is unclear at this time and is the subject of ongoing mechanistic studies.
- 20.The requisite iodide was prepared by a two-vessel process (alkynyl lithium addition followed by zirconocene-hydride addition/iodine trapping) and 58% overall yield (>98:2 E:Z) from commercially available geranyl bromide; see: Sulake RS, Lin HH, Hsu CY, Weng CF, Chen C. J Org Chem. 2015;80:6044–6051. doi: 10.1021/acs.joc.5b00345.Wipf P, Lim S. Angew Chem Int Ed Engl. 1993;32:1068–1071.). See the Supporting Information for details.
- 21.a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; b) Chemler SR, Trauner D, Danishefsky SJ. Angew Chem Int Ed. 2001;40:4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 22.Attempts at using the boronic acid derivative led to an unidentifiable mixture of compounds.
- 23.Lee K, Kim H, Hong J. Org Lett. 2011;13:2722–2725. doi: 10.1021/ol200824r.For a review on oxa-Michael processes, see: Nising CF, Bräse S. Chem Soc Rev. 2012;41:988–999. doi: 10.1039/c1cs15167c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.