

ABSTRACT
The kinesin KIF17 localizes at microtubule plus-ends where it contributes to regulation of microtubule stabilization and epithelial polarization. We now show that KIF17 localizes at cell–cell adhesions and that KIF17 depletion inhibits accumulation of actin at the apical pole of cells grown in 3D organotypic cultures and alters the distribution of actin and E-cadherin in cells cultured in 2D on solid supports. Overexpression of full-length KIF17 constructs or truncation mutants containing the N-terminal motor domain resulted in accumulation of newly incorporated GFP–actin into junctional actin foci, cleared E-cadherin from cytoplasmic vesicles and stabilized cell–cell adhesions to challenge with calcium depletion. Expression of these KIF17 constructs also increased cellular levels of active RhoA, whereas active RhoA was diminished in KIF17-depleted cells. Inhibition of RhoA or its effector ROCK, or expression of LIMK1 kinase-dead or activated cofilinS3A inhibited KIF17-induced junctional actin accumulation. Interestingly, KIF17 activity toward actin depends on the motor domain but is independent of microtubule binding. Together, these data show that KIF17 can modify RhoA–GTPase signaling to influence junctional actin and the stability of the apical junctional complex of epithelial cells.
KEY WORDS: Rho–GTPases, Actin, Cell–cell adhesion, Kinesin
Summary: The kinesin KIF17 activates RhoA signaling at cell–cell contacts to regulate junctional actin, the stability of the apical junctional complex and polarization of epithelial cells.
INTRODUCTION
Epithelia play key roles in tissue homeostasis by establishing transport systems for vectorial secretion and absorption and by forming a physical barrier between the internal milieu and the outside environment. Adherens junctions and tight junctions, formed by trans-cellular interactions of transmembrane adhesion proteins linked to the cytoskeleton, are essential for epithelial morphogenesis and function. Known collectively as the apical junctional complex (AJC), adherens junctions couple adjacent cells physically whereas tight junctions set boundaries between apical and basolateral membranes and control paracellular permeability (Guillot and Lecuit, 2013). Components of the AJC are delivered to the membrane by transport along microtubules (Chen et al., 2003; Ivanov et al., 2006; Mary et al., 2002; Nekrasova et al., 2011; Portereiko et al., 2004; Yanagisawa et al., 2004) and are anchored at adhesive sites by their association with actin and microtubule adaptors. As cell–cell adhesions mature, signaling molecules that also associate with the AJC induce changes in actin and microtubule arrays by modifying polymer dynamics and stability (Brieher and Yap, 2013; Chausovsky et al., 2000; Mège et al., 2006; Ratheesh et al., 2012). Thus, the cytoskeleton affects AJC formation and maturation, and signaling at the AJC reciprocally affects actin and microtubules; together, these processes direct morphogenetic responses to numerous cues (Brieher and Yap, 2013; Mack and Georgiou, 2014). Although many of the components involved in remodeling of AJCs and the cytoskeleton are known, the mechanisms employed to coordinate these events are still incompletely defined.
Rho family GTPases and their effectors comprise a major class of signaling molecules at the AJC (Citi et al., 2014; Fukata and Kaibuchi, 2001) and many are regulated by cell–cell adhesion. Signaling by Cdc42, Rac1 and RhoA regulates AJC formation, maturation and remodeling. They also regulate actin and microtubule arrays (Samarin and Nusrat, 2009; Wojnacki et al., 2014). Rac1 and Cdc42 regulate Arp2/3 (also known as Actr2/3) to affect branched actin filament formation (Kraemer et al., 2007; Otani et al., 2006) and RhoA regulates formins in the generation of actin cables (Carramusa et al., 2007; Kher et al., 2014; Kobielak et al., 2004). RhoA signaling, through its effector Rho-associated protein kinase (ROCK, of which there are two isoforms ROCK1 and ROCK2), also exerts indirect effects on branched actin formation by inactivating the actin-severing protein cofilin. In addition, RhoA activation of formin leads to microtubule capture and stabilization in migrating fibroblasts (Bartolini et al., 2008; Cook et al., 1998; Palazzo et al., 2001) and plays a role in regulating microtubule stability in epithelial cells (Nakaya et al., 2008). Combined, these functions allow Rho–GTPases to orchestrate the remodeling of cytoskeletal arrays and cell–cell junctions that accompanies epithelial polarization.
Rho–GTPases are regulated upstream and downstream of the AJC by guanine-nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) that control spatiotemporal activation of Rho effectors (Quiros and Nusrat, 2014). How Rho–GTPase effectors and regulators are targeted to discrete sites for selective activation is still a topic of intense study. Dynamic microtubule plus-ends can interact with proteins at the cortex and can deliver proteins associated with the microtubule plus-end that regulate cytoskeletal and junctional organization, leading to the concentration of E-cadherin at cell–cell contacts (Ligon and Holzbaur, 2007; Ligon et al., 2001; Stehbens et al., 2006). Microtubule capture and stabilization at the cortex may also provide specialized tracks for targeted delivery of cytoplasmic and membrane proteins important for junction maturation and remodeling (Waterman-Storer et al., 2000). As such, delivery of Rho–GTPase effectors and regulators by microtubule motors can be envisioned as playing a role in regulation of localized signaling cascades at the AJC.
KIF17 is a multifunctional, homodimeric microtubule motor with roles in vesicular transport (Chu et al., 2006; Jenkins et al., 2006; Setou et al., 2000), transport of RNA granules (Chennathukuzhi et al., 2003; Kotaja et al., 2006; Saade et al., 2007; Takano et al., 2007), regulation of transcriptional activators (Kotaja et al., 2005; Macho et al., 2002), and in building sensory cilia (Dishinger et al., 2010; Fan et al., 2011; Insinna et al., 2008; Jenkins et al., 2006; Ou et al., 2005; Pan et al., 2006; Snow et al., 2004). In epithelial cells, KIF17 colocalizes and interacts with components of the microtubule plus-end cortical capture machinery, promoting microtubule stabilization and cell polarization (Acharya et al., 2013; Espenel et al., 2013; Jaulin and Kreitzer, 2010). This can influence cell architecture, but it is not yet known if microtubule modification is the only means by which KIF17 contributes to epithelial polarization. In this study, we provide evidence that KIF17 activates a RhoA signaling pathway at cell–cell contacts that influences both cortical actin and cell–cell junctions. This, in concert with its effects on microtubule stabilization, may serve to integrate cytoskeletal remodeling with maturation and stabilization of the AJC. A role for KIF17 in local RhoA activation also provides an additional potential mechanism by which this kinesin-2 motor exerts effects on epithelial polarization.
RESULTS
KIF17 contributes to actin organization in epithelial cells
We showed previously that KIF17 localizes to microtubule plus-ends with EB1 (also known as MAPRE1) and contributes to microtubule stabilization and polarization of epithelial cells (Jaulin and Kreitzer, 2010). In our analysis of KIF17 distribution in MDCK and Caco-2 epithelial cells, we also identified a pool of KIF17 localized at sites of cell–cell contact that is lost after KIF17 depletion by shRNA (Fig. 1A; Fig. S1A) (Jaulin and Kreitzer, 2010). KIF17 colocalized with E-cadherin and actin at these cell–cell junctions (Fig. 1A), and with α-actinin, a junctional actin-binding protein (Fig. 1B).
Fig. 1.

KIF17 localizes at cell–cell junctions and contributes to actin remodeling during epithelial morphogenesis. (A) Colocalization of KIF17 with actin and E-cadherin at cell–cell contacts in MDCK cells. Color overlay shows an enlarged view of the boxed region of KIF17 and E-cadherin or β-actin images; the KIF17 image was shifted by five pixels to highlight corresponding staining patterns. (B) Colocalization of KIF17 and α-actinin at cell–cell junctions. Color overlay shows an enlarged view of the boxed region; the KIF17 image was shifted by five pixels. (C) Immunoblot showing KIF17 in MDCK cells transduced with control (shNC) or KIF17-targeting (shKIF17) shRNAs. Hsp90 was used as a loading control. (D) Localization of actin (phalloidin), ZO-1 and nuclei (DAPI) in shNC and shKIF17 MDCK cysts grown in Matrigel for seven days. Dotted and dashed lines highlight apical and basal membranes, respectively. Graph shows the ratio of apical to basal actin fluorescence intensity determined by line-scan analysis. n=38 and 48 cells for shNC and shKIF17, respectively. Error bars are s.e.m., significance was determined with a two-tailed, unpaired student's t-test, ****P<0.0001.
In 3D organotypic MDCK cultures, 85±2.6% (mean±s.e.m.; n=322) of cysts that form have a single layer of cells surrounding a central lumen, and actin is enriched at the apical pole of individual cells (Fig. 1C,D; shNC, short hairpin negative control). KIF17 depletion increased the percentage of cysts with either no lumens or multiple lumens as described previously (Jaulin and Kreitzer, 2010), and reduced the percentage of cysts with one lumen to 38±5.4% (n=332). We used line-scan analysis of individual cells in cysts with a single lumen to measure enrichment of actin at the apical pole (dotted line) relative to the basal pole (dashed line) in control (shNC) and KIF17-depleted cells (shKIF17, Fig. 1D). In shNC, the apical/basal actin ratio is 6.74, but only 1.93 in shKIF17, demonstrating that apical actin enrichment is compromised by KIF17 depletion. Thus, KIF17 colocalizes with and contributes to organizing the distribution of actin in epithelial cells.
KIF17 localization at cell–cell contacts is mediated by its N-terminal motor domain
To examine how KIF17 could affect actin organization, we prepared and analyzed the localization of GFP-tagged KIF17 full-length and C-terminal truncation mutants in MDCK cells (Fig. 2A). Proteins were expressed acutely by intranuclear cDNA injection and their localization was analyzed 3 h after injection. Full-length KIF17 appeared primarily as diffuse cytoplasmic fluorescence but a population of the protein localized as discrete puncta, a large proportion of which accumulated at microtubule plus-ends in protruding regions of the cells (Fig. 2A, GFP–KIF17-FL) and as described previously (Jaulin and Kreitzer, 2010). The soluble pool of KIF17-FL represents kinesin in a compact, auto-inhibited conformation (Espenel et al., 2013; Hammond et al., 2010); this auto-inhibited conformation is disrupted by a single point mutation in the hinge region between KIF17 coiled-coil domains (G754E), and when expressed in epithelial cells GFP–KIF17-FLG754E localizes robustly at microtubule plus-ends and in cell protrusions (Espenel et al., 2013; Jaulin and Kreitzer, 2010). Protein expressed from a construct in which the tail and last coiled-coil are deleted, GFP–K490, localized at microtubule plus-ends in cell protrusions (Fig. 2A; Fig. S1B), but was also detected at cell–cell contacts in 24.3% of expressing cells (Fig. 2A,C). Proteins synthesized from two shorter constructs, GFP–K370 (encoding motor and neck) and GFP–K339 (encoding motor alone) can be detected along microtubules when cells are permeabilized briefly before fixation to release soluble protein (not shown) (Jaulin and Kreitzer, 2010) and when expressed at low levels (Fig. S1C). They also localize at the centrosome with γ-tubulin (Fig. S1D). Moreover, K370 and K339 localized prominently at cell–cell contacts in 59.7% and 54.7%, respectively, of the injected cells (Fig. 2A–C). The junctional localization of K339 and K370 is also observed in other epithelial cell types such as MCF10A and Caco-2, is independent of the tag identity (GFP, mCherry, myc, HA), and is seen with both N- and C-terminal fusion constructs (Fig. S1E and not shown) (Jaulin and Kreitzer, 2010). Together, this analysis reveals that the motor domain is sufficient to target KIF17 to cell–cell contacts and that deletion of the C-terminus favors this subcellular localization. Like endogenous KIF17, K370 and K339 colocalized with actin, E-cadherin and α-actinin at cell–cell contacts (Fig. 2B and not shown). We preferentially use K370 going forward since it behaves as a dimer in vitro. K339 behaves as a monomer in vitro (Acharya et al., 2013), but had nearly identical impact in all experiments where it was tested relative to K370.
Fig. 2.

Localization of expressed, GFP-tagged KIF17 constructs. (A) Diagram showing KIF17 constructs used for these studies. Images show localization of KIF17-FL, K339, K370 and K490 in MDCK cells 3 h after cDNA injection. Arrows indicate localization on microtubules in cell protrusions. Arrowheads indicate localization at cell–cell contacts. (B) Colocalization of GFP–K370 with immunostained E-cadherin and β-actin in MDCK cells. Color overlays show an enlargement of GFP–K370 and β-actin in the boxed region. In the lower overlay, the image of K370 was shifted by seven pixels. (C) Quantification of the junctional localization of endogenous KIF17 and expressed KIF17 constructs 3 h after cDNA injection. Values were calculated as percentage of total cells expressing each construct. Results are from 3–6 independent experiments (endogenous KIF17, n=90 cells; GFP–KIF17-FL, n=60; GFP–K490, n=295; GFP–K370, n=664; GFP–K339, n=794). (D) Quantification of the percentage cells with junctional GFP–K370 or GFP–K339 in the absence and presence of co-expressed mCh–KIF17-Tail. Data are normalized to 100% in control conditions (K370, n=162; K370+KIF17-Tail, n=121; K339, n=230; K339+KIF17-Tail, n=324 cells). Results are from ≥2 independent experiments. Error bars are error margins with 95% confidence interval. Significance was determined using a two-tailed unpaired student's t-test, **P<0.01; ***P<0.001.
Auto-inhibitory interactions of the KIF17 N-terminal motor and C-terminal tail domains regulate KIF17 activity (Espenel et al., 2013; Hammond et al., 2010; Jaulin and Kreitzer, 2010). In vitro, the KIF17 tail can bind directly to the KIF17 motor and reduces its microtubule-stimulated ATPase activity (Acharya et al., 2013); as such, it may influence K370 and K339 localization at the cortex. To test this directly, we co-expressed GFP–K339 or GFP–K370 with mCh–KIF17-Tail (Fig. 2A) and analyzed their localizations 3 h after cDNA injection. Co-expression of KIF17-Tail reduced the number of cells with junctional K339 and K370 to 45% and 33%, respectively, of cells expressing these motor domains alone (Fig. 2D). This effect of KIF17-Tail on localization of K339 and K370 could result from either competition with proteins that anchor KIF17 at cell–cell contacts, or by inhibition of the motor domain ATPase activity, which would prevent movement along microtubules (Acharya et al., 2013).
KIF17 motor domain enhances incorporation of actin at cell–cell contacts independently of microtubule binding
The colocalization of KIF17 motor domains with junctional actin, and the effects of KIF17 depletion on actin distribution in cells cultured in 3D prompted us to examine if expression of K370 affects actin organization. We expressed mCh–K370 or mCh–empty vector control (mCh–EV) for 4 h after cDNA injection and analyzed the distribution of actin by immunofluorescence microscopy. In K370-expressing cells, we observed a subtle but consistent enrichment of actin in discrete foci at cell–cell contacts. This enrichment was best detected by applying a Sobel edge detection filter to images (Fig. S2A). These actin foci were not detected by phalloidin labeling, probably because phalloidin strongly labels stress fibers and bundled actin. This can mask signal from non-bundled and branched filaments, which are detected very well with actin antibodies (Lessard, 1988; Nagasaki et al., 1994).
To further examine the change in junctional actin induced by K370, we monitored incorporation of newly synthesized, fluorescently-tagged actin probes (GFP–actin or mCh–LifeAct) into actin filaments by time-lapse fluorescence microscopy. We co-injected cells with GFP–actin and mCh–K370 or mCh–EV cDNAs. One hour after injection, cells were transferred to the microscope and images of GFP–actin were acquired at 10-min intervals for 4 h at 37°C. Newly synthesized GFP–actin (Fig. 3A) and mCh–LifeAct (Fig. S2B) accumulated in discrete foci along cell–cell contacts in control and K370-expressing cells. However, co-expression of K370 accelerated the rate at which these new filaments became apparent (Movie 1, Fig. S3) and increased the number of cells displaying these junctional actin filaments. Furthermore, K370 colocalized with GFP–actin or mCh–LifeAct in these junctional foci (Fig. 3A; Fig. S2B). In the time course of these experiments, fluorescently tagged actin incorporated into more resolvable foci at cell–cell contacts than mCh–LifeAct. For this reason, we used GFP–actin or mCh–actin in experiments going forward to determine how KIF17 can impact junctional actin organization.
Fig. 3.

K370 expression stimulates accumulation of junctional actin. (A) Localization of GFP–actin 4 h after co-injection with mCherry control, empty vector (mCh–EV) or mCh–K370 cDNAs. Insets in grayscale images show mCherry expression. Color overlay shows magnified region of the boxed area of cells shown in inset. mCh–K370 image was shifted by five pixels. (B) Localization of GFP–actin 4 h after co-injection with mCh–K370 or mCh–KIF3A-motor domain (mCh–KIF3A-M). Insets show mCherry expression. (C,D) Analysis of junctional actin accumulation. (C) Sample images of GFP–actin 4 h after cDNA injection. Dashed lines on the left panel highlight regions of interest (ROIs) at cell–cell contact zones within which segmentation was applied to identify junctional actin foci. The right panel shows the segmented image. Yellow puncta highlight the segmented regions within the selected ROI analyzed. Inset shows a magnified view of discrete junctional GFP–actin foci that are identified by segmentation. (D) Box-whisker plots showing quantification of junctional actin foci identified by segmentation as percentage of total ROI selected for measurement in each condition. Plots show minimum, 25th quartile, median, 75th quartile, and maximum values. Diamond symbols indicate outliers. Results are from images of injected cells in ≥3 independent experiments. Significance was determined using a two-tailed Mann–Whitney U test. ns, not significant; ***P<0.001; ****P<0.0001.
We quantified junctional accumulation of GFP–actin from images of cells by drawing polylines along cell–cell contacts of microinjected cells. These regions of interest (ROIs) were further processed and segmented to identify actin foci within these ROIs (Fig. 3C). We then measured the percentage of each ROI that was segmented as a readout of the junctional region covered by GFP–actin foci (% segmented area/total area measured, Fig. 3D) for control and experimental data. This analysis revealed a 4.9- and 5.5-fold increase in junctional GFP–actin foci in cells expressing mCh–K339 or mCh–K370, respectively, as compared with controls expressing mCh–EV. By contrast, junctional GFP–actin foci were not increased in cells expressing the heterodimeric kinesin-2 motor KIF3A (mCh–KIF3A-M, Fig. 3B,D) as compared with controls, demonstrating a selective effect of K370 or K339 on junctional actin accumulation in MDCK cells. The effects of expressing KIF17 constructs on actin in 3D cultured cells could not be determined as they induced substantial changes in cell shape over extended times needed for cysts to develop.
We next tested if the effects of KIF17 on cortical actin accumulation are microtubule dependent. We pre-incubated cells with 33 µM nocodazole to break down microtubules prior to injecting GFP–actin and K370 cDNAs. After an additional 4 h with continuous nocodazole exposure, cells were fixed and analyzed for accumulated GFP–actin at the cell periphery. In the absence of microtubules, K370 localized at cell–cell contacts and the accumulation of junctional GFP–actin was not significantly changed by comparison with untreated controls (Fig. 4A,C). Despite not reaching statistical significance, there was a trend toward increased junctional actin foci in nocodazole-treated cells. This could reflect release of RhoGEF family protein GEF-H1 (also known as ARHGEF2) from microtubules and activation of RhoA, which affects actin organization in many cell types (Krendel et al., 2002; Ren et al., 1998). Microtubule stabilization with 10 μM Taxol also had no significant effect on junctional actin accumulation induced by K370 expression (not shown). Junctional GFP–actin foci also increased 4.8-fold in cells expressing a K370 mutant defective in microtubule binding (Fig. 4B, K370R288/294A) (Acharya et al., 2013), as compared with empty vector control. Thus, the effects of K370 on cortical actin accumulation are independent of microtubules and microtubule binding.
Fig. 4.

Junctional actin accumulation induced by K370 is independent of microtubules. (A) Localization of mCh–actin, GFP–K370 and stable (detyrosinated) microtubules in untreated and nocodazole-treated (33 µM) cells. After injection, cells were maintained in nocodazole for 4 h at 37°C before fixation. (B) Localization of GFP–actin and the microtubule-binding mutant mCh–K370G288/294A 4 h after cDNA injection. (C) Box-whisker plots showing quantification of junctional actin foci identified by segmentation as percentage of total ROI selected for measurement in each experimental condition. Results are derived from images of injected cells in ≥2 independent experiments. Significance was determined using a two-tailed Mann–Whitney U test. ns, not significant.
Effects of KIF17 on junctional actin are mediated by activation of Rho signaling to ROCK and its downstream effectors LIMK1 and cofilin
Purified K370 and K339 did not interact directly with actin in vitro (not shown), suggesting KIF17 exerts its effects on junctional actin by modifying the localization or activity of actin regulatory factors. RhoA is involved in regulating both cortical actin dynamics and cortical microtubule capture and stabilization. To determine if RhoA signaling contributes to the effects of KIF17 on junctional actin, we co-injected MDCK cells with mCh–actin, GFP–K370 and either the Rho inhibitor Clostridium botulinum toxin C3 (myc–C3), the GDP-bound, inactive mutant RhoAN19 (myc–RhoAN19), or a control myc-empty vector (myc–EV). Rho inhibition by expression of myc–C3 or myc–RhoAN19 reduced the abundance of junctional GFP–actin foci 58.8-fold and 6.2-fold, respectively, relative to controls expressing K370, and was also reduced relative to controls expressing myc–EV, by 4 h after cDNA injection (Fig. 5A,C). We could not determine if constitutively activated RhoA (RhoAV14) increased accumulation of junctional GFP–actin foci because expression of this construct led to rapid disruption of cell–cell junctions (not shown).
Fig. 5.

RhoA signaling regulates junctional actin accumulation mediated by K370. (A) MDCK cells expressing mCh–actin, GFP–K370 and either myc–C3 or myc–RhoAN19 and fixed 4 h after cDNA injection. Insets show myc-immunostaining to detect expressed C3 and RhoAN19. (B) Localization of mCh–actin and GFP–K370 in untreated MDCK cells and in cells treated with Y27632 (10 µM) or SMIFH2 (50 µM). Inhibitors were added immediately after cDNA injection and cells were fixed after 4 h. (C) Box-whisker plots showing quantification of junctional actin foci identified by segmentation as a percentage of the total ROI selected for measurement in each experimental condition. Results are from images of injected cells in 2–4 independent experiments. Significance was determined using a two-tailed Mann–Whitney U test. (D) Immunoblots showing pull-down of GTP-bound RhoA with the Rho-binding domain of Rhotekin (RBD) and total RhoA in cells expressing the indicated constructs. Graph shows relative abundance of active GTP–RhoA in each condition. Error bars are s.e.m. Statistical significance was determined using one-way Anova and Bonferroni's multiple comparison test. (E) Immunoblots showing pull-down of GTP-bound RhoA in cells treated with the Rho–GTPase activator CNF1 (0.55 µg/ml for 90 min) or transduced with shNC, shKIF17#1 or shKIF17#2. Table shows relative abundance of GTP–RhoA pulled down under each condition. ns, not significant; *P<0.05; **P<0.01; ***P<0.001.
These data suggest KIF17 modifies junctional actin through RhoA signaling. To test if KIF17 activates RhoA, we determined the levels of active GTP–RhoA in cells expressing the different KIF17 constructs. We transfected MDCK cells with myc–EV, myc–K370, myc–KIF17-FL or the conformationally extended, constitutively active mutant myc–KIF17G754E (Espenel et al., 2013; Jaulin and Kreitzer, 2010). Cell lysates were prepared 24 h after transfection and levels of active and total RhoA were determined by pull-down with the Rho-binding domain of Rhotekin fused to 6xHis (His–RBD). Expression of all KIF17 constructs resulted in an increase in Rho activity, with a significant response in cells expressing K370 (Fig. 5D); thus the KIF17 motor domain is sufficient to activate RhoA in epithelial cells. Conversely, active RhoA was reduced to 53% and 24% of control levels in MDCK cells depleted of KIF17 using two independent shRNAs targeting KIF17 (Fig. 5E).
RhoA interacts with its effectors ROCK and diaphanous-related formins to regulate actin dynamics and distribution. As such, we tested if Rho signaling to ROCK or formin contributes to the effects of KIF17 on junctional actin. First, we treated cells with the ROCK inhibitor, Y27632 (10 μM) for 4 h immediately following injection of GFP–actin and mCh–K370 cDNAs. Similar to effects of expressing C3 or dominant-negative RhoAN19, accumulation of junctional actin foci induced by GFP–K370 expression was reduced 5.9-fold in cells treated with Y27632 (Fig. 5B,C). By contrast, abundance of junctional actin foci was not affected in cells treated with the formin inhibitor SMIFH2 (50 µM; Fig. 5B,C). Thus, K370 expression affects junctional actin accumulation through Rho signaling to ROCK.
ROCK activates LIMK1/2 and inhibits myosin light chain phosphatase, which modulate actin organization by phosphorylating cofilin and inhibiting dephosphorylation of myosin light chain, respectively. This results in inactivation of cofilin-mediated actin severing and activation of myosin-II-mediated actin contraction (Riento and Ridley, 2003). We tested the effects of activating cofilin or inhibiting myosin-II on KIF17-mediated accumulation of junctional actin. Co-expression of GFP–K370 and mCh–actin with either a kinase-dead LIMK1 construct (HA–LIMK1KD) that cannot phosphorylate and inactivate cofilin (Arber et al., 1998; Yang et al., 1998), or a constitutively active cofilin mutant (FLAG–cofilinS3A; Moriyama et al., 1996) reduced the median segmented membrane area covered by actin foci to 0.85% (8.3-fold decrease) and 1.66% (4.2-fold decrease), respectively, relative to 6.84% in controls expressing only K370. In these experiments however, we noted significant heterogeneity in the intensity and density of junctional actin foci across individual cells. This reflected a high variability in the expression levels of LIMK1KD or cofilinS3A in cells triply injected with K370 and mCh–actin cDNAs. As such, we also performed a binary phenotype analysis with these samples to determine the percentage of K370-expressing cells with resolvable mCh–actin foci at cell–cell contacts, regardless of fluorescence intensity or density of foci. In this binary analysis, the number of LIMK1KD- or cofilinS3A-expressing cells with resolvable junctional actin foci was reduced to 50.3% and 60.1% of controls expressing only K370 (Fig. 6A,C). By contrast, we measured no significant effect on either the percent membrane area covered by mCh–actin foci or the fraction of cells with resolvable junctional actin foci in K370-expressing cells treated with the myosin light chain kinase inhibitor ML-7 as compared with untreated controls (Fig. 6C).
Fig. 6.

K370 promotes junctional actin accumulation by inhibiting LIMK and/or cofilin-dependent actin severing. (A) Localization of mCh–actin and GFP–K370 in MDCK cells co-expressing HA–LIMK1 kinase-dead (LIMK1KD) or FLAG–cofilinS3A 4 h after cDNA injection. Insets show HA and FLAG immunostaining to detect expressed LIMK1KD and cofilinS3A. (B) Localization of mCh–actin and myc–K370 in cells co-expressing GFP–EV control or GFP–KIF17-Tail 4 h after cDNA injection. Inset shows GFP–KIF17-Tail in injected cells. GFP–EV was detected in living cells before fixation but was frequently lost during processing for immunofluorescence analysis; thus it is not shown here. (C) Graph showing percentage cells with accumulated junctional actin foci for each experimental condition indicated normalized to GFP–K370 or myc–K370 controls. Cells were injected with K370 and mCh–actin cDNAs alone or in combination with HA–LIMK1KD (n=194), Flag–cofilinS3A (n=160) or GFP–KIF17-Tail (n=60). CK666 (100 µM, n=92) and ML7 (10 µM, n=186) were added to cells immediately following cDNA injection until fixation 4 h later. Error bars are error margins with 95% confidence interval. Significance was determined using a two-tailed unpaired student's t-test. ns, not significant, *P<0.05; **P<0.01; ****P<0.0001.
We reasoned that if KIF17 inhibits actin severing by cofilin, then inhibiting branched actin polymerization mediated by Arp2/3, the major actin-nucleator at cell–cell junctions (Verma et al., 2012), should reduce K370-mediated accumulation of junctional GFP–actin foci. We incubated cells co-injected with GFP–actin and mCh–K370 cDNAs with 100 µM CK666, a selective Arp2/3 inhibitor (Nolen et al., 2009) for 4 h prior to fixation, and analyzed junctional GFP–actin foci. CK666 treatment reduced the median membrane area covered by GFP–actin foci 17-fold to 0.40% and the percentage of cells with junctional actin foci to 47% of untreated controls (Fig. 6A,C). CK666 similarly reduced K370 localized at cell–cell contacts (Fig. 6A), suggesting there is positive feedback between KIF17 junctional localization and enhanced accumulation of junctional actin foci. Inhibition of K370-induced accumulation of junctional GFP–actin foci by CK666 is consistent with studies showing that GFP–actin is incorporated preferentially into branched actin filaments by Arp2/3 (Chen et al., 2012). From these experiments, we conclude that K370 induces accumulation of junctional actin by activating RhoA signaling, leading to inhibition of cofilin-severing activity toward branched actin at cell–cell junctions.
KIF17 tail domain inhibits effects of K370 on cortical actin accumulation
The KIF17 C-terminal tail binds to the KIF17 motor domain directly and reduces the localization of K370 at cell–cell contacts (Fig. 2D). As such, it may also influence the effect of K370 on accumulation of junctional actin. Indeed, co-expression of mCh–KIF17-Tail with myc–K370 and GFP–actin reduced the median segmented membrane area covered by actin foci 1.9-fold (to 2.61%) relative to controls expressing mCh–EV, K370 and actin (4.82%). Co-expression of KIF17-Tail also reduced the percentage of cells with resolvable junctional actin foci to 30% of controls (Fig. 6B,C). This result suggests that KIF17-Tail inhibits the effects of K370 by preventing its junctional localization (Fig. 2D), or possibly, by competing with another factor that could modify actin dynamics. These data are also consistent with the more robust effects of the tail-less K370 over KIF17-FL on RhoA activation (Fig. 5D).
KIF17 is involved in regulating the distribution of E-cadherin and adhesion strength in response to calcium depletion
Actin associates with and anchors AJC components at the plasma membrane during formation, maturation and maintenance of cell–cell adhesions. When adhesions are remodeled, due to either experimental manipulation or in response to physiological cues, transmembrane components of the AJC such as E-cadherin are endocytosed, loosening junctions. Since KIF17 colocalizes with both actin and E-cadherin (Fig. 1A), we hypothesized that, by regulating junctional actin, KIF17 could affect AJC protein localization and junction stability or remodeling. To test this, we first expressed KIF17 constructs in MDCK cells and analyzed the distribution of E-cadherin by immunostaining cells fixed 4 h after cDNA microinjection. In uninjected cells, E-cadherin localized primarily at cell–cell contacts but was also seen in cytoplasmic vesicles. These vesicles likely represent a combination of E-cadherin transport intermediates in the biosynthetic, endocytic or recycling pathways. In cells expressing K370 or K339, we observed a reduction in the number of cytoplasmic vesicles containing E-cadherin (Fig. 7A and not shown). We quantified the number of cytoplasmic E-cadherin vesicles in individual cells and measured a 3.9-fold decrease in the median number of intracellular E-cadherin puncta in cells expressing KIF17 constructs as compared with uninjected controls (Fig. 7C). By contrast, we measured a 2.53-fold increase in the median number of cytoplasmic E-cadherin puncta in KIF17-depleted cells (shKIF17) as compared with shNC controls (Fig. 7B,C). We did not detect a difference in the levels of E-cadherin or actin by immunoblot in KIF17-depleted cells (Fig. 7D) suggesting that KIF17 acts on E-cadherin by modifying its subcellular distribution.
Fig. 7.
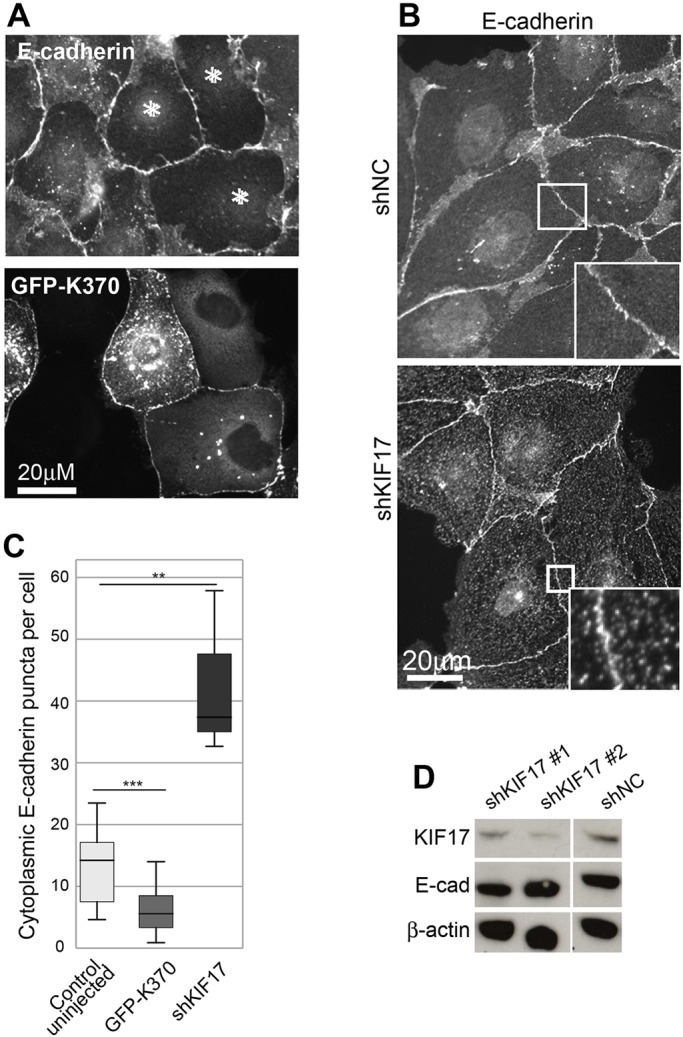
KIF17 regulates the distribution of E-cadherin. (A) Localization of E-cadherin in MDCK cells expressing GFP–K370 or GFP–KIF17-FL and fixed 4 h after cDNA injection. Asterisks mark injected cells. (B) Localization of E-cadherin in cells transduced with shNC or shKIF17. Boxed regions are magnified in insets. (C) Box-whisker plots showing quantification of cytoplasmic E-cadherin puncta in uninjected controls and in cells expressing GFP–K370 and in KIF17-depleted cells (shKIF17). Results are from images of injected cells in 2–4 independent experiments. Significance was determined using a two-tailed Mann–Whitney U test. **P<0.01; ***P<0.001. (D) Immunoblots showing KIF17, E-cadherin and β-actin in MDCK cells transduced with shNC, shKIF17#1 or shKIF17#2.
In a fluorescence pulse-chase assay to monitor biosynthetic trafficking of newly synthesized membrane proteins (Kreitzer et al., 2000), we did not measure any change in the kinetics of GFP–E-cadherin export from the Golgi or its delivery to the plasma membrane when we co-expressed either K339 or KIF17-Tail as compared with cells expressing only GFP–E-cadherin (not shown) (Jaulin et al., 2007). This suggests clearance of E-cadherin puncta in K370-expressing cells and increased abundance of E-cadherin puncta in KIF17-depleted cells result from changes in E-cadherin endocytosis or recycling. To determine if KIF17 affects E-cadherin distribution selectively, or if it also affects the internalization of membrane proteins not associated with the AJC, we loaded transferrin receptors (TfR) with Cy3–transferrin (Cy3–Tf; 25 µg/ml) and monitored TfR endocytosis. Cells were incubated with Cy3–Tf on ice for 60 min and then warmed to 37°C for 30 min and fixed for image acquisition. In these experiments, we detected no difference in TfR internalization in cells expressing GFP–K370 as compared with GFP–EV controls (Fig. S4A), showing that KIF17 does not broadly inhibit endocytosis of plasma membrane proteins.
We next determined if the effects of KIF17 perturbation on E-cadherin distribution and accumulation of junctional actin foci induced by KIF17 perturbations reflect changes in cellular function by analyzing the strength of cell–cell adhesions when challenged by calcium depletion. We expressed K370 or the control KIF3A-M (Jaulin et al., 2007) and incubated cells in media containing 1.5 mM EDTA, beginning 4 h after cDNA injection. Cells were imaged by time-lapse microscopy for 90 min following addition of EDTA. By 60 min, most of the uninjected or KIF3A-M-expressing cells detached from neighbors and had rounded up from the coverslip. Conversely, the majority of cells expressing K370 or K339 remained adhered to each other and did not exhibit significant rounding (Fig. 8A; Fig. S4B). Furthermore, E-cadherin, as well as ZO-1 and γ-catenin, were retained at cell–cell contacts in K370- or K339-expressing cells treated with EDTA (Fig. S4C). Thus, expression of K370 or K339 inhibits internalization of AJC components, attenuating cellular responses to triggers that reduce adhesiveness of cell–cell contacts. Considered together, these data support a model in which KIF17 contributes to regulation of cell–cell junction remodeling by activating RhoA signaling to reduce cofilin-mediated severing of junctional actin (Fig. 8B). We speculate that by shifting the balance between actin polymerization and severing, KIF17 reduces the internalization of AJC proteins and thereby increases the strength of cell–cell adhesions.
Fig. 8.

K370 strengthens cell–cell adhesions to challenge with calcium chelators. (A) Phase-contrast and fluorescence images showing first and last frames from a time-lapse recording of MDCK cells incubated with 1.5 mM EDTA. EDTA was added 4 h after cDNA injection and cells were imaged at 1-min intervals. Upper panels show cells expressing GFP–K370. Lower panels show cells expressing GFP–KIF3A-M. Insets show GFP fluorescence of expressed constructs at the start of the recordings. Outlined regions on phase-contrast images show the area encompassed by GFP–KIF-expressing cells. Fluorescent images on the far right show GFP–KIFs at the last frame of the time-lapse. (B) Model for KIF17 function at the AJC of epithelial cells: (1) KIF17 associates with the AJC, where it colocalizes with E-cadherin and junctional actin. (2) The KIF17 motor domain is sufficient for junctional localization and triggers activation of a RhoA signaling pathway leading to inhibition of cofilin. (3) Decreased cofilin severing activity allows for the accumulation of junctional actin, alters E-cadherin trafficking and impairs junction dissociation in response to calcium depletion. (4) KIF17 also regulates microtubule stabilization (Jaulin and Kreitzer, 2010), likely downstream of RhoA signaling. (5) KIF17 functions at the AJC and on microtubules are inhibited by intramolecular interactions between the motor and tail domains.
DISCUSSION
The data presented here provide evidence that KIF17 contributes to regulation of branched actin stability at cell–cell contacts, stabilization of E-cadherin at the plasma membrane, and to intercellular adhesion strength. Remarkably, these functions are dependent on the kinesin motor domain but independent of microtubules. In concert with EB1 and APC, components of the microtubule plus-end capture machinery, KIF17 also promotes microtubule stabilization in epithelial cells and is sufficient to stabilize microtubules in vitro (Acharya et al., 2013; Espenel et al., 2013; Jaulin and Kreitzer, 2010). Considering that KIF17 depletion also compromises apical actin recruitment and lumen formation in 3D culture (Fig. 1) (Jaulin and Kreitzer, 2010), our findings suggest KIF17 plays a central role in coordinating actin and microtubule remodeling with formation and remodeling of cell–cell junctions to promote morphogenesis and epithelial polarization.
During expansion of primordial cell–cell contacts, distinct arrays of branched and unbranched actin associate with E-cadherin as spot junctions are remodeled into mature, junctional complexes at the apicolateral membrane domain of polarized cells. Experiments monitoring actin incorporation by FRAP showed that 80–90% of filaments are very dynamic (Yamada et al., 2005; Kovacs et al., 2011) and are generated by Arp2/3-dependent branched actin nucleation (Kovacs et al., 2002; Otani et al., 2006; Tang and Brieher, 2012). This is consistent with our data in MDCK cells showing that the accumulation of GFP–actin at cell–cell contacts is attenuated by inhibiting Arp2/3. Although circumferential, formin-dependent unbranched actin arrays contribute to maturation of adherens junctions, they do not appear to be regulated by KIF17, and are likely utilized downstream of the initial establishment of cell–cell junctions. Formation and organization of branched actin and actin cables are regulated by a combination of actin nucleation, elongation and severing activities coordinated by junction-associated Rho–GTPases, their regulators and effectors; these concentrate E-cadherin at AJCs during polarization and modulate junction assembly and maintenance (Citi et al., 2014; Mack and Georgiou, 2014). As such, the effects of KIF17 on cortical actin and intercellular junctions can be attributed, at least in part, to activation of RhoA signaling as levels of active RhoA are increased by expression of KIF17 constructs and reduced by KIF17 knockdown. Although Rho has no reported role in regulating nucleation of branched actin filaments, signaling to its effector ROCK activates LIMK1, which then phosphorylates and inhibits cofilin-mediated severing of branched actin. This would be expected to shift the balance between actin polymerization and depolymerization at cell–cell contacts. In support of a role for KIF17 in this pathway, we found that pharmacological inhibitors of ROCK, or expression of either kinase-dead LIMK1 or a cofilin phospho-mimic, inhibited the effect of KIF17 expression on accumulation of junctional actin.
KIF17 may have additional functions in regulating establishment and remodeling of the AJC. Microtubule capture and cortical stabilization by KIF17 (Jaulin and Kreitzer, 2010) could generate specialized tracks comprising post-translationally modified, stable microtubules, for targeted delivery of cytoplasmic and membrane proteins important for junction formation and remodeling (Waterman-Storer et al., 2000). However, neither microtubule depolymerization nor expression of a K370 mutant that cannot bind microtubules (K370R288/294A) impinges on the ability of K370 to induce accumulation of junctional actin foci. Based on this, we believe that the effects of KIF17 on junctional actin are independent of its effects on microtubules.
In contacting, but not yet polarized cells, microtubules are organized primarily in radial arrays emanating from the MTOC, with a subset of plus-ends localizing in close proximity to the developing AJC. Kinesin-mediated transport on microtubules is used to both deliver and retrieve cadherin and other adhesion components to and from the plasma membrane (Chen et al., 2003; Ivanov et al., 2006; Krylyshkina et al., 2002; Mary et al., 2002; Nekrasova et al., 2011; Portereiko et al., 2004; Yanagisawa et al., 2004). Dynamic microtubule plus-ends, where KIF17 localizes with EB1 and APC (Jaulin and Kreitzer, 2010), can interact with proteins at the cortex and deposit microtubule plus-end-associated proteins that regulate cytoskeletal and junctional organization, such as APC, leading to the concentration of E-cadherin at cell–cell contacts (Ligon and Holzbaur, 2007; Ligon et al., 2001; Stehbens et al., 2006). We show here that KIF17 overexpression clears E-cadherin-containing vesicles from the cytoplasm, and conversely, that KIF17 depletion results in an increase in cytoplasmic E-cadherin. As there is no evidence implicating KIF17 in biosynthetic trafficking of E-cadherin, we speculate that by modifying cortical actin through RhoA signaling, KIF17 stabilizes E-cadherin in the plasma membrane and affects junction stability by enhancing anchorage of AJC proteins to the underlying cortical cytoskeleton. This would be expected to attenuate endocytosis of E-cadherin and AJC components both at steady-state, and in response to signals that induce junction remodeling and is consistent with our data showing that internalization of E-cadherin induced by calcium depletion is blocked by expression of K370 and K339 motor domain constructs.
We do not yet fully understand how KIF17 and K370 activate RhoA signaling to promote junctional actin accumulation and stabilization of cell–cell adhesions. KIF17 may activate RhoGEFs, or inhibit RhoGAPs (also known as ARHGAPs), to maintain high levels of active RhoA, either on microtubule plus-ends or at the cortex when it contacts the plasma membrane. In one possible scenario, the KIF17 motor could interact with a RhoGEF and deliver it to the cortex, where it would be activated when off-loaded from microtubules (Enomoto, 1996). Candidates for activating RhoA signaling for microtubule stabilization and/or remodeling of the AJC include GEF-H1, p115-RhoGEF and ECT2. The KIF17 motor domain does interact with several cytoplasmic proteins (our unpublished data and Jaulin and Kreitzer, 2010), thus, it is reasonable to suspect additional protein interactions could occur in this domain. Microtubule capture, stabilization, and the subsequent accumulation of post-translationally modified microtubules by KIF17 could also trigger a change in the captured microtubule that induces local GEF release from the lattice of that microtubule. Indeed, inactive GEF-H1 localizes preferentially on dynamic, unmodified microtubules and is not seen on stable, acetylated or detyrosinated microtubules (Nagae et al., 2013; Yoshimura and Miki, 2011). Because KIF17 is activated by PKC (Espenel et al., 2013), which contributes to RhoA-dependent microtubule stabilization in fibroblasts (Wen et al., 2004), we also envision a model wherein feedback signaling could amplify microtubule capture in response to initial Rho activation events at cell–cell contacts (Fig. 8B).
The effects of full-length KIF17 on junctional actin and RhoA activation are less robust than that of the motor domain alone, suggesting the KIF17 tail domain is a negative regulator of these KIF17 activities. In support of this, we showed previously that KIF17 tail interacts directly with the motor domain, decreasing its ATPase activity (Acharya et al., 2013; Espenel et al., 2013), and we show here that KIF17 tail expression abrogates accumulation of junctional actin induced by expression of K370. Our localization studies suggest that the KIF17 tail competes with a factor(s) that anchors KIF17 at cell–cell junctions, in line with our previous demonstration that the KIF17 tail domain competes with EB1 for binding to the KIF17 head (Acharya et al., 2013). Alternatively, the KIF17 tail could interfere with the motor-dependent activation of RhoA signaling by carrying a cargo that acts as a negative regulator of RhoA. A precedent for regulation of a RhoGEF and a RhoGAP by a single kinesin has been reported, although the mechanism of action may differ somewhat from that of KIF17. MKLP1 (KIF23, kinesin-6 family), a component of the centralspindlin complex, affects microtubule and actin arrays, formation of adherens junctions, and polarization of foregut epithelia in Caenorhabditis elegans through an interaction with CYK4−RhoGAP (Portereiko et al., 2004). CYK4−RhoGAP induces a conformational change in MKLP1 leading to activation of RhoA (Saade et al., 2007; Yamamoto et al., 2006). MKLP1, as part of the centralspindlin complex, also binds and recruits the RhoGEF ECT2 to cell–cell junctions, and inhibits junctional localization of p190 RhoGAP in MCF-7 cells, leading to activation of RhoA (Ratheesh et al., 2012). These data support the idea that a kinesin can influence the activities of both RhoGEFs and RhoGAPs at cell–cell junctions, fine-tuning local RhoA signaling.
Mounting evidence shows an interdependence between signaling events at the cortex of mammalian cells with cytoskeletal dynamics and organization that lead to cell polarization (Gundersen, 2002a,b; Siegrist and Doe, 2007). An emerging theme is that protein delivery to and retrieval from the cortex can influence cytoskeletal dynamics and organization. Remodeling of actin and microtubule arrays is mediated by an overlapping set of effectors that respond to cortical stimuli (Bartolini et al., 2008; Gundersen et al., 2004; Mikhailov and Gundersen, 1998; Tatebe et al., 2008; Watanabe et al., 2004). Both focal adhesions and the AJC are sites of microtubule plus-ends targeting and where cytoskeletal dynamics may be regulated locally (Chausovsky et al., 2000; Efimov et al., 2008; Ezratty et al., 2005; Waterman-Storer et al., 2000). These cortical adhesions are also sites of active membrane recycling and kinesin-dependent delivery and retrieval of transmembrane and membrane–cytoskeleton linkers (Chen et al., 2003; Ivanov et al., 2006; Krylyshkina et al., 2002). The effects of KIF17 on RhoA activity, actin and microtubule arrays, and on stability of the AJC lend support to the idea that KIF17 plays an important role in coordinating formation of nascent cell–cell adhesions with remodeling of actin and microtubules to initiate morphological polarization of epithelial cells.
MATERIALS AND METHODS
Cell culture and treatments
Madin–Darby canine kidney (MDCK) cells were cultured in DMEM (4.5 g/l glucose) with 5% FBS, 20 mM HEPEs, pH 7.2, and were tested for mycoplasma contamination prior to initiating these studies. MCF10A cells were purchased from ATCC (CRL-10317). MDCK cells were seeded on sterilized coverslips and used at ∼80% confluence 2–3 days after plating. For 3D cultures, cells were infected with lentiviral shRNAs and allowed to grow for 2–4 days. Cells were then trypsinized and suspended in complete medium containing 2% Matrigel™ (BD Biosciences) and layered onto Matrigel at 104 cells/well in 8-well chamber slides (Lab-Tek) as described (O'Brien et al., 2006). Cysts were fixed after 7 days in culture in 4% PFA for 30 min and permeabilized with 0.5% Triton X-100 for 15 min before immunostaining.
Microtubules were depolymerized completely by incubating cells on an ice-slurry in bicarbonate-free, complete media supplemented with 33 µM nocodazole (Sigma) for 30 min followed by incubation at 37°C for an additional 30 min prior to microinjection. Cells were maintained in 33 µM nocodazole during microinjections and subsequently transferred back into bicarbonate-containing complete media with 33 µM nocodazole. Cells were fixed 4 h after cDNA injection and processed for immunofluorescence analysis and imaging. Inhibitors of Arp2/3 (CK666, 100 µM), ROCK (Y27632, 10 µM), myosin light chain kinase (ML-7, 10 µM) and formins (SMIFH2, 50 µM) (Sigma), were added to cells immediately following cDNA injections.
Immunofluorescence staining and immunoblot analysis
Cells were fixed in −20°C methanol for 1–2 min or in 2% paraformaldehyde for 5 min at room temperature followed by permeabilization in either PBS-CM (PBS with 100 μM CaCl2, 1 mM MgCl2) with 0.1% Triton X-100 for 2 min or in −20°C methanol (for immunolabeling of endogenous actin). KIF17 was detected with a rabbit polyclonal antibody at 1:100 dilution described and characterized previously (Jaulin and Kreitzer, 2010). Endogenous actin was detected by indirect immunofluorescence with mouse anti-β-actin ascites fluid diluted 1:200 (clone AC-74, Sigma). Other antibodies: rat anti-E-cadherin (1:200; DECMA, Sigma), mouse anti-α-actinin (1:200; clone BM75.2, Sigma) mouse-anti-myc tag (1:200; 9B11, Sigma), mouse anti-FLAG M2 tag (1:100; F3165, Sigma), mouse anti-HA tag (1:100 dilution; MMS-101P, Covance), rabbit-anti-detyrosinated tubulin [1:400; SG, provided by Gregg Gundersen, Columbia University, USA (Gundersen et al., 1984)]. Fluorescently conjugated secondary antibodies raised in donkey and cross-adsorbed against related species were from Jackson Immunoresearch. Antibodies for immunoblots are: rabbit anti-KIF17 (1:1000; K3638, Sigma), mouse anti-E-cadherin (1:500; 610181, BD Biosciences) and mouse anti-β-actin (1:5,000; clone AC-74, A5316, Sigma).
Expression constructs
KIF17 constructs used for this study were amplified by PCR from human A549 or Caco2 cells and cloned into Gateway expression vectors (Invitrogen) as recommended by the manufacturer and as described previously (Jaulin and Kreitzer, 2010; Acharya et al., 2013). Myc–C3, GFP–RhoA WT, myc–RhoAV14, and myc–RhoAN19 were generous gifts from Dr Alan Hall (Memorial Sloan Kettering Cancer Center, NY, USA). HA-LIMK1KD and FLAG-cofilinS3A (Salvarezza et al., 2009) were provided by Enrique Rodriguez-Boulan (Weill Cornell Medical College, NY, USA).
Protein expression
Exogenous proteins were expressed by intranuclear injection of cDNAs, either individually or together (10–50 µg/ml for each plasmid), in HKCl (10 mM HEPEs, 140 mM KCl, pH 7.4) using a Narishige micromanipulator with back-loaded capillary glass needles. Protein expressed from injected cDNAs could be detected after incubation at 37°C for 60–90 min. Cells were fixed at indicated times after injection and processed for direct fluorescence or indirect immunofluorescence microscopy.
Protein knock-down
pGIPZ lentiviral shRNAs targeting KIF17 and pLKO.1 lentiviral control plasmid (Open Biosystems) were prepared and introduced by viral transduction as described previously (Jaulin and Kreitzer, 2010).
Fixed cell imaging and analysis
Fixed cell images were acquired on either a Nikon E400 Eclipse or a Nikon TiE using a 40× (NA 1.0) plan-apochromat oil immersion objective and collected with digital charge-coupled device cameras (ORCA II-ER, 6.45 μm pixels, 1 MHz for 14-bit images, Hamamatsu Photonics; or Neo sCMOS, 6.45 µm pixels, 200 MHz for 16-bit images, Andor Technology). 14–16-bit images were scaled linearly to illustrate features of interest as indicated in the Results and converted to 8-bit copies for figure assembly. Devices were controlled by either MetaMorph (Molecular Devices, Inc.) or Elements (Nikon Instruments). Post-acquisition analysis and processing were performed using MetaMorph. Images of 3D cysts were acquired with a Zeiss LSM510 scanning confocal microscope (Rockefeller University Bioimaging Resource Center).
Time-lapse imaging
After microinjection, cells were transferred to recording medium (Hanks’ balanced salt solution with 20 mM HEPES, 1% FBS, 4.5 g/l glucose, essential and non-essential amino acids) and incubated at 37°C in a thermal-controlled chamber (Harvard Apparatus) on a TE-2000U (Nikon). Time-lapse images were acquired using a 20× (NA 0.5) plan-fluor, phase contrast, dry objective and collected with a Neo sCMOS camera (6.45 µm pixels, 200 MHz for 16-bit images, Andor Technology). 14–16-bit images were scaled to illustrate features of interest and converted to 8-bit copies for figure assembly. Devices were controlled with Elements software (Nikon Instruments). Post-acquisition analysis and processing were performed with MetaMorph.
Quantitative image analysis
Image processing and analysis was performed using Python scripts using scikit-image (van der Walt et al., 2014) and OpenCV (Bradski, 2000) packages, as well as Fiji software (Schindelin et al., 2012). All source codes developed are open source and freely available at https://github.com/cespenel/image_processing.
Analysis of junctional actin foci
We developed an image segmentation method called, ‘membrane_accumulation’. Briefly, a polyline with a thickness of 4 pixels (∼700 nm) was drawn following the membrane at sites of cell–cell contact. These polylines were used as regions of interest (ROIs), averaging ∼2200 pixels2/cell, inside which the following processing was performed. Images were smoothed using a median blur filter of 5 and then images were convolved with a 3×3 Laplacian kernel with values from −1 to 8. Images were further smoothed using a median blur. For segmentation of actin foci, we use a marker-controlled watershed with two seed points (or markers), one given by an Otsu threshold and a second by 30% of the value given by the Otsu method. From these thresholded images, we measured the percentage of the ROI that was segmented.
Analysis of cytoplasmic E-cadherin puncta
Two methods were developed. The first, ‘blobs per cell’, was used for analysis of KIF17-depleted cells wherein the number of cells was calculated automatically based on the number of DAPI-stained nuclei. The second, ‘blobs_per_cell_click’, was used for analysis of cells expressing microinjected KIF17 constructs wherein we extracted the number of blobs (puncta) in injected cells and uninjected cells. We then created a mask on the injected cells images, and applied this mask to the corresponding images of E-cadherin. We then determined the number of puncta in the masked (injected cells) and non-masked (uninjected cells) areas. All images were first convolved with a 3×3 Laplacian kernel with values from −1 to 8 and then a Gaussian filter with a sigma value of 3 was applied. Blobs/puncta were identified using the difference of Gaussians approach above a defined Otsu threshold.
Graphs and statistical analysis of junctional actin foci and E-cadherin puncta
Boxplots were generated using the 2D python-plotting library Matplotlib (Hunter, 2007) and show minima, 25% quartile, median, 75th quartile, and maxima. Statistical tests were performed using a two-tailed Mann–Whitney U test (Python library SciPy, scipy.stats.mannwhitneyu). Statistical significance is defined as: ns, not significant (P>0.05); *P<0.05; **P<0.01; ***P<0.001 and ****P<0.0001.
Statistics for binary analysis
Statistical significance was determined by two-tailed unpaired Student's t-test unless noted otherwise in the figure legends. Data are presented as mean±s.e.m. for quantitative values and error margins for percentages (95% level of confidence). Sample size (n) and P-values are specified in the text or figure legends. Data were collected from at least three independent experiments.
Rho–GTP binding assay
Rhotekin-RBD was purchased from Cytoskeleton (Denver, CO) and RhoA–GTP binding was performed as recommended by the manufacturer. Briefly, MDCK were transfected with indicated constructs. After 24 h, cells were washed in PBS and lysed in 25 mM HEPES pH 7.5, 150 mM NaCl, 1% NP-40 (Igepal CA-630), 10 mM MgCl2, 1 mM EDTA and 10% glycerol, 10 µg/ml leupeptin, 10 µg/ml pepstatin, and 10 µg/ml aprotinin. For assays in KIF17-depleted cells, cells were also treated with RhoA activator, cytotoxic necrotizing factor 1 (CNF1, 55 µg/ml) as a positive control. Lysates were clarified by centrifugation at 13,000 g at 4°C for 1 min. Clarified lysates (200 µg) were divided in two; one to detect total RhoA, and one for use in pull-downs. 100 µg of lysate was incubated with Rhotekin–RBD protein beads (50 µg) at 4°C for 90 min. The beads were collected by centrifugation, washed thoroughly and resuspended in 2× Laemmeli buffer. Input samples and collected beads were analyzed by western blot using a RhoA-specific antibody. Densitometry was performed using ImageJ (NIH). The amount of RBD-bound RhoA was normalized to total RhoA in cell lysates for comparison of Rho activity (level of GTP-bound Rho) across samples.
Acknowledgements
We thank Alan Hall (Memorial Sloan Kettering Cancer Center, NY, USA) and Enrique Rodriguez-Boulan (Weill Cornell Medical College, NY, USA) for providing plasmids.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
B.R.A., C.E., F.L., J.R., J.M., F.J. and G.K. performed experiments. C.E. developed algorithms for quantitative image analysis. F.J. and G.K. conceived the project and wrote the manuscript.
Funding
This work was supported by grants from the National Institutes of Health [R01GM087575] and the Irma T. Hirschl Trust to G.K., and from the Centre national de la recherche scientifique (ATIP-AVENIR program) and the Gustave Roussy Foundation to F.J. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.173674/-/DC1
References
- Acharya B. R., Espenel C. and Kreitzer G. (2013). Direct regulation of microtubule dynamics by KIF17 motor and tail domains. J. Biol. Chem. 288, 32302-32313. 10.1074/jbc.M113.494989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S., Barbayannis F. A., Hanser H., Schneider C., Stanyon C. A., Bernard O. and Caroni P. (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805-809. 10.1038/31729 [DOI] [PubMed] [Google Scholar]
- Bartolini F., Moseley J. B., Schmoranzer J., Cassimeris L., Goode B. L. and Gundersen G. G. (2008). The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 181, 523-536. 10.1083/jcb.200709029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradski G. (2000). The OpenCV library. Dr Dobb's J. 25, 120-125. [Google Scholar]
- Brieher W. M. and Yap A. S. (2013). Cadherin junctions and their cytoskeleton(s). Curr. Opin. Cell Biol. 25, 39-46. 10.1016/j.ceb.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Carramusa L., Ballestrem C., Zilberman Y. and Bershadsky A. D. (2007). Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J. Cell Sci. 120, 3870-3882. 10.1242/jcs.014365 [DOI] [PubMed] [Google Scholar]
- Chausovsky A., Bershadsky A. D. and Borisy G. G. (2000). Cadherin-mediated regulation of microtubule dynamics. Nat. Cell Biol. 2, 797-804. 10.1038/35041037 [DOI] [PubMed] [Google Scholar]
- Chen X., Kojima S., Borisy G. G. and Green K. J. (2003). p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J. Cell Biol. 163, 547-557. 10.1083/jcb.200305137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Nag S. and Pollard T. D. (2012). Formins filter modified actin subunits during processive elongation. J. Struct. Biol. 177, 32-39. 10.1016/j.jsb.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennathukuzhi V., Morales C. R., El-Alfy M. and Hecht N. B. (2003). The kinesin KIF17b and RNA-binding protein TB-RBP transport specific cAMP-responsive element modulator-regulated mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 100, 15566-15571. 10.1073/pnas.2536695100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu P.-J., Rivera J. F. and Arnold D. B. (2006). A role for Kif17 in transport of Kv4.2. J. Biol. Chem. 281, 365-373. 10.1074/jbc.M508897200 [DOI] [PubMed] [Google Scholar]
- Citi S., Guerrera D., Spadaro D. and Shah J. (2014). Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases 5, e973760 10.4161/21541248.2014.973760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T. A., Nagasaki T. and Gundersen G. G. (1998). Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J. Cell Biol. 141, 175-185. 10.1083/jcb.141.1.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishinger J. F., Kee H. L., Jenkins P. M., Fan S., Hurd T. W., Hammond J. W., Truong Y. N.-T., Margolis B., Martens J. R. and Verhey K. J. (2010). Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat. Cell Biol. 12, 703-710. 10.1038/ncb2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A., Schiefermeier N., Grigoriev I., Ohi R., Brown M. C., Turner C. E., Small J. V. and Kaverina I. (2008). Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J. Cell Sci. 121, 196-204. 10.1242/jcs.012666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T. (1996). Microtubule disruption induces the formation of actin stress fibers and focal adhesions in cultured cells: possible involvement of the rho signal cascade. Cell Struct. Funct. 21, 317-326. 10.1247/csf.21.317 [DOI] [PubMed] [Google Scholar]
- Espenel C., Acharya B. R. and Kreitzer G. (2013). A biosensor of local kinesin activity reveals roles of PKC and EB1 in KIF17 activation. J. Cell Biol. 203, 445-455. 10.1083/jcb.201305023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty E. J., Partridge M. A. and Gundersen G. G. (2005). Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 7, 581-590. 10.1038/ncb1262 [DOI] [PubMed] [Google Scholar]
- Fan S., Whiteman E. L., Hurd T. W., McIntyre J. C., Dishinger J. F., Liu C. J., Martens J. R., Verhey K. J., Sajjan U. and Margolis B. (2011). Induction of Ran GTP drives ciliogenesis. Mol. Biol. Cell 22, 4539-4548. 10.1091/mbc.E11-03-0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M. and Kaibuchi K. (2001). Rho-family GTPases in cadherin-mediated cell–cell adhesion. Nat. Rev. Mol. Cell Biol. 2, 887-897. 10.1038/35103068 [DOI] [PubMed] [Google Scholar]
- Guillot C. and Lecuit T. (2013). Mechanics of epithelial tissue homeostasis and morphogenesis. Science 340, 1185-1189. 10.1126/science.1235249 [DOI] [PubMed] [Google Scholar]
- Gundersen G. G. (2002a). Evolutionary conservation of microtubule-capture mechanisms. Nat. Rev. Mol. Cell Biol. 3, 296-304. 10.1038/nrm777 [DOI] [PubMed] [Google Scholar]
- Gundersen G. G. (2002b). Microtubule capture: IQGAP and CLIP-170 expand the repertoire. Curr. Biol. 12, R645-R647. 10.1016/S0960-9822(02)01156-9 [DOI] [PubMed] [Google Scholar]
- Gundersen G. G., Kalnoski M. H. and Bulinski J. C. (1984). Distinct populations of microtubules: tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell 38, 779-789. 10.1016/0092-8674(84)90273-3 [DOI] [PubMed] [Google Scholar]
- Gundersen G. G., Gomes E. R. and Wen Y. (2004). Cortical control of microtubule stability and polarization. Curr. Opin. Cell Biol. 16, 106-112. 10.1016/j.ceb.2003.11.010 [DOI] [PubMed] [Google Scholar]
- Hammond J. W., Blasius T. L., Soppina V., Cai D. and Verhey K. J. (2010). Autoinhibition of the kinesin-2 motor KIF17 via dual intramolecular mechanisms. J. Cell Biol. 189, 1013-1025. 10.1083/jcb.201001057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. D. (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90-95. 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- Insinna C., Pathak N., Perkins B., Drummond I. and Besharse J. C. (2008). The homodimeric kinesin, Kif17, is essential for vertebrate photoreceptor sensory outer segment development. Dev. Biol. 316, 160-170. 10.1016/j.ydbio.2008.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I., McCall I. C., Babbin B., Samarin S. N., Nusrat A. and Parkos C. A. (2006). Microtubules regulate disassembly of epithelial apical junctions. BMC Cell Biol. 7, 12 10.1186/1471-2121-7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaulin F. and Kreitzer G. (2010). KIF17 stabilizes microtubules and contributes to epithelial morphogenesis by acting at MT plus ends with EB1 and APC. J. Cell Biol. 190, 443-460. 10.1083/jcb.201006044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaulin F., Xue X., Rodriguez-Boulan E. and Kreitzer G. (2007). Polarization-dependent selective transport to the apical membrane by KIF5B in MDCK cells. Dev. Cell 13, 511-522. 10.1016/j.devcel.2007.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins P. M., Hurd T. W., Zhang L., McEwen D. P., Brown R. L., Margolis B., Verhey K. J. and Martens J. R. (2006). Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Curr. Biol. 16, 1211-1216. 10.1016/j.cub.2006.04.034 [DOI] [PubMed] [Google Scholar]
- Kher S. S., Struckhoff A. P., Alberts A. S. and Worthylake R. A. (2014). A novel role for p115RhoGEF in regulation of epithelial plasticity. PLoS ONE 9, e85409 10.1371/journal.pone.0085409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak A., Pasolli H. A. and Fuchs E. (2004). Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat. Cell Biol. 6, 21-30. 10.1038/ncb1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaja N., Macho B. and Sassone-Corsi P. (2005). Microtubule-independent and protein kinase A-mediated function of kinesin KIF17b controls the intracellular transport of activator of CREM in testis (ACT). J. Biol. Chem. 280, 31739-31745. 10.1074/jbc.M505971200 [DOI] [PubMed] [Google Scholar]
- Kotaja N., Lin H., Parvinen M. and Sassone-Corsi P. (2006). Interplay of PIWI/Argonaute protein MIWI and kinesin KIF17b in chromatoid bodies of male germ cells. J. Cell Sci. 119, 2819-2825. 10.1242/jcs.03022 [DOI] [PubMed] [Google Scholar]
- Kovacs E. M., Goodwin M., Ali R. G., Paterson A. D. and Yap A. S. (2002). Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr. Biol. 12, 379-382. 10.1016/S0960-9822(02)00661-9 [DOI] [PubMed] [Google Scholar]
- Kovacs E. M., Verma S., Ali R. G., Ratheesh A., Hamilton N. A., Akhmanova A. and Yap A. S. (2011). N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat. Cell Biol. 13, 934-943. 10.1038/ncb2290 [DOI] [PubMed] [Google Scholar]
- Kraemer A., Goodwin M., Verma S., Yap A. S. and Ali R. G. (2007). Rac is a dominant regulator of cadherin-directed actin assembly that is activated by adhesive ligation independently of Tiam1. Am. J. Physiol. Cell Physiol. 292, C1061-C1069. 10.1152/ajpcell.00073.2006 [DOI] [PubMed] [Google Scholar]
- Kreitzer G., Marmorstein A., Okamoto P., Vallee R. and Rodriguez-Boulan E. (2000). Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat. Cell Biol. 2, 125-127. 10.1038/35000081 [DOI] [PubMed] [Google Scholar]
- Krendel M., Zenke F. T. and Bokoch G. M. (2002). Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4, 294-301. 10.1038/ncb773 [DOI] [PubMed] [Google Scholar]
- Krylyshkina O., Kaverina I., Kranewitter W., Steffen W., Alonso M. C., Cross R. A. and Small J. V. (2002). Modulation of substrate adhesion dynamics via microtubule targeting requires kinesin-1. J. Cell Biol. 156, 349-360. 10.1083/jcb.200105051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J. L. (1988). Two monoclonal antibodies to actin: one muscle selective and one generally reactive. Cell Motil. Cytoskeleton 10, 349-362. 10.1002/cm.970100302 [DOI] [PubMed] [Google Scholar]
- Ligon L. A. and Holzbaur E. L. F. (2007). Microtubules tethered at epithelial cell junctions by dynein facilitate efficient junction assembly. Traffic 8, 808-819. 10.1111/j.1600-0854.2007.00574.x [DOI] [PubMed] [Google Scholar]
- Ligon L. A., Karki S., Tokito M. and Holzbaur E. L. F. (2001). Dynein binds to beta-catenin and may tether microtubules at adherens junctions. Nat. Cell Biol. 3, 913-917. 10.1038/ncb1001-913 [DOI] [PubMed] [Google Scholar]
- Macho B., Brancorsini S., Fimia G. M., Setou M., Hirokawa N. and Sassone-Corsi P. (2002). CREM-dependent transcription in male germ cells controlled by a kinesin. Science 298, 2388-2390. 10.1126/science.1077265 [DOI] [PubMed] [Google Scholar]
- Mack N. A. and Georgiou M. (2014). The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases 5, e973768 10.4161/21541248.2014.973768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary S., Charrasse S., Meriane M., Comunale F., Travo P., Blangy A. and Gauthier-Rouvière C. (2002). Biogenesis of N-cadherin-dependent cell-cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. Mol. Biol. Cell 13, 285-301. 10.1091/mbc.01-07-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mège R.-M., Gavard J. and Lambert M. (2006). Regulation of cell–cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541-548. 10.1016/j.ceb.2006.08.004 [DOI] [PubMed] [Google Scholar]
- Mikhailov A. and Gundersen G. G. (1998). Relationship between microtubule dynamics and lamellipodium formation revealed by direct imaging of microtubules in cells treated with nocodazole or taxol. Cell Motil. Cytoskeleton 41, 325-340. [DOI] [PubMed] [Google Scholar]
- Moriyama K., Iida K. and Yahara I. (1996). Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1, 73-86. 10.1046/j.1365-2443.1996.05005.x [DOI] [PubMed] [Google Scholar]
- Nagae S., Meng W. and Takeichi M. (2013). Non-centrosomal microtubules regulate F-actin organization through the suppression of GEF-H1 activity. Genes Cells 18, 387-396. 10.1111/gtc.12044 [DOI] [PubMed] [Google Scholar]
- Nagasaki T., Liao G. and Gundersen G. G. (1994). Isolated plasma membranes induce the loss of oriented detyrosinated microtubules and other contact inhibition-like responses in migrating NRK cells. J. Cell Sci. 107, 3413-3423. [DOI] [PubMed] [Google Scholar]
- Nakaya Y., Sukowati E. W., Wu Y. and Sheng G. (2008). RhoA and microtubule dynamics control cell–basement membrane interaction in EMT during gastrulation. Nat. Cell Biol. 10, 765-775. 10.1038/ncb1739 [DOI] [PubMed] [Google Scholar]
- Nekrasova O. E., Amargo E. V., Smith W. O., Chen J., Kreitzer G. E. and Green K. J. (2011). Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J. Cell Biol. 195, 1185-1203. 10.1083/jcb.201106057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B. J., Tomasevic N., Russell A., Pierce D. W., Jia Z., McCormick C. D., Hartman J., Sakowicz R. and Pollard T. D. (2009). Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 460, 1031-1034. 10.1038/nature08231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien L. E., Yu W., Tang K., Jou T.-S., Zegers M. M. P. and Mostov K. E. (2006). Morphological and biochemical analysis of Rac1 in three-dimensional epithelial cell cultures. Methods Enzymol. 406, 676-691. 10.1016/S0076-6879(06)06053-8 [DOI] [PubMed] [Google Scholar]
- Otani T., Ichii T., Aono S. and Takeichi M. (2006). Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J. Cell Biol. 175, 135-146. 10.1083/jcb.200605012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou G., Blacque O. E., Snow J. J., Leroux M. R. and Scholey J. M. (2005). Functional coordination of intraflagellar transport motors. Nature 436, 583-587. 10.1038/nature03818 [DOI] [PubMed] [Google Scholar]
- Palazzo A. F., Cook T. A., Alberts A. S. and Gundersen G. G. (2001). mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat. Cell Biol. 3, 723-729. 10.1038/35087035 [DOI] [PubMed] [Google Scholar]
- Pan X., Ou G., Civelekoglu-Scholey G., Blacque O. E., Endres N. F., Tao L., Mogilner A., Leroux M. R., Vale R. D. and Scholey J. M. (2006). Mechanism of transport of IFT particles in C. elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J. Cell Biol. 174, 1035-1045. 10.1083/jcb.200606003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portereiko M. F., Saam J. and Mango S. E. (2004). ZEN-4/MKLP1 is required to polarize the foregut epithelium. Curr. Biol. 14, 932-941. 10.1016/j.cub.2004.05.052 [DOI] [PubMed] [Google Scholar]
- Quiros M. and Nusrat A. (2014). RhoGTPases, actomyosin signaling and regulation of the Epithelial Apical Junctional Complex. Semin. Cell Dev. Biol. 36, 194-203. 10.1016/j.semcdb.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A., Gomez G. A., Priya R., Verma S., Kovacs E. M., Jiang K., Brown N. H., Akhmanova A., Stehbens S. J. and Yap A. S. (2012). Centralspindlin and alpha-catenin regulate Rho signalling at the epithelial zonula adherens. Nat. Cell Biol. 14, 818-828. 10.1038/ncb2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Li R., Zheng Y. and Busch H. (1998). Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273, 34954-34960. 10.1074/jbc.273.52.34954 [DOI] [PubMed] [Google Scholar]
- Riento K. and Ridley A. J. (2003). Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446-456. 10.1038/nrm1128 [DOI] [PubMed] [Google Scholar]
- Saade M., Irla M., Govin J., Victorero G., Samson M. and Nguyen C. (2007). Dynamic distribution of Spatial during mouse spermatogenesis and its interaction with the kinesin KIF17b. Exp. Cell Res. 313, 614-626. 10.1016/j.yexcr.2006.11.011 [DOI] [PubMed] [Google Scholar]
- Salvarezza S. B., Deborde S., Schreiner R., Campagne F., Kessels M. M., Qualmann B., Caceres A., Kreitzer G. and Rodriguez-Boulan E. (2009). LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Mol. Biol. Cell 20, 438-451. 10.1091/mbc.E08-08-0891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarin S. and Nusrat A. (2009). Regulation of epithelial apical junctional complex by Rho family GTPases. Front. Biosci. 14, 1129-1142. 10.2741/3298 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setou M., Nakagawa T., Seog D. H. and Hirokawa N. (2000). Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science 288, 1796-1802. 10.1126/science.288.5472.1796 [DOI] [PubMed] [Google Scholar]
- Siegrist S. E. and Doe C. Q. (2007). Microtubule-induced cortical cell polarity. Genes Dev. 21, 483-496. 10.1101/gad.1511207 [DOI] [PubMed] [Google Scholar]
- Snow J. J., Ou G., Gunnarson A. L., Walker M. R. S., Zhou H. M., Brust-Mascher I. and Scholey J. M. (2004). Two anterograde intraflagellar transport motors cooperate to build sensory cilia on C. elegans neurons. Nat. Cell Biol. 6, 1109-1113. 10.1038/ncb1186 [DOI] [PubMed] [Google Scholar]
- Stehbens S. J., Paterson A. D., Crampton M. S., Shewan A. M., Ferguson C., Akhmanova A., Parton R. G. and Yap A. S. (2006). Dynamic microtubules regulate the local concentration of E-cadherin at cell-cell contacts. J. Cell Sci. 119, 1801-1811. 10.1242/jcs.02903 [DOI] [PubMed] [Google Scholar]
- Takano K., Miki T., Katahira J. and Yoneda Y. (2007). NXF2 is involved in cytoplasmic mRNA dynamics through interactions with motor proteins. Nucleic Acids Res. 35, 2513-2521. 10.1093/nar/gkm125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang V. W. and Brieher W. M. (2012). alpha-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J. Cell Biol. 196, 115-130. 10.1083/jcb.201103116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebe H., Nakano K., Maximo R. and Shiozaki K. (2008). Pom1 DYRK regulates localization of the Rga4 GAP to ensure bipolar activation of Cdc42 in fission yeast. Curr. Biol. 18, 322-330. 10.1016/j.cub.2008.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S., Schönberger J. L., Nunez-Iglesias J., Boulogne F., Warner J. D., Yager N., Gouillart E. and Yu T. (2014). scikit-image: image processing in Python. PeerJ. 2, e453 10.7717/peerj.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S., Han S. P., Michael M., Gomez G. A., Yang Z., Teasdale R. D., Ratheesh A., Kovacs E. M., Ali R. G. and Yap A. S. (2012). A WAVE2-Arp2/3 actin nucleator apparatus supports junctional tension at the epithelial zonula adherens. Mol. Biol. Cell 23, 4601-4610. 10.1091/mbc.E12-08-0574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Wang S., Noritake J., Sato K., Fukata M., Takefuji M., Nakagawa M., Izumi N., Akiyama T. and Kaibuchi K. (2004). Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev. Cell 7, 871-883. 10.1016/j.devcel.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Waterman-Storer C. M., Salmon W. C. and Salmon E. D. (2000). Feedback interactions between cell-cell adherens junctions and cytoskeletal dynamics in newt lung epithelial cells. Mol. Biol. Cell 11, 2471-2483. 10.1091/mbc.11.7.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y., Eng C. H., Schmoranzer J., Cabrera-Poch N., Morris E. J. S., Chen M., Wallar B. J., Alberts A. S. and Gundersen G. G. (2004). EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 6, 820-830. 10.1038/ncb1160 [DOI] [PubMed] [Google Scholar]
- Wojnacki J., Quassollo G., Marzolo M.-P. and Cáceres A. (2014). Rho GTPases at the crossroad of signaling networks in mammals: impact of Rho-GTPases on microtubule organization and dynamics. Small GTPases 5, e28430 10.4161/sgtp.28430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S., Pokutta S., Drees F., Weis W. I. and Nelson W. J. (2005). Deconstructing the cadherin-catenin-actin complex. Cell 123, 889-901. 10.1016/j.cell.2005.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H., Imai K., Kamegaya E., Takamatsu Y., Irago M., Hagino Y., Kasai S., Shimada K., Yamamoto T., Sora I. et al. (2006). Repeated methamphetamine administration alters expression of the NMDA receptor channel epsilon2 subunit and kinesins in the mouse brain. Ann. N. Y. Acad. Sci. 1074, 97-103. 10.1196/annals.1369.009 [DOI] [PubMed] [Google Scholar]
- Yanagisawa M., Kaverina I. N., Wang A., Fujita Y., Reynolds A. B. and Anastasiadis P. Z. (2004). A novel interaction between kinesin and p120 modulates p120 localization and function. J. Biol. Chem. 279, 9512-9521. 10.1074/jbc.M310895200 [DOI] [PubMed] [Google Scholar]
- Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E. and Mizuno K. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809-812. 10.1038/31735 [DOI] [PubMed] [Google Scholar]
- Yoshimura Y. and Miki H. (2011). Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem. Biophys. Res. Commun. 408, 322-328. 10.1016/j.bbrc.2011.04.032 [DOI] [PubMed] [Google Scholar]