

ABSTRACT
Mitochondrial morphology is regulated by fusion and fission machinery. Impaired mitochondria dynamics cause various diseases, including Alzheimer's disease. Appoptosin (encoded by SLC25A38) is a mitochondrial carrier protein that is located in the mitochondrial inner membrane. Appoptosin overexpression causes overproduction of reactive oxygen species (ROS) and caspase-dependent apoptosis, whereas appoptosin downregulation abolishes β-amyloid-induced mitochondrial fragmentation and neuronal death during Alzheimer's disease. Herein, we found that overexpression of appoptosin resulted in mitochondrial fragmentation in a manner independent of its carrier function, ROS production or caspase activation. Although appoptosin did not affect levels of mitochondrial outer-membrane fusion (MFN1 and MFN2), inner-membrane fusion (OPA1) and fission [DRP1 (also known as DNM1L) and FIS1] proteins, appoptosin interacted with MFN1 and MFN2, as well as with the mitochondrial ubiquitin ligase MITOL (also known as MARCH5) but not OPA1, FIS1 or DRP1. Appoptosin overexpression impaired the interaction between MFN1 and MFN2, and mitochondrial fusion. By contrast, co-expression of MFN1, MITOL and a dominant-negative form of DRP1, DRP1K38A, partially rescued appoptosin-induced mitochondrial fragmentation and apoptosis, whereas co-expression of FIS1 aggravated appoptosin-induced apoptosis. Together, our results demonstrate that appoptosin can interact with mitochondrial outer-membrane fusion proteins and regulates mitochondrial morphology.
KEY WORDS: Appoptosin, Apoptosis, Mitochondrial dynamics, MFN1, MFN2, MITOL
Summary: Overexpression of the pro-apoptotic protein appoptosin impairs interaction of the mitochondrial fusion proteins MFN1 and MFN2, and reduces mitochondrial fusion activity, leading to mitochondrial fragmentation.
INTRODUCTION
Mitochondria are dynamic organelles, whose shapes continually change from filament to fragment (and vice versa) through balance between the rates of mitochondrial fusion and fission. Morphology variations in the mitochondria are physiologically important for cell homeostasis. Mitochondrial fusion is necessary for the fidelity of oxidative phosphorylation, the complementation of damaged mitochondria and the repairing of small amounts of mitochondrial damage. Although mitochondrial fission is required for the formation of new mitochondria, it is also involved in mitochondrial mobility, mitophagy, cell mitosis and apoptosis (Youle and van der Bliek, 2012).
Mitochondrial fusion comprises two events – outer-membrane fusion and inner-membrane fusion. In mammalian cells, two GTPases MFN1 and MFN2, which anchor on the outer membrane of mitochondria, are responsible for the outer-membrane fusion (Chen et al., 2003; Koshiba et al., 2004). However, compared to MFN2, MFN1 is relatively specific in regulating mitochondrial fusion (Shen et al., 2007). MFN1-harboring mitochondria have a higher tethering efficiency than those with MFN2, and purified MFN1 also possesses higher GTPase activity than MFN2 (Ishihara et al., 2004). In contrast, MFN2 is more tissue specific in its expression pattern than MFN1 (Liesa et al., 2009). In addition to promoting mitochondrial fusion (Chen et al., 2003; Neuspiel et al., 2005), MFN2 is also reported to be involved in various signaling cascades and acts as a pro-apoptotic and anti-proliferative protein (Guo et al., 2007; Papanicolaou et al., 2011; Shen et al., 2007; Wang et al., 2015). Mitochondrial inner-membrane fusion is mediated by the protein OPA1, which is located in the intra-mitochondrial space (Alavi and Fuhrmann, 2013). However, there are eight OPA1 isoforms generated from alternative splicing and processing during maturation, making the mechanism of inner-membrane fusion much more complicated (Westermann, 2010).
Mitochondrial fission is primarily mediated by FIS1 and DRP1 (also known as DNM1L) in mammalian cells. During the fission process, mitochondrial outer-membrane protein FIS1 recruits the GTPase DRP1 from the cytosol to the cytosolic face of mitochondria, where oligomeric DRP1 assembles into a spiral structure and severs mitochondria membranes following GTP hydrolysis (Westermann, 2010).
Recently, MITOL (also known as MARCH5), which belongs to the membrane-associated RING-CH E3 ubiquitin ligase (MARCH) family, has been illustrated to be an important regulator of mitochondrial morphology. MITOL is a mitochondrial ubiquitin ligase and has been reported to interact with DRP1, FIS1, MFN2 (Nakamura et al., 2006) and acetylated MFN1 (Park et al., 2014). MITOL ubiquitylates FIS1 and DRP1, resulting in their degradation. Overexpression of MITOL can also promote the formation of long tubular mitochondria in a manner that is dependent on MFN2 activity (Nakamura et al., 2006); MITOL can also reverse the phenotypes caused by FIS1 overexpression (Yonashiro et al., 2006). By contrast, MITOL is also reported to be a regulator of the subcellular trafficking of DRP1 and, therefore, is required for DRP1-dependent mitochondrial fission (Karbowski et al., 2007).
Disruption of mitochondria homeostasis has been found in and proposed as the cause of diseases such as cancer, cardiovascular disease, diabetes and neurodegenerative diseases (Archer, 2013; Jheng et al., 2012). Alzheimer's disease is one of the most common neurodegenerative diseases and is primarily caused by the accumulation and deposition of neurotoxic β-amyloid (Aβ) peptides in vulnerable brain regions (Jiang et al., 2014; Lemere, 2013). Mitochondrial morphology has been found to be abnormal in degenerating dendrites in the brains of individuals with Alzheimer's disease (Saraiva et al., 1985). More recently, mitochondria have been found to be redistributed away from axons in the pyramidal neurons of Alzheimer's disease brain, accompanied by decreased levels of DRP1, OPA1, MFN1 and MFN2, and increased levels of FIS1 (Wang et al., 2009). Another research group has also confirmed the change of FIS1, OPA1, MFN1 and MFN2, except that they report increased levels of DRP1 in Alzheimer's disease brain instead (Manczak et al., 2011). Moreover, overproduction of Aβ as well as treatment with Aβ can cause mitochondrial fragmentation by affecting these mitochondrial-morphology-regulating proteins (Manczak et al., 2010; Wang et al., 2008). These results demonstrate that mitochondria dynamics are perturbed during Alzheimer's disease. However, the underlying mechanism has yet to be fully elucidated.
We have recently identified a new pro-apoptotic protein, appoptosin (also known as SLC25A38), that belongs to the mitochondria carrier family (MCP) and resides in the mitochondrial inner membrane. The physiological function of appoptosin is to transport glycine and δ-aminolevulinic acid between mitochondria and the cytosol to initiate heme biosynthesis (Guernsey et al., 2009; Kannengiesser et al., 2011). We found that overexpression of appoptosin results in the overproduction of reactive oxygen species (ROS) and caspase-dependent apoptosis. Importantly, the levels of appoptosin are upregulated in neurons that had been treated with Aβ and samples from the brain of Alzheimer's disease individuals, whereas downregulation of appoptosin abrogates mitochondrial fragmentation and caspase activation caused by insults of glutamate and Aβ in neurons (Zhang et al., 2012). These results suggest that appoptosin is a crucial regulator of Aβ-induced neuronal injury and mitochondrial dysfunction during Alzheimer's disease. In the present study, we found that overexpression of appoptosin directly induced mitochondrial fragmentation in an ROS-overproduction- and caspase-activation-independent manner. In addition, we showed that appoptosin interacted with MFN1, MFN2 and MITOL but not OPA1, FIS1 or DRP1. Overexpression of appoptosin decreased the heterotypic interaction of MFN1–MFN2 and reduced mitochondrial fusion activity. Co-expression of MFN1, MITOL and a dominant-negative form of DRP1, DRP1K38A, rescued appoptosin-induced apoptosis, whereas co-expression of FIS1 aggravated appoptosin-induced apoptosis. Finally, we found that SLC25A26, another MCP-family member that is closely related to appoptosin based on their protein sequence similarity, could interact with MITOL but not MFN1 or MFN2, and the expression of SLC25A26 did not affect mitochondrial morphology. Taken together, these results suggest that appoptosin is a unique MCP that can interact with mitochondrial outer-membrane fusion proteins and regulate mitochondrial morphology.
RESULTS
Overexpression of appoptosin induces mitochondrial fragmentation
We have previously found that downregulation of appoptosin prevents mitochondrial fragmentation and apoptosis that is induced by β-amyloid (Aβ) peptides and treatment with glutamate in neuronal cells (Zhang et al., 2012). These results imply that appoptosin modulates mitochondrial morphology. To prove this, we overexpressed appoptosin in HeLa cells (Fig. 1A) and neurons (Fig. 1B). We found that overexpressed Myc–appoptosin colocalized well with the mitochondrial marker MitoTracker Red, and this is consistent with the suggested mitochondrial inner-membrane localization of appoptosin. Notably, we also found that mitochondria in Myc–appoptosin-overexpressing cells had dot-like small punctum morphology, in contrast to the long filamentous morphology in pCMV-Myc (empty vector) transfected cells (Fig. 1A,B). These results suggest that overexpression of appoptosin leads to dramatic mitochondrial fragmentation. Interestingly, we found that mitochondrial morphology was unaffected (Fig. S1A,B) when appoptosin was downregulated (Fig. S1C), implying that only an excess of appoptosin can interfere with mitochondrial morphology.
Fig. 1.

Overexpression of appoptosin induces mitochondrial fragmentation. (A) HeLa cells and (B) mouse primary neurons were transfected with pCMV-Myc (as control, CMV) and Myc–appoptosin (appop) for 24 h. MitoTracker Red was added into medium 30 min before fixation to stain mitochondria. After immunostaining with an anti-myc antibody (to indicate appoptosin, myc), cells were counterstained with DAPI and observed under a confocal microscope. The number of cells exhibiting the different types of mitochondria morphology (normal and fragmented) were quantified (n>100). Means±s.e.m., *P<0.05, ***P<0.001 (unpaired t-test). Signals for Myc–appoptosin, mitochondria and nuclei are shown in green, red and blue, respectively. Scale bars: 5 μm (‘zoom in’ images in A); 10 μm (all other images in A); 50 μm (‘zoom in’ images in B); 100 μm (all other images in B).
Appoptosin-induced mitochondrial fragmentation is not dependent on its carrier function, ROS generation or caspase activity
Appoptosin functions as a carrier of glycine and δ-aminolevulinic acid in order to transport them across the mitochondrial inner membrane for the initiation of heme synthesis. Because overexpression of appoptosin induces caspase-dependent cell apoptosis through the promotion of heme synthesis and ROS generation (Zhang et al., 2012), oxidative stress might be one of the stimuli for appoptosin-induced mitochondrial fragmentation. Oxidative stress has been suggested to cause mitochondrial fragmentation by increasing the mitochondrial translocation of DRP1 (Iqbal and Hood, 2014; Wu et al., 2011). Herein, after transfection with appoptosin in HeLa cells, we treated cells with the ROS scavenger, N-acetyl-L-cysteine (NAC) (Fig. 2A), the anti-oxidant resveratrol (RSV) (Fig. 2B), the caspase inhibitor Z-VAD (Fig. 2C) and the heme-synthesis inhibitor succinylacetone (Fig. 2D). However, we found that none of these chemicals could block appoptosin-induced mitochondrial fragmentation (Fig. 2A–E), suggesting that this process is not dependent on ROS generation, caspase activity or the carrier function of appoptosin.
Fig. 2.

Appoptosin-induced mitochondrial fragmentation is not dependent on ROS generation or caspase activation. HeLa cells were transfected with pCMV-Myc (CMV) or Myc–appoptosin (appop). Four hours later, the medium was refreshed and the cells were treated with (A) 1 mM NAC, (B) 50 μM RSV, (C) 50 μM Z-VAD or (D) 1 μM succinylacetone (SA) for an additional 20 h. After staining with MitoTracker Red, fixation, immunostaining with an antibody against Myc (to indicate appoptosin) and staining with DAPI, cells were observed under a confocal microscope. Signals for Myc–appoptosin, mitochondria and nuclei are shown in green, red and blue, respectively. (E) The number of cells exhibiting the different types of mitochondria morphology were counted and the percentages of cells with fragmented mitochondria were compared (n>100). Means±s.e.m.; ns, not significant (unpaired t-test). Ctrl, control. Scale bars: 5 μm (for ‘zoom in’ images); 10 μm (all other images).
Appoptosin does not affect the levels of proteins involved in mitochondrial fusion and fission
Mitochondrial morphology is maintained by the balance between fusion and fission machinery. To determine the mechanism underlying appoptosin-induced mitochondrial fragmentation, we first studied whether appoptosin affects the proteins involved in mitochondrial morphology regulation. However, we found that overexpression of appoptosin did not affect the protein levels of DRP1, FIS1, MFN1, MFN2 or OPA1 (Fig. 3A,B). Because translocation of DRP1 from the cytosol to mitochondria is a requisite event in DRP1-dependent mitochondrial fragmentation, we studied whether appoptosin affects DRP1 translocation. We transfected HeLa cells with pCMV-Myc or Myc–appoptosin, and then separated the cytosolic and mitochondrial fractions. Western blot results showed that overexpression of appoptosin did not alter DRP1 content in either cytosolic or mitochondrial fractions (Fig. 3C,D), suggesting that the translocation of DRP1 is not required for appoptosin-induced mitochondrial fragmentation.
Fig. 3.
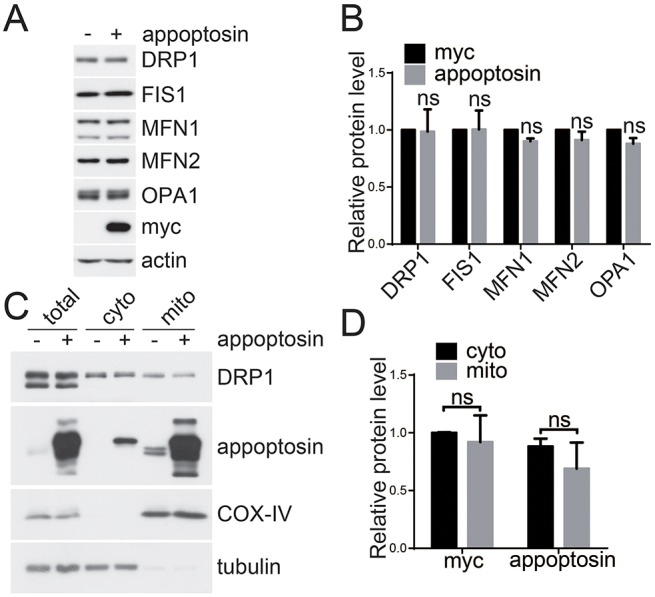
Appoptosin does not affect the levels of mitochondrial fusion and fission proteins nor the mitochondrial translocation of DRP1. (A) HeLa cells were transfected with pCMV-Myc (−) and Myc–appoptosin (+) for 24 h before western blot analysis. (B) Protein levels of DRP1, FIS1, MFN1, MFN2 and OPA1 were quantified by using densitometry and normalized to those of β-actin for comparison, n=3. (C) Cells were transfected with Myc–appoptosin (+) or control vector (−) for 24 h. Mitochondria (mito) and the cytosol (cyto) were then separated, and subsequently subjected to western blot analysis. (D) Protein levels of DRP1 in experiments described in C were quantified by using densitometry and normalized to those of tubulin (cytosolic fractions) or COX-IV (mitochondrial fractions) for comparison, n=3. ns, not significant.
Appoptosin interacts with MFN1, MFN2 and MITOL
Next, we studied whether appoptosin interacts with the mitochondrial morphology-regulating proteins. We co-expressed Myc–appoptosin with hemagglutinin (HA)-tagged important regulators of mitochondrial morphology in HEK 293T cells and performed a co-immunoprecipitation study. We found that an antibody against Myc immunoprecipitated appoptosin, as well as HA-tagged MFN1 (Fig. 4C) and MFN2 (Fig. 4D), but not DRP1 (Fig. 4A), FIS1 (Fig. 4B) or OPA1 (Fig. 4E). By contrast, although an antibody against HA immunoprecipitated all HA-tagged proteins (Fig. 4A–E), it only immunoprecipitated appoptosin from HA–MFN1- (Fig. 4C) and HA–MFN2-expressing (Fig. 4D) cells. In addition, we found that endogenous appoptosin could also be pulled down by antibodies against MFN1 (Fig. 4G) and MFN2 (Fig. 4H). These results indicate that appoptosin can interact with MFN1 and MFN2 but not DRP1, FIS1 or OPA1. MITOL has been reported to be an important regulator of mitochondrial morphology in recent years. Here, in cells co-expressing Myc–appoptosin and HA–MITOL, we found that antibodies against Myc and HA immunoprecipitated appoptosin and MITOL (Fig. 4F), indicating that appoptosin also interacts with MITOL.
Fig. 4.
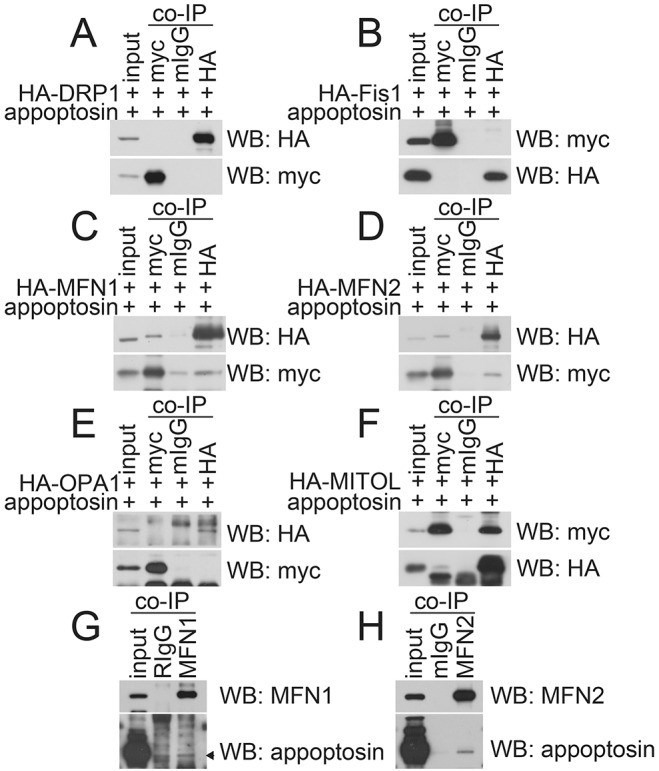
Appoptosin interacts with MFN1, MFN2 and MITOL. Myc–appoptosin was co-transfected with (A) HA–DRP1, (B) HA–FIS1, (C) HA–MFN1, (D) HA–MFN2, (E) HA–OPA1 or (F) HA–MITOL into HEK 293T cells for 24 h. Cell lysates were subjected to co-immunoprecipitation (co-IP) with mouse IgG (mIgG), mouse antibody against Myc or mouse antibody against HA, and then western blot (WB) analysis. (G) HEK 293T cell lysates were subjected to co-IP with rabbit IgG (RIgG) or a rabbit antibody against MFN1 and then western blot analysis. (H) HEK 293T cell lysates were subjected to co-IP with mIgG or a mouse antibody against MFN2 and then western blot analysis. Images are representative of three biological replicates.
Because overexpression of appoptosin promotes ROS production and apoptosis, we studied whether interactions between appoptosin and MFN1, MFN2 and MITOL were mediated by ROS production and apoptosis. However, appoptosin still interacted with MFN1, MFN2 and MITOL upon treatment with NAC or Z-VAD (Fig. S2A,B), implying that ROS levels and apoptosis do not contribute to such interactions.
Overexpression of appoptosin reduces MFN1–MFN2 hetero-oligomer formation and mitochondrial fusion
It has been demonstrated that MFN1 and MFN2 regulate fusion of the mitochondrial outer membrane by forming homo- or hetero-oligomers in cis or in trans (Detmer and Chan, 2007). We tested whether overexpression of appoptosin could affect the formation of MFN1 and MFN2 homo-oligomers, and MFN1–MFN2 hetero-oligomers. As shown in Fig. 5A, overexpression of appoptosin reduced the hetero-interaction of MFN1–MFN2 but not the formation of MFN1–MFN1 and MFN2–MFN2 homo-dimers.
Fig. 5.

Overexpression of appoptosin impairs the MFN1–MFN2 interaction and mitochondrial fusion. HEK 293T cells were co-transfected with (A) HA–MFN1+Myc–MFN1, HA–MFN1+Myc–MFN2 or HA–MFN2+Myc–MFN2. After equal splitting, cells were transfected with control or appoptosin-expressing plasmids for 24 h. Cell lysates were subjected to co-immunoprecipitation (IP) with the mouse anti-Myc antibody 9E10 and then western blot (WB) analysis for the indicated proteins. (B) After co-culture of HeLa cells expressing pCMV-myc+mtGFP with those expressing pCMV-myc+mtRFP (left panels, CMV), or cells expressing Myc–appoptosin+mtGFP with those expressing Myc–appoptosin+mtRFP (right panels, appoptosin), cells were treated with 40% PEG 1500 for 5 min and then incubated in cycloheximide-containing medium for 7 h. Cells were then fixed, counterstained with DAPI and observed by using microscopy. Bottom panels are zoomed images of the areas indicated in the upper panels. Scale bars: 20 μm (upper panels); 5 μm (zoomed images). The number of fusing cell clusters exhibiting different mitochondrial fusion status (full or partial) were counted for comparison, n≥100 cells, **P<0.01 (unpaired t-test). Means±s.e.m. are shown.
To measure mitochondrial fusion activity, we generated mitochondria-targeted GFP (mtGFP) and mitochondria-targeted RFP (mtRFP) plasmids by fusing GFP and RFP with COX8. When cells expressing mtGFP were mixed with cells expressing mtRFP and treated with polyethylene glycol (PEG) for 7 h to induce fusion, we found that 100% of fused cells contained fully fused mitochondria in controls, as demonstrated by colocalization of red and green fluorescent signals (Fig. 5B). However, only about 33% of fused cells contained fully fused mitochondria, and 67% of fused cells contained partially fused mitochondria in appoptosin-overexpressing cells (Fig. 5B), indicating decreased fusion efficiency upon appoptosin overexpression.
Appoptosin-mediated mitochondrial fragmentation is alleviated by overexpression of MFN1, MITOL and DRP1K38A
MFN1 and MFN2 regulate fusion of the mitochondrial outer membrane, whereas FIS1 and DRP1 regulate mitochondrial fission (Knott et al., 2008; Westermann, 2010). We first ascertained the effects of these proteins on mitochondrial morphology. As shown in Fig. 6A, transient overexpression of DRP1 had no effect on mitochondrial morphology, and this was consistent with previous studies (Pitts et al., 1999; Smirnova et al., 1998). DRP1K38A is a dominant-negative mutant of DRP1 and its overexpression has been shown to inhibit mitochondrial fission and block cell death (Frank et al., 2001; James et al., 2003). We found that transient overexpression of DRP1K38A led mitochondria to fuse into an inter-connected network. By contrast, transient overexpression of FIS1 resulted in the fragmentation of mitochondria into dot-like puncta (Gomes and Scorrano, 2008). Conversely, mitochondria in MFN1- and MFN2-expressing cells hyper-fused into ‘grape-like’ clusters. Mitochondria in MITOL-overexpressing cells appeared to be inter-connected and elongated filaments, and this is consistent with previous reports (Nakamura et al., 2006; Yonashiro et al., 2006).
Fig. 6.

Appoptosin-induced mitochondrial fragmentation is alleviated by promoting mitochondrial fusion. (A) HeLa cells that had been transfected with pCMV-HA (CMV), HA–DRP1 (DRP1), HA–FIS1 (FIS1), HA–DRP1K38A (K38A), HA–MFN1 (MFN1), HA–MFN2 (MFN2) and HA–MITOL (MITOL) were treated with MitoTracker Red, immunofluorescently stained and observed under a confocal microscope. Mitochondria (MitoTracker) and HA signals are shown in red and green, respectively. (B) Myc–appoptosin was co-transfected with HA–MFN1, HA–MITOL or HA–DRP1K38A into HeLa cells. 24 h later, cells were subjected to immunofluorescent staining and counterstaining with DAPI. Myc–appoptosin, HA and DAPI signals are shown in green, red and blue, respectively. (C) Quantitative analysis of the numbers of transfected cells with normal, fragmented or aggregated and/or fused mitochondria in experiments detailed in B; n≥100 cells. Scale bars: 5 μm (‘zoom in’ images); 10 μm (all other images). *P<0.05; **P<0.01 (unpaired t-test). Means±s.e.m. are shown.
To investigate whether appoptosin-induced mitochondria fragmentation can be affected by factors involved in mitochondria fusion, we co-expressed Myc–appoptosin and HA–MFN1 or HA–MITOL in HeLa cells. We found that overexpression of MFN1 reversed appoptosin-induced mitochondria fragmentation, resulting in mitochondria with a highly fused morphology (Fig. 6B–C). Overexpression of MITOL also dramatically rescued appoptosin-induced mitochondrial fragmentation (Fig. 6B–C). In addition, we found that co-expression of DRP1K38A, which was reported to block mitochondrial fission and promote mitochondrial fusion (Frank et al., 2001; James et al., 2003), also markedly ameliorated appoptosin-induced mitochondrial fragmentation (Fig. 6B–C).
Appoptosin-induced apoptosis is inhibited by DRP1K38A, MFN1 and MITOL, and is aggravated by FIS1
Because mitochondrial fission is directly involved in the initiation of apoptosis (Frank et al., 2001; Karbowski et al., 2002) and overexpression of appoptosin induces apoptosis, we studied whether these regulators of mitochondrial morphology affect appoptosin-induced apoptosis. As expected, overexpression of appoptosin alone induced dramatic polyADP ribose polymerase (PARP) cleavage (indicative of apoptosis) in HeLa cells (Fig. 7A–E). In addition, we found that overexpression of FIS1 alone induced marked PARP cleavage and that co-expression of FIS1 with appoptosin further enhanced appoptosin-induced PARP cleavage (Fig. 7A,F). In contrast, overexpression of DRP1K38A (Fig. 7B,F), MFN1 (Fig. 7C,F) or MITOL (Fig. 7E,F) alone had no notable effect on PARP cleavage, but their co-expression significantly reduced appoptosin-induced PARP cleavage. Interestingly, although MFN2 is a mitochondrial fusion protein, its overexpression alone resulted in marked PARP cleavage (Fig. 7D,F). PARP cleavage resulting from co-expression of MFN2 with appoptosin was comparable to that from overexpression of MFN2 alone but more intensive than that from overexpression of appoptosin alone (Fig. 7D,F). However, because co-expression of MFN2 seemed to interfere with appoptosin overexpression (Fig. 7D), the exact role of MFN2 in appoptosin-induced apoptosis requires further determination.
Fig. 7.
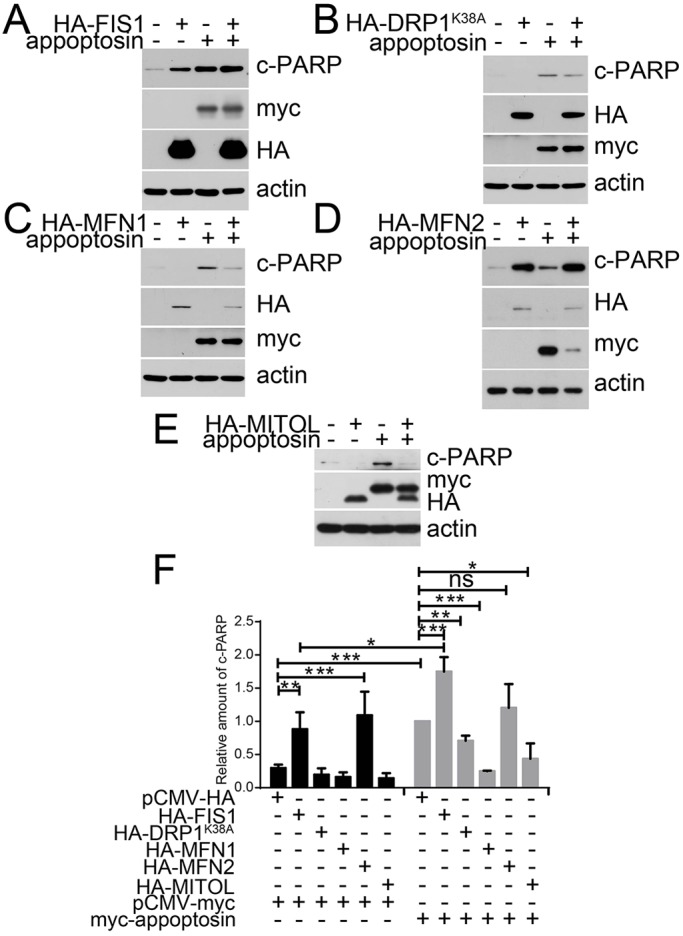
Appoptosin-induced apoptosis is aggravated by expression of FIS1 and alleviated by expression of DRP1K38A, MFN1 and MITOL. Myc–appoptosin was co-transfected with (A) HA–FIS1, (B) HA–DRP1K38A, (C) HA–MFN1, (D) HA–MFN2 or (E) HA–MITOL into HeLa cells for 24 h. Cell lysates were subjected to western blot analysis in order to study the levels of cleaved PARP (c-PARP). (F) Densitometry was used to quantify blots (A–E); n=3; ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (unpaired t-test). Means±s.e.m. are shown.
Moreover, we studied whether co-expression of mitochondrial fusion and/or fission proteins affects appoptosin-promoted ROS production. However, co-expression of these proteins did not affect ROS production in cells overexpressing appoptosin (Fig. S3), even though co-expression of MFN1, MITOL and DRP1K38A attenuated mitochondrial fragmentation (Fig. 6) and apoptosis (Fig. 7). This suggests that mitochondrial fragmentation is not necessarily required for increased ROS production.
SLC25A26 does not affect mitochondrial morphology
Another mitochondrial-carrier-family member, SLC25A26, is closely related to appoptosin based on their protein sequence similarity and has been identified as mitochondrial S-adenosylmethionine transporter (Agrimi et al., 2004; Haitina et al., 2006). In order to study whether the mitochondrial-morphology-regulating effect of appoptosin applies to other mitochondrial-carrier-family members, we transiently overexpressed SLC25A26 in HeLa cells. However, we found that overexpression of SLC25A26 had no obvious effect on mitochondrial morphology (Fig. 8A,B). We also found that SLC25A26 could interact with MITOL but not with MFN1 or MFN2 (Fig. 8C,D).
Fig. 8.

SLC25A26 does not affect mitochondrial morphology. (A) HeLa cells were transfected with pCMV-Myc (CMV), Myc–appoptosin (appop) or Myc–SLC25A26 (25A26) for 24 h, and then treated with MitoTracker Red and immunofluorescently stained for observation with a confocal microscope. Scale bars: 5 μm (‘zoom in’ images); 10 μm (all other images). (B) Quantitative analysis of the numbers of transfected cells with fragmented mitochondria in experiments described in A; n=80 cells; ns, not significant; **P<0.01 (unpaired t-test). (C) Myc–SLC25A26 was co-transfected with HA–MFN1 or HA–MFN2 into HEK 293T cells. 24 h later, cell lysates were subjected to co-immunoprecipitation with mouse IgG (mIgG), a mouse antibody against Myc (myc) or mouse antibody against HA (HA), and then western blot analysis for the indicated proteins. (D) Myc-appoptosin was co-transfected with HA–MITOL in HEK 293T cells. 24 h later, cell lysates were subjected to co-immunoprecipitation with mIgG, mouse antibody against Myc or mouse antibody against HA, and then western blot analysis for the indicated proteins.
DISCUSSION
Mitochondrial morphology is regulated by a series of proteins that are involved in mitochondrial fusion and fission. Dysregulation of these proteins might impair mitochondria homeostasis and lead to the pathogenesis of various diseases, including Alzheimer's disease. However, the detailed mechanism underlying mitochondrial morphology regulation is still obscure and requires further elucidation. We have previously found that the MCP member appoptosin can mediate Aβ neurotoxicity during Alzheimer's disease and that a reduction of appoptosin can abolish Aβ-induced mitochondrial fragmentation. Here, we have further shown that overexpression of appoptosin directly resulted in mitochondrial fragmentation. However, although overexpression of appoptosin can cause ROS overproduction and caspase activation, we found that neither reduction of ROS nor inhibition of caspase activity affected appoptosin-induced mitochondrial fragmentation. These results confirm that mitochondrial fragmentation is an early event during cell death (Karbowski et al., 2002).
Although the levels of mitochondrial fission (DRP1 and FIS1) and fusion (OPA1, MFN1 and MFN2) proteins have been shown to be altered in the brains of individuals with Alzheimer's disease (Manczak et al., 2011; Wang et al., 2009), we found that overexpression of appoptosin did not affect their protein levels. Rather, we found that appoptosin interacted with MFN1 and MFN2, as well as with the mitochondrial ubiquitin ligase MITOL but not DRP1, FIS1 or OPA1. In addition, we found that overexpression of appoptosin decreased the hetero-interaction between MFN1 and MFN2, and caused a reduction of mitochondrial fusion activity. Because the fusion activity of MFN1–MFN2 heterotypic oligomers is comparable to those of homo-oligomers of MFN1 or MFN2 (Detmer and Chan, 2007), overexpression of appoptosin might interfere with the fusion machinery by impairing the interaction between MFN1 and MFN2, thus leading to mitochondrial fragmentation. Interestingly, considerable efforts have been devoted to studying the fission machinery deficits in Alzheimer's disease, with a focus on DRP1, and findings show that excessive ROS increases DRP1 activity (Iqbal and Hood, 2014; Wu et al., 2011), that Aβ interacts with DRP1 and increases its activity (Manczak et al., 2011), that S-nitrosylation of DRP1 enhances its activity (Cho et al., 2009) and that phosphorylation of DRP1 at specific sites promotes its activity (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Merrill et al., 2011; Meuer et al., 2007; Rambold et al., 2011; Rehman et al., 2012; Taguchi et al., 2007). In contrast, research on the fusion machinery during Alzheimer's disease is limited. Hence, our findings suggest that a defect in the fusion machinery could be as equally important during Alzheimer's disease, which deserves further scrutiny.
Appoptosin-overexpression-induced mitochondrial fragmentation and apoptosis were at least partially reversed upon co-expression with MFN1, MITOL or the dominant-negative form of DRP1, DRP1K38A. These results further support the idea of targeting mitochondria dynamics as a therapeutic strategy for Alzheimer's disease. Interestingly, although appoptosin also interacted with the mitochondrial fusion protein MFN2, co-expression with MFN2 had a stronger effect on apoptosis induction than appoptosin overexpression alone. This is possibly because MFN2 is also involved in various signaling cascades and acts as a pro-apoptotic and anti-proliferative protein (Guo et al., 2007; Papanicolaou et al., 2011; Shen et al., 2007; Wang et al., 2015). Indeed, here, we also found that overexpression of MFN2 alone resulted in apoptosis.
Appoptosin belongs to the MCP family, whose members are primarily located in the inner membrane of mitochondria and shuttle metabolites, nucleotides, and cofactors between cytoplasm and mitochondrial matrix (Palmieri, 1994). However, to our knowledge, we are the first to show that a member of the MCP family (i.e. appoptosin) can regulate mitochondrial morphology. To study whether other MCPs also regulate mitochondrial morphology, we investigated SLC25A26, another MCP that is closely related to appoptosin based on their protein sequence similarity. However, although we found that SLC25A26 interacted with MITOL, overexpression of SLC25A26 had no effect on mitochondrial morphology. Because SLC25A26 does not interact with MFN1 or MFN2, it is possible that interaction with MFN1 and/or MFN2 is a prerequisite for appoptosin-induced mitochondrial fragmentation. To address this, we generated several appoptosin deletion constructs of its mitochondrial carrier domains (Fig. S4A,B). However, although these appoptosin deletion mutants did not interact with MFN1 and/or MFN2 (data not shown), their overexpression also resulted in mitochondrial fragmentation and apoptosis (Fig. S4C–E). We noticed that these overexpressed appoptosin deletion mutants localized all over the cell body rather than specifically in mitochondria; therefore, we speculate that these deletion mutants induce mitochondrial fragmentation and cell death through a different mechanism (possibly by overwhelming the ubiquitin–proteasome system) than that of full-length appoptosin. This deserves further scrutiny.
Taken together, our study newly identifies a function of the pro-apoptotic protein appoptosin in regulating mitochondrial morphology, which might strengthen our understanding of the interplay between mitochondrial morphology and apoptosis, as well as the pathology of Alzheimer's disease.
MATERIALS AND METHODS
Cells, plasmids, antibodies and reagents
Maintenance of HeLa cells (Wang et al., 2006), HEK 293T cells and primary neurons derived from mouse at embryonic day 17.5 (Zhang et al., 2012) have been described previously. HeLa cells and HEK 293T cells were originally purchased from American Type Culture Collection (ATCC). The Myc–appoptosin expression plasmid has also been described previously (Zhang et al., 2012). MFN1, MFN2, DRP1, FIS1, MITOL and DRP1K38A plasmids were constructed using pCMV-HA, pCMV-Myc or pEGFP-C1 as backbones. mtGFP and mtRFP were generated by fusing GFP or RFP to mitochondria-localized COX8, following the previously reported procedure (Partikian et al., 1998).
Anti-appoptosin antibody (ab133614) and rabbit anti-HA antibody (ab9110) were from Abcam, UK; anti-Myc antibody (clone 9E10, SC-40) was from Santa Cruz Biotechnology; mouse anti-HA antibody was from Abmart (26D11), China; anti-MFN1 (13798-1-AP) and anti-FIS1 (10956-1-AP) antibodies were from Proteintech; anti-MFN2 (WH0009927M3-100UG) antibody was from Sigma-Aldrich; anti-actin (4970), anti-cleaved PARP (5625), anti-COX IV (4850) and anti-DRP1 antibodies (8570) were from Cell Signaling. All the primary antibodies were used at 1:1000 dilution. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H+L) secondary antibody (31460), HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody (31430), Alexa-Fluor-488-conjugated goat anti-mouse IgG (A-11017) and Alexa-Fluor-594-conjugated goat anti-rabbit IgG (A-11012) were purchased from Thermo Fisher Scientific.
RSV was from Bio Basic, Canada; NAC, Z-VAD, 2,7-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), propidium iodide and 4′,6-diamidino-2-phenylindole (DAPI) were from Sigma-Aldrich; MitoTracker Red was purchased from Life Technologies; protease and phosphatase inhibitor cocktails were obtained from Roche.
Plasmid transfection
Plasmids were transfected into cells by using Turbofect (Thermo Fisher Scientific) by following the manufacturer's instructions.
PEG cell fusion assay
HeLa cells were first transfected with (i) pCMV-Myc+mtGFP, (ii) pCMV-Myc+mtRFP, (iii) Myc–appoptosin+mtGFP or (iv) Myc–appoptosin+mtRFP. Then pCMV-Myc+mtGFP-transfected cells were mixed with pCMV-Myc+mtRFP-expressing cells, and Myc–appoptosin+mtGFP-expressing cells were mixed with Myc–appoptosin+mtRFP-expressing cells. After co-culture on cover slips for 24 h, cells were washed with PBS once, treated with pre-warmed 40% PEG 1500 for 5 min, washed with PBS five times and cultured in cycloheximide-containing medium for 7 h. Cells were then fixed with 4% paraformaldehyde, counterstained with DAPI and observed by using microscopy.
Mitochondria isolation
Fractionation of mitochondria and the cytosol was performed by using a cell mitochondria isolation kit (Beyotime, China), following the manufacturer's instructions. Briefly, cells were pelleted by centrifuging at 600 g for 5 min at 4°C; cells were then re-suspended in mitochondria isolation solution with protease inhibitor cocktail and homogenized on ice using a tight-fitting pestle attached to a homogenizer. The homogenate was then centrifuged at different speeds to isolate intact cells, mitochondria and cytosol fractions.
Immunofluorescence microscopy
MitoTracker Red was added into cell medium 30 min before fixation. Cells were then fixed in 4% paraformaldehyde saline for 15 min at room temperature, permeabilized, immunostained with the indicated antibodies, incubated with fluorescence-conjugated secondary antibodies, counterstained with DAPI and visualized under a two-photon microscope (FV10-MEP, Olympus, Japan).
Statistics
Results were expressed as means±s.e.m. using GraphPad Prism 5 software. Unpaired t-test was used to assess statistical significance in two groups.
Acknowledgements
We thank all members at the Facility Core of College of Medicine, Xiamen University for their technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
C.Z., P.E.F., H.X. and Y.Z. conceived and designed the experiments; C.Z., Z.S., L.Z., Z.Z., X.Z. and G.L. performed the experiments; C.Z., G.B., H.X. and Y.Z. analyzed and interpreted the data; C.Z. and Y.Z. wrote the paper.
Funding
This work was supported, in part, by grants from National Natural Science Foundation of China [grant numbers 81225008, 81161120496, 91332112, 91332114 and U1405222]; Canadian Institutes of Health China-Canada Alzheimer's disease Initiative [grant number TAD-117950]; the National Institutes of Health [grant numbers R01AG021173, R01AG038710, R01AG044420 and R01NS046673]; Fujian Provincial Department of Science and Technology [grant number 2015Y4008]; and Xiamen University President Fund [grant number 20720150170]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.176792/-/DC1
References
- Agrimi G., Di Noia M. A., Marobbio C. M. T., Fiermonte G., Lasorsa F. M. and Palmieri F. (2004). Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 379, 183-190. 10.1042/bj20031664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi M. V. and Fuhrmann N. (2013). Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol. Neurodegener. 8, 32 10.1186/1750-1326-8-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S. L. (2013). Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236-2251. 10.1056/NEJMra1215233 [DOI] [PubMed] [Google Scholar]
- Chang C.-R. and Blackstone C. (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583-21587. 10.1074/jbc.C700083200 [DOI] [PubMed] [Google Scholar]
- Chen H., Detmer S. A., Ewald A. J., Griffin E. E., Fraser S. E. and Chan D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189-200. 10.1083/jcb.200211046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D.-H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z. and Lipton S. A. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102-105. 10.1126/science.1171091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs J. T. and Strack S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939-944. 10.1038/sj.embor.7401062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer S. A. and Chan D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405-414. 10.1083/jcb.200611080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S., Gaume B., Bergmann-Leitner E. S., Leitner W. W., Robert E. G., Catez F., Smith C. L. and Youle R. J. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515-525. 10.1016/S1534-5807(01)00055-7 [DOI] [PubMed] [Google Scholar]
- Gomes L. C. and Scorrano L. (2008). High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta 1777, 860-866. 10.1016/j.bbabio.2008.05.442 [DOI] [PubMed] [Google Scholar]
- Guernsey D. L., Jiang H., Campagna D. R., Evans S. C., Ferguson M., Kellogg M. D., Lachance M., Matsuoka M., Nightingale M., Rideout A. et al. (2009). Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 41, 651-653. 10.1038/ng.359 [DOI] [PubMed] [Google Scholar]
- Guo X., Chen K.-H., Guo Y., Liao H., Tang J. and Xiao R.-P. (2007). Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ. Res. 101, 1113-1122. 10.1161/CIRCRESAHA.107.157644 [DOI] [PubMed] [Google Scholar]
- Haitina T., Lindblom J., Renström T. and Fredriksson R. (2006). Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 88, 779-790. 10.1016/j.ygeno.2006.06.016 [DOI] [PubMed] [Google Scholar]
- Iqbal S. and Hood D. A. (2014). Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell Physiol. 306, C1176-C1183. 10.1152/ajpcell.00017.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Eura Y. and Mihara K. (2004). Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 117, 6535-6546. 10.1242/jcs.01565 [DOI] [PubMed] [Google Scholar]
- James D. I., Parone P. A., Mattenberger Y. and Martinou J.-C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 278, 36373-36379. 10.1074/jbc.M303758200 [DOI] [PubMed] [Google Scholar]
- Jheng H.-F., Tsai P.-J., Guo S.-M., Kuo L.-H., Chang C.-S., Su I.-J., Chang C.-R. and Tsai Y.-S. (2012). Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 32, 309-319. 10.1128/MCB.05603-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Li Y., Zhang X., Bu G., Xu H. and Zhang Y. W. (2014). Trafficking regulation of proteins in Alzheimer's disease. Mol. Neurodegener. 9, 6 10.1186/1750-1326-9-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannengiesser C., Sanchez M., Sweeney M., Hetet G., Kerr B., Moran E., Fuster Soler J. L., Maloum K., Matthes T., Oudot C. et al. (2011). Missense SLC25A38 variations play an important role in autosomal recessive inherited sideroblastic anemia. Haematologica 96, 808-813. 10.3324/haematol.2010.039164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M., Lee Y. J., Gaume B., Jeong S. Y., Frank S., Nechushtan A., Santel A., Fuller M., Smith C. L. and Youle R. J. (2002). Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159, 931-938. 10.1083/jcb.200209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M., Neutzner A. and Youle R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71-84. 10.1083/jcb.200611064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott A. B., Perkins G., Schwarzenbacher R. and Bossy-Wetzel E. (2008). Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505-518. 10.1038/nrn2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T., Detmer S. A., Kaiser J. T., Chen H., McCaffery J. M. and Chan D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858-862. 10.1126/science.1099793 [DOI] [PubMed] [Google Scholar]
- Lemere C. A. (2013). Immunotherapy for Alzheimer's disease: hoops and hurdles. Mol. Neurodegener. 8, 36 10.1186/1750-1326-8-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M., Palacin M. and Zorzano A. (2009). Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 89, 799-845. 10.1152/physrev.00030.2008 [DOI] [PubMed] [Google Scholar]
- Manczak M., Mao P., Calkins M. J., Cornea A., Reddy A. P., Murphy M. P., Szeto H. H., Park B. and Reddy P. H. (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 20 Suppl. 2, S609-S631. 10.3233/JAD-2010-100564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M., Calkins M. J. and Reddy P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495-2509. 10.1093/hmg/ddr139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill R. A., Dagda R. K., Dickey A. S., Cribbs J. T., Green S. H., Usachev Y. M. and Strack S. (2011). Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 9, e1000612 10.1371/journal.pbio.1000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuer K., Suppanz I. E., Lingor P., Planchamp V., Göricke B., Fichtner L., Braus G. H., Dietz G. P. H., Jakobs S., Bähr M. et al. (2007). Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 14, 651-661. 10.1038/sj.cdd.4402087 [DOI] [PubMed] [Google Scholar]
- Nakamura N., Kimura Y., Tokuda M., Honda S. and Hirose S. (2006). MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 7, 1019-1022. 10.1038/sj.embor.7400790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M., Zunino R., Gangaraju S., Rippstein P. and McBride H. (2005). Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 280, 25060-25070. 10.1074/jbc.M501599200 [DOI] [PubMed] [Google Scholar]
- Palmieri F. (1994). Mitochondrial carrier proteins. FEBS Lett. 346, 48-54. 10.1016/0014-5793(94)00329-7 [DOI] [PubMed] [Google Scholar]
- Papanicolaou K. N., Khairallah R. J., Ngoh G. A., Chikando A., Luptak I., O'Shea K. M., Riley D. D., Lugus J. J., Colucci W. S., Lederer W. J. et al. (2011). Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 31, 1309-1328. 10.1128/MCB.00911-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.-Y., Nguyen O. T. K., Kang H. and Cho H. (2014). MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 5, e1172 10.1038/cddis.2014.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partikian A., Ölveczky B., Swaminathan R., Li Y. and Verkman A. S. (1998). Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J. Cell Biol. 140, 821-829. 10.1083/jcb.140.4.821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts K. R., Yoon Y., Krueger E. W. and McNiven M. A. (1999). The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 10, 4403-4417. 10.1091/mbc.10.12.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold A. S., Kostelecky B., Elia N. and Lippincott-Schwartz J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 108, 10190-10195. 10.1073/pnas.1107402108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman J., Zhang H. J., Toth P. T., Zhang Y., Marsboom G., Hong Z., Salgia R., Husain A. N., Wietholt C. and Archer S. L. (2012). Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26, 2175-2186. 10.1096/fj.11-196543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva A. A., Borges M. M., Madeira M. D., Tavares M. A. and Paula-Barbosa M. M. (1985). Mitochondrial abnormalities in cortical dendrites from patients with Alzheimer's disease. J. Submicrosc. Cytol. 17, 459-464. [PubMed] [Google Scholar]
- Shen T., Zheng M., Cao C., Chen C., Tang J., Zhang W., Cheng H., Chen K.-H. and Xiao R.-P. (2007). Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 282, 23354-23361. 10.1074/jbc.M702657200 [DOI] [PubMed] [Google Scholar]
- Smirnova E., Shurland D.-L., Ryazantsev S. N. and van der Bliek A. M. (1998). A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143, 351-358. 10.1083/jcb.143.2.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N., Ishihara N., Jofuku A., Oka T. and Mihara K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521-11529. 10.1074/jbc.M607279200 [DOI] [PubMed] [Google Scholar]
- Wang R., Zhang Y.-W., Zhang X., Liu R., Zhang X., Hong S., Xia K., Xia J., Zhang Z. and Xu H. (2006). Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 20, 1275-1277. 10.1096/fj.06-5839fje [DOI] [PubMed] [Google Scholar]
- Wang X., Su B., Siedlak S. L., Moreira P. I., Fujioka H., Wang Y., Casadesus G. and Zhu X. (2008). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 105, 19318-19323. 10.1073/pnas.0804871105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Su B., Lee H.-g., Li X., Perry G., Smith M. A. and Zhu X. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 29, 9090-9103. 10.1523/JNEUROSCI.1357-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xie Q., Zhou X., Yao J., Zhu X., Huang P., Zhang L., Wei J., Xie H., Zhou L. et al. (2015). Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 358, 47-58. 10.1016/j.canlet.2014.12.025 [DOI] [PubMed] [Google Scholar]
- Westermann B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872-884. 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- Wu S., Zhou F., Zhang Z. and Xing D. (2011). Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 278, 941-954. 10.1111/j.1742-4658.2011.08010.x [DOI] [PubMed] [Google Scholar]
- Yonashiro R., Ishido S., Kyo S., Fukuda T., Goto E., Matsuki Y., Ohmura-Hoshino M., Sada K., Hotta H., Yamamura H. et al. (2006). A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 25, 3618-3626. 10.1038/sj.emboj.7601249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R. J. and van der Bliek A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062-1065. 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Zhang Y.-w., Chen Y., Huang X., Zhou F., Wang W., Xian B., Zhang X., Masliah E., Chen Q. et al. (2012). Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J. Neurosci. 32, 15565-15576. 10.1523/JNEUROSCI.3668-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]