

Abstract
Background and Purpose
N‐arachidonyl dopamine (NADA) has been identified as a putative endocannabinoid, but there is little information about which signalling pathways it activates. The purpose of this study was to identify the signalling pathways activated by NADA in vitro.
Experimental Approach
Human or rat cannabinoid CB1 receptors were expressed in AtT20, CHO or HEK 293 cells. NADA displacement of radiolabelled cannabinoids, and CB1 receptor mediated activation of K channels or ERK phosphorylation, release of intracellular calcium ([Ca]i) and modulation of adenylyl cyclase were measured in addition to NADA effects on CB1 receptor trafficking.
Key Results
At concentrations up to 30 μM, NADA failed to activate any signalling pathways via CB1 receptors, with the exception of mobilization of [Ca]i. The elevations of [Ca]i were insensitive to pertussis toxin, and reduced or abolished by blockers of Gq/11‐dependent processes including U73122, thapsigargin and a peptide antagonist of Gq/11 activation. Prolonged NADA incubation produced modest loss of cell surface CB1 receptors. The prototypical cannabinoid agonist CP55940 signalled as expected in all assays.
Conclusions and Implications
NADA is an ineffective agonist at most canonical cannabinoid receptor signalling pathways, but did promote mobilization of [Ca]i via Gq‐dependent processes and some CB1 receptor trafficking. This signalling profile is distinct from that of any known cannabinoid, and suggests that NADA may have a unique spectrum of effects in vivo. Our results also indicate that it may be possible to identify highly biased CB1 receptor ligands displaying a subset of the pharmacological or therapeutic effects usually attributed to CB1 ligands.
Abbreviations
- 2‐AG
2‐arachidonoyl glycerol
- AtT20‐rCB1
mouse pituitary tumour cells stably transfected with HA‐tagged rat CB1 receptors
- [Ca]i
intracellular calcium
- CHO‐hCB1
CHO cells stably transfected with HA‐tagged human CB1 receptors
- FSK
forskolin
- GIRK (Kir3)
G protein gated inwardly rectifying K channel
- HA‐hCB1
haemagglutinin‐tagged human CB1 receptor
- HA‐rCB1
haemagglutinin‐tagged rat CB1 receptor
- PBS‐T
PBS supplemented with 0.2% Tween
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| β2‐adrenoceptor | Adenylyl cyclase (AC) |
| CB1 receptor | ERK1 |
| CB2 receptor | ERK2 |
| Ion channels b | |
| GIRK (Kir3) | |
| TRPV1 |
| LIGANDS | |
|---|---|
| 2‐AG | Forskolin (FSK) |
| Arachidonic acid | NADA |
| ATP | SR141716A |
| Bradykinin | Thapsigargin |
| Capsaicin | U73122 |
| CP55940 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,2013b,2013c).
Introduction
Cannabinoid CB1 and CB2 receptors have important roles in modulating neuronal and immune system function. CB1 receptors are expressed throughout the brain in presynaptic compartments and generally act there to modify the release of other neurotransmitters (Mackie, 2005; Lovinger, 2008). Endogenous cannabinoid tone controlling moment‐to‐moment communication has been recognized at many synapses, but persistent elevation of endocannabinoid levels is also observed following significant pathophysiological stimuli (Hohmann et al., 2005). The principle endocannabinoids are 2‐arachidonoyl glycerol (2‐AG) and anandamide, although candidate molecules such as virodhamine, noladin ether and NADA have also been identified (Alexander and Kendall, 2007).
CB1 receptors are unusually pleiotropic in their signalling. Although principally exerting their actions through Gi/Go‐type G proteins, CB1 receptors can also couple to Gs to stimulate AC activity (Glass and Felder, 1997), and to Gq to mobilize intracellular calcium ([Ca]i) via phospholipase C (PLC) (Lauckner et al., 2005). The physiological consequences of this abundance of potential signalling avenues for CB1 receptors remains an area of intense investigation, particularly in light of the idea that different ligands may preferentially activate one set of effectors over another (Hudson et al., 2010; Laprairie et al., 2014; Khajehali et al., 2015)
NADA was identified as a CB1 agonist in vitro (Bisogno et al., 2000) and subsequently identified in vivo (Huang et al., 2002; Bradshaw et al., 2006), although much subsequent work has focussed on its activity as an agonist of TRPV1, ion channels found in both brain and peripheral sensory neurons. In brain, NADA is most abundant in regions containing the cell bodies or terminals of dopaminergic neurons (Huang et al., 2002; Bradshaw et al., 2006; Hu et al., 2009), and NADA has been reported to modulate GABAergic neurotransmission via both CB1‐ and TRPV1‐dependent mechanisms (Marinelli et al., 2007, Fawley et al., 2014; Freestone et al., 2014).
The initial report of NADA activity at CB1 receptors measured changes in [Ca]i in N18TG2 neuroblastoma cells, with NADA producing an elevation of [Ca]i similar to that of the synthetic cannabinoid HU‐210. The elevations of [Ca]i were blocked by the CB1 antagonist SR141716A. However, the mechanism of NADA‐induced elevations of [Ca]i in N18 cells was not determined. Surprisingly, the report of Bisogno et al. (2000) remains the only cellular study of the signalling mechanisms potentially underlying NADA actions. In this study, we have examined NADA signalling through recombinant CB1 receptors expressed in several cell lines. Intriguingly, we found that NADA did not modulate K channels, AC or ERK phosphorylation in cell lines expressing CB1 receptors; in fact, the only signalling pathway activated by NADA was an elevation of [Ca]i mediated by pertussis toxin‐insensitive G proteins. Thus, the endocannabinoid NADA appears to have a highly pathway‐biased CB1 receptor signalling profile, unique amongst the cannabinoid agonists described to date.
Methods
Cell culture
Experiments utilized human or rat CB1 tagged at the N‐terminus with three haemagglutinin sequences [haemagglutinin‐tagged human CB1 receptor (HA‐hCB1) and haemagglutinin‐tagged rat CB1 receptor (HA‐rCB1)] stably transfected into HEK 293, CHO or AtT20 cells. HEK 293‐hCB1 (Cawston et al., 2013) cells were used for AC, phosphoERK, internalization and binding assays. CHO cells stably transfected with HA‐tagged human CB1 receptors (CHO‐hCB1) (Grimsey et al., 2010) were used for measurements of changes in [Ca]i, cAMP and receptor internalization. AtT20‐rCB1 cells (Mackie et al., 1995) were used for assays of G protein‐gated inwardly rectifying K channel (GIRK)‐mediated hyperpolarization. HEK 293 FLPIn‐TREx cells transfected with hTRPV1 were used to assess the efficacy of NADA at a known effector. N18TG2 cells (obtained from the European Collection of Cell Cultures) were used for assays of [Ca]i. N18TG2, HEK 293 and AtT20 cells were cultivated in DMEM supplemented with 10% FBS, 100 U penicillin and 100 µg streptomycin ⋅mL−1. HEK 293‐hCB1 cell media contained zeocin, 250 µg⋅mL−1, media for AtT20 cells was supplemented with 400 µg⋅mL−1 G418. HEK 293‐hTRPV1 cells were grown in media supplemented with hygromycin (150 µg⋅mL−1) and blasticidin (10 µg⋅mL−1). CHO‐hCB1 cells were cultivated in DMEM/F12‐HAM media supplemented as for HEK 293‐HA‐hCB1. Cells were maintained in 5% CO2 at 37°C in a humidified atmosphere. Cells were grown in 75 mm2 flasks and passaged when 80–90% confluent.
Binding assays
HEK 293‐hCB1 cells were grown to 90–100% confluence in 175 cm2 flasks and harvested in ice‐cold PBS with 5 mM EDTA. Cells were centrifuged at 200 × g for 10 min and the pellet frozen at −80°C until required. Pellets were thawed with Tris‐sucrose buffer (50 mM Tris‐HCl, pH 7.4, 200 mM sucrose, 5 mM MgCl2, 2.5 mM EDTA) and homogenized with a glass homogenizer. The homogenate was centrifuged at 1000 × g for 10 min at 4°C and the pellet discarded. The supernatant was then centrifuged at 27 000 × g for 30 min at 4°C. The final pellet was resuspended in a minimal volume of Tris‐sucrose buffer, aliquoted and stored at −80°C. Protein concentration was determined using the DC protein assay kit (Bio‐Rad, Hercules, CA, USA) following the manufacturer's protocol. Membranes (20 µg per point) were resuspended in binding buffer (50 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, 0.2% (w v‐1) BSA; ICP Bio, New Zealand, pH 7.4) and incubated with either [3H]‐CP55940 (2.5 nM) or [3H]‐SR141716A (1 nM, both PerkinElmer, Waltham, MA, USA) and a range of NADA concentrations at 30°C for 60 min. Non‐specific binding was determined in the presence of 1 μM SR141716A. GF/C Harvest Plates (PerkinElmer) were pre‐soaked in 0.1% polyethylenimine and then washed with 100 μL ice‐cold wash buffer (50 mM HEPES pH 7.4 500 mM NaCl, 0.1% BSA) before filtration of samples, which were then subjected to three additional 200 μL washes in ice‐cold wash buffer. Harvested plates were dried overnight at 24°C, 50 μL of scintillation fluid was added to each well and plates were read 30 min later for 2 min per well in a Microbeta Trilux (PerkinElmer).
Intracellular calcium measurements
[Ca]i was measured with the Calcium 5 kit from Molecular Devices (Sunnyvale, CA, USA) using a FlexStation 3 Microplate Reader (Molecular Devices), as outlined in Redmond et al. (2014). Briefly, N18TG2, CHO‐hCB1 or HEK 293‐hTRPV1 cells from an 80–90% confluent 75 mm2 flask were resuspended in L‐15 medium supplemented with 1% FBS, 100 U penicillin and 100 µg streptomycin mL−1 and plated in 96‐well black‐walled plates (Corning, Castle Hill, Australia). Cells were incubated overnight in humidified room air at 37°C. hTRPV1 expression was induced with 1 µg⋅mL−1 tetracycline at least 4 h before the assay. Calcium 5 dye dissolved in a HBS containing (in mM): NaCl 140, KCl 5.33, CaCl2 1.3, MgCl2 0.5, HEPES 22, Na2HPO4 0.338, NaHCO3 4.17, KH2PO4 0.44, MgSO4 0.4, glucose 10, probenicid 2.5 (pH to 7.3, osmolarity 330 ± 5 mosmol) was loaded into each well and incubated at 37°C for at least 1 h before the assay. Fluorescence was measured every 2 s (λexcitation = 485 nm, λemission = 525 nm). Drugs were dissolved in HBS and added after at least 2 min of baseline recording in volumes of 20–50 μL. Assays were carried out at 37°C unless otherwise noted. NADA produced small elevations of [Ca]i in untransfected CHO cells. These changes in [Ca]i were similar to those produced by equivalent concentrations of arachidonic acid in both wild‐type and CHO‐hCB1 cells, and were not sensitive to inhibitors of PLC (Felder et al., 1993; data not shown). When group responses are expressed as % change in RFU, data were corrected by subtracting the non‐specific changes in [Ca]i seen in wild‐type CHO cells. These effects were less than 30% of the equivalent response following CB1 receptor activation. On rare occasions, CHO‐hCB1 cells failed to respond to cannabinoids with an elevation of [Ca]i. The reasons for this are unknown, but may reflect unusually high densities of plated cells.
K channel measurements in AtT20 cells
Changes in membrane potential were determined using the blue membrane potential dye (Molecular Devices) in a FlexStation 3, as outlined in Knapman et al. (2013). AtT20‐rCB1 cells from an 80–90% confluent 75 mm2 flask were resuspended in L‐15 medium supplemented with 1 % FBS, 100 U penicillin and 100 µg streptomycin mL−1 and plated in 96‐well black‐walled plates in a volume of 100 μL per well. Cells were incubated overnight in humidified room air at 37°C. Membrane potential dye was dissolved in a modified HBS where KCl was omitted and loaded into each well and incubated at 37°C for at least 1 h before the assay. Fluorescence was measured every 2 s (λexcitation = 530 nm, λemission = 565 nm). Assays were carried out at 37°C, and drugs were added in volumes of 25–50 μL after at least 2 min of baseline recording.
cAMP measurement
Cellular cAMP levels were measured as previously described (Cawston et al., 2013). Briefly, the pcDNA3L‐His‐CAMYEL plasmid (ATCC, Manassas, VA, USA) was transfected into HEK 293‐hCB1 cells using linear polyethyleneimine (m.w. 25 kDa) (Polysciences, Warrington, PA, USA). Twenty‐four hours after transfection cells were re‐plated in poly‐l‐lysine (0.2 mg⋅mL−1 in PBS) (Sigma‐Aldrich, St Louis, MO, USA) coated with CulturPlate™‐96 (PerkinElmer) at a density of 55 000–80 000 cells per well. After 24 h, cells were serum‐starved in HBSS containing 1 mg⋅mL−1 BSA, pH 7.4 for 30 min before assay. Five minutes before the addition of drug or vehicle dissolved in HBSS plus 1 mg⋅mL−1 BSA cells were treated with 5 μM coelenterazine‐h (Promega, Madison, WI, USA). Emission signals were detected simultaneously at 460/25 nM (RLuc) and 560/25 nM (YFP), immediately following drug addition with a Victor‐Lite plate reader (PerkinElmer) at 37°C. Raw data are presented as an inverse BRET ratio of emission at 460/535 so that an increase in ratio correlates with an increase in cAMP production.
Modulation of forskolin (FSK)‐stimulated AC activity in CHO‐hCB1 cells was measured as described in detail in Knapman et al. (2014). In CHO cells, stimulation of AC hyperpolarizes cells, this can be reversed by agonists for Gi/o‐coupled receptors. Briefly, CHO‐hCB1 cells were prepared for membrane potential measurements using the blue membrane potential dye as outlined above. After 2 min of baseline recording, FSK was added to the cells in a volume of 20 μL, either with or without NADA or CP55940. Changes in membrane potential were measured 5 min after drug addition. In some experiments, cells were treated overnight with 200 ng⋅mL−1 pertussis toxin.
CB1 receptor cell surface expression
Surface hCB1 receptor expression was determined by utilizing a live cell antibody feeding technique and quantified via the ImageXpress Micro XLS automated fluorescent microscope (Molecular Devices) as previously described (Grimsey et al., 2008). In brief, HEK 293‐hCB1 or CHO‐hCB1 cells were seeded at 30 000 or 22 000 cells per well (respectively) in poly‐l‐lysine treated 96‐well, flat bottom clear plates (Nunc, Roskilde, Denmark). Untransfected HEK 293 and CHO cells and HEK 293 cells transfected with HA‐tagged human β2 adrenoceptors were also used in control experiments and treated equivalently. Approximately 24 h later, cells were equilibrated in DMEM (HEK 293‐hCB1) or DMEM/F12‐HAM (CHO‐hCB1) supplemented with 1 mg⋅mL−1 BSA (assay media) for 30 min at 37°C. For experiments assessing CB1 receptor internalization with 1 h stimulation, cells were subsequently incubated with anti‐mouse monoclonal HA11 primary antibody (MMS‐101P, Covance, Princeton, NJ, USA) diluted 1:500 in assay media at 37°C for 30 min. After one wash with assay media, NADA at various concentrations and/or CP55940 at its approximate EC80 for internalization (1 nM for HEK 293‐hCB1, 3.2 nM for CHO‐hCB1; data not shown) were applied for 60 min at 37°C. Following drug incubation, plates were cooled rapidly on ice to prevent any further receptor trafficking, then incubated with Alexa Fluor® 488‐conjugated goat anti‐mouse secondary antibody (Life Technologies, Mulgrave, Victoria, Australia) diluted 1:300 in assay media at room temperature for 30 min. Cells were washed once in assay media, fixed with 4% paraformaldehyde and stained with Hoechst 33258 (Life Technologies) diluted 1:500 in PBS with 0.2% Triton‐X (PBS‐T). For 6 h concentration‐response and timecourse experiments, cells were treated as above (specific time points and drug concentrations noted in the text and figures) with the exception that primary antibody incubation was not carried out before drug incubation. Instead, the primary antibody was applied at the conclusion of drug incubation and incubated for 30 min at room temperature after rapidly cooling of the plates on ice to prevent further receptor trafficking. After being washed with assay media and fixed with paraformaldehyde, secondary antibody was incubated for 3 h at room temperature (diluted 1:400 in immunobuffer, PBS‐T with 1% normal goat serum and 0.4 mg⋅mL−1 merthiolate; Merck, Darmstadt, Germany). Cells were then washed with PBS‐T before Hoechst staining as described above.
Images of the cells were acquired with a ImageXpress Micro XLS microscope (10× objective, four images per well) and experimental effects quantified using MetaMorph (v.7.8.0.0, Molecular Devices) by calculating the intensity of fluorescent labelling above background per cell (Grimsey et al., 2008).
ERK1/2 measurements
AlphaScreen® SureFire® Phospho(p)ERK1/2(Thr202/Tyr204) assay kits (PerkinElmer) were utilized following the manufacturer's protocol; 40 000 HEK 293‐hCB1 cells per well were seeded into poly‐l‐lysine treated 96‐well plates (Corning) and incubated at 37°C, 5% CO2 and 95% humidity for 24 h. Cells were serum‐starved in 50 μL DMEM supplemented with 1 mg⋅mL−1 BSA overnight before drug treatment. All drugs were added at 2× concentration in DMEM‐BSA and incubated for the times indicated. Assay plates were put onto ice, media/drug removed and 30 μL of lysis buffer added followed by agitation of the plates for 10 min at RT. Cell lysate (5 μL) was transferred into a white 96‐well low volume plate (PerkinElmer) and 7 μL detection mix was added. Plates were sealed, wrapped with foil and incubated for 2–4 h at RT, and fluorescent signals detected on an EnSpire reader (PerkinElmer) using the manufacturer‐defined AlphaScreen settings.
Data analysis
Analysis for all experiments was performed with GraphPad Prism (Version 5.02, GraphPad Software, Inc., La Jolla, CA, USA) and SigmaPlot (v.11.0, Systat Software, Chicago, IL, USA). Binding assay data were analysed by fitting a one‐site competition curve, and K i was calculated using the previously determined K d of 2.5 nM [3H]‐CP55940 or 1 nM [3H]‐SR141716A. For ERK 1/2 phosphorylation data, statistical significance was determined at 5 min. Raw data for the cAMP assays were usually fitted with one‐phase association curves for each replicate, and plateau values were obtained. In the case of NADA attenuation of FSK‐stimulated cAMP by CP55940, data were analysed using ‘area under the curve’ analysis in GraphPad Prism. Paired t‐tests were used when comparing two datapoints, one‐way anova repeated measures for more than two data points with one independent variable. The response to drugs in assays of [Ca]i and membrane potential was expressed as a percentage change over the baseline averaged for 30 s immediately before drug addition. Changes produced by parallel solvent blanks were subtracted before normalization. Concentration–effect data were fit to a four‐parameter logistic Hill equation to derive the EC50 values. A two‐way anova was used to analyse the effect of NADA on CP55940 concentration–effect curves generated in this assay. Unless otherwise noted, data represent the mean ± SEM of at least five independent experiments, each conducted in duplicate or triplicate.
Statistical significance was defined as P < 0.05.
Drugs and reagents
HEK 293 FlpInTRex‐hTRPV1 cells were kind gift from Peter McIntyre and were generated as described for cells expressing rTRPV1 (Veldhuis et al., 2012). Drugs were made up in ethanol or DMSO and diluted to give a final concentration of solvent of 0.05–0.1%. U73122 and Gq Palpeptide were made fresh before each use and were diluted in DMSO and water respectively. NADA and other endocannabinoids were purchased from Biomol (Plymouth Meeting, PA, USA), Cayman Chemicals (Ann Arbor, MI, USA) or Ascent Scientific, (Bristol, UK). Palpeptides were custom synthesized by Genscript (Piscataway, NJ, USA). Other drugs were from Tocris Cookson (Bristol, UK) or Cayman Chemicals unless otherwise noted. Cell culture media, buffers, antibiotics and general chemicals were from Life Technologies, Sigma‐Aldrich or InvivoGen (San Diego, CA, USA).
Results
NADA has been reported to act as a CB1 agonist in tissue and cell lines derived from both rats and mice (Bisogno et al., 2000; Marinelli et al., 2007; Fawley et al., 2014). Therefore, we initially examined its ability to modify canonical CB1 receptor Gi/o‐mediated signalling. The classical CB1 agonist CP55940‐induced concentration‐dependent GIRK‐mediated hyperpolarization (pEC50 8.2 ± 0.3) in AtT20‐rCB1 cells. However, when NADA (30–100 μM) was applied to AtT20‐rCB1, it did not produce a change in membrane potential by itself, nor did it modify the GIRK‐mediated hyperpolarization produced by the CB1 agonist CP55940 (pEC50 8.0 ± 0.3 in the presence of 30 μM NADA, Figure 1, two‐way anova, significant effect of CP55940 concentration, P < 0.0001, no effect of NADA, P = 0.36).
Figure 1.
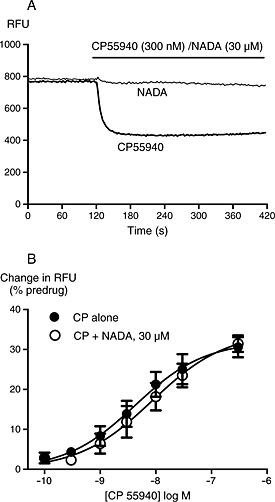
NADA does not activate K channels in AtT20 cells. GIRK activation was determined as described in the Methods. (A) Raw traces showing changes in fluorescent signal from the membrane potential dye (raw fluorescence units, RFU) in AtT20‐rCB1 cells during application of CP55940 but not NADA. Drug was added for the duration of the bar; the traces are representative of at least five independent experiments. (B) Concentration‐response curves for CP55940 activation of GIRK in AtT20‐rCB1 cells in the presence and absence of 30 μM NADA. Data represent the mean ± SEM of at least five independent experiments performed in duplicate; pooled data were fit with a four‐parameter logistic equation. There was no difference in the potency or maximal effect of CP55940 between control conditions or in the presence of NADA.
CB1 receptors couple very efficiently to inhibition of AC activity, and in some circumstances to stimulation of the enzyme (Glass and Felder, 1997; Bonhaus et al., 1998). Therefore, we examined the ability of NADA to modify cAMP in HEK 293‐hCB1 cells. At concentrations up to 10 μM, NADA did not modify basal (P = 0.777–0.940, n = 7) or FSK‐stimulated (P = 0.390–0.976, n = 3) cAMP production in HEK 293‐hCB1 cells. At 32 μM, NADA did not modify basal cAMP production (Figure 2A); however, when co‐applied with 10 μM FSK, it modestly but significantly increased cAMP levels above that of FSK alone (P < 0.001, n = 10) (Figure 2B). To ensure this was receptor‐mediated, NADA was tested in HEK 293 wild‐type cells; in these cells, NADA (32 μM) also produced a small increase in FSK‐stimulated cAMP accumulation (P = 0.011, n = 3), suggesting that these effects were occurring via a mechanism unrelated to CB1 receptor activity. NADA was then tested for its ability to antagonize CP55940‐mediated inhibition of cAMP. Consistent with a weak interaction of NADA with hCB1 receptors in these conditons, 32 μM NADA blocked the inhibition of FSK‐stimulated cAMP activity by CP55940 (10 nM) (P = 0.003, n = 4) (Figure 2C), while 10 μM NADA was without effect (P = 0.15). It is, however, difficult to know how much of the effect of 32 μM NADA was due to reversal of CB1 receptor‐mediated inhibition of FSK‐stimulated AC activity by CP55940 or due to the modest CB1 receptor‐independent stimulation of AC activity by NADA. Finally, we examined if NADA could inhibit SR141716A‐mediated inverse agonism. SR141716A (40 nM) produced an increase in cAMP in the presence of FSK which was not affected by inclusion of NADA (1–32 μM, P = 0.627–0.985, n = 5).
Figure 2.
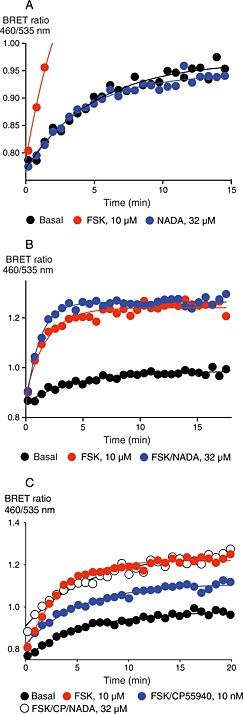
NADA does not modulate cAMP accumulation via CB1 receptors in HEK 293‐hCB1 cells. Cellular cAMP levels were determined as outlined in the Methods. Figures are representative time plots of BRET emissions expressed as a ratio. Each point represents the mean of duplicate determinations. (A) NADA does not affect basal levels of cAMP, while FSK produces a robust increase (note truncated axis). Data are representative of four independent experiments. (B) NADA (32 μM) modestly but significantly enhances FSK‐stimulated cAMP levels (P < 0.001). Data are representative of 10 independent experiments. (C) CP55940 (10 nM) inhibits FSK‐stimulated cAMP accumulation, and this inhibition was occluded by co‐application of NADA (32 μM).
Another ubiquitous signalling pathway for CB1 receptors is stimulation of ERK1/2 phosphorylation. While generally CB1 agonists produce a pertussis toxin‐sensitive pERK1/2 response, CB1 receptor‐mediated G‐protein independent responses have also been measured (Ahn et al., 2013); therefore, we examined if NADA could activate either of these pathways. As expected, in HEK 293‐hCB1 cells, CP55940 produced a robust stimulation of pERK, with an EC50 at 5 min of 1.7 ± 0.7 nM (n = 4), administration of CP55940 at approximate EC90 (15 nM) produced a significant increase in ERK1/2 phosphorylation between 2 and 10 min after application, with a peak increase at 4–5 min (Figure 3, n = 4). NADA (30 μM) did not stimulate ERK 1/2 phosphorylation at any time (Figure 3, repeated measures anova showed that at 4 min, there was a significant difference in pERK phosphorylation between CP55940 and vehicle (P = 0.033) and CP55940 and NADA (P = 0.021), but not between NADA and vehicle P = 0.503).
Figure 3.
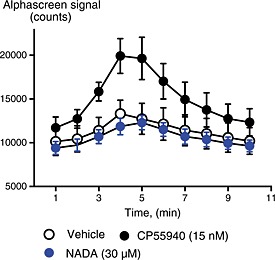
NADA does not modulate ERK phosphorylation via CB1 receptors in HEK 293‐hCB1 cells. ERK1/2 phosphorylation was determined as outlined in the Methods. The time plot represents the fluorescent signal arising from phosphorylated ERK1/2 measured every minute after drug addition (T = 0). CP55940 (P = 0.0.33) but not NADA (P = 0.503) produced a significant increase in ERK1/2 phosphorylation compared with vehicle at 5 min. The data presented are the mean ± SEM of four experiments, each performed in duplicate.
These data from multiple cell types suggested a weak interaction of NADA with human CB1 receptors, so we confirmed that NADA did indeed bind to hCB1 receptors using radioligand binding assays. NADA displaced both [3H]‐CP55940 (K i 780 ± 240 nM) and [3H]‐SR141716A (K i 230 ± 36 nM) from hCB1 receptors (n = 4), with an affinity similar to that previously reported for rat brain receptors (Bisogno et al., 2000).
Our data suggest that NADA does not readily activate Gi/o or Gs‐coupled signalling pathways via CB1 receptors. However, some cannabinoids can couple to mobilization of [Ca]i via Gq (Lauckner et al., 2005), so more in hope than expectation we examined this. We were able to detect small elevations of [Ca]i by NADA in HEK 293‐rCB1 cells (not shown), but the modest nature of the effect led us to examine a range of other cell lines. In CHO‐hCB1 cells, NADA elevated [Ca]i in a concentration‐dependent manner, with a maximum increase in fluorescence of 138 ± 21% at the highest concentration of NADA tested (100 μM, Figure 4, n = 8). The elevation of [Ca]i by NADA (100 μM) was completely prevented by pre‐incubation with SR141716A (1 μM, Figure 4, n = 5). High concentrations of the prototypic synthetic cannabinoid agonists CP55940 (300 nM, 37 ± 12%) and WIN55212 (10 μM, 40 ± 8%) also produced elevations of [Ca]i in CHO‐hCB1 cells, but to a much lesser extent. As this is a considerably less potent effect than reported in N18TG2 cells, we attempted to reproduce the original report of NADA‐mediated elevations of [Ca]i in these cells (Bisogno et al., 2000). NADA (30 μM) produced a maximum change in fluorescence of 6 ± 5% in N18TG2 cells when experiments were performed at 37°C (Figure 4, n = 6). By contrast, ATP (100 μM) and bradykinin (1 μM) produced increases of 51 ± 32% and 44 ± 36% respectively (Figure 4, n = 5–6). When experiments were performed at 25°C, a temperature similar to that of the original report, the increases in fluorescence produced by NADA, ATP and BK were 5 ± 6%, 56 ± 19% and 30 ± 9% respectively (n = 3–5). NADA robustly elevated [Ca]i in HEK 293‐hTRPV1 cells, indicating that NADA was active under the experimental conditions used to probe activity at CB1 receptors (Supporting Information Fig. S1).
Figure 4.
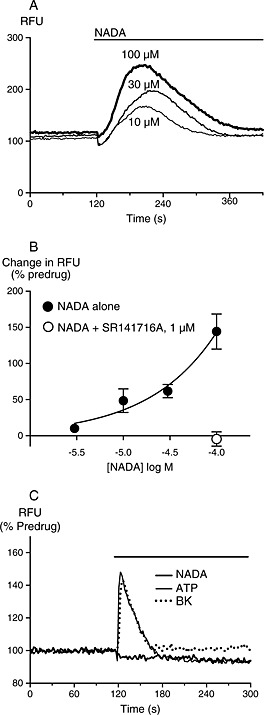
NADA elevates [Ca]i in CHO‐hCB1 but not N18TG2 cells. Changes in [Ca]i were determined as outlined in the Methods. (A) Raw traces showing increases in the fluorescent signal from the calcium 5 dye (raw fluorescence units, RFU) in CHO‐hCB1 cells during application of NADA. Drug was added for the duration of the bar, and the traces are representative of at least five independent experiments. (B) Concentration‐response relationship for NADA elevation of [Ca]i in CHO‐hCB1 cells. Data represent the mean ± SEM of at least five independent experiments performed in duplicate or triplicate. (C) Traces illustrating the effects of NADA, ATP and bradykinin on [Ca]i in N18TG2 cells, drugs were added for the duration of the bar and the traces are representative of at least five independent experiments.
The increase in [Ca]i elicited by NADA in CHO‐hCB1 cells was independent of Gi/o as it was unaffected in pertussis toxin‐treated hCB1 cells (Figure 5A and B). Pretreating the CHO‐hCB1 cells with the PLC‐pathway inhibitor U73122 (3 μM) strongly inhibited the elevation of [Ca]i by NADA 100 μM (P < 0.05, Figure 5A and B). Pre‐incubation of the CHO‐hCB1 cells with thapsigargin (10 μM), a sarco/endoplasmic reticulum Ca pump inhibitor which depletes intracellular Ca pools, completely occluded any elevation of [Ca]i by NADA (100 μM), confirming that NADA is mobilizing Ca from intracellular pools.
Figure 5.
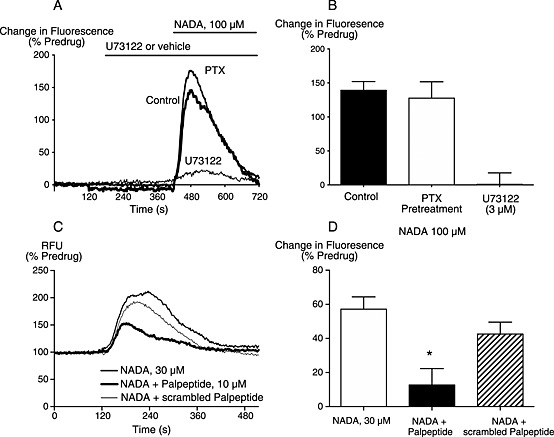
NADA elevates [Ca]i in a manner consistent with activation of Gq. Changes in [Ca]i were measured as described in the Methods. (A) Traces from a representative experiment showing that elevations of [Ca]i were inhibited by U73122, but not affected by pretreatment with pertussis toxin overnight. (B) Summary of the data from five similar experiments, expressed as mean ± SEM. (C) Traces from a representative experiment showing that elevations of [Ca]i were attenuated by a peptide antagonist of Gq (palpeptide) but not by the scrambled palpeptide control. (D) Summary of the data from five similar experiments, the Gq palpeptide significantly inhibited the elevations of [Ca]i in response to 30 μM NADA, (P = 0.006).
There are few readily available compounds to pharmacologically disrupt the coupling of GPCRs to Gq. We used a palmitoylated peptide (palmitoyl‐QLNLKEYNLV) (Robbins et al., 2006), corresponding to the last 10 amino acids of Gq, to test the involvement of Gq in the NADA‐evoked elevation of [Ca]i. Pre‐incubation of the Gq palpeptide (10 μM) for an hour before the administration of NADA (30 μM) resulted in significant inhibition of the NADA elevation of [Ca]I (P = 0.006), (Figure 5C and D). Pre‐incubation with a scrambled version of the palpeptide (palmitoyl‐NLVLNEKLYQ) did not significantly affect the increase in [Ca]i produced by NADA (30 μM, Figure 5C and D, P = 0.18).
As the NADA effects were only readily observed in CHO cells, we carried out additional experiments to determine if NADA could signal through Gi/o‐type G proteins in CHO‐hCB1. We examined the CB1 receptor‐mediated inhibition of FSK‐stimulated AC activity in intact CHO cells using an assay of membrane potential. We have previously reported that CHO cells hyperpolarize in response to AC activation, and this hyperpolarization can be reversed by activation of the Gi/Go‐coupled μ‐opioid receptor (Knapman et al., 2014). FSK‐hyperpolarized CHO‐hCB1 cells with a pEC50 of 6.7 ± 0.1, and a maximum change in fluorescence of 23 ± 2% (Figure 6A, n = 4). CP55940 (30 nM) completely inhibited the hyperpolarization produced by FSK (1 μM); cells hyperpolarized by 20 ± 1% in response to FSK, and by −3 ± 2% in response to FSK and CP55940 together (n = 11, Figure 6B). By contrast, NADA (30 μM) did not affect the FSK‐induced hyperpolarization in CHO‐hCB1 cells (Figure 6C, P = 0.4, n = 6). The effects of CP55940 were abolished after treatment of the cells overnight with pertussis toxin (200 ng⋅mL−1, Figure 6D).
Figure 6.
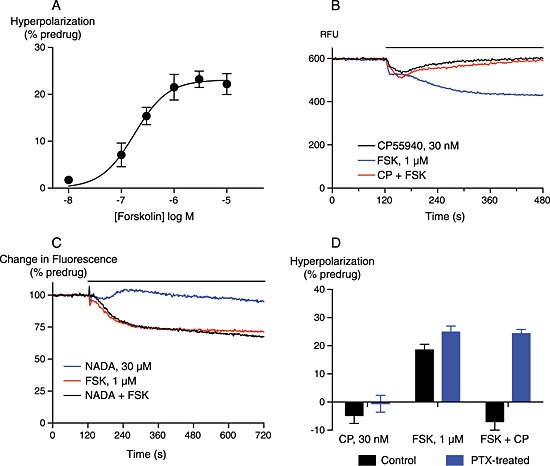
NADA does not modulate adenylyl cyclase activity in CHO‐hCB1 cells. Adenylyl cyclase activity was determined using a membrane potential assay, as outlined in the Methods. (A) A concentration‐response relationship for FSK hyperpolarization of CHO‐hCB1 cells. FSK‐hyperpolarized cells with a pEC50 of 6.7. Data were fit with a four‐parameter logistic equation, and each point represents the mean ± SEM of four independent experiments performed in duplicate. (B) A representative experiment showing raw fluorescence traces (RFU) of FSK‐induced hyperpolarization of CHO‐hCB1 cells, and its reversal by CP55940. Drugs were added for the duration of the bar. (C) Normalized traces from a representative experiment (of 12) showing the lack of effect of NADA (30 μM) on the FSK‐induced hyperpolarization. The solvent blank trace has been subtracted from this data. (D) A bar chart summarizing the inhibitory activity of CP55940 (30 nM) on FSK‐stimulated hyperpolarization of CHO‐hCB1 cells, in control conditions and after overnight treatment of cells with 200 ng⋅mL−1 pertussis toxin (PTX). The bars represent the mean ± SEM of five or six independent experiments, each performed in duplicate or quadruplicate.
Receptor internalization is a signalling pathway which is generally activated by agonists, regardless of which signalling pathway they are activating; therefore, we examined whether NADA modified cell surface hCB1 receptor levels. At concentrations up to 32 μM, NADA failed to change the levels of cell surface hCB1 receptors in HEK 293 cells over 60 min (P = 0.137–0.936, n = 3, Figure 7A). Although 100 μM NADA induced significant CB1 receptor internalization (29 ± 2%, P < 0.006, n = 3), this was not blocked by CB1 antagonist (SR141716A 100 nM, P = 0.602, n = 3) indicating that this was unlikely to represent a CB1 receptor‐mediated effect. As previously reported (Grimsey et al., 2008), incubation with an approximate EC80 concentration of CP55940 (1 nM) stimulated internalization of CB1 receptors, with only 17 ± 7% receptors remaining on the cell surface after 60 min. Co‐application of NADA with CP55940 antagonized CP55940‐induced internalization of CB1 receptors, with a notional pIC50 of 4.4 ± 0.2 (n = 3; sigmoidal curves fitted with upper plateau constrained to 100%, Figure 7A), consistent with the ability of NADA to displace CP55940 at CB1 receptors. When applied for 6 h, NADA down‐regulated surface hCB1 receptors in HEK 293 cells with pEC50 4.49 ± 0.01 and maximum reduction in surface expression of 76 ± 5% at 100 μM NADA (Figure 7B). To investigate whether this reduction in surface expression was specific to CB1 receptors, we assessed the effect of NADA on HEK 293 cells expressing the hβ2 adrenoceptor, which is not expected to bind NADA. NADA at 32 μM produced a change in hβ2 adrenoceptor expression (P = 0.03, n = 3); however, the extent of the down‐regulation was small in comparison with the effect on CB1 receptors at the same concentration (β2 adrenoceptor 13 ± 3%, CB1 receptor 54 ± 5%, n = 3). A substantial reduction was observed at 100 μM NADA (48 ± 3%; P = 0.018, n = 3, Figure 7B). We also noted a small but significant reduction in cell count (14 ± 3%, P = 0.006, n = 3) indicating that cell viability may have been adversely affected at this high concentration; 100 μM NADA produced a similar effect on cell number in untransfected HEK 293 cells (11 ± 2% reduction in cell number). These data suggest that 100 μM NADA has non‐specific effects on cell viability and receptor trafficking. However, as it appeared that 32 μM NADA induced a specific reduction of cell surface CB1 receptors, evident following 6 h but not 1 h of stimulation, considerably slower than would be typical for agonist stimulation (Grimsey et al., 2008), we were curious to determine the rate at which this occurred. Time course experiments (Figure 7C) revealed that the initiation of down‐regulation of surface CB1 receptors appeared to be considerably delayed, and the subsequent rate of CB1 receptor loss was also slower in comparison with that stimulated by 10 nM CP55940. Qualitatively, similar results were found in CHO‐hCB1 cells indicating that the stimulation of Gq in these cells did not alter the internalization of the receptor (data not shown).
Figure 7.
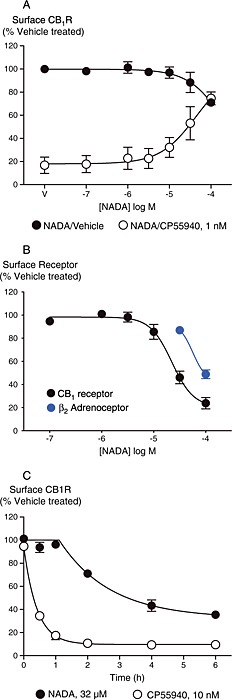
NADA modulates CB1 receptor surface expression in HEK 293‐hCB1 cells. CB1 surface expression was determined as outlined in the Methods. Data represent the mean ± SEM of three independent experiments performed in duplicate and normalized to vehicle control (V). (A) When incubated for 1 h, NADA alone induced significant internalization only at the highest concentration tested (100 μM, P < 0.006). 1 nM CP55940 induced internalization which was inhibited with co‐incubation of NADA (pIC50 4.4 ± 0.2). (B) Incubation for 6 h with NADA induced a down‐regulation of surface CB1 receptors (pEC50 4.49 ± 0.01), and also loss of cell surface β2‐adrenoceptors. (C) Timecourse of 32 μM NADA and 10 nM CP55940 effects on the expression of surface CB1 receptors.
Discussion
The principal finding of this study is that NADA, a putative endogenous agonist for CB1 receptors, couples effectively to a limited range of effectors in cells expressing recombinant human or rat CB1 receptors. We were unable to detect any coupling to Gi/Go‐ or Gs‐mediated processes, but we did observe elevations of [Ca]i consistent with Gq‐mediated stimulation of phospholipase C and subsequent mobilization of thapsigargin‐sensitive intracellular Ca stores. Recombinant rCB1 receptors can couple through Gq in HEK 293 cells (Lauckner et al., 2005), and while WIN55212 has been reported to be the most effective agonist at this pathway, no CB ligands with appreciable Gq‐selectivity have previously been identified. The only previous CB1 receptor‐mediated signalling pathway identified for NADA was an elevation of [Ca]i following activation of native mouse CB1 receptors in N18TG2 cells (Bisogno et al., 2000). Although the G protein mediating this effect was not identified in that study, others using related NG108‐15 neuroblastoma cells indicate that this elevation of [Ca]i is probably mediated by PTX‐sensitive G proteins (Sugiura et al., 1997). We were unable to reproduce the NADA‐mediated elevations of [Ca]i reported by Bisogno et al. (2000); this probably reflects the quite different assay conditions. In the original study, the N18TG2 cells were acutely treated with trypsin and resuspended in a continuously stirred cuvette where drugs were added and measurements of [Ca]i made. By contrast, the cells in our study were attached to a tissue culture plate and drugs added very gently after at least an hour of equilibration between dye and cells within the Flexstation. Although we do not know which differences are crucial, we note that it has been previously reported that CB1 receptors can couple to elevations of [Ca]i when added under conditions where a Gq‐coupled receptor is activated (Marini et al., 2009). Stirring cells in a cuvette has also been reported to produce a release of substances such as ATP which act in this manner to permit Gi/Go‐coupled receptor elevations of [Ca]i (Okajima et al., 1993). It is thus possible that NADA was acting as a CB1 agonist in a situation where there was an ongoing Gq‐coupled receptor activity permissive for pertussis toxin‐sensitive elevations of [Ca]i.
The effects of NADA on [Ca]i were not very potent, with robust elevations only occurring at concentrations of 30 μM and above. In N18TG2 cells, NADA mobilized Ca via CB1 receptors with an EC50 of about 700 nM (Bisogno et al., 2000), while in brain slices significant effects of NADA on synaptic transmission have been reported at concentrations between 1 and 10 μM (Marinelli et al., 2007, Fawley et al., 2014). The affinity of NADA for native rCB1 was reported to be 250 nM (vs. [3H]‐SR141716A, Bisogno et al., 2000); in the present study, the K i for NADA was 230 nM versus [3H]‐SR141716A, and 780 nM for the agonist [3H]‐CP55940. Thus, the mobilization of [Ca]i we observed occurred at much higher concentrations than effects in brain slices and in radioligand binding assays, notwithstanding the potential differences arising from the different assays. Given that 30 μM NADA had no effect at all on other signalling pathways we measured, our data suggest either that NADA couples more efficiently to Gq in nerve terminals than in whole CHO cells (or that the consequences of small changes in [Ca]i in nerve terminals are magnified as effects on neurotransmitter release), or that the NADA modulation of neurotransmitter release in brain slices does not involve modulation of AC, ERK or GIRK activity and proceeds through other mechanisms.
We have provided evidence consistent with NADA utilizing Gq‐family G proteins to mediate elevation of [Ca]i, but we do not have direct evidence. The elevation of [Ca]i was not mediated via pertussis toxin‐sensitive G proteins, ruling out mechanisms observed in NG108‐15 cells. The NADA‐mediated elevations of [Ca]i were disrupted by U73122, an antagonist of Gq‐PLC signalling in a number of systems, including CB1 receptor‐mediated Gq coupling (Lauckner et al., 2005). The elevations of [Ca]i produced by NADA were also occluded by thapsigargin, an agent which depletes intracellular Ca stores. Finally, the elevations of [Ca]i were largely occluded by a Gq palpeptide, a palmitoylated peptide corresponding to the last 10 amino acids of Gq that is purported to act as a competitive inhibitor of Gq‐PLC interactions (Robbins et al., 2006). Taken together, these data strongly suggest an involvement of Gq in NADA signalling via CB1 receptors, at least in CHO cells.
Mobilization of Ca by native CB1 receptors has been observed occasionally (Sugiura et al., 1997; Bisogno et al., 2000; McIntosh et al., 2007; Marini et al., 2009), but the physiological consequences of this remain largely unknown. Ca is a second messenger that can modulate many cellular processes through activation of protein kinases, phosphatases and transcription factors, while changes in [Ca]i can also activate or inhibit ion channels to alter cellular excitability. The mechanism(s) of action of NADA modulation of neurotransmitter release in native neurons are unknown, beyond demonstration of the involvement of CB1 receptors and/or TRPV1 (Price et al., 2004; Marinelli et al., 2007, Fawley et al., 2014; Freestone et al., 2014). Given that NADA does not seem to readily couple to conventional second messengers such as AC, ERK or K channels, it is an intriguing possibility that the effects of NADA can in some cases be mediated by NADA/CB1/Gq coupling, perhaps by 2‐AG or AEA generated as a result of the elevations of [Ca]i or liberation of fatty acids. Evidence to support the idea of NADA‐mediated elevations of [Ca]i being relevant in actions on neurons comes from the observation that NADA‐stimulated CGRP release from trigeminal ganglion neurons in conditions where NADA‐mediated TRPV1 activation is completely blocked (Price et al., 2004). However, putative NADA‐dependent cannabinoid tone in the substantia nigra was not sensitive to inhibition of 2‐arachidonoyl glycerol synthesis (Freestone et al., 2014), indicating that NADA did not elevate 2‐AG levels via a Gq‐dependent process in this brain region. In preliminary experiments in CHO‐hCB1 cells, we found that brief NADA exposure (up to 10 min) did indeed cause a modest elevation in levels of 2‐AG; however, the effects of NADA were not sensitive to SR141716A and persisted in wild‐type CHO cells (data not shown). These actions could reflect NADA inhibition of monoacylglycerol lipase, a principle catabolic enzyme for 2‐AG (Bjorkland et al., 2010).
NADA acts via cannabinoid receptor‐dependent mechanisms in non‐neuronal cells (O'Sullivan et al., 2004; Wilhelmsen et al., 2014), but there is also no information as to the signalling pathways used by NADA in these situations. A plethora of non‐CB receptor, non‐TRPV1‐mediated effects of NADA have been reported, including direct actions on ion channels, lipid‐modifying enzymes, transcription factors and possibly other GPCR (e.g. Ross et al., 2009; Bjorkland et al., 2010; Soler‐Torronteras et al., 2014; reviewed in Connor et al., 2010). None of these effects are likely to lead to rapid elevations of [Ca]i, nor are they likely to acutely interfere with conventional CB1 receptor signalling pathways.
Evidence is emerging for ligand bias at CB1 receptors, particularly with respect to arrestin recruitment and activation of pathways downstream of this such as ERK (Laprairie et al., 2014; Khajehali et al., 2015). The only coupling we could observe with NADA appeared to be via Gq, suggesting that NADA is a highly biased agonist. Although WIN55212 has been reported to be the most efficient cannabinoid at promoting elevations of [Ca]i via Gq (Lauckner et al., 2005; McIntosh et al., 2007), unlike NADA, WIN55212 also couples effectively to Gi/Go and Gs (Bonhaus et al., 1998; Glass and Northrup, 1999; Laprairie et al., 2014). In some cell types, all CB1 agonists can promote Gq‐dependent signalling (Laprairie et al., 2014), although this is not always observed (McIntosh et al., 2007). However, it is not possible to quantify bias for NADA, as we were unable to detect coupling to other pathways. The lack of any other in vitro reports of NADA coupling to CB1 receptors suggests measuring them may not be straightforward, and we await confirmation of our results with interest. It remains possible that NADA can couple to more commonly observed CB1 receptor signal transduction cascades, perhaps in the presence of as yet unidentified regulators of CB1 receptor coupling.
It is important to note that positive controls were used in all experiments, and the robust activity of the cannabinoid agonists CP55940 or WIN55212 indicated that the CB1 receptors were functional during these experiments, notwithstanding the many papers we (and others) have published using these receptor constructs and signalling assays. Furthermore, we measured NADA activation of hTRPV1 under the same conditions as our assays of [Ca]i in CHO‐hCB1, HEK 293‐hCB1 and N18TG2 cells, and NADA activated the channel in a manner consistent with previous reports (Huang et al., 2002; Sutton et al., 2005), indicating that it was not metabolized too rapidly to act. The assays we used are well characterized and can reliably detect the activity of lower efficacy CB1 agonists such as Δ9 tetrahydrocannabinol and anandamide (Banister et al., 2013; Cawston et al., 2013).
Despite the apparent lack of coupling to canonical CB1 receptor signalling pathways and low potency for elevating elevation of [Ca]i, the ability of NADA to displace orthosteric CB1 ligands (Bisogno et al., 2000, this study) indicates that NADA may be able to influence the activity of other cannabinoids by acting as a competitive antagonist. Indeed, NADA inhibited internalization of CB1 receptors induced by CP55940. Although this effect also occurred with fairly low potency, it illustrates the potential for NADA to subtly influence endocannabinoid tone by antagonizing canonical signalling pathways while simultaneously inducing Ca signalling. Interestingly, although NADA did not stimulate rapid internalization of CB1 receptors as usually expected of CB1 agonists (Grimsey et al., 2008), the delayed down‐regulation of surface CB1 receptors was induced with a similar potency to the inhibition of CP55940‐stimulated internalization. Thus, as well as the potential for competitive inhibition, the prolonged presence of NADA down‐regulates surface CB1 receptors and is likely to functionally desensitize surface CB1 receptor‐mediated signalling. The apparent lag to the start of down‐regulation and subsequent slow decay is reminiscent of what is observed with application of low concentrations of efficacious agonists (Grimsey et al., 2008). NADA may inefficiently stabilize a CB1 receptor conformation that is recognized by internalization machinery, resulting in a slow internalization rate. Alternatively, NADA may influence other aspects of cell function or endocannabinoid tone, as discussed previously. A very high NADA concentration (100 μM) produced non‐CB1 receptor‐mediated reductions in surface expression of the β2 adrenoceptor and cell density, both of which probably reflect reduced cell viability.
In summary, NADA appears to represent a CB1 ligand that preferentially accesses conformations of the receptor which activate Gq, suggesting that it might be possible to develop analogous drugs. NADA may also represent a useful tool for investigating the consequences of CB1 receptor‐Gq coupling in native systems. Our data do not directly address the importance of NADA as an endogenous cannabinoid, but the high concentrations of NADA required to stimulate detectable activation of any conventional G protein‐coupled pathways suggests that if endogenous NADA acts widely as an endocannabinoid, it does so through as yet unidentified signalling mechanisms. Despite evidence for the endogenous production of NADA (Huang et al., 2002; Bradshaw et al., 2006, Freestone et al., 2014), its role as an endocannabinoid remains unclear.
Author contributions
All authors performed experiments and contributed to the analysis and interpretation the data. W. J. R. and M. C. largely wrote the paper; all authors have seen a final copy of the Ms.
Conflict of interest
The authors declare that they have no conflicts of interest related to this work.
Supporting information
Figure S1 NADA activates hTRPV1 expressed in HEK 293 cells. Changes in [Ca]i were determined as outlined in the Methods. A) Traces showing increases in the fluorescent signal from the calcium 5 dye in HEK 293‐hTRPV1 cells during application of NADA or capsaicin. Drug was added for the duration of the bar, the traces are representative of at least 7–8 independent experiments. B) Concentration response relationship for NADA and capsaicin elevation of [Ca]i in HEK 293‐hTRPV1 cells. Data represents the mean ± SEM of 7–8 independent experiments performed in duplicate or triplicate.
Supporting info item
Redmond, W. J. , Cawston, E. E. , Grimsey, N. L. , Stuart, J. , Edington, A. R. , Glass, M. , and Connor, M. (2016) Identification of N‐arachidonoyl dopamine as a highly biased ligand at cannabinoid CB1 receptors. British Journal of Pharmacology, 173: 115–127. doi: 10.1111/bph.13341.
References
- Ahn KW, Mahmoud MM, Shim J‐Y, Kendall DA (2013). Distinct roles of β‐arrestin 1 and β‐arrestin 2 in ORG27569‐induced biased signalling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem 288: 9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al. (2013b). The concise guide to pharmacology 2013/14: ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013c). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kendall DA (2007). The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol 152: 602–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banister S, Wilkinson S, Longworth M, Stuart J, Apetz N, English K et al. (2013). The synthesis and pharmacological evaluation of adamantane‐derived indoles: novel cannabimimetic drugs of abuse. ACS Chem Neurosci 4: 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Melck D, Bobrov MY, Gretskaya NM, Bezuglov VV, De Petrocellis L et al. (2000). N‐acyl‐dopamines: novel synthetic CB1 cannabinoid‐receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo . Biochem J 351: 817–814. [PMC free article] [PubMed] [Google Scholar]
- Bjorkland E, Noren E, Nilsson J, Fowler CJ (2010). Inhibition of monoacylglycerol lipase by troglitazone, N‐arachidonoyl dopamine and the irreversible inhibitor JZL184: comparison of two different assays. Br J Pharmacol 161: 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR (1998). Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist‐specific trafficking of intracellular responses. J Pharmacol Exp Ther 287: 884–888. [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Krey JF, Walker JM (2006). Sex and hormonal cycle differences in rat brain levels of pain‐related cannabimimetic lipid mediators. Am J Physiol Regul Integr Comp Physiol 291: R349–R358. [DOI] [PubMed] [Google Scholar]
- Cawston EE, Redmond WJ, Breen C, Grimsey N, Connor M, Glass M (2013). Real‐time characterisation of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol 170: 893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Vaughan CW, Vandenberg R (2010). N‐Acyl amino acids and N‐acyl neurotransmitter conjugates: neuromodulators and probes for new drug targets. Br J Pharmacol 160: 1857–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley JA, Hofmann ME, Andresen MC (2014). Cannabinoid 1 and transient receptor potential vanilloid 1 receptors discretely modulate evoked glutamate separately from spontaneous glutamate transmission. J Neurosci 34: 8324–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA (1993). Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor‐mediated signal transduction. Proc Natl Acad Sci U S A 90: 7656–7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freestone PS, Guatteo E, Piscitelli F, di Marzo V, Lipski J, Mercuri N (2014). Glutamate spillover drives endocannabinoids production and inhibits GABAergic transmission in the Substantia Nigra pars compacta. Neuropharmacology 79: 467–475. [DOI] [PubMed] [Google Scholar]
- Glass M, Felder CC (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17: 5327–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Northrup JK (1999). Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol 56: 1362–1369. [DOI] [PubMed] [Google Scholar]
- Grimsey NL, Narayan PJ, Dragunow M, Glass M (2008). A novel high‐throughput assay for the quantitative assessment of receptor trafficking. Clin Exp Pharm Physiol 35: 1377–1382. [DOI] [PubMed] [Google Scholar]
- Grimsey NL, Graham ES, Dragunow M, Glass M (2010). Cannabinoid receptor 1 trafficking and the role of the intracellular pool: implications for therapeutics. Biochem Pharmacol 80: 1050–1062. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R et al. (2005). An endocannabinoid mechanism for stress‐induced analgesia. Nature 435: 1108–1112. [DOI] [PubMed] [Google Scholar]
- Hu SS, Bradshaw HB, Benton VM, Chen JS, Huang SM, Minassi A et al. (2009). The biosynthesis of N‐arachidonoyl dopamine (NADA), a putative endocannbinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot Essential Fatty Acids 81: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al‐Hayani A, de Petrocellis L, Fezza F et al. (2002). An endogenous capsaicin‐like substance with high potency at recombinant and native vanilloid receptors. Proc Natl Acad Sci U S A 99: 8400–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Hebert TE, Kelly MEM (2010). Ligand‐and heterodimer‐directed signalling of the CB1 cannabinoid receptor. Mol Pharmacol 77: 1–9. [DOI] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K (2015). Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol Pharmacol 88: 368–379. [DOI] [PubMed] [Google Scholar]
- Knapman A, Santiago M, Du YP, Bennallack PR, Christie MJ, Connor M (2013). A continuous, fluorescence‐based assay of mu‐opioid receptor activation in AtT20 cells. J Biomol Screen 18: 269–276. [DOI] [PubMed] [Google Scholar]
- Knapman A, Abogadie F, McIntyre P, Connor M (2014). A real time, fluorescence‐based assay for measuring μ‐opioid receptor modulation of adenylyl cyclase activity in Chinese hamster ovary cells. J Biomol Screen 19: 223–231. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly MEM, Dupre DJ, Denovan‐Wright EM (2014). Type 1 cannabinoid receptor ligands display functional selectivity in cell culture model of striatal medium spiny projection neurons. J Biol Chem 289: 24845–24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K (2005). The cannabinoid agonist WIN55,212‐2 increases intracellular calcium via CB1 receptor coupling to Gq proteins. Proc Natl Acad Sci U S A 102: 19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM (2008). Presynaptic modulation by endocannabinoids In: Sudhoff TC. (ed). Pharmacology of Neurotransmitter release Starke K. Handb Exp Pharm 184. Springer: Berlin, pp. 435–477. [DOI] [PubMed] [Google Scholar]
- Mackie K (2005). Distribution of cannabinoid receptors in the central and peripheral nervous system In: Pertwee RG. (ed). Cannabinoids Handb Exp Pharm 168. Springer: Berlin, pp. 299–325. [DOI] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R (1995). Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q‐type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptors. J Neurosci 15: 6552–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Florenzano F, Fezza F, Viscomi MT, van der Selt M et al. (2007). N‐Arachidonoyl‐dopamine tunes synaptic transmission onto dopaminergic neurons by activating both cannabinoid and vanilloid receptors. Neuropsychopharmacology 32: 298–308. [DOI] [PubMed] [Google Scholar]
- Marini P, Moriello AS, Cristino L, Palmery M, De Petrocellis L, Di Marzo V (2009). Cannabinoid CB1 receptor elevation of intracellular calcium in neuroblastoma SH‐SY5Y cells: interactions with muscarinic and δ‐opioid receptors. Biochem Biophys Acta 1793: 1289–1303. [DOI] [PubMed] [Google Scholar]
- McIntosh BT, Hudson B, Yergorova S, Jollimore CAB, Kelly MEM (2007). Agonist‐dependent cannabinoid receptor signalling in human trabecular network cells. Br J Pharmacol 152: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okajima F, Tomura H, Kondo Y (1993). Enkephalin activates the phospholipase C/Ca2 + system through cross‐talk between opioid receptors and P2‐purinergic or bradykinin receptors in NG108‐15 cells. Biochem J 290: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD (2004). Characterization of the vasorelaxant properties of the novel endocannabinoid N‐arachidonoyl dopamine (NADA). Br J Pharmacol 141: 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM (2004). Modulation of trigeminal sensory neuron activity by the dual cannabinoid‐vanilloid agonists anandamide, N‐arachidonoyl dopamine and arachidonoyl‐2‐chlorethylamide. Br J Pharmacol 141: 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond WJ, Gu L, Camo M, McIntyre P, Connor M (2014). Ligand determinants of fatty acid activation of the pronociceptive ion channel TRPA1. PeerJ 2: e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, Marsh SJ, Brown DA (2006). Probing the regulation of M (Kv7) potassium channels in intact neurons with membrane‐targeted peptides. J Neurosci 26: 7950–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HR, Gilmore AJ, Connor M (2009). Inhibition of human recombinant T‐type calcium channels by the endocannabinoid arachidonyl dopamine. Br J Pharmacol 156: 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler‐Torronteras R, Lara‐Chica M, Garcia V, Calzado MA, Munoz E (2014). Hypoximimetic activity of N‐acyl‐dopamines. N‐arachidonoyl dopamine stabilizes HIF1α protein through a SIAH2‐dependent pathway. Biochem Biophys Acta 1843: 2730–2743. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kodak T, Kondo S, Tonegawa T, Nakane S, Kishimoto S et al. (1997). 2‐Arachidonoylglycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free Ca2+ in neuroblastoma x glioma hybrid NG108‐15 cells. Biochem Biophys Res Comm 229: 58–64. [DOI] [PubMed] [Google Scholar]
- Sutton KG, Garret EM, Rutter AR, Bonnert TP, Jarolimek W, Seabrook GR (2005). Functional characterization of the S512Y mutant vanilloid human TRPV1 receptor. Br J Pharmacol 146: 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhuis NA, Lew MJ, Abogadie FC, Poole DP, Jennings EA, Ivanusic JJ et al. (2012). N‐glycosylation determiens the ionic permeability and desensitization of the TRPV1 capsaicin receptor. J Biol Chem 287: 21765–21672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Khakpour S, Tran A, Sheehan K, Schumacher M, Xu F et al. (2014). The endocannabinoid/endovanilloid N‐arachidonoyl dopamine (NADA) and synthetic cannabinoid WIN55,212‐2 abate the inflammatory activation of human endothelial cells. J Biol Chem 289: 13079–13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 NADA activates hTRPV1 expressed in HEK 293 cells. Changes in [Ca]i were determined as outlined in the Methods. A) Traces showing increases in the fluorescent signal from the calcium 5 dye in HEK 293‐hTRPV1 cells during application of NADA or capsaicin. Drug was added for the duration of the bar, the traces are representative of at least 7–8 independent experiments. B) Concentration response relationship for NADA and capsaicin elevation of [Ca]i in HEK 293‐hTRPV1 cells. Data represents the mean ± SEM of 7–8 independent experiments performed in duplicate or triplicate.
Supporting info item