

Abstract
Background and Purpose
Hepatitis C virus (HCV) infection is responsible for various chronic inflammatory liver diseases. Here, we have identified a naturally occurring compound with anti‐HCV activity and have elucidated its mode of antiviral action.
Experimental Approach
Luciferase reporter and real‐time RT‐PCR assays were used to measure HCV replication. Western blot, fluorescence‐labelled HCV replicons and infectious clones were employed to quantitate expression levels of viral proteins. Resistant HCV mutant mapping, in vitro NS3 protease, helicase, NS5B polymerase and drug affinity responsive target stability assays were also used to study the antiviral mechanism.
Key Results
A resveratrol tetramer, vitisin B from grapevine root extract showed high potency against HCV replication (EC50 = 6 nM) with relatively low cytotoxicity (EC50 >10 μM). Combined treatment of vitisin B with an NS5B polymerase inhibitor (sofosbuvir) exhibited a synergistic or at least additive antiviral activity. Analysis of a number of vitisin B‐resistant HCV variants suggested an NS3 helicase as its potential target. We confirmed a direct binding between vitisin B and a purified NS3 helicase in vitro. Vitisin B was a potent inhibitor of a HCV NS3 helicase (IC50 = 3 nM). In vivo, Finally, we observed a preferred tissue distribution of vitisin B in the liver after i.p. injection in rats, at clinically attainable concentrations.
Conclusion and Implications
Vitisin B is one of the most potent HCV helicase inhibitors identified so far. Vitisin B is thus a prime candidate to be developed as the first HCV drug derived from natural products.
Abbreviations
- CC50
concentration causing 50% cell toxicity
- DAAs
direct‐acting antivirals
- DARTS
drug affinity responsive target stability
- GO
graphene oxide
- GRE
grapevine root extract
- GT
genotype
- HCV
hepatitis C virus
- IRES
internal ribosome entry site
- NS
nonstructural
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| HO‐1, haem oxygenase‐1 |
| Other proteins b |
| TNF‐α |
| LIGANDS |
|---|
| Boceprevir |
| IL‐6 |
| Resveratrol |
| Sofosbuvir |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (
Introduction
Hepatitis C virus (HCV) infection is responsible for many chronic inflammatory liver diseases, including liver cirrhosis and hepatocellular carcinoma. A recent epidemiological study suggested that approximately 170 million people are infected with HCV (Shepard et al., 2005). In addition, almost half of all liver transplantations performed in the USA are directly related to HCV infection (Mukherjee and Sorrell, 2008). Therefore, HCV‐associated morbidity and mortality impose a severe burden on the healthcare systems of countries with a high rate of HCV infection.
The HCV is a hepatotropic single‐stranded RNA virus. It is classified as a hepacivirus belonging to the Flaviviridae family. Upon entry into a host hepatocyte, internal ribosome entry site (IRES)‐dependent translation of its RNA genome leads to the expression of a ~3000 amino acid polyprotein. This polyprotein undergoes subsequent cleavage into 10 individual viral proteins by host and virus‐encoded proteases (Grakoui et al., 1993b, 1993a). Viral structural proteins such as E1, E2 and core serve as structural components for a mature virion. On the other hand, viral nonstructural (NS) proteins such as NS2, NS3, NS4A, NS4B, NS5A and NS5B build a functional replication complex around ER membranes (Lohmann et al., 1999; Blight et al., 2000; Moradpour et al., 2007). Several HCV replication inhibitors were developed to target NS viral proteins and are in various stages of clinical development. These include NS3 protease (Dvory‐Sobol et al., 2012) helicase inhibitors (Gemma et al., 2011), NS4B inhibitors (Bryson et al., 2010; Cho et al., 2010), NS5A inhibitors (Lee, 2011; Lee et al., 2011) and NS5B polymerases inhibitors (Watkins et al., 2010).
Combined treatment with pegylated interferon (IFN)‐α and ribavirin has served as the standard of care for most HCV patients (Wilby et al., 2012). However, undesirable side effects, including flu‐like symptoms, anaemia, depression and suicidal thoughts, have been consistently associated with this combination therapy. Because of the recent approval of direct‐acting antivirals (DAAs) – including NS3 protease inhibitors (telaprevir and boceprevir), NS5A inhibitors (daclatasvir and ledipasvir) and an NS5B polymerase inhibitor (sofosbuvir) – the current standard of care for HCV patients has moved towards a multiple combination regimen composed of one DAA plus pegylated IFN‐α and ribavirin (Lee, 2013a). In addition, several promising clinical results have been revealed recently, leading to FDA approval of IFN‐free combination treatment using only ledipasvir and sofosbuvir (Everson et al., 2014). However, despite their high efficacy and good safety profiles, DAAs alone are unlikely to play a major role in combating HCV infection in the near future due to their exorbitant cost. There has been heated criticism regarding the extreme high cost of this IFN‐free therapy among HCV patients and activists. Therefore, to develop a more affordable and accessible regimen for the treatment of HCV infection, a new class of anti‐HCV therapeutics with a novel mechanism of action is urgently needed.
The HCV NS3 protein plays an essential role in the viral life cycle. Its N‐terminally encoded protease activity together with a viral scaffold protein, NS4A, is required for efficient cleavage of a viral polyprotein. Therefore, much attention has been devoted to this NS3 protease domain to identify pharmacological inhibitors. Two NS3 protease inhibitors including telaprevir (Matthews and Lancaster, 2012) and boceprevir (Berman and Kwo, 2009) have been already actively used in the clinics as an important component of combination therapy with IFN‐α and ribavirin. In addition, studies using whole animal (Kolykhalov et al., 2000) and replicon models (Lam and Frick, 2006; Mackintosh et al., 2006) have also demonstrated a NS3 helicase as another indispensable component for HCV replication. An HCV helicase resides in the C‐terminal two‐thirds of the HCV NS3 protein. It rearranges nucleic acid duplexes by ATP hydrolysis (Belon and Frick, 2009). The NS3 helicase seems to assist HCV RNA replication by resolving double‐stranded RNA intermediates formed during viral RNA replication (Belon and Frick, 2009). Therefore, much effort has been made to find low MW compounds with a specific inhibitory activity against an HCV NS3 helicase. However, development of HCV NS3 helicase inhibitors has been far more difficult and slower than it has been for other HCV drug targets possibly due to its high homology with other host helicase proteins (Belon and Frick, 2009).
Because plants lack mobility, they need to produce a range of secondary metabolites for defence against invading external pathogens. The grape vine also produces numerous phenolic compounds including one of the best known plant metabolite, resveratrol, for its own protection. Many of grapevine metabolites also turned out to be active against various human viruses including adenovirus (Matias et al., 2010), cytomegalovirus (CMV) (Evers et al., 2004), hepatitis viruses (Sharaf et al., 2012), noroviruses (Li et al., 2012) and rotaviruses (Steven et al., 2011). The mechanism of antiviral action of these polyphenolic compounds mainly depends on their diverse abilities to neutralize cytotoxic oxidants, to inhibit essential host or viral enzymes, to inhibit viral binding and penetration into cells and to trigger the host immune system.
To identify new HCV drug candidates from natural products, we performed a cell‐based screening of Asian herbal plants. We found that a resveratrol tetramer, vitisin B from the grapevine root, exhibited the highest anti‐HCV replication activity. Analysis of several vitisin B‐resistant HCV variants as well as in vitro binding and helicase assays suggested inhibition of the viral helicase NS3 as its mode of action.
Methods
Animal studies
All animal care and experimental procedures followed the guidelines for animal care and protection in Korea and were approved by the Ethics Review Board of the College of Pharmacy, The Catholic University of Korea. Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
An Asian herbal plant library
An Asian herbal plant library, which is composed of 344 traditional plants, was obtained from the Korean plant extract bank (https://extract.kribb.re.kr).
Preparation of enriched oligostilbene extract from Vitis vinifera
The roots of V. vinifera (600 g) were pulverized and extracted with ethanol (5 L) and evaporated under reduced pressure to give an ethanolic extract (43 g). This ethanolic extract was suspended in water and successively partitioned with n‐hexane, ethyl acetate and n‐butanol. The ethyl acetate soluble extract (14.1 g) was subjected to silica gel column chromatography (CC) eluting with chloroform–methanol mixture [chloroform–methanol; 50:1 (Fraction EA‐A), 20:1 (Fr. EA‐B), 10:1 (Fr. EA‐C), 5:1 (Fr. EA‐D), 2:1 (Fr. EA‐E) and 1:1 (Fr. EA‐F)]. Fr. EA‐B (6.3 g) was chromatographed on silica gel CC [chloroform–methanol, 25:1 (v/v)] to give Fr. EA‐Ba–EA‐Bh. The Fr. EA‐Bd (150 mg) was subjected to flow‐rate gradient HPCC chromatography using two‐phase solvent system composed of n‐hexane–ethyl acetate–methanol–water [4:8:4:10 (v/v), reversed phase mode, mobile phase flow rate: 4 mL·min−1 in 0–70 min, 8 mL·min−1 in 70–250 min] to yield compounds 1 (10.2 mg), 2 (8.9 mg), 3 (2.9 mg), 4 (3.1 mg) and 5 (34.3 mg). The structure of compounds 1–5 were identified as ampelopsin A (1), (+)‐ε‐viniferin (2), vitisin A (3), wilsonol C (4) and vitisin B (5), respectively, by comparing their 1H NMR, 13C NMR and Q‐TOF/MS spectroscopic data with published values (Chen and Wang, 2009; do Ha et al., 2009; Wang et al., 2011; Jiang et al., 2012).
Cell culture
Cells of the human hepatoma cell line Huh7.5 were cultured in monolayers as described previously (Blight et al., 2002; Sklan et al., 2007).
Plasmids
Rluc‐J6/JFH1 (FL‐J6/JFH‐5′C19Rluc2AUbi) (Tscherne et al., 2006) is a monocistronic, full‐length HCV genome that expresses a Renilla luciferase and was derived from the previously described infectious GT2a HCV genome J6/JFH1 (Lindenbach et al., 2005). Bart79I is a high‐efficiency bicistronic subgenomic replicon of HCV derived from the HCV GT1b Con1 sequence that harbours a neomycin phosphotransferase gene in the first cistron and the HCV NS proteins in the second cistron under the translational control of an EMCV IRES (Blight et al., 2002). FL‐J6/JFH‐5′C19Rluc2Aubiand Bart79I were gifts from Dr Charles Rice of Rockefeller University. Cell culture‐derived infectious HCV (HCVcc) expressing an HCV NS5A‐GFP fusion protein was described elsewhere (Hwang et al., 2012).
In vitro transcription for production of HCV RNA genomes
In vitro transcription for production of HCV RNA genomes was performed as previously described (Lee, 2013b).
Generation of stable HCV replicon cell lines
The establishment of Huh7.5 cells stably maintaining a Bart79I subgenomic replicon in the presence of G418 selection has been described elsewhere (Cho et al., 2010).
Cell viability and HCV replication assay
In vitro transcribed RLuc‐J6/JFH1 RNAs were transfected into Huh7.5 cells by using a lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) as described by the manufacturer. Transfected cells were plated onto a white 96‐well plate (Costar 3610, Corning, NY, USA) and supplemented with DMSO or 1, 10, 100 nM, 1, 10 and 100 μM of vitisin B. At 3 days after incubation, cells were incubated for 3 h at 37°C in the presence of EZ‐CYTOX (10% tetrazolium salt; Dogen, Seoul, Korea) reagent to assess the cytotoxicity. Renilla luciferase activities were measured by using a luciferase reagent (1 mM coelenterazine in methanol–HCL; Goldbio, St.Louis, MO, USA). A time‐response curve was also determined by measuring Renilla luciferase activities as well as cell viabilities at 24, 48 and 72 h after treating HCV RNA‐transfected cells with 0.5 μM of vitisin B.
Vitisin B and sofosbuvir combination analysis using a luciferase assay
The 2 × 104 cells (Huh7.5‐Rluc‐J6/JFH1) were plated onto a 96‐well plate (Costar 3610) and supplemented with DMSO or 10, 100 pM 1, 10, 100 nM, 1 μM of sofosbuvir. After 3 h incubation, add DMSO or 10, 100 pM, 1, 10, 100 nM and 1 μM of vitisin B. At 3 days after incubation, cells were incubated for 3 h at 37°C in the presence of EZ‐CYTOX (10% tetrazolium salt; Dogen) reagent to assess the cytotoxicity. Renilla luciferase activities were measured by using a luciferase reagent (1 mM coelenterazine in methanol–HCL; Goldbio).
Analysis of vitisin B and sofosbuvir combination data
The MacSynergy II programme was used to analyse data according to the Bliss independence model (Prichard and Shipman, 1990; Prichard and Shipman, 1993). The combination's effect is determined by subtracting the experimental values from theoretical additive values (Prichard and Shipman, 1990). Matrix data sets in four replicates were assessed at the 95% confidence level for each experiment (Prichard and Shipman, 1990; Prichard and Shipman, 1993; Prichard and Shipman, 1996).
Quantitative real‐time RT‐PCR (qRT‐PCR) analysis
The qRT‐PCR analysis was performed as previously described (Lee, 2013b).
Confocal microscopic analyses
Huh7.5‐JFH‐1‐NS5A‐GFP or Bart79I‐NS5A‐yellow fluorescent protein (YFP) cells were plated onto coverslips in 24‐well plate. Coverslips were rinsed in PBS three times. Cells were fixed at room temperature for 10 min in 4% paraformaldehyde followed by three rinses with PBS. The coverslips were mounted onto slide using Prolong Gold anti‐fade reagent with DAPI (Invitrogen) fluorescence were examined and captured by Nikon confocal laser scanning microscopic system.
Graphene oxide (GO)‐based NS3 helicase inhibition assay
The GO‐based NS3 helicase assay was performed as described previously (Jang et al., 2013).
ATP hydrolysis assay
ATP hydrolysis assay was performed as described previously (Lee et al., 2009; Yu et al., 2012).
Infection and transfection assay
Infection and transfection assay to establish a vaccinia virus‐based HCV replicase assembly system was performed as previously described (Elazar et al., 2003).
In vitro HCV NS5B RNA polymerase assay
Recombinant HCV NS5B protein with an N‐terminal hexa‐histidine tag was expressed in Escherichia coli and purified, as described previously (Kim et al., 2004). In vitro RNA polymerase activity assays were performed with 50 ng of recombinant HCV NS5B lacking the C‐terminal 21 hydrophobic amino acids (NS5B∆21), as described previously (Kim et al., 2004; Kim et al., 2009; Ahn et al., 2011).
Nrf2 reporter assay
Nrf2 reporter plasmid (Kratschmar et al., 2012) were transfected into Huh7.5 cells by using a lipofectamine 2000 transfection reagent (Invitrogen) as described by the manufacturer. Transfected cells were plated onto a white 96‐well plate (Costar 3610) and supplemented with DMSO or 100 pM, 1, 10, 100 nM, 1 and 10 μM of vitisin B. At 48 h after incubation, cells were incubated for 3 h at 37°C in the presence of EZ‐CYTOX (10% tetrazolium salt; Dogen) reagent to assess the cytotoxicity. Firefly luciferase activities were measured by using a luciferase reagent (Luciferase assay system; Promega, Madison, WI, USA).
Data analysis
Values in graphs represent the mean ± SD of representative experiments performed in triplicate or quadruplicate, calculated using prism v5.0c software. For Student's t‐test, P < 0.05 was considered as statistically significant. The resulting data were fit to the Hill equation using prism v5.0c software to calculate EC50, cytotoxic concentration 50 (CC50) and IC50 values.
Other experiments
All the relevant information regarding instrumentations, reagents and plant materials, antiviral activity in the JFH‐1‐GFP infectious HCV cell culture system, Western blot analyses, analysis of vitisin B‐resistant HCV variants, expression and purification of recombinant NS3 proteins, drug affinity responsive target stability (DARTS) assay, fluorometric in vitro NS3/4A protease assay, pharmacokinetic study of vitisin B, cytokine assay, HCV pseudo‐particles (HCVpp) entry assay and assembly assay are described in detail in the Supporting Information.
Results
Vitisin B inhibits HCV replication
To search for new drug candidates for HCV infection from natural sources, we obtained an herbal library composed of 344 traditional Asian plants from a Korean plant extract bank. For antiviral and cell viability screenings, Huh7.5 hepatocarcinoma cells were first transfected with in vitro‐transcribed Rluc‐J6/JFH1 RNAs [HCV genotype (GT) 2a] harbouring Renilla luciferase (Huh7.5‐Rluc‐J6/JFH1 cells) (Tscherne et al., 2006). The luciferase activity was used as a surrogate measure for HCV RNA replication levels. A 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide‐based cell proliferation assay was conducted in parallel to access cell viability. When 344 ethanol extracts at 10 µg·mL−1 were tested for 72 h, the grapevine root extract (GRE) exhibited the highest ratio of cell viability to viral replication (44.7) (Supporting Information Fig. S1a). A dose‐dependent inhibition of HCV replication by GRE, with no cytotoxicity, was confirmed at the concentrations examined (EC50 = 0.168 µg·mL−1; CC50 >10 µg·mL−1) (Supporting Information Fig. S1b).
To identify the chemical nature of the active compounds responsible for the suppression of HCV replication by GRE, grapevine roots were extracted with ethanol and sequentially partitioned with various solvents of different polarities, including n‐hexane, ethyl acetate, n‐butanol and water. Among them, the ethyl acetate extract showed the highest antiviral activity as measured using Huh7.5‐Rluc‐J6/JFH1 cells (EC50 = 0.093 µg·mL−1) (Supporting Information Fig. S1c). We sub‐fractionated this ethyl acetate fraction for two more rounds until we identified chemical structures of five oligostilbene compounds by using HPLC‐assisted separation, Q‐TOF MS and 1H and 13C NMR spectroscopic analyses (Supporting Information Fig. S1c). They included two resveratrol dimers, ampelopsin A and (+)‐ε‐viniferin, and three resveratrol tetramers, wilsonol C, vitisin A, and vitisin B (Figure 1A). When their antiviral activities were compared using Huh7.5‐Rluc‐J6/JFH1 cells, vitisin B was found to be more potent (EC50 = 0.006 μM) than any oligostilbene compound identified previously, including wilsonol C (EC50 = 0.016 μM), vitisin A (EC50 = 0.035 μM), (+)‐ε‐viniferin (EC50 = 0.159 μM) and ampelopsin A (EC50 = 5.740 μM) (Figure 1B). A dramatic time‐dependent reduction in HCV replication was confirmed by application of 0.5 μM vitisin B (T1/2 = 6.28 h) (Figure 1C). Because of its considerable antiviral potency, we focused on vitisin B in subsequent experiments.
Figure 1.
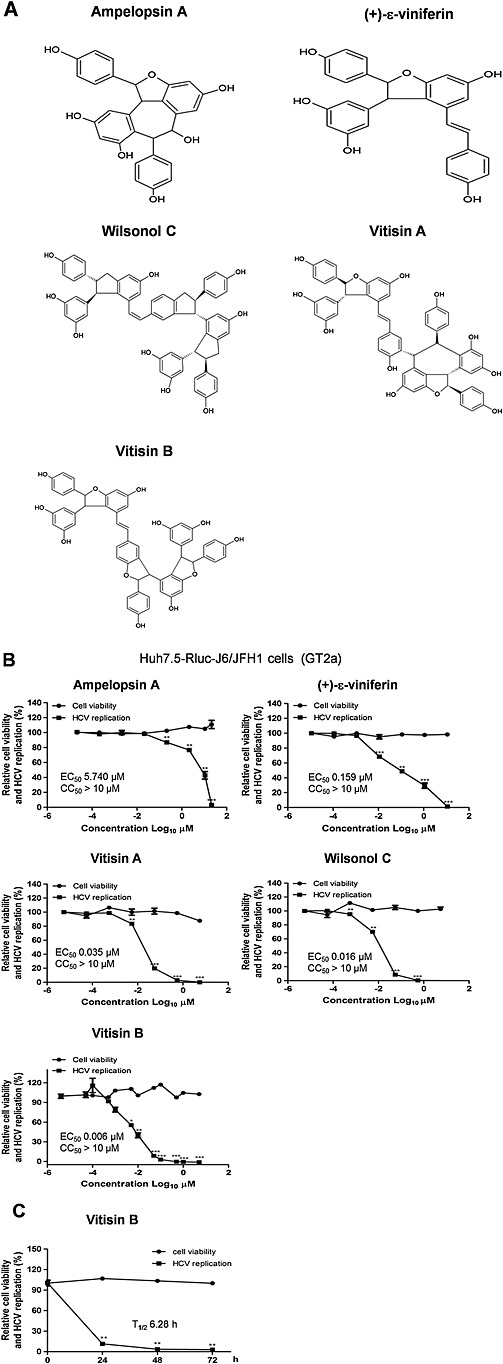
Identification of vitisin B as an HCV replication inhibitor. A Chemical structures of two resveratrol dimers, ampelopsin A and (+)‐ε‐viniferin, and three resveratrol tetramers, vitisin A, wilsonol C and vitisin B, identified from GRE. B Concentration–response curves were determined by measuring relative luciferase activities and cell viabilities of Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of either ampelopsin A, (+)‐ε‐viniferin, vitisin A, wilsonol C or vitisin B for 72 h. C A time‐response curve was determined by measuring the relative luciferase activities and cell viabilities of Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with 0.5 μM vitisin B for increasing periods of time. * P < 0.05, ** P < 0.01, significantly different from control.
To test the feasibility of vitisin B as a new component of combination therapy with other clinically available HCV drugs, we examined the antiviral activity of the combination of vitisin B with the NS5B polymerase inhibitor sofosbuvir using Huh7.5‐Rluc‐J6/JFH1 cells. As shown in Figure 2A, the vitisin B and sofosbuvir combination resulted in a synergistic – or at least additive – effect on HCV replication. When a three‐dimensional differential surface was plotted to demonstrate synergy as peaks above a theoretical additive plane and antagonism as depressions below it (Prichard and Shipman, 1990), a high level of synergism was evident at 1 nM to 1 μM vitisin B and 0.01 to 100 nM sofosbuvir (Figure 2B). We also calculated synergy index by using the Prichard method. The CompuSyn 3.0.1 programme was used to analyse data according to the Bliss independence model (Chou, 2010). The combined effect is determined by subtracting the experimental values from theoretical additive values (Chou, 2010). Matrix data sets in three replicates were assessed at the 95% confidence level for each experiment. According to this analysis, we found that most of Cl values were smaller than 1, which indicates there is a synergic effect between vitisin B and sofosbuvir.
Figure 2.
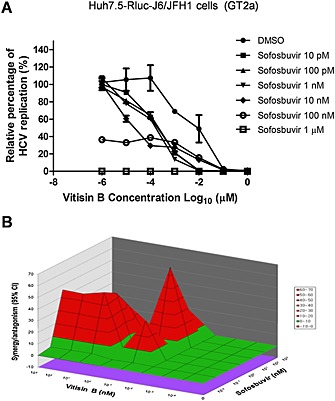
Combined effect of vitisin B and sofosbuvir on HCV replication. A Combined effect of vitisin B with an NS5B polymerase inhibitor, sofosbuvir on HCV replication. A dose–response curve was determined by measuring the relative luciferase activities in Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of vitisin B in the presence of different doses of sofosbuvir for 72 h. B MacSynergy analysis of the combined antiviral effect of vitisin B with sofosbuvir. Differential surface plots at the 95% confidence interval (CI) are shown. The peaks above the theoretical additive plane indicate synergy, whereas depressions below it indicate antagonism.
To rule out the possibility of an effect of the inserted Renilla luciferase on HCV replication, we retested the antiviral effect of vitisin B on two other reporter‐free HCV systems. We first transfected Huh7.5 cells with either full‐length infectious J6/JFH1 RNAs (Huh7.5‐J6/JFH1) (GT2a) (Lindenbach et al., 2005) or sub‐genomic Bart79I RNAs (Huh7.5‐Bart79I) (GT1b) harbouring a neomycin‐resistant gene (Cho et al., 2010). Next, we treated the cells with an increasing concentration of vitisin B for 72 h or with 0.5 μM vitisin B for an increasing period of time. Real‐time RT‐PCR analyses confirmed dose‐dependent and time‐dependent block of HCV replication by vitisin B (EC50 = 0.003 μM, T1/2 = 4.7 h for GT2a; EC50 = 0.001 μM, T1/2 = 17.9 h for GT1b) (Figure 3A and B). Vitisin B showed considerably more rapid viral RNA decay kinetics than daclatasvir, which is an NS5A inhibitor (Gao et al., 2010) (T1/2 = 14.8 h for GT2a; 27.6 h for GT1b) (Figure 3C). Collectively, these data suggest that vitisin B is a major active compound responsible for the inhibition of HCV replication by the crude GRE.
Figure 3.
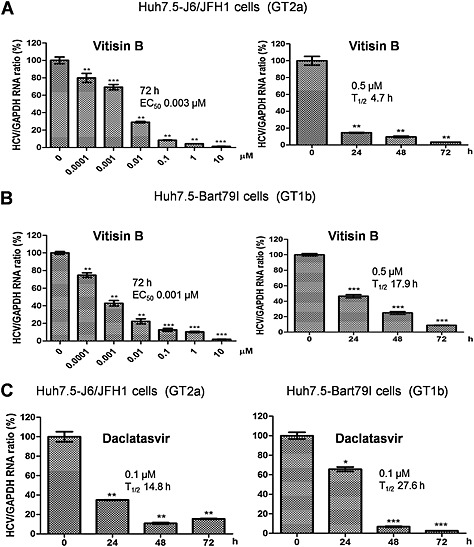
Inhibition of HCV replication by vitisin B. Dose–response graphs were generated by measuring the relative HCV and GAPDH RNA levels via real‐time qRT‐PCR analyses of either A Huh7.5‐J6/JFH1 or B Huh7.5‐Bart79I cells treated with increasing concentrations of vitisin B for 72 h. Time‐response graphs were also determined by measuring the relative HCV and GAPDH RNA levels via real‐time qRT‐PCR analyses of either A Huh7.5‐J6/JFH1 or B Huh7.5‐Bart79I cells treated with 0.5 μM vitisin B for increasing periods of time. C Time‐response graphs were determined by measuring the relative HCV and GAPDH RNA levels via real‐time qRT‐PCR analyses of either Huh7.5‐J6/JFH1 or Huh7.5‐Bart79I cells treated with 0.1 μM daclatasvir for increasing periods of time. * P < 0.05, ** P < 0.01, significantly different from control.
Vitisin B diminishes the expression levels of HCV proteins
To determine whether the inhibition of HCV replication by vitisin B could reduce HCV protein production, we first plated Huh7.5 cells and incubated them with increasing concentrations of vitisin B for 2 h, followed by inoculation with a JFH1 virus expressing an NS5A‐GFP fusion protein (Huh7.5‐JFH1‐NS5A‐GFP cells) (GT2a) (Hwang et al., 2013). At 72 h post infection, the HCV infection rates were determined by fully automated confocal microscopic analysis. As shown in Figure 4A, a dose‐dependent reduction in the number of HCV‐replicating cells was observed. The total cell number was unaffected (EC50 = 0.002 μM and CC50 >30 μM). Unlike the DMSO‐treated control, a significantly reduced number of GFP‐positive cells were evident upon treatment with 10, 0.1 and 0.002 μM vitisin B (Figure 4B). To confirm the antiviral activity of vitisin B against other HCV GTs, we first transfected Huh7.5 cells with sub‐genomic Bart79I‐NS5A‐YFP RNAs expressing an NS5A‐GFP fusion protein (Huh7.5‐Bart79I‐NS5A‐YFP cells) (GT1b) (Jin et al., 2014). Next, we treated the cells with increasing concentrations of vitisin B for 72 h or with 0.5 μM vitisin B for an increasing period of time. FACS and confocal microscopic analyses clearly showed a dose‐dependent and time‐dependent reduction in YFP‐positive cell numbers (EC50 = 0.004 μM and T1/2 = 35.9 h) (Figures 4C–E). To rule out the possibility of an effect of the inserted fluorescent proteins on the expression of HCV proteins, we assessed the effect of vitisin B on Huh7.5‐J6/JFH1 and Huh7.5‐Bart79I cells. A similar dose‐dependent and time‐dependent reduction in the expression levels of viral proteins, including NS3 and NS5A, was confirmed by Western blot analyses (EC50 = 0.006 μM, T1/2 = 29.6 h for GT2a; EC50 = 0.006 μM, T1/2 = 25.3 h for GT1b) (Figures 4F and G). Collectively, these data suggest that vitisin B can reduce the production of HCV proteins through its inhibition of viral genome replication.
Figure 4.
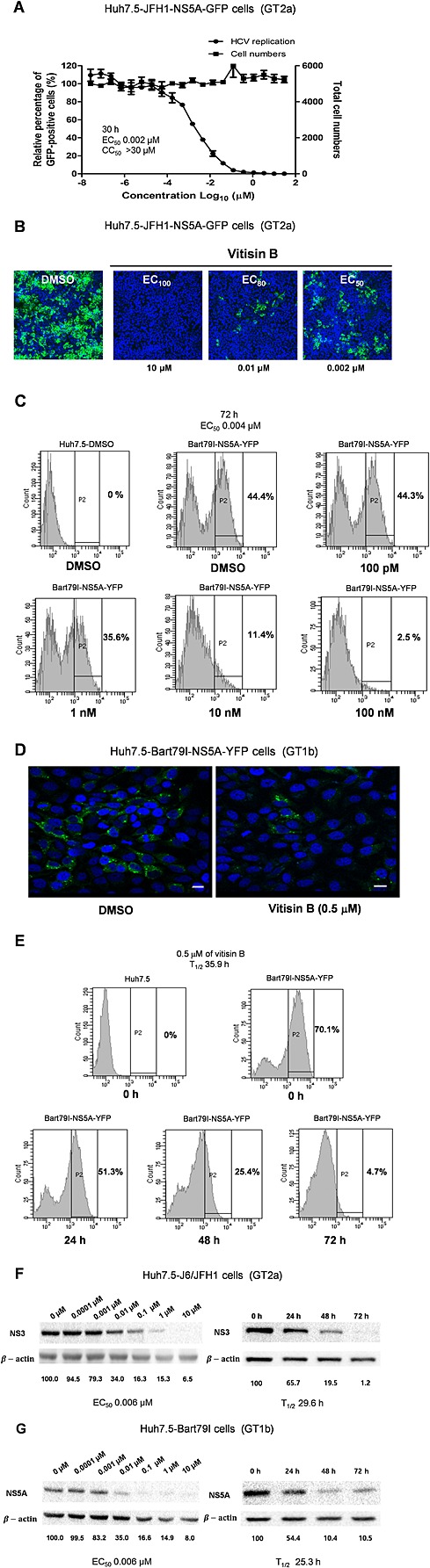
Vitisin B diminishes the expression of HCV proteins. A The dose‐dependent effect of vitisin B on the levels of NS5A protein was determined using HCVcc JFH‐1 cells expressing an NS5A‐GFP fusion protein. The abundance of NS5A protein was assessed by determining the number of NS5A‐GFP‐positive cells, and the total cell number was used as a marker for cytotoxicity. Experiments were performed in duplicate. B Huh‐7.5 cells were pretreated with either DMSO or 10, 0.01 and 0.002 μM vitisin B followed by inoculation with HCVcc for 30 h. The effect of vitisin B on the expression NS5A was determined by visualizing the relative level of NS5A‐GFP via confocal microscopy. Nuclei were stained in blue with DAPI. The images were obtained using a Nikon confocal laser scanning microscope. C The dose‐dependent effect of vitisin B on the levels of NS5A protein was determined by measuring the relative percentages of NS5A‐YFP‐positive cells via FACS analysis of Huh7.5‐Bart79I‐NS5A‐YFP cells treated with increasing concentrations of vitisin B for 72 h. D The effect of vitisin B on the subcellular localization of NS5A protein was determined by visualizing the relative levels of NS5A‐GFP via confocal microscopic analysis of Huh7.5‐Bart79I‐NS5A‐YFP cells treated with either 1% DMSO or 0.5 μM vitisin B for 30 h. Nuclei were stained in blue with DAPI. The images were obtained using a Nikon confocal laser scanning microscope. The scale bar represents 15 µm. E The time‐dependent effect of vitisin B on the levels of NS5A protein was determined by measuring the relative levels of HCV NS5A‐YFP‐positive cells via FACS analysis of Huh7.5‐Bart79I‐NS5A‐YFP cells treated with 0.5 μM vitisin B for increasing periods of time. EC50 and T1/2 values were determined based on each response curve. F and G Dose‐independent and time‐dependent effects of vitisin B on the levels of viral proteins, as well as β‐action proteins, were determined by Western blot analyses of F Huh7.5‐J6/JFH1 or G Huh7.5‐Bart79I cells treated with either increasing concentrations of vitisin B for 72 h or 0.5 μM vitisin B for increasing periods of time.
Identification of the NS3 helicase as a potential target for vitisin B
To gain insight into the potential antiviral mechanism of action of vitisin B, we hypothesized that inhibition of HCV replication by vitisin B might be due to its targeting one of the viral proteins. To test this hypothesis, we selected vitisin B‐resistant HCV mutant variants. For this purpose, we cultured Huh7.5‐Bart79I cells harbouring a neomycin‐resistant gene in the presence of 1 μM vitisin B for 4 weeks. Next, we isolated and propagated five vitisin B‐resistant colonies designated M1 to M5 respectively. When total RNA was extracted from each of these colonies and the entire HCV coding region was sequenced, several mutations within the NS3‐encoding and NS5B‐encoding regions of HCV were identified (Figure 5A). In particular, most of the variant residues conferring resistance to vitisin B were heavily concentrated at the C‐terminal NS3 helicase domain, which consists of amino acids 166 through 631 in the case of the GT1b HCV NS3 (Figure 5B). Among the five vitisin‐B‐resistant variants, M4 containing both the E177G and I249T mutations showed the greatest resistance to the inhibition of HCV replication in the presence of 1 μM vitisin B (Figure 5C). While the replication level of wild‐type HCV was reduced to almost less than 20% upon treatment with 0.004 μM vitisin B, M4 maintained almost 100% replication even upon exposure to 10 μM vitisin B (Figure 5D). A more than 2500‐fold difference in the EC50 values between wild‐type and M4 variant replicons was observed. Therefore, these data strongly suggest the NS3 helicase as a potential antiviral target for vitisin B.
Figure 5.
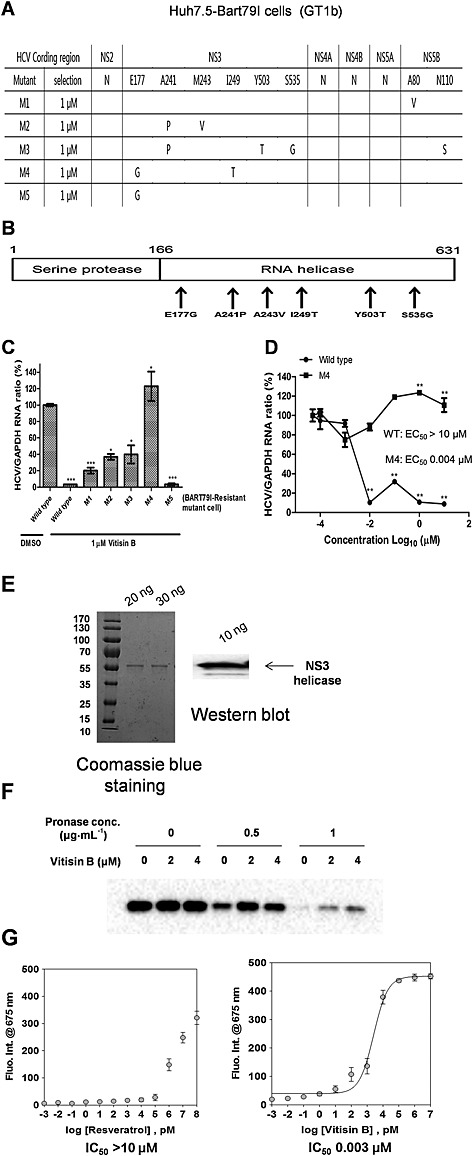
Identification of NS3 helicase as a potential target for vitisin B. A Selection of vitisin B‐resistant HCV mutant variants. Huh7.5‐Bart79I cells harbouring a neomycin‐resistant gene were incubated with 1 μM vitisin B for 4 weeks. Five vitisin B‐resistant colonies designated M1 to M5, respectively, were propagated. Total RNA was extracted, and viral RNAs were sequenced by RT‐PCR analysis. B Location of mutated amino acids isolated from vitisin B‐resistant HCV variants. All of the variant residues conferring resistance to vitisin B were located at the C‐terminal NS3 helicase domain. C Five vitisin B‐resistant colonies were treated with 1 μM vitisin B for 72 h, and their relative HCV and GAPDH RNA levels were compared by real‐time RT‐PCR analysis. D A dose–response curve was determined by measuring the relative HCV and GAPDH RNA levels via real‐time qRT‐PCR analysis of either wild‐type or vitisin B‐resistant M4 cells treated with increasing concentrations of vitisin B for 72 h. EC50 values were determined based on each response curve. E Coomassie staining and Western blot analysis of a purified recombinant NS3 helicase of 20, 30 and 10 ng. M, protein molecular mass markers in kDa F Recombinant NS3 helicase protein was treated with either DMSO or 2 or 4 μM vitisin B in vitro, followed by digestion with 0.5 or 1 µg·mL−1 pronase. The remaining amount of recombinant NS3 helicase protein was measured by Western blot. G The effect of either resveratrol or vitisin B on in vitro NS3 helicase activity was determined by fluorescence intensity measurement of Cy5‐labelled double‐stranded DNA at excitation wavelength/emission wavelengths of 650/675 nm at increasing concentrations of either resveratrol or vitisin B. The IC50 values were determined based on the corresponding response curve. * P < 0.05, ** P < 0.01, significantly different from control.
To determine whether vitisin B could interact with the NS3 helicase directly, we first purified an NS3 helicase recombinant protein from bacterial cells, which were transformed with an NS3 helicase expression vector. Quality of the purified NS3 helicase was confirmed by Coomassie staining and Western blot analysis (Figure 5E). Then, we performed a DARTS assay. In this assay, a drug‐free protein is more susceptible to pronase digestion, whereas a drug‐bound protein is more resistant. As shown in Figure 5F, a concentration‐dependent protective effect of the in vitro‐purified NS3 helicase against pronase digestion was observed in the presence of 2 or 4 μM vitisin B. Based on this result, we hypothesized that vitisin B might inhibit the NS3 helicase activity through its direct binding. To test this hypothesis, we utilized a recently published GO‐based NS3 helicase assay (Jang et al., 2013). In this assay, when Cy5‐labelled double‐stranded DNA is dissociated into single‐stranded DNA by an HCV NS3 helicase, the released Cy5‐labelled single‐stranded DNA loses its fluorescent activity through its binding to GO. Therefore, block of the HCV NS3 helicase by an inhibitor will lead to restoration of fluorescent activity. As shown in Figure 5G, vitisin B increased the fluorescence in a dose‐dependent manner at a concentration below its EC50 value [half maximal inhibitory concentration (IC50) = 0.003 μM], whereas the negative control resveratrol at up to 10 μM did not.
After confirmation of a negative effect of vitisin B on HCV replication, we wished to examine whether vitisin B affected any other aspects of the HCV life cycle including entry, assembly and particle production. For an HCV entry experiment, we generated HCVpp coated with E1 and E2 glycoproteins by using a retroviral system (McGivern et al., 2014). We were able to detect expression of a luciferase reporter gene by infection of naïve Huh7.5 cells with these HCVpp (Figure 6A). We also comfirmed that this infection was potently repressed by a recently discovered HCV E2 entry inhibitor (Bush et al., 2014) (Figure 6A). However, vitisin B failed to induce any effect on HCV entry by using HCVpp system (Figure 6B). When we studied effects of vitisin B on intracellular viral RNA abundance and assembly/infectious particle production using Huh7.5‐J6/JFH1 cells, we found that HCV replication and assembly/infectious particle production were negatively affected by vitisin B in a very similar manner when they were treated for 24, 48 and 72 h (Figure 6C). The EC50 differences between suppression of infectious virus release versus viral RNA abundance by vitisin B at 24, 48 and 72 h all stayed around onefold, which were 0.5909, 0.6250 and 0.7466, respectively. On the other hand, vitisin B was able to suppress the infectious particle production with twofold less concentration (0.0019 μM) than its EC50 (0.0038 μM) for inhibition of HCV RNA replication when treated for 12 h. These EC50‐fold differences increased dramatically when vitisin B was used for much shorter periods of time (11.551 for 6 h and 320.87 for 3 h) (Figure 6C and table). This suggests that vitisin B has an ability to regulate HCV assembly/production in a negative way, independent of its effect on HCV replication.
Figure 6.
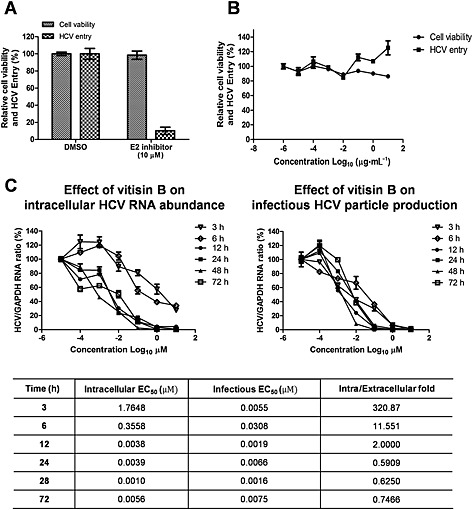
Effects of vitisin B on other aspects of HCV life cycle. A Generation of HCVpp. HCVpp coated with E1 and E2 glycoproteins were generated by using a retroviral system. Expression of a luciferase reporter gene by infection of naïve Huh7.5 cells with HCVpp were detected in the absence or presence of the 10 μM of an HCV E2 inhibitor. B Effect of vitisin B on HCV entry. Naïve Huh7.5 cells were infected with HCVpp in the presence of increasing concentrations of vitisin B. Luciferase activities were measured to test effect of vitisin B on HCV entry. C Effect of vitisin B on HCV replication, assembly and particle production. Huh7.5‐J6/JFH‐1 cells were incubated with either DMSO or increasing concentrations of vitisin B for 3, 6, 12, 24, 48 and 72 h. Total RNAs were extracted from treated cells, and virus‐containing media were harvested at each time point. Naïve Huh7.5 cells were infected with these virus‐containing media. Total RNAs were extracted from Huh7.5 cells infected with virus‐containing media. qRT‐PCR analysis was performed to measure effect of vitisin B on HCV replication and virus particle production. Intracellular EC50 (vitisin B concentration required to reduce the intracellular viral RNA abundance in half) and infectious EC50 values (vitisin B concentration required to reduce the infectious particle production in half) obtained from each time points, and their fold differences were summarized as a separate table.
To rule out the possibility of the negative regulation of HCV NS3/4A protease activity by vitisin B, we utilized the vaccinia virus‐based HCV replicase assembly system (Elazar et al., 2003). In this system, individual viral proteins are constitutively expressed due to continuous transcription of HCV genomes by a T7 RNA polymerase (Elazar et al., 2003). We failed to find any effect of vitisin B on the expression levels of NS3 protein at up to 10 μM, whereas an NS3/4A protease inhibitor, telaprevir, completely abolished its expression (Figure 7A). In a fluorometric in vitro HCV NS3/4A protease assay, no anti‐NS3 protease activity was detected following incubation in the presence of vitisin B at up to 100 μM (Figure 7B). These data suggest that vitisin B does not affect polyprotein cleavage by the NS3/4A protease. In addition, we found no significant effect of vitisin B on the subcellular localization of NS5A in relationship with ER marker protein, calnexin, in the vaccinia virus‐based HCV replicase assembly system (Elazar et al., 2003) (Figure 7C). In order to test potential effects of vitisin B on enzymatic activity of HCV RNA polymerase, we first obtained a bacterially purified NS5B protein and confirmed its quality by coomassie staining and western blot analysis (Figure 7D). Then, we measured the RNA polymerase activity in the presence of various subfractions of GRE. We found that RNA‐dependent RNA polymerase activity of NS5B was unaltered by GRE treatment (Figure 7E). Based on these results, we concluded that vitisin B could inhibit HCV replication by specifically targeting an NS3 helicase activity.
Figure 7.
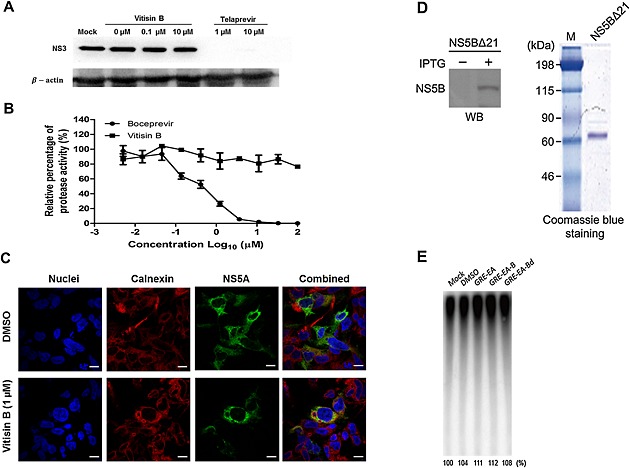
Effect of vitisin B on functions of other HCV proteins. A Effect of vitisin B on viral polyprotein processing. Individual viral proteins were constitutively expressed by the vaccinia virus‐based HCV replicase assembly system in the presence of increasing concentrations of either vitisin B or the NS3 protease inhibitor telaprevir. Western blot analysis was conducted to determine the expression levels of NS3 and β‐actin. B Effect of vitisin B on HCVNS3/4A protease activity. Inhibition of NS3/4A protease activity was determined by fluorometric readout. The viral protease was pretreated with various concentrations of vitisin B or boceprevir, and the efficacy was determined as described in the Methods. The data are presented as the mean percentages of protease activity by measuring fluorescence intensity. Data were normalized to the DMSO control. C Effect of vitisin B on the subcellular localization of NS5A. Immunofluorescence analysis was conducted to examine the subcellular localization of NS5A protein in the presence of either DMSO or 1 μM vitisin B in the vaccinia virus‐based HCV replicase assembly system. D Coomassie blue staining and Western blot analysis of a purified recombinant NS5B polymerase. E. coli cells transformed with an expression vector for NS5B∆21 were grown at 37°C, and overexpression of NS5B∆21 was induced with isopropyl‐beta‐d‐thiogalactopyranoside (IPTG) in log phase for 16 h at 25°C. Total cellular lysates were analysed by Western blotting (WB) using an antibody against the (His)6‐tag fused to the N‐terminus of HCV NS5B protein. Visualization of the purified NS5B∆21 (500 ng) by Coomassie blue staining following SDS‐PAGE. M, protein molecular mass markers in kDa E Effect of GRE on the NS5B RNA polymerase. NS5B RNA‐dependent RNA polymerase activity was measured in the presence of either mock, DMSO, GRE–EA, GRE–EA‐B or GRE–EA‐Bd.
Induction of haem oxygenase‐1 (HO‐1) has been shown to play an important role in suppression of HCV replication through activation of the IFN pathway and subsequent inhibition of HCV NS3/4A (Wuestenberg et al., 2014) because transcriptional activation of HO‐1 is mediated by stabilization and binding of Nrf2 to HO‐1 promoter region. We used Nrf2 reporter system to see if vitisin B has any effect on Nrf2‐dependent transcriptional activation. As shown in Supporting Information Fig. S1d, we did not see any effect of vitisin B on transcriptional activation by Nrf2. Based on this result, we concluded that induction of HO‐1 does not play any role in inhibition of HCV replication by vitisin B.
Tissue distribution of vitisin B after i.p. injection
In a preliminary study, we observed almost no peaks for vitisin B at all blood sampling times after oral administration of up to 250 mg·kg−1 of vitisin B to rats (Choi et al., 2015). After i.p. injection of vitisin B at doses of 5, 10 and 25 mg·kg−1, Tmax was reached after 240–360 min, suggesting continuous absorption of vitisin B from the peritoneal cavity into the systemic circulation. Furthermore, the bioavailability after i.p. dosing (10 mg·kg−1) was high, 80.9%, indicating that i.p. injection may be a favourable route of administration to obtain high exposure of vitisin B in plasma, relative to oral administration (Choi et al., 2015). Thus, in the tissue distribution study, vitisin B was injected i.p. Figure 8A shows the quantitative tissue distribution of vitisin B at 60 and 480 min after i.p. injection at a dose of 10 mg·kg−1. The amounts of vitisin B recovered from each tissue (µg·mL−1 for plasma or µg·g−1 for other tissues) were determined. Compared with the corresponding plasma concentrations of vitisin B, only the liver exhibited extremely high distributions. of the seven tested tissues, in which the liver‐to‐plasma ratios were over 1. The calculated liver‐to‐plasma ratios at 60 and 480 min were 2.48 ± 1.54 and 2.77 ± 0.47 respectively. These suggest that the hepatic concentrations of vitisin B were higher than those plasma levels. The distributions of vitisin B in lung and spleen were all lower than those in plasma at 60 min. However, the corresponding concentrations in lung and spleen of vitisin B were higher than those in the plasma with corresponding tissue‐to‐plasma ratios of 1.44 ± 0.46 and 1.72 ± 0.51 at 480 min. Trace amounts of vitisin B were found in brain, fat, heart and kidney in both times. In order to maximize the therapeutic potential of vitisin B, we wished to see if treatment of vitisin B causes any acute adverse response, which is usually manifested by rapid induction of pro‐inflammatory cytokines. We decided to examine whether pro‐inflammatory cytokines such as TNF‐α and IL‐6 are induced after i.p. injection of vitisin B (10 mg·kg−1) into rats. As shown in Figure 8B, we did not see any significant increase in the plasma levels of either TNF‐α or IL‐6 by i.p. administration of vitisin B.
Figure 8.
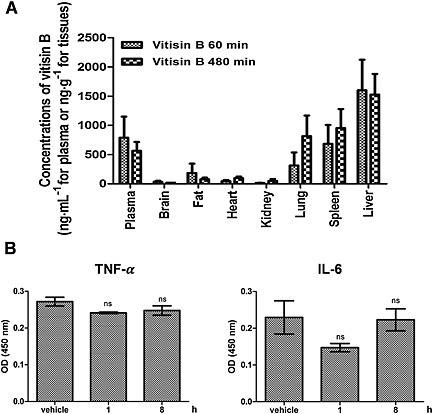
Pharmacokinetic study of vitisin B A Mean amount of vitisin B recovered from each tissue (ng·mL−1 for plasma or ng·g−1 for other tissues) at 60 min (n = 5) and 480 min (n = 5) after i.p. injection of vitisin B at a dose of 10 mg·kg−1 to rats. B Effects of vitisin B on induction of pro‐inflammatory cytokines. Serum from a vitisin B‐treated rat (10 mg·kg−1) after 1 h (n = 4) and 8 h (n = 4) was collected. Serum concentration of TNF and IL‐6 were measured using elisa kits.
Discussion and conclusion
Analysis of resistant HCV variants strongly suggested the NS3 helicase as a potential target for vitisin B (Figure 5A and B). Consistent with this hypothesis, we confirmed direct binding of vitisin B with a purified HCV NS3 helicase in vitro using a DARTS assay (Figure 5F). Interestingly, the same DARTS methodology was also applied to identify eIF4A (eukaryotic translation initiation factor 4A), which is a DEAD‐box RNA helicase, as a direct host target for resveratrol (Lomenick et al., 2009). This host RNA helicase plays an essential role in the translation initiation process in eukaryotic cells. Resveratrol was shown to inhibit specifically the host cell translation catalysed by eIF4A, whereas translation from the eIF4A‐independent HCV IRES was unaffected (Lomenick et al., 2009). In addition, highly conserved homology between eIF4A and HCV NS3 helicase was also reported (Du et al., 2002). Therefore, it is tempting to speculate that oligostilbene compounds may have evolved to protect the grapevine against plant pathogens by inhibiting a specific member of non‐plant helicase family proteins. Particularly, the resveratrol tetramer vitisin B may have evolved to increase its substrate specificity for a viral helicase. However, to our knowledge, this hypothesis has never been tested or reported in the literature
The HCV helicase can strip RNA‐binding proteins from viral RNA, assist translation or contribute to coordination of translation and polyprotein processing. The NS3 helicase is a Y‐shaped protein composed of three domains. Domains 1 and 2 are RecA‐like domains. The ATP‐binding site is located between domains 1 and 2. Oligonucleotides are thought to be located between two RecA‐like domains and domain 3. Adaptive mutations in HCV replicons frequently arise in the helicase region (Grobler et al., 2003). Vitisin B‐resistant HCV variants from the Bart79I replicon possess E177G and I249T mutations (Figure 5A). Because both of these residues are located in close proximity to the ATP binding site, there is the possibility of inhibiting NS3 helicase activity by competing with its natural substrate, ATP. Considering its relatively bulky structure with a molecular weight of 906, direct binding of vitisin B to the active site of the NS3 helicase seems unlikely. Both E177 and I249 residues are located in domain 1 of the HCV NS3 helicase (Frick, 2006). The residue E177 is a part of the α‐helical structure. Because I249 is located in very close proximity to the ATP‐binding site (distance of 10–20 Å), this residue may play a role in loading ATP onto the NS3 helicase active site. Vitisin B might interfere with this ATP loading process by I249, leading to inhibition of NS3 helicase activity. I249T substitution may decrease the affinity of vitisin B for the NS3 helicase due to its lower hydrophobicity than isoleucine. We are now testing the possibility of suppressing ATP hydrolysis of NS3 helicase by vitisin B. In addition, we are engaged in generating HCV variants containing either the E177G or I249T mutation to evaluate the effect of substituted amino acids on alleviating the inhibition of HCV replication by vitisin B. It would be of great interest to determine which mutation affects the binding of vitisin B to the NS3 helicase by using recombinant NS3 helicase protein containing either the E177G or I249T mutation.
One of the best characterized NS5A inhibitors, daclatasvir, was recently shown to inhibit both viral RNA replication and assembly (McGivern et al., 2014). In this paper, they studied kinetics of antiviral suppression by different classes of DAAs including NS3 protease and NS5A and NS5B polymerase inhibitors. They found that there were no difference in the kinetics of suppression of viral RNA abundance and infectious particle production by the NS5B polymerase inhibitor regardless of periods of incubation times, suggesting that most of its anti‐HCV activity comes from direct inhibition of HCV RNA replication. However, NS5A inhibitors showed marked disparity in the kinetics of suppression of infectious virus release versus viral RNA abundance at early time point (12 h), suggesting that NS5A inhibitors may block viral assembly. When we employed the same strategy to study effect of vitisin B on viral assembly as well as replication, we found that vitisin B exerted a dramatic negative effect on viral replication, assembly as well as secretion (Figure 6A–C). Especially, vitisin B also showed severe disparity in the kinetics of suppression of infectious virus release versus viral RNA abundance at early time points (3, 6 and 12 h) (Figure 6C and table). This negative regulation of secretion by vitisin B seems to be a combined consequence of its inhibitory activity against both HCV replication and assembly as the NS3 protease/helicase was shown to have a dual role in both HCV replication and assembly by other groups (Ma et al., 2008; Kohlway et al., 2014; McGivern et al., 2015). Therefore, we expect that targeting NS3 helicase by vitisin B will be a much better strategy to combat HCV infection, compared with targeting NS5B polymerase, by attacking multiple stages of the HCV life cycle, which are mediated by a common NS3 helicase activity.
Grapevines are one of the best known plants in the genus Vitis and are widely distributed worldwide. Various stilbene compounds (oligomers of resveratrol) have been found in grapevines. In particular, the inedible parts (root and stems) of the grapevine have been used as traditional medicines in Asian countries. Resveratrol tetramers, including vitisin A, wilsonol C and vitisin B, were reported to be enriched in these parts. Several biological activities for vitisins A and B have been described to date. These include suppression of lipopolysaccharide‐induced NO production (Mi Jeong et al., 2009), blockade of cell migration and enhancement of cell adhesiveness (Ong et al., 2011), blockade of adipocyte differentiation (Kim et al., 2008), regulation of myocyteapoptosis (Seya et al., 2009), inhibition of osteoclast differentiation (Chiou et al., 2014), prevention of bone loss (Huang et al., 2013) and inhibition of the 3‐hydroxy‐3‐methylglutaryl‐CoA enzyme (Koo et al., 2008). To explain these diverse pharmacological properties of vitisins A and B, various modes of action have been suggested. These include inhibition of ERK, p38 and NF‐κB activation (Mi Jeong et al., 2009), cell cycle arrest (Kim et al., 2008), inhibition of platelet‐derived growth factor signalling (Ong et al., 2011) and suppression of NF of activated T‐cells, cytoplasmic 1 activation (Chiou et al., 2014). However, all of these biological activities of vitisins A and B were observed only at concentrations considerably higher than 10 μM in cell culture condition. None of these reports described biological activity of either vitisin A or vitisin B at sub‐micromolar concentrations. We also failed to detect an inhibitory effect of vitisin B on either ERK or p38 kinase at concentrations that inhibit HCV replication (data not shown). Therefore, we do not believe that all of the biological activities described earlier for vitisins A or B play a significant role in their suppression of HCV replication.
Because of its numerous health‐related beneficial properties, including the prevention of several cancers and inflammatory diseases, resveratrol may be one of the most extensively investigated polyphenol compounds isolated from natural plants (Santos et al., 2011). Its superior antioxidant capacity has been described as one of the main mechanisms underlying its many desirable biological properties (Udenigwe et al., 2008). However, most of these favourable in vitro activities of resveratrol have failed to translate into the expected clinical outcome due to its poor physicochemical characteristics, including low stability, increased oxidation on heat and light exposure, low water solubility and high hepatic metabolism (Santos et al., 2011; Walle, 2011). Therefore, its oral bioavailability is usually less than 1%. In addition, considering its relatively high EC50 (usually around 50 μM) needed to exhibit its biological activities, it would be extremely difficult to saturate the target tissue with such a high concentration of resveratrol in vivo. However, resveratrol has shown very high levels of oral absorption, hepatic uptake and volume of distribution, particularly in hepatic tissue. Although no pharmacokinetic data for vitisin B are at present available, its 8000‐fold lower EC50 value (0.006 μM) for the suppression of HCV replication in vitro compared with that of resveratrol might underlie its antiviral efficacy in vivo. Because the liver is a major organ for HCV replication, a favourable pharmacokinetic property of vitisin B strongly supports explicit consideration of liver exposure for vitisin B through this route (Figure 8A). We also confirmed that there was no acute adverse response to treatment of vitisin B including up‐regulation of pro‐inflammatory cytokines such as TNF‐α and IL‐6 (Figure 8B). We are currently engaged in investigating an in vivo efficacy of vitisin B using various HCV animal models to determine its feasibility as a therapeutic agent against HCV infection.
By using an unbiased cell‐based screening system, one group of researchers was able to identify pterostilbene as a potent HCV inhibitor (Gastaminza et al., 2010). Its structural similarity to our oligostilbene compound, vitisin B, implies a possibility of existence of common antiviral mechanism of actions shared by these two molecules. However, pterostilbene was shown to implement its antiviral actions by inhibiting infectious particle assembly as well as secretion without affecting viral replication (Gastaminza et al., 2010). This suggests that vitisin B may have a distinct mode of action against HCV replication, regardless of its structural relatedness to pterostilbene.
Regarding its effect on viruses, resveratrol was reported to inhibit the replication of several major viruses including CMV (Evers et al., 2004), varicella zoster (Docherty et al., 2006), influenza A (Palamara et al., 2005) and herpes simplex virus (HSV) (Faith et al., 2006). Negative effect of resveratrol on CMV infection involves block of virus‐induced activation of the EGF receptor and PI3‐kinase signal transduction as well as NF‐κB and Sp1 transcription factor activation (Evers et al., 2004). On the other hand, blockade of the nuclear‐cytoplasmic translocation of influenza virus ribonucleoproteins was shown to play a role in suppression of influenza A replication by resveratrol (Palamara et al., 2005). Inhibition of HSV‐induced activation of NF‐κB by resveratrol was also reported to impair expression of essential immediate–early, early and late HSV genes and synthesis of viral DNA (Faith et al., 2006). In addition, natural stilbenoids isolated from grapevines, including ampelopsin A and vitisin B, exhibit inhibitory effects against HIV‐1 integrase (Pflieger et al., 2013). Interestingly, one study described a positive effect of resveratrol on HCV replication (Nakamura et al., 2010). In this report, there was an approximately fourfold increase in luciferase reporter gene activity from the O strain of GT1b HCV upon treatment with 10 μM resveratrol. Based on this result, the authors recommended against consumption of resveratrol‐containing red wine by HCV patients. However, we found no significant effects of up to 20 μM resveratrol on HCV replication in our experiments using either J6/JFH1 (GT2a) or Bart79I (GT1b) replicon cells (data not shown). Although we do not know the exact reason for this discrepancy, the difference in HCV strains used in the experiments (O and Con1 strains of GT1b HCV) may have played a role. When we performed the sequence alignment analysis of NS3 regions between Con1 and Japan O, we were able to detect many different amino acid residues of NS3 between Con1 isolates of GT1b and Japan O. Especially, we noticed that isoleucine at 249 position was changed into valine in the case of NS3 of Japan O. Because I249T substitution played an important role in development of resistance to vitisin B according to our data (Figure 5A–D), we speculate that this I249V amino acid change may contribute to development of resistance to vitisin B by Japan O isolate. This hypothesis needs to be tested in the further study.
In spite of superior biological properties of vitisin B including a nanomolar antiviral potency, a novel mechanism of action targeting the NS3 helicase, and a preferential delivery into liver, there are many hurdles to be overcome in order to develop vitisin B as an anti‐HCV therapeutic agent. First, due to its complicated chemical structure, it will be very hard to produce vitisin B in large quantities by chemical synthesis. Therefore, we may need to rely on purification of vitisin B from grapevine roots, and this may be a very challenging task. Second, it is well known that polyphenolic compounds are chemically unstable. Vitisin B does not seem to be an exception to this. We noticed a change of colour in vitisin B when exposed to a room temperature for a long period of time. We may need to perform chemical modifications to increase its stability. Third, due to its relatively large molecular size, vitisin B is not able to penetrate the gastrointestinal tract. Because most of HCV therapeutic agents are already developed as orally available forms, it would be less attractive to develop vitisin B derivatives as non‐oral drugs.
In conclusion, we identified the crude GRE as a potent HCV inhibitor from a plant extract library. We determined which extract had the maximal antiviral activity and minimal cytotoxicity, and identified the chemical structures of active compounds within the GRE–EA‐Bd. We identified vitisin B to have the greatest activity against HCV replication. Finally, direct binding to and inhibition of HCV NS3 helicase by vitisin B may play an important role in its superior suppression of HCV replication. The current work provides a molecular basis for the development of a new class of plant extract inhibitors targeting HCV replication. In spite of its essential role in the life cycle of HCV, understanding of the detailed molecular mechanisms underlying NS3 helicase‐induced unwinding of duplex RNA intermediates and its significance in overall HCV RNA replication is still lacking. We believe that this newly identified potent NS3 helicase inhibitor will facilitate elucidation of these unknown molecular functions of the HCV NS3 helicase.
Author contributions
K. D. Y., T‐H. H. and C. L. designed the research. S. L., K. D. Y., G. C., Y. C., M. L., H. J., B. S. K., D‐H. J., J‐G. O., G‐W. K. and S. K. B. performed experiments. J‐W. O., Y‐J. J., H. J. K., D‐H. M., M. P. W., T‐H. H. and C. L. analysed data. S. L., K. D. Y., T‐H. H. and C. L. wrote the manuscript.
Conflict of interest
None.
Supporting information
Figure S1 (A) Grapevine root extract (GRE) inhibits HCV replication. (A) Three hundred and forty‐four ethanol extracts from an Asian herbal plant library were prepared, and their effects on HCV replication were evaluated using Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells at 10 µg/ml for 72 h. Their effects on cell viability were measured in parallel using an MTT‐based cytotoxicity assay. The y‐axis indicates the relative percentage cell viability divided by relative HCV replication as measured by Renilla luciferase activity. (B) Dose‐dependent effects of GRE on HCV replication and cell viability. A dose‐response curve was determined by measuring the relative luciferase activities and cell viabilities in Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of GRE for 72 h. EC50 and CC50 are the mean 50% effective and cytotoxic concentrations, respectively. (C) Fractionation of GRE and EC50 and CC50 values in each GRE fractions. Dose‐response curves for each GRE fraction were determined by measuring relative luciferase activities and cell viabilities of Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of each GRE fraction for 72 h. (D) Effect of vitisin B on Nrf2‐dependent transactivation. Nrf2 reporter plasmid was transfected into Huh7.5 cells. Transfected cells were plated onto a white 96‐well plate (Costar 3610) and supplemented with DMSO or 100 pM, 1 nM, 10 nM, 100 nM, 1 μM, and 10 μM of vitisin B. At 48 hr after incubation, firefly luciferase activities were measured by using a luciferase reagent.
supporting Information
supporting Information
supporting Information
supporting Information
Acknowledgements
This work was supported by the GRRC programme of Gyeonggi Province [(GRRC‐DONGGUK2014‐A01), Development of therapeutics for hepatitis C]. This research was also supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009‐0083522, 2010‐0017984, 2013R1A1A2009176, 2012M3A9C1053532, 2013R1A1A1011851 and 2014K1A4A7A01074644).
Lee, S. , Yoon, K. D. , Lee, M. , Cho, Y. , Choi, G. , Jang, H. , Kim, B. , Jung, D.‐H. , Oh, J.‐G. , Kim, G.‐W. , Oh, J.‐W. , Jeong, Y.‐J. , Kwon, H. J. , Bae, S. K. , Min, D.‐H. , Windisch, M. P. , Heo, T.‐H. , and Lee, C. (2016) Identification of a resveratrol tetramer as a potent inhibitor of hepatitis C virus helicase. British Journal of Pharmacology, 173: 191–211. doi: 10.1111/bph.13358.
References
- Ahn DG, Shim SB, Moon JE, Kim JH, Kim SJ, Oh JW (2011). Interference of hepatitis C virus replication in cell culture by antisense peptide nucleic acids targeting the X‐RNA. J. Viral Hepat. 18: e298–306. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br. J. Pharmacol. 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br. J. Pharmacol. 170: 1449–1458.24528237 [Google Scholar]
- Belon CA, Frick DN (2009). Helicase inhibitors as specifically targeted antiviral therapy for hepatitis C. Future Virol 4: 277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman K, Kwo PY (2009). Boceprevir, an NS3 protease inhibitor of HCV. Clin. Liver Dis. 13: 429–439. [DOI] [PubMed] [Google Scholar]
- Blight KJ, Kolykhalov AA, Rice CM (2000). Efficient initiation of HCV RNA replication in cell culture. Science 290: 1972–1974. [DOI] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM (2002). Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76: 13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson PD, Cho NJ, Einav S, Lee C, Tai V, Bechtel J et al. (2010). A small molecule inhibits HCV replication and alters NS4B's subcellular distribution. Antiviral Res. 87: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush CO, Pokrovskii MV, Saito R, Morganelli P, Canales E, Clarke MO et al. (2014). A small‐molecule inhibitor of hepatitis C virus infectivity. Antimicrob. Agents Chemother. 58: 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LG, Wang CC (2009). Preparative separation of oligostilbenes from Vitis thunbergii var. taiwaniana by centrifugal partition chromatography followed by Sephadex LH‐20 column chromatography. Sep. Purif. Technol. 66: 65–70. [Google Scholar]
- Chiou WF, Huang YL, Liu YW (2014). (+)‐Vitisin A inhibits osteoclast differentiation by preventing TRAF6 ubiquitination and TRAF6‐TAK1 formation to suppress NFATc1 activation. PLoS One 9: e89159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NJ, Dvory‐Sobol H, Lee C, Cho SJ, Bryson P, Masek M et al. (2010). Identification of a class of HCV inhibitors directed against the nonstructural protein NS4B. Sci. Transl. Med. 2: 15–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WK, Yoon KD, Lee JK, Park JB, Heo TH, Lee C et al. (2015). Development and validation of a liquid chromatography‐tandem mass spectrometry method for quantification of vitisin B, a resveratrol tetramer, in rat plasma and urine. J. Sep. Sci. 38: 1872–1880. [DOI] [PubMed] [Google Scholar]
- Chou TC (2010). Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res. 70: 440–446. [DOI] [PubMed] [Google Scholar]
- Docherty JJ, Sweet TJ, Bailey E, Faith SA, Booth T (2006). Resveratrol inhibition of varicella‐zoster virus replication in vitro . Antiviral Res. 72: 171–177. [DOI] [PubMed] [Google Scholar]
- Du MX, Johnson RB, Sun XL, Staschke KA, Colacino J, Wang QM (2002). Comparative characterization of two DEAD‐box RNA helicases in superfamily II: human translation‐initiation factor 4A and hepatitis C virus non‐structural protein 3 (NS3) helicase. Biochem. J. 363 (Pt 1): 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvory‐Sobol H, Wong KA, Ku KS, Bae A, Lawitz EJ, Pang PS et al. (2012). Characterization of resistance to the protease inhibitor GS‐9451 in hepatitis C virus‐infected patients. Antimicrob. Agents Chemother. 56: 5289–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar M, Cheong KH, Liu P, Greenberg HB, Rice CM, Glenn JS (2003). Amphipathic helix‐dependent localization of NS5A mediates hepatitis C virus RNA replication. J. Virol. 77: 6055–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers DL, Wang X, Huong SM, Huang DY, Huang ES (2004). 3,4′,5‐Trihydroxy‐trans‐stilbene (resveratrol) inhibits human cytomegalovirus replication and virus‐induced cellular signaling. Antiviral Res. 63: 85–95. [DOI] [PubMed] [Google Scholar]
- Everson GT, Sims KD, Rodriguez‐Torres M, Hezode C, Lawitz E, Bourliere M et al. (2014). Efficacy of an interferon‐ and ribavirin‐free regimen of daclatasvir, asunaprevir, and BMS‐791325 in treatment‐naive patients with HCV genotype 1 infection. Gastroenterology 146: 420–429. [DOI] [PubMed] [Google Scholar]
- Faith SA, Sweet TJ, Bailey E, Booth T, Docherty JJ (2006). Resveratrol suppresses nuclear factor‐kappaB in herpes simplex virus infected cells. Antiviral Res. 72: 242–251. [DOI] [PubMed] [Google Scholar]
- Frick DN (2006). Hepatitis C viruses: genomes and molecular biology. edn. Horizon Bioscience. [PubMed]
- Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA et al. (2010). Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaminza P, Whitten‐Bauer C, Chisari FV (2010). Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. U. S. A. 107: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma S, Butini S, Campiani G, Brindisi M, Zanoli S, Romano MP et al. (2011). Discovery of potent nucleotide‐mimicking competitive inhibitors of hepatitis C virus NS3 helicase. Bioorg. Med. Chem. Lett. 21: 2776–2779. [DOI] [PubMed] [Google Scholar]
- Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM (1993a). A second hepatitis C virus‐encoded proteinase. Proc. Natl. Acad. Sci. U. S. A. 90: 10583–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM (1993b). Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67: 1385–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobler JA, Markel EJ, Fay JF, Graham DJ, Simcoe AL, Ludmerer SW et al. (2003). Identification of a key determinant of hepatitis C virus cell culture adaptation in domain II of NS3 helicase. J. Biol. Chem. 278: 16741–16746. [DOI] [PubMed] [Google Scholar]
- do Ha T, Chen QC, Hung TM, Youn UJ, Ngoc TM, Thuong PT et al. (2009). Stilbenes and oligostilbenes from leaf and stem of Vitis amurensis and their cytotoxic activity. Arch. Pharm. Res. 32: 177–183. [DOI] [PubMed] [Google Scholar]
- Huang YL, Liu YW, Huang YJ, Chiou WF (2013). A special ingredient (VtR) containing oligostilbenes isolated from Vitis thunbergii prevents bone loss in ovariectomized mice: in vitro and in vivo study. Evid Based Complement Alternat Med 2013: 409421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Kim HY, Jo S, Park E, Choi J, Kong S et al. (2013). Synthesis and evaluation of hexahydropyrimidines and diamines as novel hepatitis C virus inhibitors. Eur. J. Med. Chem. 70: 315–325. [DOI] [PubMed] [Google Scholar]
- Hwang JY, Windisch MP, Jo S, Kim K, Kong S, Kim HC et al. (2012). Discovery and characterization of a novel 7‐aminopyrazolo[1,5‐a]pyrimidine analog as a potent hepatitis C virus inhibitor. Bioorg. Med. Chem. Lett. 22: 7297–7301. [DOI] [PubMed] [Google Scholar]
- Jang H, Ryoo SR, Kim YK, Yoon S, Kim H, Han SW et al. (2013). Discovery of hepatitis C virus NS3 helicase inhibitors by a multiplexed, high‐throughput helicase activity assay based on graphene oxide. Angew. Chem. Int. Ed. Engl. 52: 2340–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, He S, Sun C, Pan Y (2012). Selective (1)O(2) quenchers, oligostilbenes, from Vitis wilsonae: structural identification and biogenetic relationship. Phytochemistry 77: 294–303. [DOI] [PubMed] [Google Scholar]
- Jin G, Lee S, Choi M, Son S, Kim GW, Oh JW et al. (2014). Chemical genetics‐based discovery of indole derivatives as HCV NS5B polymerase inhibitors. Eur. J. Med. Chem. 75: 413–425. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park HS, Lee MS, Cho YJ, Kim YS, Hwang JT et al. (2008). Vitisin A inhibits adipocyte differentiation through cell cycle arrest in 3T3‐L1 cells. Biochem. Biophys. Res. Commun. 372: 108–113. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim JH, Kim YG, Lim HS, Oh JW (2004). Protein kinase C‐related kinase 2 regulates hepatitis C virus RNA polymerase function by phosphorylation. J. Biol. Chem. 279: 50031–50041. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim JH, Sun JM, Kim MG, Oh JW (2009). Suppression of hepatitis C virus replication by protein kinase C‐related kinase 2 inhibitors that block phosphorylation of viral RNA polymerase. J. Viral Hepat. 16: 697–704. [DOI] [PubMed] [Google Scholar]
- Kohlway A, Pirakitikulr N, Ding SC, Yang F, Luo D, Lindenbach BD et al. (2014). The linker region of NS3 plays a critical role in the replication and infectivity of hepatitis C virus. J. Virol. 88: 10970–10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM (2000). Hepatitis C virus‐encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo . J. Virol. 74: 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo M, Kim SH, Lee N, Yoo MY, Ryu SY, Kwon DY et al. (2008). 3‐Hydroxy‐3‐methylglutaryl‐CoA (HMG‐CoA) reductase inhibitory effect of Vitis vinifera . Fitoterapia 79: 204–206. [DOI] [PubMed] [Google Scholar]
- Kratschmar DV, Calabrese D, Walsh J, Lister A, Birk J, Appenzeller‐Herzog C et al. (2012). Suppression of the Nrf2‐dependent antioxidant response by glucocorticoids and 11beta‐HSD1‐mediated glucocorticoid activation in hepatic cells. PLoS One 7: e36774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AM, Frick DN (2006). Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 80: 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Lee JM, Lee NR, Jin BS, Jang KJ, Kim DE et al. (2009). Aryl diketoacids (ADK) selectively inhibit duplex DNA‐unwinding activity of SARS coronavirus NTPase/helicase. Bioorg. Med. Chem. Lett. 19: 1636–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Ma H, Hang JQ, Leveque V, Sklan EH, Elazar M et al. (2011). The hepatitis C virus NS5A inhibitor (BMS‐790052) alters the subcellular localization of the NS5A non‐structural viral protein. Virology 414: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C (2011). Discovery of hepatitis C virus NS5A inhibitors as a new class of anti‐HCV therapy. Arch. Pharm. Res. 34: 1403–1407. [DOI] [PubMed] [Google Scholar]
- Lee C (2013a). Daclatasvir: potential role in hepatitis C. Drug Des. Devel. Ther. 7: 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C (2013b). Interaction of hepatitis C virus core protein with Janus kinase is required for efficient production of infectious viruses. Biomol. Ther. 21: 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Baert L, Zhang D, Xia M, Zhong W, Van Coillie E et al. (2012). Effect of grape seed extract on human norovirus GII.4 and murine norovirus 1 in viral suspensions, on stainless steel discs, and in lettuce wash water. Appl. Environ. Microbiol. 78: 7572–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC et al. (2005). Complete replication of hepatitis C virus in cell culture. Science 309: 623–626. [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285: 110–113. [DOI] [PubMed] [Google Scholar]
- Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S et al. (2009). Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U. S. A. 106: 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Yates J, Liang Y, Lemon SM, Yi M (2008). NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82: 7624–7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackintosh SG, Lu JZ, Jordan JB, Harrison MK, Sikora B, Sharma SD et al. (2006). Structural and biological identification of residues on the surface of NS3 helicase required for optimal replication of the hepatitis C virus. J. Biol. Chem. 281: 3528–3535. [DOI] [PubMed] [Google Scholar]
- Matias AA, Serra AT, Silva AC, Perdigao R, Ferreira TB, Marcelino I et al. (2010). Portuguese winemaking residues as a potential source of natural anti‐adenoviral agents. Int. J. Food Sci. Nutr. 61: 357–368. [DOI] [PubMed] [Google Scholar]
- Matthews SJ, Lancaster JW (2012). Telaprevir: a hepatitis C NS3/4A protease inhibitor. Clin. Ther. 34: 1857–1882. [DOI] [PubMed] [Google Scholar]
- McGivern DR, Masaki T, Lovell W, Hamlett C, Saalau‐Bethell S, Graham B (2015). Protease inhibitors block multiple functions of the NS3/4A protease–helicase during the hepatitis C virus life cycle. J. Virol. 89: 5362–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivern DR, Masaki T, Williford S, Ingravallo P, Feng Z, Lahser F, et al (2014). Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 147: 453–462 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br. J. Pharmacol. 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Jeong S, Davaatseren M, Kim W, Sung Kwang P, Kim SH, Haeng Jeon H et al. (2009). Vitisin A suppresses LPS‐induced NO production by inhibiting ERK, p38, and NF‐kappaB activation in RAW 264.7 cells. Int. Immunopharmacol. 9: 319–323. [DOI] [PubMed] [Google Scholar]
- Moradpour D, Penin F, Rice CM (2007). Replication of hepatitis C virus. Nat. Rev. Microbiol. 5: 453–463. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Sorrell MF (2008). Controversies in liver transplantation for hepatitis C. Gastroenterology 134: 1777–1788. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Saito H, Ikeda M, Hokari R, Kato N, Hibi T et al. (2010). An antioxidant resveratrol significantly enhanced replication of hepatitis C virus. World J. Gastroenterol. 16: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong ET, Hwang TL, Huang YL, Lin CF, Wu WB (2011). Vitisin B, a resveratrol tetramer, inhibits migration through inhibition of PDGF signaling and enhancement of cell adhesiveness in cultured vascular smooth muscle cells. Toxicol. Appl. Pharmacol. 256: 198–208. [DOI] [PubMed] [Google Scholar]
- Palamara AT, Nencioni L, Aquilano K, De Chiara G, Hernandez L, Cozzolino F et al. (2005). Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 191: 1719–1729. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflieger A, Waffo Teguo P, Papastamoulis Y, Chaignepain S, Subra F, Munir S et al. (2013). Natural stilbenoids isolated from grapevine exhibiting inhibitory effects against HIV‐1 integrase and eukaryote MOS1 transposase in vitro activities. PLoS One 8: e81184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MNAK, Shipman C Jr (1993). MacSynergy II manual. University of Michigan: Ann Arbor, Michigan. [Google Scholar]
- Prichard MN, Shipman C Jr (1996). Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir. Ther. 1: 9–20. [PubMed] [Google Scholar]
- Prichard MN, Shipman C Jr (1990). A three‐dimensional model to analyze drug–drug interactions. Antiviral Res. 14: 181–205. [DOI] [PubMed] [Google Scholar]
- Santos AC, Veiga F, Ribeiro AJ (2011). New delivery systems to improve the bioavailability of resveratrol. Expert Opin. Drug Deliv. 8: 973–990. [DOI] [PubMed] [Google Scholar]
- Seya K, Kanemaru K, Sugimoto C, Suzuki M, Takeo T, Motomura S et al. (2009). Opposite effects of two resveratrol (trans‐3,5,4′‐trihydroxystilbene) tetramers, vitisin A and hopeaphenol, on apoptosis of myocytes isolated from adult rat heart. J. Pharmacol. Exp. Ther. 328: 90–98. [DOI] [PubMed] [Google Scholar]
- Sharaf M, El‐Deeb NM, EL‐Adawi HI (2012). The potentiality of grape seed extract as a novel anti‐hepatitis C virus agent. J. Med. Sci. 12: 107–113. [Google Scholar]
- Shepard CW, Finelli L, Alter MJ (2005). Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5: 558–567. [DOI] [PubMed] [Google Scholar]
- Sklan EH, Staschke K, Oakes TM, Elazar M, Winters M, Aroeti B et al. (2007). A Rab‐GAP TBC domain protein binds hepatitis C virus NS5A and mediates viral replication. J. Virol. 81: 11096–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven M, Lipson REG, Ozen FS, Karthikeyan L, Kirov N, Stotzky G (2011). Cranberry and grape juices affect tight junction function and structural integrity of rotavirus‐infected monkey kidney epithelial cell monolayers. Food Environ. Virol. 3: 46–54. [DOI] [PubMed] [Google Scholar]
- Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM (2006). Time‐ and temperature‐dependent activation of hepatitis C virus for low‐pH‐triggered entry. J. Virol. 80: 1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udenigwe CC, Ramprasath VR, Aluko RE, Jones PJ (2008). Potential of resveratrol in anticancer and anti‐inflammatory therapy. Nutr. Rev. 66: 445–454. [DOI] [PubMed] [Google Scholar]
- Walle T (2011). Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 1215: 9–15. [DOI] [PubMed] [Google Scholar]
- Wang KT, Chen LG, Tseng SH, Huang JS, Hsieh MS, Wang CC (2011). Anti‐inflammatory effects of resveratrol and oligostilbenes from Vitis thunbergii var. taiwaniana against lipopolysaccharide‐induced arthritis. J. Agric. Food Chem. 59: 3649–3656. [DOI] [PubMed] [Google Scholar]
- Watkins WJ, Ray AS, Chong LS (2010). HCV NS5B polymerase inhibitors. Curr. Opin. Drug Discov. Devel. 13: 441–465. [PubMed] [Google Scholar]
- Wilby KJ, Partovi N, Ford JA, Greanya E, Yoshida EM (2012). Review of boceprevir and telaprevir for the treatment of chronic hepatitis C. Can. J. Gastroenterol. 26: 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuestenberg A, Kah J, Singethan K, Sirma H, Keller AD, Rosal SR et al. (2014). Matrix conditions and KLF2‐dependent induction of heme oxygenase‐1 modulate inhibition of HCV replication by fluvastatin. PLoS One 9: e96533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu MS, Lee J, Lee JM, Kim Y, Chin YW, Jee JG et al. (2012). Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 22: 4049–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Grapevine root extract (GRE) inhibits HCV replication. (A) Three hundred and forty‐four ethanol extracts from an Asian herbal plant library were prepared, and their effects on HCV replication were evaluated using Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells at 10 µg/ml for 72 h. Their effects on cell viability were measured in parallel using an MTT‐based cytotoxicity assay. The y‐axis indicates the relative percentage cell viability divided by relative HCV replication as measured by Renilla luciferase activity. (B) Dose‐dependent effects of GRE on HCV replication and cell viability. A dose‐response curve was determined by measuring the relative luciferase activities and cell viabilities in Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of GRE for 72 h. EC50 and CC50 are the mean 50% effective and cytotoxic concentrations, respectively. (C) Fractionation of GRE and EC50 and CC50 values in each GRE fractions. Dose‐response curves for each GRE fraction were determined by measuring relative luciferase activities and cell viabilities of Rluc‐J6/JFH1 RNA‐transfected Huh7.5 cells treated with increasing concentrations of each GRE fraction for 72 h. (D) Effect of vitisin B on Nrf2‐dependent transactivation. Nrf2 reporter plasmid was transfected into Huh7.5 cells. Transfected cells were plated onto a white 96‐well plate (Costar 3610) and supplemented with DMSO or 100 pM, 1 nM, 10 nM, 100 nM, 1 μM, and 10 μM of vitisin B. At 48 hr after incubation, firefly luciferase activities were measured by using a luciferase reagent.
supporting Information
supporting Information
supporting Information
supporting Information