

Abstract
The triggering receptor expressed on myeloid cells (TREM) 2 is a member of the immunoglobulin superfamily of receptors and mediates signaling in immune cells via engagement of its co-receptor DNAX-activating protein of 12 kDa (DAP12). Homozygous mutations in TREM2 or DAP12 cause Nasu-Hakola disease, which is characterized by bone abnormalities and dementia. Recently, a variant of TREM2 has also been associated with an increased risk for Alzheimer disease. The selective expression of TREM2 on immune cells and its association with different forms of dementia indicate a contribution of this receptor in common pathways of neurodegeneration.
Keywords: Alzheimer disease, cell signaling, microglia, neurodegenerative disease, neuroinflammation, protein processing
Introduction
The triggering receptors expressed on myeloid cells (TREMs)2 are members of the immunoglobulin-lectin-like superfamily of receptors. They represent type I membrane proteins with a single immunoglobulin-like domain in their N-terminal ectodomain, one transmembrane domain, and a short C-terminal intracellular tail (1, 2) (Fig. 1).
FIGURE 1.

TREM2-DAP12 dependent intracellular signaling pathways. TREM2 associates with DAP12 via electrostatic interaction within the transmembrane domains. Ligand binding to TREM2 results in phosphorylation of tyrosine residues within an ITAM motif of the DAP12 cytoplasmic domain, as well as recruitment of several signaling proteins, including Syk, Dok3, Sos1, and Grb2. The related signaling pathways regulate Ca2+ mobilization, cell cytoskeletal remodeling, and gene transcription. The regulation of TREM2-DAP12 via PI3K and RAS allows cross-talk with and modulation of TLR signaling pathways. See text for details. P, phosphorylation; PLC, phospholipase C; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol; LEF, lymphoid enhancer-binding factor; MEF, myocyte enhancer factor; MKK, mitogen-activated protein kinase kinase.
In humans, two homologous genes on chromosomes 6p21 encode two similar proteins, TREM1 and TREM2 (1, 3). Despite their high homology, TREM1 and TREM2 have divergent expression patterns and signaling functions. Although activation of TREM1 increases the secretion of pro-inflammatory cytokines, TREM2 instead has anti-inflammatory activity (4–7). However, both receptors lack signaling motifs in their cytoplasmic domains and require association with the co-receptor DNAX-activating protein of 12 kDa (DAP12) to mediate intracellular signal transduction. The cytoplasmic domain of DAP12 contains a characteristic immunoreceptor tyrosine-based activation motif (ITAM). The interaction of TREMs and DAP12 is mediated via electrostatic interactions within their transmembrane domains. Stimulation of TREMs results in phosphorylation of critical tyrosine residues in the ITAM motif of DAP12, thereby regulating different intracellular signaling pathways in monocytes and monocyte-derived cells (1, 2, 3, 8) (Fig. 1).
Mutations in TREM2 or DAP12 are associated with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, also called Nasu-Hakola disease (NHD), which is characterized by bone abnormalities and dementia (6, 9, 10). Recent genetic studies also indicate an association of TREM2 variants with Alzheimer disease (AD) and other neurodegenerative disorders (6, 11). Disease-associated variants of TREM1 have not been identified so far. Thus, TREM2 might represent a common modulator in the pathogenesis of different neurodegenerative diseases. These findings also support a functional involvement of neuroinflammatory processes in the pathways to neurodegeneration.
Neuroinflammation is a common feature of neurodegenerative diseases and characterized by micro- and astrogliosis and increased levels of proinflammatory cytokines (12, 13). The inflammatory process is likely promoted by the accumulation of protein aggregates and cell damage. Microglia resemble resident macrophages in the brain and express several cell surface receptors that recognize danger or pathogen-associated molecular patterns (14–16). Their activation results in several cellular responses, including the synthesis and secretion of cytokines, migration, and phagocytosis. However, persistent activation of microglia and chronic neuroinflammation could also exert detrimental effects to brain function and might promote neurodegeneration (14, 17, 18).
Under physiological conditions, microglia constantly scan their environment and interact with other cell types, such as neurons and astrocytes, and with the extracellular matrix and blood vessels. They contribute to brain homeostasis and synaptic plasticity and mediate repair processes during brain injury (17, 18, 19).
Expression, Metabolism, and Signaling of TREMs
The first gene of the TREM family, TREM1, was identified in natural killer cells and shown to be also expressed in different monocytes and monocyte-derived macrophages (20). The homologous TREM2 was initially cloned from dendritic cells (21). Although certain macrophages and neutrophils express both TREM homologs, dendritic cells, osteoclasts, and microglia show predominant expression of TREM2 (1, 4).
Both receptors require engagement of the co-receptor DAP12 for transmembrane signaling. DAP12 is also a type I membrane protein. In contrast to TREMs, DAP12 has no globular ectodomain and might not be involved in binding of ligands (Fig. 1). The extracellular sequence of about 20 amino acids contains cysteine residues that mediate homodimer formation by disulfide bridges. The interaction of TREMs with DAP12 is mediated by a salt bridge linkage of an arginine and aspartic acid residue within the respective transmembrane domains (22) (Fig. 1).
Cell biological studies revealed that TREM2 is transported in the secretory pathway and shuttles between the plasma membrane and cytoplasmic vesicles (23, 24). It is unclear whether DAP12 already associates with TREMs in early secretory compartments or whether interaction is induced by ligand binding and clustering of TREMs at the cell surface.
During biosynthesis and secretory transport, TREM2 also undergoes maturation by complex glycosylation (24, 25, 26), but the biological relevance of these modifications remains to be determined. TREM2 is also subjected to sequential proteolytic processing by ectodomain shedding and intramembranous cleavage (24, 25, 26) (Fig. 2). The ectodomain shedding results in the release of the soluble TREM2 from cells into extracellular fluids and involves proteases of the a disintegrin and metalloprotease (ADAM) family. Soluble TREM2 has also been detected in cerebrospinal fluid (CSF) and blood plasma of humans (26, 27), and levels in CSF were significantly reduced in samples of AD and FTD patients, suggesting altered proteolytic processing during neurodegeneration (26). In CSF and plasma of a patient with FTD-like symptoms carrying a homozygous TREM T66M mutation, soluble TREM2 was not detected, suggesting that this mutation decreases shedding of TREM2 (26). Whether the recently identified AD-associated R47H variant of TREM2 also affects protein levels in CSF and plasma remains to be determined.
FIGURE 2.
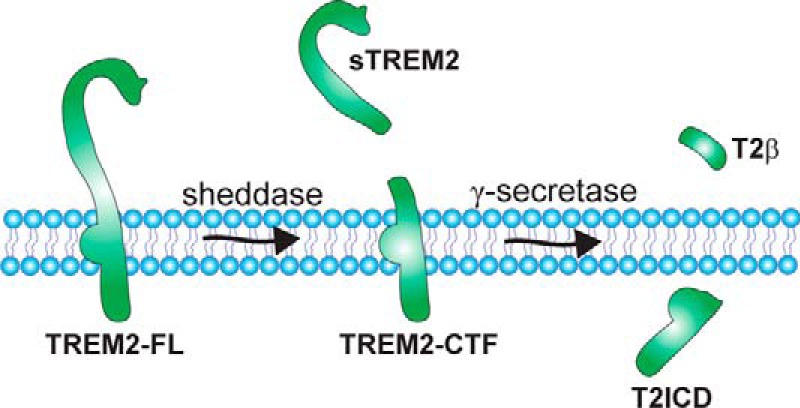
Sequential proteolytic processing of TREM2. The full-length TREM2 receptor (TREM2-FL) can be cleaved by a shedding protease of the ADAM family (sheddase), resulting in the secretion of a soluble ectodomain (sTREM2) and generation of a membrane-bound C-terminal fragment (TREM2-CTF). This C-terminal fragment represents a substrate for γ-secretase-dependent intramembrane proteolysis. The putative cleavage products resulting from γ-secretase-dependent cleavage are indicated as T2β (TREM2-A β-like peptides) and T2ICD (TREM2 intracellular domain). Potential implications of the proteolytic processing for TREM2 function are discussed in the text.
Soluble TREM2 variants have also been found to be altered in multiple sclerosis. However, in contrast to the decreased levels found in CSF of AD and FTD patients, soluble TREM2 was increased in CSF of multiple sclerosis cases and other inflammatory neurological diseases, but was unchanged in non-inflammatory neurological diseases (27). Thus, it will be interesting to further explore the detection of soluble TREM2 in biological fluids as a biomarker for different neurological diseases.
Whether the soluble variants of TREM2 in biological fluids exclusively derive from proteolytic processing of the full-length receptor is unclear. For both TREM1 and TREM2, alternative mRNA transcripts have been identified that could also result in the synthesis and secretion of truncated variants (28, 29).
It has been shown that soluble TREM1 competes with cell surface-bound TREM1 for ligands and thereby affects ligand-induced signaling (30, 31, 27). In addition, soluble TREM1 might also exert paracrine and autocrine effects upon binding to cell surface molecules. However, a functional role of soluble TREM2 in extracellular fluids remains to be determined.
The C-terminal fragment (CTF) of TREM2 that results from ectodomain shedding remains membrane-tethered via its transmembrane domain and could be subjected to intramembranous proteolysis by the γ-secretase complex that is also involved in the generation of the AD-associated amyloid-β (Aβ) peptide (24–26, 32) (Fig. 2). Interestingly, inhibition of γ-secretase also impaired the phosphorylation of DAP12 and downstream metabolism of phosphatidylinositides (24), cellular rearrangement, and phagocytosis (33), suggesting that TREM2-DAP12-mediated signaling is affected by γ-secretase activity. Overexpression of the TREM2 CTF in microglial BV2 cells also decreased LPS-induced pro-inflammatory cytokine production (32), further supporting a role of the TREM2 CTF and its processing by γ-secretase in signaling.
Although both TREM1 and TREM2 signal via DAP12, their stimulation has different effects on LPS-induced signaling. While TREM1 activation promotes pro-inflammatory signaling and cytokine production in response to LPS, TREM2 activation instead suppresses these LPS-induced effects (7). In addition to TREMs, more than 20 different surface receptors associate with DAP12 to regulate several intracellular signaling pathways. In general, receptor ligation results in Src kinase-dependent phosphorylation of DAP12 at critical tyrosine residues and the recruitment of spleen tyrosine kinases (Syk) and the tyrosine kinase ζ-chain-associated protein 70 (ZAP70) (1, 4). Whether both kinases are also involved in the phosphorylation of DAP12 is unclear. Syk and ZAP70 can activate additional signaling proteins including PI3K, guanine nucleotide exchange factors (GEF), and phosphotyrosine kinase 2 (Pyk2), and can also regulate membrane-proximal signaling of other cell surface receptors, including TLR4, IFNαR1/2, and Plexin-A1. The complex regulation and interplay of multiple pathways allow fine-tuned responses of immune cells, including cytokine production and release, migration, phagocytosis, proliferation and differentiation, and cell survival (Fig. 1). Signaling via DAP12 has been described in detail in several recent reviews (2, 7, 34, 35). It is important to note that most studies on the signaling of TREMs and DAP12 have been performed with peripheral monocytes or monocyte-derived cells.
Functional studies with microglia from mouse brain revealed that TREM2 also signals via DAP12 and regulates several pathways (4). Because a specific ligand for TREM2 is unknown, Takahashi et al. (36) used overexpression of tagged variants of TREM2 in primary microglia that could be activated by cross-linking with antibodies against the epitope tag. Activation of TREM2 resulted in increased phosphorylation of DAP12. Cross-linking of TREM2 also increased phagocytosis of damaged neurons by primary mouse microglia (36). In addition, activation of TREM2 resulted in remodeling of the actin cytoskeleton and promoted migratory activity toward chemokines accompanied with increased phosphorylation of ERK. Knockdown of endogenous TREM2 in primary microglia decreased phagocytic activity and increased transcription of pro-inflammatory cytokines and nitric oxide synthase-2 (36). These findings were consistent with previous observations obtained with macrophages from TREM2 knock-out mice that also suggested an anti-inflammatory role of this receptor (31, 38). TREM2 KO cells had an elevated cytokine response to TLR stimulation by LPS (38). In addition, primary microglia of TREM2 KO cells show decreased proliferation in culture and elevated cell death (39).
Homozygous and Compound Mutations of TREM2 or DAP12 in Nasu-Hakola Disease and Frontotemporal Dementia
Interestingly, mutations in DAP12 and TREM2 are associated with autosomal recessively inherited NHD, also called polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy or membranous lipodystrophy. This rare disease is characterized by impaired bone metabolism leading to cysts and fractures, and by progressive dementia with early onset between 15 and 30 years of age (6, 9). In most cases, osseous lesions occur before neurological symptoms. Early in the neurological stage, patients develop changes in their personality followed by memory deficits. The neurological symptoms closely resemble those of frontotemporal dementias (FTDs), neurodegenerative disorders that are much more common in patients below the age of 65 years and that are characterized by inappropriate behavior and changes in personality and language impairment (6, 40). Brains of FTD cases show atrophy of frontal and temporal lobes and degeneration of spindle neurons. Based on different inclusions of aggregated proteins, FTD is neuropathologically subdivided into three groups depending on the occurrence of tau, α-synuclein, and TAR DNA-binding protein 43 (TDP43) or fused in sarcoma (FUS) proteins in neurons and glial cells (42, 43). Mutations in some of these proteins cause familial dominantly inherited forms of FTD (44, 45). Disease onset of FTD is typically between the ages of 50 and 65. Interestingly, rare homozygous or compound mutations in TREM2 have been identified in cases with FTD-like symptomatic presentation in the absence of associated lesions in the bone system, therefore differing in the clinical symptoms from NHD (40, 45, 47–49) (Table 1). Another rare variant of TREM2 with an R47H substitution has recently also been associated with an increased risk for late-onset AD (50, 51).
TABLE 1.
Disease-associated mutations in TREM2
| Mutation/polymorphism | Effect | Disease | Genotype | References |
|---|---|---|---|---|
| c.40G→T | E14X, truncated, nonfunctional protein | NHD | Homozygous | 91 |
| c.40 + 3delAGG | Altered splicing, decreased protein | FTD-L | Homozygous | 92 |
| c.97C→T | Q33X, truncated, nonfunctional protein (also FTD-like syndrome) | NHD, FTD-L | Homozygous | 41, 93 |
| c.113A→G | Y38C | NHD, FTD-L | Homozygous, homozygous compound heterozygous | 40, 41, 93 |
| c.132G→A | W44X, truncated, nonfunctional protein | NHD | Homozygous | 91 |
| c.140G→A | R47H | AD risk | Heterozygous | 50, 51 |
| c.185G→A | R62H | AD risk | Heterozygous | 46 |
| c.197C→T | T66M | NHD, FTD-L | Homozygous | 41, 93 |
| c.233G→A | W78X, truncated, nonfunctional protein | NHD | Homozygous | 91 |
| c.257A→T | D86V | FTD-L | Compound heterozygous | 40 |
| c.267delG | Frame-shift, nonfunctional protein | NHD | Homozygous | 91 |
| c.313delG | Frame-shift, nonfunctional protein | NHD | Homozygous | 91 |
| c.377T→G | V126G | NHD | Homozygous | 93 |
| c.401A→G | D134G | NHD | Homozygous | 93 |
| c.482 + 2T→C | Impaired splicing, decreased/nonfunctional protein | NHD | Homozygous | 93 |
| c.558G→A | K186N, no interaction with DAP12 | NHD | Homozygous | 91 |
| c.594G→A | W198X | FTD-L | Homozygous | 37 |
Molecular Genetics of Alzheimer Disease
AD is the most common form of dementia and is characterized at the neuropathological level by the accumulation of extracellular plaques and intraneuronal neurofibrillary tangles that are composed of Aβ peptides and the microtubule-associated protein tau, respectively (52–54). Aβ derives from the amyloid precursor protein (APP) by sequential proteolytic processing by β- and γ-secretase (55, 56). In an alternative processing pathway, APP can be cleaved by α-secretase within the Aβ domain, which precludes the subsequent generation of Aβ. α-Secretase activity is exerted by members of the ADAM family (57). As described above, ADAM proteases and γ-secretase are also involved in the proteolytic processing of TREM2.
In the vast majority of cases (>95%), AD manifests beyond the age of 65 years. However, rare familial cases with an age of onset below 65 years also occur. Genetic analyses of familial early-onset AD cases led to the identification of autosomal dominant mutations in three different genes (11, 58, 59). The first gene identified to bear AD-associated mutations was the APP gene itself. The different mutations within APP are localized within or close to the Aβ domain and promote the generation or aggregation of the peptide. Interestingly, a mutation within the Aβ domain of APP has also been shown be associated with a decreased risk for AD (60). In contrast to the disease-causing mutations of APP, the preventive variant decreased the proteolytic generation of Aβ and its propensity to aggregate (61, 62). Besides the APP gene, only two additional genes have been identified to contain mutations that also cause dominantly inherited early-onset familial AD. The two genes encode homologous proteins called presenilin (PS)-1 and PS-2 (63, 64). PS proteins are the catalytic components of the γ-secretase complex and thus, are critically involved in the proteolytic generation of Aβ (65, 66). Mutations in the PS proteins commonly favor the accumulation of Aβ42 variants that have increased propensity to aggregate. Thus, the genetics of familial early-onset AD provide strong support for a critical role of Aβ peptides in the pathogenesis of AD. However, mutations in APP and PS genes are very rare and only represent 1–3% of all AD cases.
By far the most common risk factor for late-onset AD is the ϵ4 allele of the apoE gene (67–69). The apolipoprotein (apo) E4 variant is one of three major isoforms occurring in humans that only differ in two amino acids at position 112 and 158, which could either be Cys or Arg residues (Cys115–Cys158 for apoE2, Cys115–Arg158 for apoE3, and Arg115–Arg158 for apoE4) (68–70). Since its initial discovery in 1993 as a risk factor for late-onset AD, apoE4 has been confirmed in numerous follow-up studies (59, 71, 72). However, the functional role of apoE in the pathogenesis of AD is still enigmatic. ApoE can interact with and affect the aggregation and clearance of Aβ in an isoform- and lipidation-state dependent manner (73, 74). Several receptors of the low density lipoprotein receptor (LDLR) family have been shown to bind apoE, Aβ, and APP, and thereby could affect both generation and degradation of Aβ (67, 75, 76). Interestingly, genome-wide association studies of common single nucleotide polymorphisms have identified additional genes related to lipid and lipoprotein metabolism associated with an increased risk of AD. These include the bridging integrator 1 (BIN1), clusterin (CLU), ATP-binding cassette A7 (ABCA7), phosphatidylinositol-binding clathrin assembly protein (PICALM), and CD2-associated protein (CD2AP) (11, 77, 78). However, the functional roles of the respective proteins in the pathogenesis of AD remain to be characterized in more detail. It is important to note that the odds ratios of the additional factors, that is, their effect on risk for developing AD, is much lower than that of apoE. Although the apoE4 allele increases the risk between 3- and 10-fold, the individual effects of the other factors range between 0.15- and 0.2-fold. Another set of genes with similarly low odds ratios could be related to vesicular trafficking and inflammatory pathways, including the complement receptor 1 (CR1), cluster of differentiation 33 (CD33), membrane-spanning 4-domains, and subfamily A, member 6A and 6E (MS4A6A, MS4A6E) (11, 77, 78).
Potential Role of TREM2 in the Pathogenesis of AD
The TREM2 R47H variant showed a significant association with late-onset AD with odds ratios between 2 and 4, suggesting that it increases the risk for AD (50, 51). It is currently under debate whether the TREM2 R47H or other variants in the heterozygous state are also associated with other neurodegenerative diseases such as frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson disease. A meta-analysis across these diseases indicates that the R47H variant of TREM2 is specifically associated with AD and probably with frontotemporal lobar degeneration, but not with the other tested disorders (79). However, additional missense mutations could also contribute to the risk for these neurodegenerative diseases.
It is important to note that the TREM2 R47H is rare with allele frequencies below 1% (50, 51). Thus, even if the effect size on individual AD risk for carriers of the TREM2 R47H variant is comparable with that of apoE4, the overall effect on AD prevalence in the general population is much smaller (80). However, understanding the physiological and pathophysiological functions of TREM2 in AD is important to further explore its potential as a target for therapy, prevention, and diagnosis.
Even before the identification of AD-associated mutations of TREM2, it has been shown that microglia surrounding extracellular plaques in APP transgenic mouse models have increased expression of TREM2 protein (81). Up-regulation of TREM2 mRNA in brain samples of APP transgenic mice was observed upon active vaccination with Aβ (82), and immunohistochemical analysis of human brains with TREM2 antibodies also showed reactivity in microglia and neurons in different brain regions (51). In addition, the co-receptor DAP12 was identified in an integrative network analysis as an important factor in the pathogenesis of late-onset AD (83).
The functional role of TREM2, and of microglia in general, in AD pathogenesis is currently under debate (12–18). Effects of TREM2 on Aβ deposition have been assessed by crossing heterozygous or homozygous TREM2 KO mice with APP transgenic mouse models of AD. The deletion of one TREM2 allele did not significantly affect Aβ plaque load up to the age of seven months (84). However, the number and size of microglia in the vicinity of Aβ deposits were reduced in heterozygous TREM2 KO mice (84). Crossing of homozygous TREM2 KO mice with APP transgenic models resulted in controversial results on Aβ plaque load in two independent studies. Although Jay et al. (85) showed decreased Aβ deposition, Wang. et al. (86) found increased Aβ load in TREM2 KO mice as compared with TREM2 WT mice. The latter study also showed significantly increased levels of insoluble Aβ in heterozygous TREM2 KO mice at higher ages. It will be important to assess whether these discrepancies might result from the usage of different TREM2 KO and APP transgenic mouse models, different analytical methods, or other factors. Both studies, however, revealed increased expression of TREM2 in plaque-associated myeloid cells. Whether these TREM2-positive cells represented resident microglia or were derived from infiltrated peripheral monocytes is currently under debate (85, 86). As observed already in heterozygous TREM2 KO mice (84), homozygous TREM2 KO led to a decreased number of monocyte-derived cells in the vicinity of extracellular plaques. These observations are consistent with a role of TREM2 in the regulation of cell migration, proliferation, and survival (85, 86).
How Could Mutations in TREM2 Contribute to the Risk of AD?
The TREM2 mutations found in NHD patients are deletion, nonsense, or frameshift mutations that result in the complete lack of protein or synthesis of truncated variants (6) (Table 1). Interestingly, missense mutations that decrease the interaction with DAP12 have also been identified. Mutations of DAP12 in NHD also result in the generation of non-functional protein or no protein, indicating that the disease is caused by complete loss of function in TREM2-DAP12 signaling (9, 10). Mutations in other downstream signaling components or other receptors that associate with DAP12 have not been identified. The homozygous or compound FTD- and NHD-associated mutations Y38C and T66M strongly impair the transport of TREM2 from the ER to the Golgi, and thus, its glycosylation and expression at the cell surface, also supporting a loss-of-function mechanism (25, 26). These mutations also showed decreased solubility, accumulation in the ER, and induced ER stress (25). Thus, TREM2 mutations might not only impair receptor signaling activity, but also cause cellular stress and thereby affect cell function and viability.
When compared with the NHD- and FTD-associated missense mutations, the AD-associated TREM2 R47H variant has weaker effects on its transport to the cell surface, as well as its secretion into conditioned media (26). The localization of the AD-associated R47H in the ectodomain of TERM2 could also suggest it affects the interaction with ligands. Interestingly, the R47H variant showed reduced binding to anionic and zwitterionic lipids in vitro (86). Thus, the AD-associated TREM2 R47H mutation might interfere with the interaction of microglia with membranes of damaged neurons and subsequent clearance by phagocytosis. Indeed, the TREM2 R47H variant reduced the phagocytic activity of different cell types in vitro (26, 86). Whether the R47H variant could also affect functions of microglia or peripheral cells in vivo is currently unknown.
The decreased phagocytic activity could contribute to neurodegeneration by impairing the clearance of damaged neurons and aggregated proteins, which could promote chronic pro-inflammatory reactions in the brain. Interestingly, apoE has been identified as a ligand for TREM2 in vitro (87, 88), suggesting that apoE might also regulate TREM2-DAP12-mediated signaling in microglia. Although the different apoE isoforms E2, E3, and E4 showed very similar binding to TREM2, the TREM2 R47H mutation strongly decreased this interaction, suggesting that the effects of apoE4 on AD risk might not involve differential binding to TREM2. It will now be important to investigate the functional role of the apoE-TREM2 interaction in the regulation of microglia during neurodegeneration.
TREM2 has also been linked to the hyperphosphorylation and accumulation of tau. Silencing of TREM2 by lentivirus-mediated knockdown exacerbated tau hyperphosphorylation and tau pathology, neurodegeneration, and learning deficits in a mouse model of tauopathy (89). Because these mice did not overexpress APP or develop amyloid plaques, the observed effects upon silencing of TREM2 are likely independent of Aβ. Together, these studies indicate that loss of TREM2 could trigger tau and Aβ pathology independently. It is interesting to note that senile plaques and neurofibrillary tangles have been observed in a brain autopsy of a 48-year-old patient clinically diagnosed with NHD and a homozygous mutation in TREM2 (90, 91), suggesting that impaired TREM2 signaling might be sufficient to induce the two major neuropathological hallmarks of AD. However, the relative occurrence of AD characteristic neuropathology in NHD cases remains to be determined. NHD is also associated with leukodystrophy, which is not characteristic for AD or FTD. Thus, it will be interesting to further dissect the molecular mechanisms that contribute to the different pathologies associated with TREM2 mutations.
Conclusions
The association of TREM2 mutations with different diseases, including NHD, FTD, and AD, strongly indicates an important role of this immune receptor and innate immunity during neurodegeneration. However, the molecular mechanisms underlying the pathogenic processes in these diseases remain largely unclear. Even complete loss of TREM2 in NHD allows development into adulthood with manifestation of clinical symptoms of dementia in the second or third decade of life. The mutations associated with FTD also cause early clinical symptoms at ages between 30 and 50 years. These mutations either result in premature stop of protein synthesis or strongly impair the transport and expression of TREM2 at the cell surface, also indicating a loss-of-function mechanism. The asymptomatic phase of 20–50 years even upon complete loss of function of TREM2 could suggest that TREM2 is dispensable during development, but exerts important functions in the regulation of brain homeostasis during aging, infection, trauma, or other detrimental processes in the brain.
Although the functional implication of the TREM2 R47H mutation in the pathogenesis of AD remains to be determined, the binding of apoE and the proteolytic processing by γ-secretase already links TREM2 to two major AD-related proteins. Thus, it will be interesting to further assess the functional connection of these proteins. However, because loss of TREM2 can cause neurodegeneration in other dementias, the effects on AD pathogenesis could also be independent on direct interaction with known AD-associated factors. In any case, TREM2 and DAP12 represent interesting targets to modulate an important signaling pathway commonly involved in neurodegeneration.
Acknowledgments
I am grateful to my colleagues Drs. H. Neumann, M. Heneka, and P. St. George-Hyslop, and to all lab members for interesting and helpful discussions on TREM2 and neuroinflammation. I also thank Dr. K. Glebov for artwork.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) and the Federal Ministry of Education and Research (BMBF). The author declares that he has no conflicts of interest with the contents of this article.
- TREM
- triggering receptor expressed on myeloid cells
- DAP12
- DNAX activating protein of 12 kDa
- NHD
- Nasu-Hakola disease
- FTD
- frontotemporal dementia
- AD
- Alzheimer disease
- PS
- presenilin
- APP
- amyloid precursor protein
- Syk
- spleen tyrosine kinase
- ITAM
- immunoreceptor tyrosine-based activation motif
- CTF
- C-terminal fragment
- ADAM
- a disintegrin and metalloprotease
- Aβ
- amyloid-β
- CSF
- cerebrospinal fluid
- TLR
- Toll-like receptor
- ER
- endoplasmic reticulum
- apoE
- apolipoprotein E.
References
- 1. Colonna M. (2003) TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453 [DOI] [PubMed] [Google Scholar]
- 2. Paradowska-Gorycka A., and Jurkowska M. (2013) Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum. Immunol. 74, 730–737 [DOI] [PubMed] [Google Scholar]
- 3. Sharif O., and Knapp S. (2008) From expression to signaling: roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology 213, 701–713 [DOI] [PubMed] [Google Scholar]
- 4. Neumann H., and Takahashi K. (2007) Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184, 92–99 [DOI] [PubMed] [Google Scholar]
- 5. Sessa G., Podini P., Mariani M., Meroni A., Spreafico R., Sinigaglia F., Colonna M., Panina P., and Meldolesi J. (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 20, 2617–2628 [DOI] [PubMed] [Google Scholar]
- 6. Xing J., Titus A. R., and Humphrey M. B. (2015) The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res. Rep. Biochem. 5, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turnbull I. R., and Colonna M. (2007) Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 7, 155–161 [DOI] [PubMed] [Google Scholar]
- 8. Colonna M. (2003) DAP12 signaling: from immune cells to bone modeling and brain myelination. J. Clin. Invest. 111, 313–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bianchin M. M., Capella H. M., Chaves D. L., Steindel M., Grisard E. C., Ganev G. G., da Silva Júnior J. P., Neto Evaldo S., Poffo M. A., Walz R., Carlotti Júnior C. G., and Sakamoto A. C. (2004) Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy–PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell. Mol. Neurobiol. 24, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaneko M., Sano K., Nakayama J., and Amano N. (2010) Nasu-Hakola disease: The first case reported by Nasu and review: The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology 30, 463–470 [DOI] [PubMed] [Google Scholar]
- 11. Guerreiro R., and Hardy J. (2014) Genetics of Alzheimer's disease. Neurotherapeutics 11, 732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heneka M. T., Kummer M. P., and Latz E. (2014) Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477 [DOI] [PubMed] [Google Scholar]
- 13. Cameron B., and Landreth G. E. (2010) Inflammation, microglia, and Alzheimer's disease. Neurobiol. Dis. 37, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., and Kummer M. P. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Landreth G. (2007) The immunology of Alzheimer's disease: prospects towards harnessing disease mechanisms for therapeutic ends. J. Neuroimmune Pharmacol. 2, 131–133 [DOI] [PubMed] [Google Scholar]
- 16. Lee C. Y. D., and Landreth G. E. (2010) The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. (Vienna) 117, 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ransohoff R. M., and El Khoury J. (2015) Microglia in health and disease. Cold Spring Harb. Perspect. Biol. 8, a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heppner F. L., Ransohoff R. M., and Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 [DOI] [PubMed] [Google Scholar]
- 19. Czirr E., and Wyss-Coray T. (2012) The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouchon A., Dietrich J., and Colonna M. (2000) Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 164, 4991–4995 [DOI] [PubMed] [Google Scholar]
- 21. Bouchon A., Hernández-Munain C., Cella M., and Colonna M. (2001) A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Call M. E., Schnell J. R., Xu C., Lutz R. A., Chou J. J., and Wucherpfennig K. W. (2006) The structure of the ζζ transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127, 355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prada I., Ongania G. N., Buonsanti C., Panina-Bordignon P., and Meldolesi J. (2006) Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience 140, 1139–1148 [DOI] [PubMed] [Google Scholar]
- 24. Wunderlich P., Glebov K., Kemmerling N., Tien N. T., Neumann H., and Walter J. (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park J.-S., Ji I. J., An H. J., Kang M.-J., Kang S.-W., Kim D.-H., and Yoon S.-Y. (2015) Disease-associated mutations of TREM2 alter the processing of N-linked oligosaccharides in the Golgi apparatus. Traffic 16, 510–518 [DOI] [PubMed] [Google Scholar]
- 26. Kleinberger G., Yamanishi Y., Suárez-Calvet M., Czirr E., Lohmann E., Cuyvers E., Struyfs H., Pettkus N., Wenninger-Weinzierl A., Mazaheri F., Tahirovic S., Lleó A., Alcolea D., Fortea J., Willem M., Lammich S., Molinuevo J. L., Sánchez-Valle R., Antonell A., Ramirez A., Heneka M. T., Sleegers K., van der Zee J., Martin J.-J., Engelborghs S., Demirtas-Tatlidede A., Zetterberg H., Van Broeckhoven C., Gurvit H., Wyss-Coray T., Hardy J., Colonna M., and Haass C. (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra86. [DOI] [PubMed] [Google Scholar]
- 27. Piccio L., Buonsanti C., Cella M., Tassi I., Schmidt R. E., Fenoglio C., Rinker J. 2nd, Naismith R. T., Panina-Bordignon P., Passini N., Galimberti D., Scarpini E., Colonna M., and Cross A. H. (2008) Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131, 3081–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gingras M.-C., Lapillonne H., and Margolin J. F. (2002) TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol. Immunol. 38, 817–824 [DOI] [PubMed] [Google Scholar]
- 29. Schmid C. D., Sautkulis L. N., Danielson P. E., Cooper J., Hasel K. W., Hilbush B. S., Sutcliffe J. G., and Carson M. J. (2002) Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bouchon A., Facchetti F., Weigand M. A., and Colonna M. (2001) TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410, 1103–1107 [DOI] [PubMed] [Google Scholar]
- 31. Piccio L., Buonsanti C., Mariani M., Cella M., Gilfillan S., Cross A. H., Colonna M., and Panina-Bordignon P. (2007) Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 37, 1290–1301 [DOI] [PubMed] [Google Scholar]
- 32. Zhong L., Chen X.-F., Zhang Z.-L., Wang Z., Shi X.-Z., Xu K., Zhang Y.-W., Xu H., and Bu G. (2015) DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glebov K., Wunderlich P., Karaca I., and Walter J. (2016) Functional involvement of γ-secretase in signaling of the triggering receptor expressed on myeloid cells-2 (TREM2). J. Neuroinflammation 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Painter M. M., Atagi Y., Liu C.-C., Rademakers R., Xu H., Fryer J. D., and Bu G. (2015) TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ford J. W., and McVicar D. W. (2009) TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 21, 38–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takahashi K., Rochford C. D. P., and Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giraldo M., Lopera F., Siniard A. L., Corneveaux J. J., Schrauwen I., Carvajal J., Muñoz C., Ramirez-Restrepo M., Gaiteri C., Myers A. J., Caselli R. J., Kosik K. S., Reiman E. M., and Huentelman M. J. (2013) Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol. Aging 34, 2077.e11–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turnbull I. R., Gilfillan S., Cella M., Aoshi T., Miller M., Piccio L., Hernandez M., and Colonna M. (2006) Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 [DOI] [PubMed] [Google Scholar]
- 39. Otero K., Shinohara M., Zhao H., Cella M., Gilfillan S., Colucci A., Faccio R., Ross F. P., Teitelbaum S. L., Takayanagi H., and Colonna M. (2012) TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J. Immunol. 188, 2612–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guerreiro R., Bilgic B., Guven G., Brás J., Rohrer J., Lohmann E., Hanagasi H., Gurvit H., and Emre M. (2013) A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol. Aging 34, 2890.e1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guerreiro R. J., Lohmann E., Brás J. M., Gibbs J. R., Rohrer J. D., Gurunlian N., Dursun B., Bilgic B., Hanagasi H., Gurvit H., Emre M., Singleton A., and Hardy J. (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kövari E. (2009) Neuropathological spectrum of frontal lobe dementias. Front. Neurol. Neurosci. 24, 149–159 [DOI] [PubMed] [Google Scholar]
- 43. Irwin D. J., Cairns N. J., Grossman M., McMillan C. T., Lee E. B., Van Deerlin V. M., Lee V. M.-Y., and Trojanowski J. Q. (2015) Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 129, 469–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goedert M., Ghetti B., and Spillantini M. G. (2012) Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lattante S., Ciura S., Rouleau G. A., and Kabashi E. (2015) Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 31, 263–273 [DOI] [PubMed] [Google Scholar]
- 46. Jin S. C., Benitez B. A., Karch C. M., Cooper B., Skorupa T., Carrell D., Norton J. B., Hsu S., Harari O., Cai Y., Bertelsen S., Goate A. M., and Cruchaga C. (2014) Coding variants in TREM2 increase risk for Alzheimer's disease. Hum. Mol. Genet. 23, 5838–5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bianchin M. M., Abujamra A. L., and Izquierdo I. (2013) TREM2, frontotemporal dementia-like disease, Nasu-Hakola disease, and Alzheimer dementia: a chicken and egg problem? JAMA Neurol. 70, 805–806 [DOI] [PubMed] [Google Scholar]
- 48. Cuyvers E., Bettens K., Philtjens S., Van Langenhove T., Gijselinck I., van der Zee J., Engelborghs S., Vandenbulcke M., Van Dongen J., Geerts N., Maes G., Mattheijssens M., Peeters K., Cras P., Vandenberghe R., De Deyn P. P., Van Broeckhoven C., Cruts M., and Sleegers K., BELNEU Consortium (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e11–9 [DOI] [PubMed] [Google Scholar]
- 49. Le Ber I., De Septenville A., Guerreiro R., Bras J., Camuzat A., Caroppo P., Lattante S., Couarch P., Kabashi E., Bouya-Ahmed K., Dubois B., and Brice A. (2014) Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35, 2419.e23–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M. A., van Duijn C. M., Thorsteinsdottir U., Kong A., and Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S. K., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J.-C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., and Hardy J., Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holtzman D. M., Mandelkow E., and Selkoe D. J. (2012) Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2, a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. LaFerla F. M. (2010) Pathways linking Aβ and tau pathologies. Biochem. Soc. Trans. 38, 993–995 [DOI] [PubMed] [Google Scholar]
- 54. Walter J., Kaether C., Steiner H., and Haass C. (2001) The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr. Opin. Neurobiol. 11, 585–590 [DOI] [PubMed] [Google Scholar]
- 55. Vassar R., and Kandalepas P. C. (2011) The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer's disease. Alzheimers Res. Ther. 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. De Strooper B., Vassar R., and Golde T. (2010) The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fahrenholz F. (2007) α-Secretase as a therapeutic target. Curr. Alzheimer Res. 4, 412–417 [DOI] [PubMed] [Google Scholar]
- 58. Kennedy J. L., Farrer L. A., Andreasen N. C., Mayeux R., and St George-Hyslop P. (2003) The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science 302, 822–826 [DOI] [PubMed] [Google Scholar]
- 59. Bertram L., Lill C. M., and Tanzi R. E. (2010) The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281 [DOI] [PubMed] [Google Scholar]
- 60. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R. R., Huttenlocher J., Bjornsdottir G., Andreassen O. A., Jönsson E. G., Palotie A., Behrens T. W., Magnusson O. T., Kong A., Thorsteinsdottir U., Watts R. J., and Stefansson K. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 61. Benilova I., Gallardo R., Ungureanu A.-A., Castillo Cano V., Snellinx A., Ramakers M., Bartic C., Rousseau F., Schymkowitz J., and De Strooper B. (2014) The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 289, 30977–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maloney J. A., Bainbridge T., Gustafson A., Zhang S., Kyauk R., Steiner P., van der Brug M., Liu Y., Ernst J. A., Watts R. J., and Atwal J. K. (2014) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 289, 30990–31000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rogaev E. I., Sherrington R., Rogaeva E. A., Levesque G., Ikeda M., Liang Y., Chi H., Lin C., Holman K., and Tsuda T. (1995) Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775–778 [DOI] [PubMed] [Google Scholar]
- 64. Sherrington R., Rogaev E. I., Liang Y., Rogaeva E. A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K., Tsuda T., Mar L., Foncin J. F., Bruni A. C., Montesi M. P., Sorbi S., Rainero I., Pinessi L., Nee L., Chumakov I., Pollen D., Brookes A., Sanseau P., Polinsky R. J., Wasco W., Da Silva H. A., Haines J. L., Perkicak-Vance M. A., Tanzi R. E., Roses A. D., Fraser P. E., Rommens J. M., and St George-Hyslop P. H. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760 [DOI] [PubMed] [Google Scholar]
- 65. De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494 [DOI] [PubMed] [Google Scholar]
- 66. Selkoe D. J., and Wolfe M. S. (2007) Presenilin: running with scissors in the membrane. Cell 131, 215–221 [DOI] [PubMed] [Google Scholar]
- 67. Holtzman D. M., Herz J., and Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Walter J. (2012) γ-Secretase, apolipoprotein E and cellular cholesterol metabolism. Curr. Alzheimer Res. 9, 189–199 [DOI] [PubMed] [Google Scholar]
- 69. Michaelson D. M. (2014) APOE ϵ4: the most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement. 10, 861–868 [DOI] [PubMed] [Google Scholar]
- 70. Verghese P. B., Castellano J. M., and Holtzman D. M. (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., and Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 72. Strittmatter W. J., Saunders A. M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G. S., and Roses A. D. (1993) Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jiang Q., Lee C. Y. D., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., Tontonoz P., and Landreth G. E. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee C. Y. D., Tse W., Smith J. D., and Landreth G. E. (2012) Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schmidt V., Carlo A.-S., and Willnow T. E. (2014) Apolipoprotein E receptor pathways in Alzheimer disease. Wiley Interdiscip. Rev. Sys. Biol. Med. 6, 255–270 [DOI] [PubMed] [Google Scholar]
- 77. Bertram L., and Tanzi R. E. (2012) The genetics of Alzheimer's disease. Prog. Mol. Biol. Transl. Sci. 107, 79–100 [DOI] [PubMed] [Google Scholar]
- 78. Lambert J.-C., and Amouyel P. (2011) Genetics of Alzheimer's disease: new evidences for an old hypothesis? Curr. Opin. Genet. Dev. 21, 295–301 [DOI] [PubMed] [Google Scholar]
- 79. Lill C. M., Rengmark A., Pihlstrøm L., Fogh I., Shatunov A., Sleiman P. M., Wang L.-S., Liu T., Lassen C. F., Meissner E., Alexopoulos P., Calvo A., Chio A., Dizdar N., Faltraco F., Forsgren L., Kirchheiner J., Kurz A., Larsen J. P., Liebsch M., Linder J., Morrison K. E., Nissbrandt H., Otto M., Pahnke J., Partch A., Restagno G., Rujescu D., Schnack C., Shaw C. E., Shaw P. J., Tumani H., Tysnes O.-B., Valladares O., Silani V., van den Berg L. H., van Rheenen W., Veldink J. H., Lindenberger U., Steinhagen-Thiessen E., SLAGEN Consortium, Teipel S., Perneczky R., Hakonarson H., Hampel H., von Arnim C. A. F., Olsen J. H., Van Deerlin V. M., Al-Chalabi A., Toft M., Ritz B., and Bertram L. (2015) The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. 11, 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tanzi R. E. (2015) TREM2 and risk of Alzheimer's disease: friend or foe? N. Engl. J. Med. 372, 2564–2565 [DOI] [PubMed] [Google Scholar]
- 81. Melchior B., Garcia A. E., Hsiung B.-K., Lo K. M., Doose J. M., Thrash J. C., Stalder A. K., Staufenbiel M., Neumann H., and Carson M. J. (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro 2, e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fisher Y., Nemirovsky A., Baron R., and Monsonego A. (2010) T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer's disease. PLoS One 5, e10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang B., Gaiteri C., Bodea L.-G., Wang Z., McElwee J., Podtelezhnikov A. A., Zhang C., Xie T., Tran L., Dobrin R., Fluder E., Clurman B., Melquist S., Narayanan M., Suver C., Shah H., Mahajan M., Gillis T., Mysore J., MacDonald M. E., Lamb J. R., Bennett D. A., Molony C., Stone D. J., Gudnason V., Myers A. J., Schadt E. E., Neumann H., Zhu J., and Emilsson V. (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ulrich J. D., Finn M. B., Wang Y., Shen A., Mahan T. E., Jiang H., Stewart F. R., Piccio L., Colonna M., and Holtzman D. M. (2014) Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jay T. R., Miller C. M., Cheng P. J., Graham L. C., Bemiller S., Broihier M. L., Xu G., Margevicius D., Karlo J. C., Sousa G. L., Cotleur A. C., Butovsky O., Bekris L., Staugaitis S. M., Leverenz J. B., Pimplikar S. W., Landreth G. E., Howell G. R., Ransohoff R. M., and Lamb B. T. (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang Y., Cella M., Mallinson K., Ulrich J. D., Young K. L., Robinette M. L., Gilfillan S., Krishnan G. M., Sudhakar S., Zinselmeyer B. H., Holtzman D. M., Cirrito J. R., and Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Atagi Y., Liu C.-C., Painter M. M., Chen X.-F., Verbeeck C., Zheng H., Li X., Rademakers R., Kang S. S., Xu H., Younkin S., Das P., Fryer J. D., and Bu G. (2015) Apolipoprotein E is a ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bailey C. C., DeVaux L. B., and Farzan M. (2015) The Triggering Receptor Expressed on Myeloid Cells 2 binds Apolipoprotein E. J. Biol. Chem. 290, 26033–26042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jiang T., Tan L., Zhu X.-C., Zhou J.-S., Cao L., Tan M.-S., Wang H.-F., Chen Q., Zhang Y.-D., and Yu J.-T. (2015) Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol. Aging 36, 3176–3186 [DOI] [PubMed] [Google Scholar]
- 90. Bird T. D., Koerker R. M., Leaird B. J., Vlcek B. W., and Thorning D. R. (1983) Lipomembranous polycystic osteodysplasia (brain, bone, and fat disease): a genetic cause of presenile dementia. Neurology 33, 81–86 [DOI] [PubMed] [Google Scholar]
- 91. Paloneva J., Manninen T., Christman G., Hovanes K., Mandelin J., Adolfsson R., Bianchin M., Bird T., Miranda R., Salmaggi A., Tranebjaerg L., Konttinen Y., and Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chouery E., Delague V., Bergougnoux A., Koussa S., Serre J.-L., and Mégarbané A. (2008) Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 29, E194–204 [DOI] [PubMed] [Google Scholar]
- 93. Klünemann H. H., Ridha B. H., Magy L., Wherrett J. R., Hemelsoet D. M., Keen R. W., De Bleecker J. L., Rossor M. N., Marienhagen J., Klein H. E., Peltonen L., and Paloneva J. (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64, 1502–1507 [DOI] [PubMed] [Google Scholar]