

Abstract
DNA damage must be repaired in an accurate and timely fashion to preserve genome stability. Cellular mechanisms preventing genome instability are crucial to human health because genome instability is considered a hallmark of cancer. Collectively referred to as the DNA damage response, conserved pathways ensure proper DNA damage recognition and repair. The function of numerous DNA damage response components is fine-tuned by posttranslational modifications, including ubiquitination. This not only involves the enzyme cascade responsible for conjugating ubiquitin to substrates but also requires enzymes that mediate directed removal of ubiquitin. Deubiquitinases remove ubiquitin from substrates to prevent degradation or to mediate signaling functions. The Saccharomyces cerevisiae deubiquitinase Ubp7 has been characterized previously as an endocytic factor. However, here we identify Ubp7 as a novel factor affecting S phase progression after hydroxyurea treatment and demonstrate an evolutionary and genetic interaction of Ubp7 with DNA damage repair pathways of homologous recombination and nucleotide excision repair. We find that deletion of UBP7 sensitizes cells to hydroxyurea and cisplatin and demonstrate that factors that stabilize replication forks are critical under these conditions. Furthermore, ubp7Δ cells exhibit an S phase progression defect upon checkpoint activation by hydroxyurea treatment. ubp7Δ mutants are epistatic to factors involved in histone maintenance and modification, and we find that a subset of Ubp7 is chromatin-associated. In summary, our results suggest that Ubp7 contributes to S phase progression by affecting the chromatin state at replication forks, and we propose histone H2B ubiquitination as a potential substrate of Ubp7.
Keywords: deubiquitylation (deubiquitination), DNA damage response, DNA replication, molecular evolution, yeast genetics, deubiquitination
Introduction
Genome stability is constantly challenged by exogenous agents or by DNA damage that occurs as a result of endogenous processes such as cellular metabolism or replication (reviewed in Ref. 1). If DNA damage is not detected and properly repaired in a timely fashion, then it can result in cell death or, in the case of erroneous repair, lead to mutations, chromosomal aberrations, and genome instability. Preventing genome instability is essential to human cells because it is considered an enabling characteristic of cancer (2). Therefore, in a normal cell, multiple complex pathways function to detect and repair diverse types of DNA damage, collectively referred to as the DNA damage response (DDR).3 During S phase, replicative stress activates a signaling cascade, called the intra-S phase checkpoint, that inhibits cell cycle progression, late-origin firing, and homologous recombination (HR) while promoting the function of specific checkpoint and replication stress response factors (1). In budding yeast, hyperphosphorylation of the kinase Rad53 is central for checkpoint activation and mediates the downstream response.
The DDR and multiple other cellular pathways depend on posttranslational modification of substrates with ubiquitin (3). Ubiquitination is facilitated by a sequential cascade of E1, E2, and E3 enzymes, which catalyzes the covalent attachment of ubiquitin to a lysine residue or the N terminus of a substrate protein (4, 5). Ubiquitination can target proteins to the 26S proteasome for degradation, have signaling functions, alter the subcellular localization, or change protein-protein interactions (5). Ubiquitination, like phosphorylation, is a reversible posttranslational modification whereby ubiquitin is removed from cellular substrate proteins by specialized ubiquitin proteases or deubiquitinating enzymes (DUB) (6).
DUBs function in numerous cellular processes, such as the regulation of protein stability, cell cycle control, regulation of gene expression, membrane trafficking, and DNA repair (6, 7). In addition to cleaving and activating proubiquitin, DUBs recycle ubiquitin and, importantly, reverse substrate ubiquitination. Regulated ubiquitination and deubiquitination is crucial for the DDR. One well studied example is the deubiquitination of proliferating cell nuclear antigen, which is fulfilled by budding yeast Ubp10 during S phase (8). There are five main DUB protein families: the ubiquitin C-terminal hydrolases, ovarian tumor, Josephin domain, JAB1/MPN/Mov34 metalloenzyme, and the largest group, the ubiquitin-specific protease class (reviewed in Refs. 6, 7). In budding yeast, 16 DUBs of the Ubp family (ubiquitin-specific protease in mammals) have been identified (9), among them Ubp7. Although overall the function of Ubp7 is not fully understood, it has been characterized as a late-arriving endocytic protein (10) that interacts with the endosomal sorting complex member Hse1 (11) and can deubqiuitinate the early endocytic protein Ede1 (10). Additionally, there is some evidence that Ubp7 may be involved in the DDR. First, Ubp7 has been found to interact with the Cdc48 cofactor Doa1/Ufd3 upon MMS treatment and to partially rescue the DNA damage sensitivity of doa1Δ mutants (12). Second, UBP7 has been identified as a weak high-copy suppressor of srs2Δ elg1Δ (13).
Here we demonstrate that deletion of UBP7 sensitizes yeast cells toward the DNA-damaging agents hydroxyurea (HU) and cisplatin, suggesting that Ubp7 plays a role in the DDR. Furthermore, we find that ubp7 mutants have evolutionary and genetic interactions with the DNA repair subpathways of HR and nucleotide excision repair (NER). Finally, we provide evidence that Ubp7 is involved in the control of S phase progression following HU treatment and the intra-S phase checkpoint, likely by contributing to replication fork speed through alteration of the surrounding chromatin state.
Experimental Procedures
Yeast Strains and Plasmids
The yeast strains used in this study are listed in supplemental Table S1. Unless noted otherwise, all strains were derivatives of W303 (14, 15). Media and plates were prepared according to standard protocols but with twice the amount of leucine (16). Yeast transformation and epitope tagging were carried out as described previously (17, 18).
Stress Plates and Serial Dilutions
The indicated amounts of hydroxyurea (Sigma-Aldrich) were added directly to SC-agar, whereas cisplatin (Sigma-Aldrich) was dissolved at 100 mg/ml in DMSO prior to addition to cooled SC-agar (pH 5.8). Cultures of the indicated yeast strains were grown at 30 °C in SC (for dilution on SC plates) or YPD to log phase and diluted to a starting A600 of 0.2. 5-Fold serial dilutions in H2O were plated on the appropriate plates and incubated at 30 °C for 2 (YPD) or 3 (SC + HU) days. UV and γ irradiation were performed after dilutions of yeast cultures had been spotted on plates. Cisplatin plates were stored in the dark.
Cell Viability Assay
WT, ubp7Δ, and rad51Δ cells were grown overnight to stationary phase in 3 ml of YPD medium at 30 °C, diluted to an A600 of 0.2, and grown for 4 h at 30 °C. The culture was diluted to an A600 of 0.5 and subsequently diluted 1:2000. 250 μl was plated onto YPD medium or YPD medium containing 10, 50, or 100 mm HU. The plates were incubated at 30 °C for 48 h and photographed. The surviving colonies were counted and compared with their corresponding untreated YPD control plates. The experiment was done in triplicate, with three individual clones per genotype analyzed per trial.
Evolutionary Rate Covariation (ERC) Analysis
ERC values compare sequence evolution rates for two proteins along all branches of a multispecies phylogenetic tree. Specifically, the ERC statistic is calculated between each protein pair as the correlation coefficient of their branch-specific rates. Rate of change is calculated using a projection operation to normalize branch lengths against the underlying species tree (19). ERC values were calculated using protein sequences from 18 yeast species (20). The Ubp7 protein was compared with genes within diverse DNA repair pathways by calculating the ERC values between it and their constituent genes as annotated in the Gene Ontology through YeastMine (19, 21). The ERC values for these pathways can be found in their entirety in supplemental Table S2. Statistical significance of a relationship between Ubp7 and a pathway was determined by comparing Ubp7 ERC values with a specific DNA repair pathway to the background genome distribution using the non-parametric Wilcoxon rank-sum test.
Mitotic Recombination and CAN1 Mutation Frequency Assays
The direct repeat assay was performed as described previously (22, 23). In brief, nine single yeast colonies of each strain were grown in 2 ml of SC medium and plated on SC plates and SC plates lacking leucine. The average recombination rate and standard deviation over five independent trials were calculated according to Ref. 24. The assays with HU (35 mm) and cisplatin (10 μg/ml) treatment were conducted in an identical fashion, but the cultures were treated with the indicated drug doses for 26 h before plating. The experiments were performed in quadruplicate. For the CAN1 mutation assay, five individual colonies of each genotype were grown in 2 ml of SC medium and plated on SC plates and SC plates lacking arginine supplemented with 60 mg/liter L-canavanine (Sigma-Aldrich). The median mutation frequency per trial was used to determine the depicted mean frequency ± S.D. from three independent trials.
Microscopy
The respective yeast cultures were grown in SC with adenine (100 mg/l) at room temperature and, when applicable, treated with 100 mm HU (Sigma-Aldrich) for 2 h. Prior to microscopy, 1 ml of yeast culture was harvested (1500 g, 2 min, room temperature), mixed with an equal volume of liquid 1.4% agarose (in SC medium), and mounted on slides. Images were acquired using a Nikon TiE inverted live-cell system with a ×100 oil immersion objective (1.45 numerical aperture), a Photometrics HQ2 camera, and motorized Prior Z stage. For individual strains, stacks of 11 0.3-μm sections were captured using the following exposure times for the respective fluorophore: differential interference contrast, 60 ms; Rfa1-YFP, 1 s. Nikon Elements software (Nikon Instruments) was used for acquisition and analysis. Images were exported from Nikon Elements as 8-bit RGB TIF files and enhanced for contrast and brightness using Photoshop (Adobe Systems Inc.). All images depicted in one panel were adjusted in an identical fashion. For analysis of focus formation, all 11 z slices of each image (3 μm in total) were analyzed manually for foci. To be counted as a focus, the signal had to be visible in at least two to three consecutive planes. Single z slices are depicted in the figures.
Western Blotting Analyses
Yeast whole-cell lysates were prepared by TCA precipitation as described in Ref. 18. Lysates were loaded on 8%, 10%, or 15% Tris/glycine gels or 4–12% BisTris gradient gels (Invitrogen) in MOPS buffer and transferred to a PVDF membrane (Millipore). Membranes were blocked in 5% skim milk and incubated with primary antibody overnight. Exposed films of Western blotting analyses were scanned and adjusted for contrast and brightness using Photoshop (Adobe Systems Inc.). Alternatively, the chemiluminescent signal was captured with a charge-coupled device camera (Bio-Rad, Geldoc XR+). Quantification of the scanned images was performed using ImageJ (25).
Antibodies
The following antibodies were used at the indicated dilutions: Myc (mouse monoclonal, Santa Cruz Biotechnology, catalog no. sc-40, 1:500, WB), Clb2 (rabbit polyclonal, Santa Cruz Biotechnology, catalog no. sc-9071, 1:2000, WB), HA (rabbit polyclonal, catalog no. sc-805, and mouse monoclonal, catalog no. sc-7392 Santa Cruz Biotechnology, 1:1000, WB), histone H3 total (rabbit polyclonal, Cell Signaling Technology, catalog no. 9715, 1:1000, WB), histone H2B total (rabbit polyclonal, Active Motif, catalog no. 39237, 1:5000, WB), ubiquityl-histone H2B (rabbit polyclonal, Cell Signaling Technology, catalog no. 5546, 1:1000, WB), Kar2 (rabbit polyclonal, Santa Cruz Biotechnology, catalog no. sc-33630, 1:5000, WB), Adh1 (rabbit, Chemicon, catalog no. Ab1202, 1:20,000, WB), GAPDH (mouse monoclonal, UBP Bio, catalog no. Y1040, 1:1000), anti-mouse HRP (Jackson ImmunoResearch Laboratories, catalog no. 115-035-003), anti-rabbit HRP (Jackson ImmunoResearch Laboratories, catalog no. 111-035-045), and anti-goat HRP (Jackson ImmunoResearch Laboratories, catalog no. 705-035-003).
Chromatin-binding Assay
Essentially, this assay was performed according to Refs. 26, 27. Cultures of an UBP7–3HA strain were grown to log phase and, for the cell cycle-synchronized samples, arrested for 3 h with 10 μm α factor (Genescript), 200 mm HU (Sigma-Aldrich), or 15 μg/ml nocodazole (Enzo Life Sciences). Approximately 30 A were harvested, resuspended, and incubated in prespheroplast buffer (100 mm PIPES/KOH (pH 9.4), 10 mm DTT, and 0.1% NaN3) for 10 min at room temperature. The pellet was then spheroplasted using Zymolyase (20 mg/ml, 100T, Amsbio) in spheroplast buffer (50 mm K2HPO4/KH2PO4 (pH 7.5), 0.6 m sorbitol, and 10 mm DTT) for 40 min at 37 °C. Harvested spheroplasts were washed, resuspended in an equal volume of extraction buffer (50 mm HEPES/KOH (pH 7.5), 100 mm KCl, 2.5 mm MgCl2, 0.4 m sorbitol, 1 mm DTT, protease inhibitors (Roche complete), and 2 mm PMSF) and lysed on ice by increasing the Triton X-100 concentration to 1% and vortexing. A sample for the whole-cell extract was taken prior to adding the extract on the half volume of sucrose cushion (extraction buffer, 0.25% Triton X-100, and 30% sucrose). Soluble and chromatin fractions were separated by centrifugation (20,000 × g, 20 min).
FACS and Two-dimensional Gel Electrophoresis
FACS and two-dimensional gel analysis were performed as described previously (28) following treatment of yeast cultures with 200 mm HU as indicated.
Results
UBP7 Is Linked Evolutionarily and Genetically to the DNA Damage Response
To investigate the role of Ubp7 in the DDR, we first used ERC to infer which DNA repair pathways have evolutionary histories resembling Ubp7 (Fig. 1a). ERC is a statistic of correlation between the species-specific evolutionary rates of two proteins as measured across several species (20, 29). Typically, functionally related protein pairs, such as those in a shared pathway or complex, have elevated ERC values relative to random pairs, and such ERC signatures have guided DNA repair research in previous studies (30, 31). We compiled sets of proteins participating in specific DNA damage response pathways: HR, NER, base excision repair, mismatch repair, and non-homologous end joining. We also analyzed endocytosis because of the reported role of Ubp7 in this process. When comparing the evolutionary rates of Ubp7 to these gene sets across 18 yeast species, it is clear that Ubp7 has a strong evolutionary association with proteins involved in HR and NER (Fig. 1a; HR, p = 6.01 × 10−5; NER, p = 9.92 × 10−5; Wilcoxon rank-sum test; supplemental Table S2).
FIGURE 1.
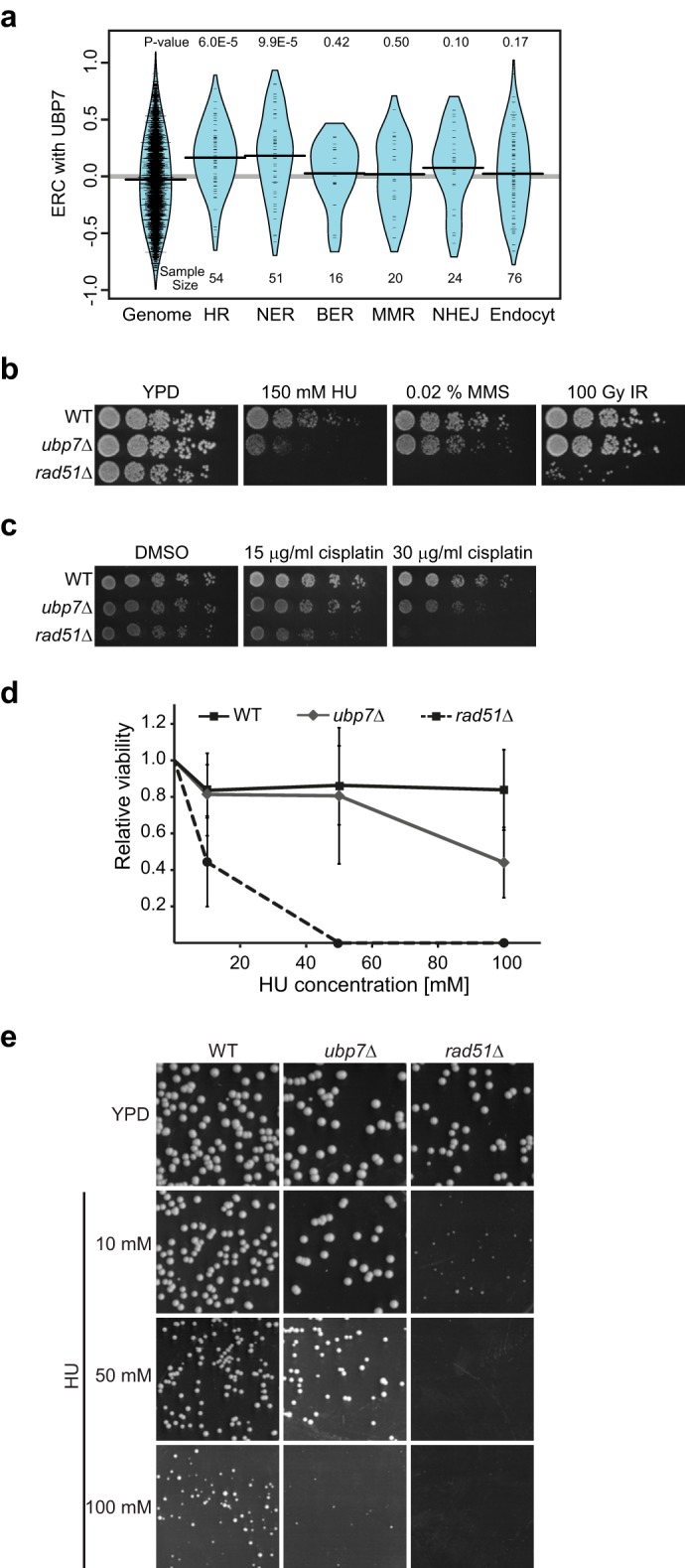
Ubp7 is linked to the DNA damage response. a, ERC values between the Ubp7 and DNA repair pathways show statistically significant correlations with HR and NER but not with base excision repair (BER), mismatch repair (MMR), nonhomologous end joining (NHEJ), or endocytosis (Endocyt). Violin plots show the distribution of ERC values between the UBP7 and DNA repair pathways. Curves show a smoothed representation of their density, whereas tick marks show the exact values. p values reflect the significance of elevation above the genome-wide distribution. b, 5-fold serial dilutions of wild-type, ubp7Δ, and rad51Δ (control) strains grown on YPD, YPD with 150 mm HU, YPD with 0.02% MMS, or YPD and irradiated with 100 Gy of γ radiation (IR). c, 5-fold serial dilutions of the same strains as in a grown on SC with 0.03% DMSO or the indicated cisplatin (in DMSO) concentrations. d, WT, ubp7Δ, and rad51Δ cells were measured for cell viability by plating on YPD medium containing the indicated doses of HU and compared with their corresponding untreated YPD control. e, wild-type, ubp7Δ, and rad51Δ cultures were grown in SC medium and plated on SC plates containing the indicated concentrations of HU. Plates were imaged 24 h after plating.
Because Ubp7 is evolutionarily correlated with the HR and NER DNA repair pathways (Fig. 1a), we next tested whether deletion of UBP7 would sensitize yeast cells to different forms of DNA damage. Although ubp7Δ mutants are HU-sensitive, they do not exhibit a pronounced growth defect on plates containing MMS (Fig. 1b). The HU sensitivity of ubp7-null cells was also evident in viability assays (Fig. 1d) and by decreased colony size (Fig. 1e). Interestingly, HU and MMS interfere with replication fork progression differently. HU is thought to cause prolonged stalling and reduce replication fork speed, whereas MMS is an alkylating agent (reviewed in Ref. 32). This suggests that Ubp7 may be involved in the cellular response to replication fork stalling but not to other forms of replicative damage that could also induce double strand break formation. In line with a specific function of Ubp7 in a DNA damage response subpathway is our finding that the ubp7-null strain is also not sensitive to γ or UV radiation (Figs. 1b and 2a).
FIGURE 2.

Ubp7 is not an NER or HR component. a, 5-fold serial dilutions of the indicated strains on YPD plates exposed to 0, 50, or 100 J/m2 UV radiation. b, 5-fold serial dilutions of the strains as in a, spotted on SC plates with DMSO or the indicated cisplatin concentrations. c, comparison of total recombination rates in a wild-type and ubp7Δ strain chromosomally harboring the direct repeat recombination assay. Cells were grown in SC medium or SC medium containing DMSO as a control compared with cells exposed to 35 mm HU or 10 μg/ml cisplatin for 26 h and then plated. The graph depicts the mean recombination rate from four to five independent trials ± S.D. The recombination rate in WT and ubp7Δ cells is not significantly different by Student's t test for each condition tested. d, spontaneous CAN1 mutation frequency in wild-type, ubp7Δ, rad51Δ, and ubp7Δ rad51Δ. The graph depicts the mean recombination frequency from three independent trials ± S.D. Only the difference between rad51Δ and the wild-type is statistically significant by Student's t test (*, p < 0.05). e, genetic analysis with the HR genes RAD51 and SGS1. 5-Fold serial dilutions were spotted on SC plates containing 25 and 100 mm HU and incubated at 30 °C for 3 days. f, genetic analysis of ubp7Δ with a deletion of MUS81. Serial dilutions of the indicated strains were performed as in a.
In cells, HR, NER, and translesion synthesis (TLS) function together to enable the repair of complex types of DNA damage such as intra- and interstrand cross-links. Because we found that UBP7 coevolved with two of these particular pathways, we determined the sensitivity of ubp7Δ toward a DNA cross-linking agent. Indeed, we found that ubp7Δ is modestly sensitive to cisplatin (Fig. 1c). Overall, these observed sensitivities support the in silico evolutionary analysis and suggest that Ubp7 affects specific DNA damage response and/or repair pathways.
Ubp7 Is Not an NER or HR Factor
Given the role of NER in repairing cisplatin-induced DNA lesions, we next tested whether Ubp7 is a component of the NER pathway. To do so, we analyzed the UV sensitivity of a UBP7 deletion (Fig. 2a) and the genetic interactions of ubp7Δ with two mutants of the NER pathway: rad4Δ and rad7Δ (Fig. 2b). Rad4 (Xeroderma pigmentosum complementation group C) and Rad23 comprise the nuclear excision repair factor 2 (NEF2) complex, which functions to detect and repair UV-damaged DNA (33, 34). NEF2 is required for the early incision step of NER, and, consistently, rad4Δ mutants are very sensitive to UV irradiation (35, 36) (Fig. 2a). Rad7, together with Rad16, forms the NEF4 complex, which binds to NEF2 and enhances its activity. In contrast to NEF2, NEF4 is not strictly required for the incision step of NER and likely acts on post-incision steps (37), consistent with its less pronounced UV sensitivity (36) (Fig. 2a).
Most deletion mutants of NER pathway members are UV-sensitive to varying degrees. We did not detect any UV sensitivity of ubp7Δ alone (Fig. 2a), indicating that Ubp7 is likely not a central component of UV-induced NER. However, rad4Δ ubp7Δ and rad7Δ ubp7Δ mutants do exhibit a very slight but reproducible synthetic sensitivity on cisplatin-containing medium (Fig. 2b). Therefore, Ubp7 may function in an additional pathway required for processing cisplatin-induced damage in the absence of Rad4/Rad7.
One of the complex lesions that arise from cisplatin treatment is interstrand cross-links (ICLs). In contrast to humans, where the Fanconi anemia pathway has been well characterized to act on this type of DNA damage, yeast has fewer known factors that are specialized in repairing this type of lesion (38). However, in both yeast and humans, HR plays an important role in the repair of interstrand cross-links. Therefore, we next tested whether Ubp7 is a functional component of the HR pathway, to which Ubp7 is related evolutionarily (Fig. 1a). Deletion of HR genes often leads to an alteration of recombination, which can result in increased utilization of other repair pathways such as TLS and, therefore, increased mutation rates (23, 39). We analyzed direct repeat recombination rates and the CAN1 mutation frequency in an ubp7Δ background. The direct repeat recombination assay strain contains a functional URA3 gene flanked by two differently mutated versions of leu2. Recombination can yield a functional LEU2 gene either via Rad51-dependent gene conversion or single-strand annealing (22, 23, 31). Using this assay, we did not find a significant increase in the overall recombination rate in ubp7Δ compared with wild-type cells in either untreated cultures or those treated with HU or cisplatin (Fig. 2c). Similarly, when we determined the spontaneous CAN1 mutation frequency in ubp7Δ, we did not observe a significant alteration of the mutation frequency (Fig. 2d). Surprisingly, even when we tested an ubp7Δ rad51Δ strain, in which HR is greatly reduced, we did not observe a significant increase in the spontaneous CAN1 mutation frequency (Fig. 2d). In contrast, the mutation frequency is reduced compared with the rad51 single mutant. In a rad51Δ cell, the elevated mutation frequency (40) is likely attributed to increased utilization of error-prone TLS because multiple pathways such as HR, TLS, NER, and base excision repair all contribute to the repair of spontaneous damage in wild-type cells (41). The contribution of each pathway likely depends on several cellular factors, including the cell cycle stage and the context of the lesion (41). Therefore, it is possible that deletion of UBP7 in the rad51-null background reduces the relative contribution of TLS to repair.
This raises the possibility that, although Ubp7 is not a direct NER or HR pathway component, deletion of UBP7 may nonetheless affect the DNA damage sensitivity of HR deletion mutants. Therefore, we next tested the genetic interaction of ubp7Δ with rad51Δ and sgs1Δ (Fig. 2e). Rad51 is the central HR filament protein that facilitates strand invasion during HR-mediated repair of double strand breaks (42, 43). Sgs1, on the other hand, is a budding yeast RecQ-like helicase that functions at several steps during HR, including initial long-range resection, processing of recombination intermediates, and dissolution of D-loops and double Holliday junctions (44, 45). Surprisingly, ubp7Δ mutants exhibited a strong negative genetic interaction with sgs1Δ on HU, whereas the ubp7Δ rad51Δ double mutant did not (Fig. 2e). This intriguing result suggests that the function of Ubp7 may be required in the absence of a Rad51-independent role of Sgs1. In addition to its role in HR, Sgs1 also functions to stabilize the replication fork and to promote checkpoint signaling upon HU treatment (46–48). Interestingly, we also observed a negative genetic interaction of ubp7Δ with deletion of the endonuclease MUS81, which is also involved in mediating replication fork restart (49) (Fig. 2f). Taken together, we hypothesize that Ubp7 may affect replication fork stalling and stability. One possible explanation for our findings is that, in the absence of Ubp7, factors that stabilize the replication fork are required upon fork stalling; for example, because of HU treatment. Replication is monitored by the intra-S phase checkpoint, and, therefore, we wanted to investigate whether Ubp7 directly affects S phase progression and/or intra-S phase checkpoint signaling.
S Phase Progression Is Perturbed in ubp7Δ Cells
The intra-S phase checkpoint ensures that replication is complete before onset of mitosis. This checkpoint is activated upon replicative stress (for example, replication fork stalling), and a cascade of phosphorylation events ultimately leads to cell cycle arrest, the inhibition of late firing origins, and the expression of checkpoint response genes such as RNR (1). As replication forks stall, increased amounts of single-stranded DNA are exposed and coated rapidly by the single-stranded DNA-binding protein replication protein A. This can be visualized by assessing the focus formation of the large subunit of RPA, Rfa1-YFP. Interestingly, the percentage of cells with one or more Rfa1 foci increases significantly in a HU-treated ubp7Δ strain (Fig. 3, a and b, p < 0.005). This indicates that there is an increased amount of single-stranded DNA in an ubp7-null cell upon HU-induced replication fork stalling and suggests that UBP7 may be involved in properly stalling and/or stabilizing the replication fork.
FIGURE 3.

S phase progression is altered in ubp7 cells. a, live-cell microscopy of wild-type and ubp7Δ Rfa1-YFP cells left untreated or after 2 h with 100 mm HU. A representative z plane of differential interference contrast (DIC)/YFP for each strain is shown. Arrowheads mark Rfa1 foci. b, quantification of a. The graph depicts the mean percentage of cells with an Rfa1-YFP focus ± S.E.M. (n (WT, untreated) = 1309, n (ubp7Δ, untreated) = 1359, n (WT, HU) = 1007, n (ubp7Δ, HU) = 1059). The indicated p values were calculated by Fischer's exact test. n.s., not significant. c, FACS analysis of WT and ubp7Δ cells in log phase (log), after α factor arrest (aF), and after release from 4 h of treatment with 200 mm HU (10, 20, 30, 40, 50, and 60 min). d, two-dimensional gel electrophoresis analysis with a probe recognizing the early origin of replication, ARS305, following HU treatment of α factor-arrested cells. The arrowhead indicates where bubbles are accumulating in ubp7Δ cells.
To determine whether deletion of UBP7 alters recovery from HU-induced replication fork stalling, we performed flow cytometry of wild-type and ubp7 cultures synchronized in G1 phase and treated with 200 mm HU (Fig. 3c). Consistent with the hypothesis from our microscopic analysis, upon release from HU treatment, ubp7 mutants exhibited a delay in S phase progression, evident at the 20- and 30-min time points (Fig. 3c).
We next tested whether the S phase delay in HU-treated ubp7Δ cells is due to altered replication fork dynamics and, therefore, analyzed replication fork fidelity by two-dimensional gel electrophoresis (Fig. 3d). After HU treatment, we observed a modest but reproducible increase in bubbles in ubp7Δ cells, most evident after 2 h (Fig. 3d, arrowhead). This indicates slower fork progression and, possibly, an increased amount of single-stranded DNA upon HU treatment, which is consistent with the increase in Rfa1 foci we observed in ubp7Δ (Fig. 3, a and b). These results suggest that Ubp7 affects replication fork dynamics and cell cycle progression after HU-induced replication fork stalling.
Epistasis Analysis of UBP7 with Intra-S Phase Checkpoint Genes
Because we found that deletion of UBP7 affects S phase progression upon HU treatment, we next wanted to determine whether Ubp7 may itself be a checkpoint component. Because we found that ubp7-null cells do not exhibit synthetic phenotypes with a deletion mutant of RAD53 or the rad53-K227A checkpoint activation-deficient mutant on HU (data not shown), we tested the genetic interaction with further checkpoint components. In budding yeast, replication fork stalling and DNA damage (such as ionizing radiation, ultraviolet radiation, and MMS) invoke distinct cellular responses that, however, have several common components (32), such as the activation of Mec1, Rad53, and Dun1 (Fig. 4a). Therefore, to differentiate which part of the checkpoint pathway Ubp7 affects, we determined the phenotype of double mutants of ubp7Δ with rad17Δ, mrc1Δ, and rad9Δ (Fig. 4b). Using this analysis, we observe a modest synthetic growth defect only for the ubp7Δ mrc1Δ double mutant on HU (Fig. 4b). The phenotypes of ubp7Δ rad17Δ and ubp7Δ rad9Δ on HU reflect the sensitivity of the ubp7 single mutant and, therefore, do not indicate a synthetic genetic interaction. Mrc1 (Claspin) is required for unperturbed replication and checkpoint signaling (50, 51). Therefore, one possibility is that Mrc1 is required for replication fork stabilization in the absence of UBP7. This requirement is consistent with the severe growth defect we observed for ubp7Δ sgs1Δ (Fig. 2e), and we believe that this also reflects a requirement of the function of Sgs1 in stabilizing the replication fork and not its canonical HR role.
FIGURE 4.
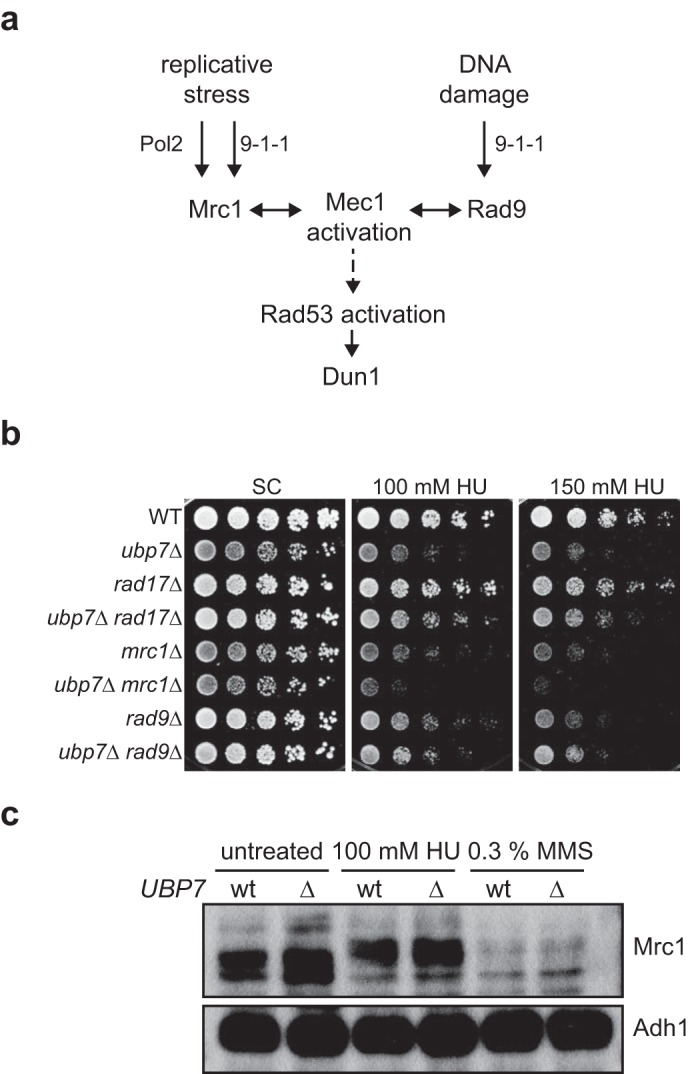
Epistasis analysis of UBP7 with intra-S phase checkpoint genes. a, schematic of relevant factors for Mec1 and Rad53 activation. b, 5-fold serial dilutions of the indicated strains spotted on plates containing HU and incubated at 30 °C for 3 d. c, Mrc1 protein levels and phosphorylation status in wild-type and ubp7Δ MRC1–9myc strains. Cultures were left untreated or exposed to 100 mm HU or 0.3% MMS for 2 h before whole-cell lysates were prepared. Shown is a Western blotting analysis against myc (Mrc1) and Adh1 (loading).
To determine whether Ubp7 directly affects checkpoint activation at the step of Mrc1, we next analyzed Mrc1 protein levels and phosphorylation status in wild-type and ubp7Δ strains. In untreated cells, Mrc1 couples the replicative helicase and polymerase, whereas it forms a pausing complex with Tof1 to stabilize Pol2 upon replication fork stalling. In the latter scenario, Mrc1 is hyperphosphorylated by Mec1. When cultures are treated with HU, Mrc1 phosphorylation is induced in both wild-type and ubp7Δ cells (Fig. 4c), indicating that this stage of intra-S phase checkpoint signaling is functional in ubp7Δ cells. We therefore conclude that the intra-S phase checkpoint is functional upon UBP7 deletion and that UBP7 likely functions in a similar pathway.
UBP7 Deletion Alters Histone Modifications
Chromatin remodeling, including the ubiquitination of histone H2B at lysine 123, is involved in intra-S phase checkpoint signaling (52, 53). Interestingly, in our initial ERC analysis, we found a highly significant correlation of Ubp7 with histone modification proteins (p = 0.0007861, Wilcoxon rank-sum test, Fig. 5a). Therefore, we hypothesized that Ubp7 could potentially affect the modification of a histone or histone-associated factor during replication. To test this, we first analyzed the genetic interactions of ubp7Δ with deletion mutants of the histone chaperone ASF1 and of the E2 enzyme for H2B, RAD6. Neither double mutant exhibited a synthetic growth defect on HU (Fig. 5b and supplemental Fig. S3). Given our finding that Ubp7 is important for an unperturbed S phase upon HU treatment, this could imply that Ubp7 functions in the same epistasis group as ASF1 and RAD6, factors that are also required for this pathway.
FIGURE 5.
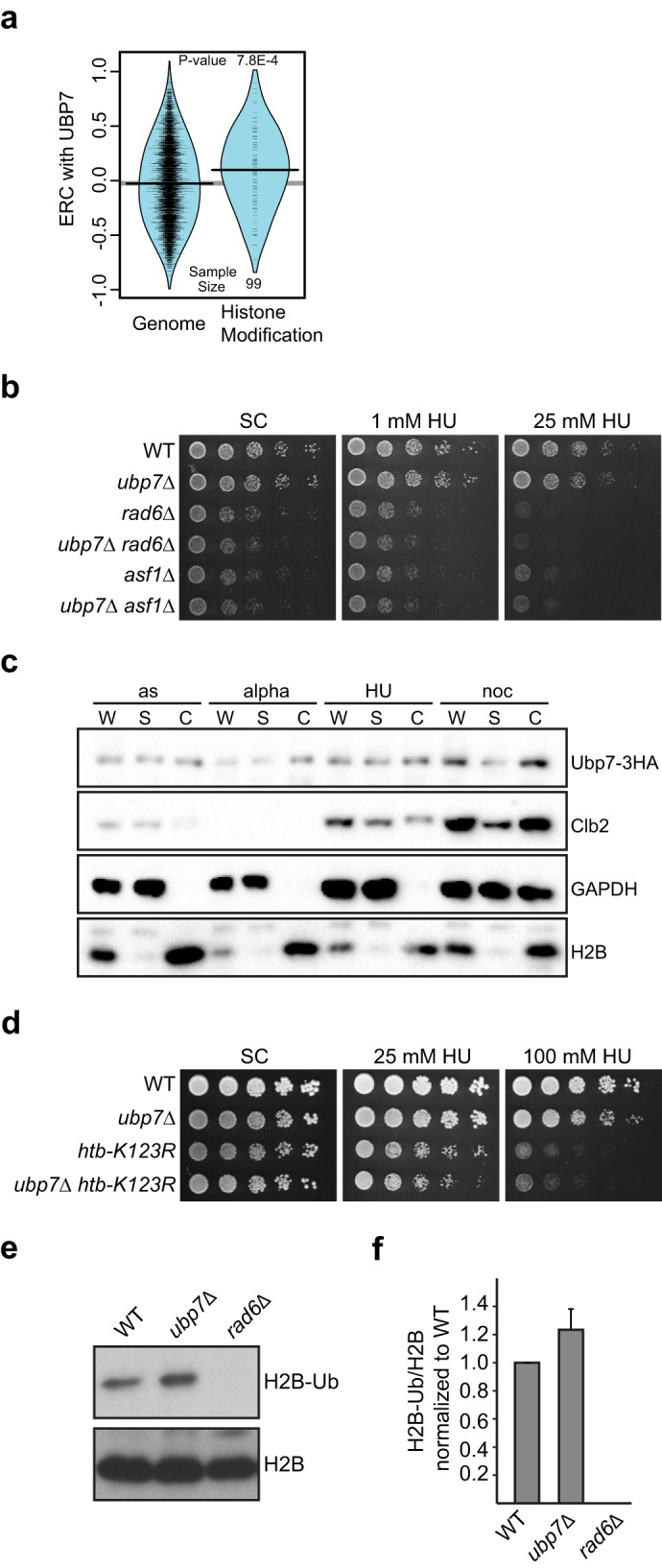
Deletion of UBP7 affects H2B ubiquitination. a, ERC values between Ubp7 and histone modification genes are elevated significantly compared with the genome-wide distribution. The violin plot shows the actual ERC values (tick marks) and their smoothed densities. b, genetic interaction of ubp7Δ with the histone chaperone ASF1 and histone-modifying E2 RAD6. Shown are 5-fold serial dilutions of the indicated strains on plates containing 1 and 25 mm HU, respectively. c, chromatin-binding assay for Ubp7–3HA. Shown are Western blotting analyses of whole-cell extract (W), supernatant (S), and chromatin-bound (C) fractions from asynchronous cultures (as) or those synchronized in G1 (alpha), S (HU), and G2/M phase (noc). Antibodies against HA, Clb2, GAPDH, and H2B were used. d, genetic interaction of ubp7Δ with htb-K123R. Shown are 5-fold serial dilutions of the indicated strains on plates containing the respective amounts of HU. Note that the strains used in this figure were in the FY2 genetic background, and, therefore, we observed a modest variation in the DNA damage sensitivity of ubp7Δ cells. e, steady-state H2B ubiquitination at Lys-123 in untreated asynchronous wild-type and ubp7Δ cultures. Shown is a Western blotting analysis for ubiquitinated H2B and total H2B. f, quantification of e by determining the ratio of H2B-Ub to total H2B and normalizing to the wild type. The graph represents the mean of four independent trials ± S.D.
If Ubp7 acts on histones or histone-associated factors, then its localization should be chromatin-associated. However, to date, Ubp7 has only been found to function in endocytosis and to localize close to the cell wall (10). Therefore, we wanted to ensure that a fraction of Ubp7 is indeed able to access chromatin. A fluorescent fusion of Ubp7 was extremely difficult to detect using fluorescent microscopy and live-cell imaging (data not shown), which is likely due to the very low expression level of Ubp7 (54). To nonetheless show that Ubp7 is chromatin-associated, we performed chromatin-binding assays using a strain expressing C-terminally 3HA-tagged Ubp7 from its endogenous locus. Indeed, we could clearly detect Ubp7 (Fig. 5c) in the chromatin fraction marked by histone H2B. In addition, although we observed a slight increase in overall Ubp7 protein levels upon nocodazole treatment, the chromatin association of Ubp7 was not dependent on the cell cycle stage or HU treatment (Fig. 5c). This further supports our hypothesis that Ubp7 may be acting on a histone or histone-associated factor.
Given the specific sensitivity of ubp7Δ cells to the replication fork stalling agent HU and the epistasis with rad6Δ and asf1Δ (Fig. 5b), one possible substrate of Ubp7 during replication could be histone H2B lysine 123 monoubiquitination. Although other DUBs have been shown to remove this modification during transcription (Ubp8 (55)) and telomere silencing (Ubp6 (56) and Ubp10 (57–59)), replication or replicative damage may trigger alternative pathways. Consistent with this hypothesis, we found that the double mutant of ubp7Δ and htb-K123R does not exhibit a synergistic defect on HU (Fig. 5d).
To further test this possibility, we used modification-specific antibodies and quantified H2B ubiquitination in asynchronous untreated wild-type and ubp7Δ cultures (Fig. 5, e and f). In line with our hypothesis, we did observe a reproducible 1.3-fold increase in H2B ubiquitination in the ubp7Δ mutant (Fig. 5, e and f). Because replication fork stalling and checkpoint activation lead to an increase in H2B ubiquitination, the observed increased H2B-Ub levels in ubp7Δ could result from at least two things. First, H2B-Ub could be a direct Ubp7 substrate, or, second, UBP7 deletion could indirectly lead to increased H2B ubiquitination as a result of checkpoint activation and/or replicative stress. Given our genetic analysis and the chromatin association of Ubp7, we favor the possibility of a chromatin-associated substrate of Ubp7, which could be H2B (Fig. 6).
FIGURE 6.
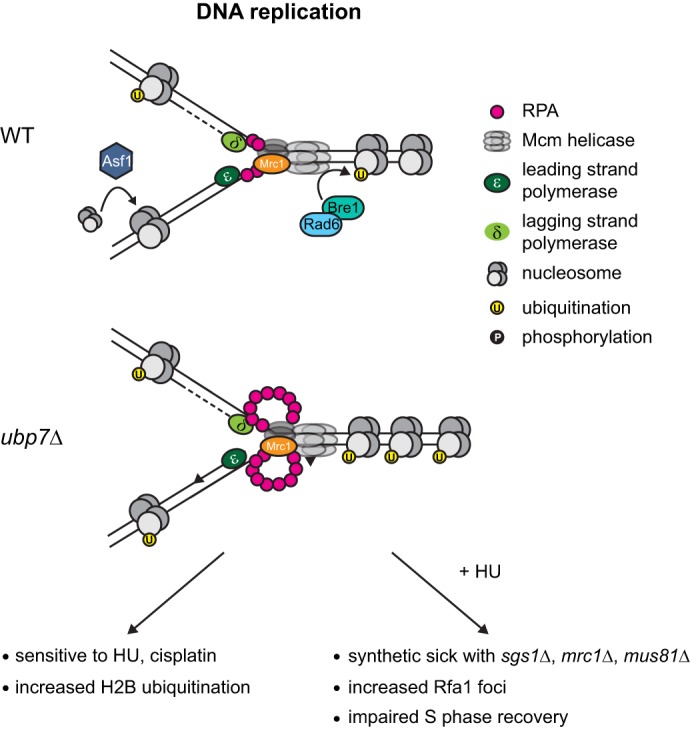
Schematic of the effect of UBP7 deletion. During DNA replication in a wild-type cell, a small amount of single-stranded DNA is exposed by the replicative helicase, and this single-stranded DNA is coated rapidly by RPA. The histone chaperone Asf1 deposits nucleosomes onto the newly synthesized DNA, and Rad6-Bre1 is responsible for H2B monoubiquitination at Lys-123. In a ubp7Δ cell, the replication fork is not stabilized properly, and this leads to HU and cisplatin sensitivity and elevated H2B ubiquitination. If an ubp7Δ strain is treated with HU, then there is an increase in single-stranded DNA coated by RPA and a requirement for SGS1, MRC1, and MUS81 for viability.
Discussion
In this study, we provide the first extensive characterization of the deubiquitinase Ubp7 in the context of the DNA damage response and propose Ubp7 as a novel factor important for S phase progression following replication fork stalling by HU. Although Ubp7 has been shown previously to act during endocytosis (10, 11), we provide several lines of evidence that Ubp7 is also needed during recovery from replication stress. UBP7 deletion specifically sensitizes cells to the DNA-damaging agents cisplatin and HU, and mutant cells exhibit characteristics of an activated intra-S phase checkpoint and recover from HU treatment more slowly than wild-type cells. This is in line with our finding that replication fork-stabilizing factors such as Sgs1 are required for HU tolerance in the absence of UBP7. Therefore, we propose a model in which Ubp7 acts to ensure replication fidelity, possibly by controlling the chromatin state around the replication fork.
In an unperturbed cell, the replication fork is coupled to the replicative helicase Mcm, thereby ensuring that only limited stretches of DNA in front of the replication fork are unwound (Fig. 6, top panel). One of the factors that ensures this association and also acts as a checkpoint component is Mrc1 (Claspin in mammals) (50, 51, 60). Among other factors, such as Mus81 (49), Sgs1 (BLM) is recruited to the stalled replication fork, and this process is required for fork stability and to enable fork restart (46–48). Sgs1 and Mrc1 function in the same genetic pathway to mediate Rad53 activation upon replication fork stalling by HU treatment, and, importantly, this role of Sgs1 is Rad51-independent (46). We find that ubp7Δ exhibits negative synthetic interactions with deletions of MRC1, SGS1, and MUS81 (Figs. 2, e and f, and 4b), all components required for replication fork stability. Therefore, we hypothesize that, upon deletion of UBP7, there is a requirement for factors involved in the stabilization of stalled replication forks. Further supporting this hypothesis is our finding that an ubp7Δ rad51Δ mutant does not exhibit a synthetic phenotype (Fig. 2e), whereas sgs1Δ ubp7Δ does. This indicates that the function of Sgs1 that is required upon HU treatment in a ubp7-null background is Rad51-independent and, therefore, likely attributed to the role of Sgs1 in maintaining replication fork stability (46). This, together with the finding that ubp7 mutants do not display altered rates of recombination or mutation frequencies (Fig. 2, c and d), argues that Ubp7 is not involved in canonical HR but, rather, functions in parallel to Sgs1 and Mus81 to stabilize stalled replication forks. The conclusions drawn from this genetic analysis are further supported by the fact that we observed a significant increase in Rfa1 foci (Fig. 3, a and b) and replication bubbles (Fig. 3d) upon HU treatment, indicating an increase in single-stranded DNA upon replication fork stalling in ubp7Δ.
An additional factor important for replication fork stability is the histone chaperone Asf1, which also possibly acts to prevent uncoupling of Mcm proteins from stalled forks (61). In contrast to the genetic interactions discussed above for mrc1 and sgs1, we did not observe a synthetic phenotype of ubp7Δ with a deletion of ASF1 even though it is similarly important for replication fork stability (61). Because we provided several lines of evidence showing that Ubp7 plays a role in ensuring S phase progression upon HU-exposure, we conclude that Asf1 likely functions in the same genetic pathway as Ubp7 to ensure replication fork stability.
Ubp7 is a deubiquinating enzyme that removes ubiquitin from substrate protein lysine residues (9). There are several potential substrates for Ubp7 in the context of replication and the response to replicative stress because many factors involved in the DDR are known to be modified posttranslationally with ubiquitin (3). In budding yeast, H2B is ubiquitinated predominantly at lysine 123 by Rad6, Bre1, and Lge1 (62–64), and there are several reports that demonstrate a function of H2B ubiquitination in DNA repair (52). In the context of replication fork stalling, H2B ubiquitination has been proposed to enable the retention of Sgs1 at a stalled fork and to enhance Rad53 phosphorylation (53). Both an htb-K123R strain and sgs1Δ exhibit reduced replication fork stability in HU (53). Because we only observe a severe synthetic phenotype of ubp7Δ with sgs1Δ and not htb-K123R (Figs. 2e and 5d), we hypothesize that Ubp7 may act in the same genetic pathway as H2B, which, in turn, acts in parallel to Sgs1 to ensure replication fork stability. This is consistent with an Sgs1-independent role of H2B in Rad53 phosphorylation and checkpoint activation that has been published previously (53) and that, in turn, raises the possibility that monoubiquitinated H2B could be an Ubp7 substrate. We attempted to directly address this option and used FLAG-H2B and Ubp7–3HA purified from yeast for in vitro deubiquitination assays. Unfortunately, because of the low expression level of endogenous Ubp7, the yield of active enzyme was too low to reliably detect a significant change in H2B ubiquitination. Further experiments, possibly with recombinant Ubp7, will be required to answer this question.
A better understanding of the cellular mechanisms that regulate replication and, thereby, ensure genetic fidelity (39) is of crucial importance to human health. Genome instability is considered a hallmark of cancer (2), and, although the importance of coordinated ubiquitination for DNA repair has been well studied, the role of DUBs has only shifted into focus recently (65). Intriguingly, there have also been reports that H2B ubiquitination is associated with cancer (66) and that ubiquitin pathways, including DUBs, are potential therapeutic targets for human diseases, including cancer (67). Future studies are needed to identify a functional orthologue of Ubp7 in mammals and to determine whether its role in S phase progression is conserved from yeast to humans.
Author Contributions
B. W. H. performed the experiments shown in Figs. 1, d and e, and 2c. B. S. performed the experiments shown in Fig. 3, c and d. M. R. S. helped with manuscript revision. M. J. M. performed the experiments shown in Figs. 2, d and f, and 3, a and b. F. F. K. helped with Figure 2c. D. B. designed and analyzed the experiments shown in Fig. 3, c and d, and wrote the paper. N. L. C. designed, performed, and analyzed the experiments shown in Figs. 1a and 5a and wrote the paper. S. B. designed, performed, and analyzed the experiments shown in Figs. 1, b and c; 2, a, b, and e; 4, b and c; and 5, b–f and wrote the paper. K. A. B. conceived and coordinated the study, analyzed the experiments, and wrote the paper. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Alexander Buchberger for laboratory space, materials and helpful discussions, Karen Arndt for the htb-K123R strain, Margaret Shirra and Branden Van Oss for valuable suggestions, Rodney Rothstein for yeast strains, and David Allis for the FLAG-H2B strain. We also thank Julieta Martino for critical reading of the manuscript and Chris Bakkenist and Patricia Opresko for reagents.
This work was supported by National Institutes of Health Grants ES024872 and GM088413) and Ellison Medical Foundation Grant AG-NS-0935-12 (to K. A. B.), by Charles E. Kaufman Award KA2014-73920 through The Pittsburgh Foundation, by a CURE grant from the Pennsylvania Department of Health, by the Center for Causal Discovery (National Institutes of Health Grant U54HG008540) (to N. L. C.), by Associazione Italiana per la Ricerca sul Cancro Italian Association for Cancer Research Grant IG14171, and by Fondazione Telethon Grant GGP12160 (to D. B.). This project used the University of Pittsburgh Cancer Institute Cytometry Facility that is supported in part by National Institutes of Health Award P30CA047904. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2.
- DDR
- DNA damage response
- SC
- synthetic complete
- YPD
- yeast extract peptone dextrose
- MMS
- methyl methanesulfonate
- HR
- homologous recombination
- DUB
- deubiquitinase
- HU
- hydroxyurea
- NER
- nucleotide excision repair
- DMSO
- dimethyl sulfoxide
- ERC
- evolutionary rate covariation
- WB
- Western blotting
- TLS
- translesion synthesis
- Ub
- ubiquitin.
References
- 1. Branzei D., and Foiani M. (2010) Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3. Bergink S., and Jentsch S. (2009) Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458, 461–467 [DOI] [PubMed] [Google Scholar]
- 4. Ciechanover A., and Ben-Saadon R. (2004) N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 14, 103–106 [DOI] [PubMed] [Google Scholar]
- 5. Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 6. Amerik A. Y., and Hochstrasser M. (2004) Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207 [DOI] [PubMed] [Google Scholar]
- 7. Reyes-Turcu F. E., Ventii K. H., and Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallego-Sánchez A., Andrés S., Conde F., San-Segundo P. A., and Bueno A. (2012) Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 8, e1002826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amerik A. Y., Li S. J., and Hochstrasser M. (2000) Analysis of the deubiquitinating enzymes of the yeast Saccharomyces cerevisiae. Biol. Chem. 381, 981–992 [DOI] [PubMed] [Google Scholar]
- 10. Weinberg J. S., and Drubin D. G. (2014) Regulation of clathrin-mediated endocytosis by dynamic ubiquitination and deubiquitination. Curr. Biol. 24, 951–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ren J., Kee Y., Huibregtse J. M., and Piper R. C. (2007) Hse1, a component of the yeast Hrs-STAM ubiquitin-sorting complex, associates with ubiquitin peptidases and a ligase to control sorting efficiency into multivesicular bodies. Mol. Biol. Cell 18, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lis E. T., and Romesberg F. E. (2006) Role of Doa1 in the Saccharomyces cerevisiae DNA damage response. Mol. Cell Biol. 26, 4122–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gazy I., Liefshitz B., Bronstein A., Parnas O., Atias N., Sharan R., and Kupiec M. (2013) A genetic screen for high copy number suppressors of the synthetic lethality between elg1Δ and srs2Δ in yeast. G3 3, 917–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomas B. J., and Rothstein R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell 56, 619–630 [DOI] [PubMed] [Google Scholar]
- 15. Zhao X., Muller E. G., and Rothstein R. (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2, 329–340 [DOI] [PubMed] [Google Scholar]
- 16. Sherman F., Fink G. R., and Hicks J. B. (1986) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 17. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 18. Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., and Schiebel E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963–972 [DOI] [PubMed] [Google Scholar]
- 19. Sato T., Yamanishi Y., Kanehisa M., and Toh H. (2005) The inference of protein-protein interactions by co-evolutionary analysis is improved by excluding the information about the phylogenetic relationships. Bioinformatics 21, 3482–3489 [DOI] [PubMed] [Google Scholar]
- 20. Clark N. L., Alani E., and Aquadro C. F. (2012) Evolutionary rate covariation reveals shared functionality and coexpression of genes. Genome Res. 22, 714–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balakrishnan R., Park J., Karra K., Hitz B. C., Binkley G., Hong E. L., Sullivan J., Micklem G., and Cherry J. M. (2012) YeastMine: an integrated data warehouse for Saccharomyces cerevisiae data as a multipurpose tool-kit. Database 2012, bar062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Godin S., Wier A., Kabbinavar F., Bratton-Palmer D. S., Ghodke H., Van Houten B., VanDemark A. P., and Bernstein K. A. (2013) The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic Acids Res. 41, 4525–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alvaro D., Lisby M., and Rothstein R. (2007) Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 3, e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lea D. E., and Coulson C. A. (1949) The distribution of the numbers of mutants in bacterial populations. J. Genet. 49, 264–285 [DOI] [PubMed] [Google Scholar]
- 25. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang C., and Stillman B. (1997) Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev. 11, 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang A. Y., Schulze J. M., Skordalakes E., Gin J. W., Berger J. M., Rine J., and Kobor M. S. (2009) Asf1-like structure of the conserved Yaf9 YEATS domain and role in H2A.Z deposition and acetylation. Proc. Natl. Acad. Sci. U.S.A. 106, 21573–21578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szakal B., and Branzei D. (2013) Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 32, 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark N. L., and Aquadro C. F. (2010) A novel method to detect proteins evolving at correlated rates: identifying new functional relationships between coevolving proteins. Mol. Biol. Evol. 27, 1152–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clark N. L., Alani E., and Aquadro C. F. (2013) Evolutionary rate covariation in meiotic proteins results from fluctuating evolutionary pressure in yeasts and mammals. Genetics 193, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Godin S. K., Meslin C., Kabbinavar F., Bratton-Palmer D. S., Hornack C., Mihalevic M. J., Yoshida K., Sullivan M., Clark N. L., and Bernstein K. A. (2015) Evolutionary and functional analysis of the invariant SWIM domain in the conserved Shu2/SWS1 protein family from Saccharomyces cerevisiae to Homo sapiens. Genetics 199, 1023–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hustedt N., Gasser S. M., and Shimada K. (2013) Replication checkpoint: tuning and coordination of replication forks in S phase. Genes 4, 388–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guzder S. N., Sung P., Prakash L., and Prakash S. (1998) Affinity of yeast nucleotide excision repair factor 2, consisting of the Rad4 and Rad23 proteins, for ultraviolet damaged DNA. J. Biol. Chem. 273, 31541–31546 [DOI] [PubMed] [Google Scholar]
- 34. Jansen L. E., Verhage R. A., and Brouwer J. (1998) Preferential binding of yeast Rad4-Rad23 complex to damaged DNA. J. Biol. Chem. 273, 33111–33114 [DOI] [PubMed] [Google Scholar]
- 35. Bankmann M., Prakash L., and Prakash S. (1992) Yeast RAD14 and human xeroderma pigmentosum group A DNA-repair genes encode homologous proteins. Nature 355, 555–558 [DOI] [PubMed] [Google Scholar]
- 36. Cox B. S., and Parry J. M. (1968) The isolation, genetics and survival characteristics of ultraviolet light-sensitive mutants in yeast. Mutat. Res. 6, 37–55 [DOI] [PubMed] [Google Scholar]
- 37. Reed S. H., You Z., and Friedberg E. C. (1998) The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision repair: in vitro and in vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J. Biol. Chem. 273, 29481–29488 [DOI] [PubMed] [Google Scholar]
- 38. McHugh P. J., Ward T. A., and Chovanec M. (2012) A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 11, 3739–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aguilera A., and Gómez-González B. (2008) Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217 [DOI] [PubMed] [Google Scholar]
- 40. Huang M. E., Rio A. G., Nicolas A., and Kolodner R. D. (2003) A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl. Acad. Sci. U.S.A. 100, 11529–11534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swanson R. L., Morey N. J., Doetsch P. W., and Jinks-Robertson S. (1999) Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol. Cell Biol. 19, 2929–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ogawa T., Yu X., Shinohara A., and Egelman E. H. (1993) Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 259, 1896–1899 [DOI] [PubMed] [Google Scholar]
- 43. Hays S. L., Firmenich A. A., and Berg P. (1995) Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc. Natl. Acad. Sci. U.S.A. 92, 6925–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernstein K. A., Gangloff S., and Rothstein R. (2010) The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 44, 393–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giannattasio M., Zwicky K., Follonier C., Foiani M., Lopes M., and Branzei D. (2014) Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 21, 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bjergbaek L., Cobb J. A., Tsai-Pflugfelder M., and Gasser S. M. (2005) Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 24, 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hegnauer A. M., Hustedt N., Shimada K., Pike B. L., Vogel M., Amsler P., Rubin S. M., van Leeuwen F., Guénolé A., van Attikum H., Thomä N. H., and Gasser S. M. (2012) An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J. 31, 3768–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cobb J. A., Bjergbaek L., Shimada K., Frei C., and Gasser S. M. (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22, 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pepe A., and West S. C. (2014) MUS81-EME2 promotes replication fork restart. Cell Rep. 7, 1048–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Osborn A. J., and Elledge S. J. (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 17, 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alcasabas A. A., Osborn A. J., Bachant J., Hu F., Werler P. J., Bousset K., Furuya K., Diffley J. F., Carr A. M., and Elledge S. J. (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3, 958–965 [DOI] [PubMed] [Google Scholar]
- 52. Giannattasio M., Lazzaro F., Plevani P., and Muzi-Falconi M. (2005) The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem. 280, 9879–9886 [DOI] [PubMed] [Google Scholar]
- 53. Lin C. Y., Wu M. Y., Gay S., Marjavaara L., Lai M. S., Hsiao W. C., Hung S. H., Tseng H. Y., Wright D. E., Wang C. Y., Hsu G. S., Devys D., Chabes A., and Kao C. F. (2014) H2B mono-ubiquitylation facilitates fork stalling and recovery during replication stress by coordinating Rad53 activation and chromatin assembly. PLoS Genet. 10, e1004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 55. Daniel J. A., Torok M. S., Sun Z. W., Schieltz D., Allis C. D., Yates J. R. 3rd, and Grant P. A. (2004) Deubiquitination of histone H2B by a yeast acetyltransferase complex regulates transcription. J. Biol. Chem. 279, 1867–1871 [DOI] [PubMed] [Google Scholar]
- 56. Qin S., Wang Q., Ray A., Wani G., Zhao Q., Bhaumik S. R., and Wani A. A. (2009) Sem1p and Ubp6p orchestrate telomeric silencing by modulating histone H2B ubiquitination and H3 acetylation. Nucleic Acids Res. 37, 1843–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rhie B. H., Song Y. H., Ryu H. Y., and Ahn S. H. (2013) Cellular aging is associated with increased ubiquitylation of histone H2B in yeast telomeric heterochromatin. Biochem. Biophys. Res. Commun. 439, 570–575 [DOI] [PubMed] [Google Scholar]
- 58. Emre N. C., Ingvarsdottir K., Wyce A., Wood A., Krogan N. J., Henry K. W., Li K., Marmorstein R., Greenblatt J. F., Shilatifard A., and Berger S. L. (2005) Maintenance of low histone ubiquitylation by Ubp10 correlates with telomere-proximal Sir2 association and gene silencing. Mol. Cell 17, 585–594 [DOI] [PubMed] [Google Scholar]
- 59. Gardner R. G., Nelson Z. W., and Gottschling D. E. (2005) Ubp10/Dot4p regulates the persistence of ubiquitinated histone H2B: distinct roles in telomeric silencing and general chromatin. Mol. Cell Biol. 25, 6123–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Katou Y., Kanoh Y., Bando M., Noguchi H., Tanaka H., Ashikari T., Sugimoto K., and Shirahige K. (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 61. Franco A. A., Lam W. M., Burgers P. M., and Kaufman P. D. (2005) Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 19, 1365–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robzyk K., Recht J., and Osley M. A. (2000) Rad6-dependent ubiquitination of histone H2B in yeast. Science 287, 501–504 [DOI] [PubMed] [Google Scholar]
- 63. Wood A., Krogan N. J., Dover J., Schneider J., Heidt J., Boateng M. A., Dean K., Golshani A., Zhang Y., Greenblatt J. F., Johnston M., and Shilatifard A. (2003) Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 11, 267–274 [DOI] [PubMed] [Google Scholar]
- 64. Song Y. H., and Ahn S. H. (2010) A Bre1-associated protein, large 1 (Lge1), promotes H2B ubiquitylation during the early stages of transcription elongation. J. Biol. Chem. 285, 2361–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nishi R., Wijnhoven P., le Sage C., Tjeertes J., Galanty Y., Forment J. V., Clague M. J., Urbe S., and Jackson S. P. (2014) Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 16, 1016–1026, 1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Johnsen S. A. (2012) The enigmatic role of H2Bub1 in cancer. FEBS Lett. 586, 1592–1601 [DOI] [PubMed] [Google Scholar]
- 67. Popovic D., Vucic D., and Dikic I. (2014) Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.