

Abstract
The prion protein (PrP) seems to exert both neuroprotective and neurotoxic activities. The toxic activities are associated with the C-terminal globular parts in the absence of the flexible N terminus, specifically the hydrophobic domain (HD) or the central region (CR). The wild type prion protein (PrP-WT), having an intact flexible part, exhibits neuroprotective qualities by virtue of diminishing many of the cytotoxic effects of these mutant prion proteins (PrPΔHD and PrPΔCR) when coexpressed. The prion protein family member Doppel, which possesses a three-dimensional fold similar to the C-terminal part of PrP, is also harmful to neuronal and other cells in various models, a phenotype that can also be eliminated by the coexpression of PrP-WT. In contrast, another prion protein family member, Shadoo (Sho), a natively disordered protein possessing structural features similar to the flexible N-terminal tail of PrP, exhibits PrP-WT-like protective properties. Here, we report that, contrary to expectations, Sho expression in SH-SY5Y or HEK293 cells induces the same toxic phenotype of drug hypersensitivity as PrPΔCR. This effect is exhibited in a dose-dependent manner and is also counteracted by the coexpression of PrP-WT. The opposing effects of Shadoo in different model systems revealed here may be explored to help discern the relationship of the various toxic activities of mutant PrPs with each other and the neurotoxic effects seen in neurodegenerative diseases, such as transmissible spongiform encephalopathy and Alzheimer disease.
Keywords: cell death, cell surface protein, drug resistance, lentivirus, neuroblastoma, prion, prion disease, transposable element (TE), Shadoo, zeocin
Introduction
The prion protein (PrP)2 is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein ubiquitously expressed in vertebrates, reaching its highest levels in the central nervous system (CNS) and the heart (1, 2). It is notorious for its role in neurodegenerative diseases, such as transmissible spongiform encephalopathies (TSEs) (3) and Alzheimer disease (4–6).
Numerous functions have been attributed to PrP, and its involvement in various physiological processes has been proposed (7), notably normal olfactory behavior and physiology (8), hippocampus-dependent spatial learning (9), and peripheral myelin maintenance (10). Especially interesting are its neuroprotective and neurotoxic functions that might be associated with its role in neurodegenerative diseases (11–13).
This seemingly dual role might be revealed by the expression of PrP deletion constructs lacking most of the N-terminal domain (“Shmerling mutants”; mPrPΔ32–121 and mPrPΔ32–134) (14) or only either the hydrophobic domain (“PrPΔHD”; mPrPΔ111–134) (15) or the central region (“PrPΔCR”; mPrPΔ105–125) (16) in transgenic animal models. Expression of these proteins in mice on a PrP null background causes various symptoms of neurodegeneration, including severe ataxia, dramatic reduction of the granular cell layer of the cerebellum, and vacuolization and astrogliosis in the white matter, leading to the early death of animals in a few weeks after birth in the case of PrPΔCR, which causes the most severe symptoms (7, 16). A spontaneous cytotoxic effect is also apparent in cerebellar granular neuron (CGN) cultures expressing these mutant PrPs in the absence of wild type PrP (17). The presence/coexpression of PrP-WT can partially or completely eliminate these spontaneous cytotoxic phenotypes in both animal and primary cell culture models in a dose-dependent manner (18).
The other members of the prion protein family, Doppel (Dpl) and Shadoo (Sho), exert neurotoxic and neuroprotective effects, respectively, similar to those of PrP (17, 19, 20). Doppel and Shadoo show similarity/analogy in both structural and functional terms to either the C-terminal structured (Doppel) or the N-terminal unstructured, flexible domain (Sho) of PrP. All three members of the prion protein family localize predominantly to the cell surface, being attached to the outer leaflet of the plasma membrane via a GPI anchor (21, 22).
The ectopic expression of Doppel in the CNS causes severe neurodegeneration, ataxia, and the loss of Purkinje cells (23–25). Its expression in CGN culture likewise triggers increased cell death (17, 26), akin to mutant PrPs. Interestingly, these neurotoxic effects are also counteracted by the coexpression of PrP-WT.
Shadoo is the most recently discovered mammalian PrP paralog (27). Just like PrP, Shadoo appears, among other tissues, in the central nervous system (17). There is no extended sequence similarity between Sho and PrP, except in their hydrophobic domains (aa 62–77 in mouse Sho and aa 113–133 in mouse PrP). Nevertheless, both PrP flexible N-terminal part and Sho that are natively unstructured contain repeat regions with periodically reoccurring positively charged amino acids: histidines in the octarepeat region of PrP and arginines in the (RXXX)n motif of Sho (28). This structural similarity parallels functional analogy; coexpression of Shadoo counteracts the neurotoxic effects of Doppel and of PrPΔ32–121 in CGN culture, and of PrPΔHD in human neuroblastoma SH-SY5Y cells in a manner similar to that of PrP-WT (17, 20). Interestingly, the latter group also reported that PrP-WT and Sho, unlike their HD-deleted mutant variants, decrease the excitotoxic effect of glutamate in SH-SY5Y cells, emphasizing the neuroprotective feature of Sho that is also characteristic of PrP bearing an intact N-terminal part (20). Furthermore, it was found that both Doppel and PrPΔCR cause increased sensitivity to certain drugs (hygromycin, G418, and Zeocin) in several types of immortalized cell lines, a phenotype that was also eliminated by PrP coexpression (29).
In addition, the same mutant PrPs in various cells with distinct origins are reported to induce inward cationic currents detected in whole cell patch clamp experiments (30). This interesting phenotype was also diminished by the coexpression of PrP-WT.
Apparently, several neurotoxic and neuroprotective activities are associated with PrP and its mutant forms. However, it is not clear whether the manifestation of these various phenotypes associated with PrP-WT and mutant PrPs with N-terminal deletion in different model systems involve identical or different pathways.
In one approach, Harris and colleagues (30, 31) examined several PrP variants bearing familial TSE-associated point mutations in or next to the central region for a correlation between the appearance of spontaneous inward currents and drug hypersensitivity. Their results seem to support the existence of overlapping pathways 1) for the pathomechanisms of some forms of familial TSE and 2) for drug hypersensitivity and for the emergence of spontaneous inward currents.
As a different approach, the interference of Sho expression with various toxic phenotypes related to PrP may also help to distinguish activities that involve different pathways. To explore this approach, we set out to learn whether the neuroprotective potential of Sho, seen both in CGN culture and SH-SY5Y cells expressing N-terminal deletion mutant PrPs or Doppel and in SH-SY5Y cells by decreasing the toxic effect of glutamate, is also manifested in reverting the drug hypersensitivity phenotype caused by a deletion mutant PrP.
Experimental Procedures
Chemicals, Reagents, Antibodies
Restriction endonucleases, T4 DNA ligase, Pfu DNA polymerase, isopropyl β-d-thiogalactopyranoside, and TurboFect transfection reagent were purchased from Thermo Scientific. DNA oligonucleotides were from Microsynth AG. High-glucose Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Life Technologies/Gibco, and penicillin/streptomycin was from Lonza. 4′,6-Diamidino-2-phenylindole (DAPI), proteinase inhibitor mixture, calpain inhibitor I, G418, puromycin, etoposide, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)were obtained from Sigma-Aldrich. Bradford reagent was from Bio-Rad. Polyvinylidene difluoride (PVDF) transfer membrane and chemiluminescent substrate (Immobilon ECL substrate) were from Millipore. PNGase F was purchased from New England Biolabs. PI-PLC, Zeocin, and PrestoBlue reagent were obtained from Life Technologies. The following primary antibodies were used: SAF32 anti-PrP mouse IgG (Cayman Chemical, 189720), purified anti-H2AX.phospho antibody (Biolegend, 613402), anti-Shadoo rabbit polyclonal antibody (Abgent, AP4754b), and anti-β-actin chicken IgG (Sigma, GW23014). Secondary antibodies used were goat anti-mouse IgG (H+L), Alexa Fluor 594- or Alexa Fluor 647-conjugated (Life Technologies, Inc., A11005 and A21235). Horseradish peroxidase (HRP)-conjugated anti-mouse, anti-rabbit, and anti-chicken IgG were from Jackson ImmunoResearch (catalog no. 715-035-151), Pierce (catalog no. 31460), and Sigma-Aldrich (catalog no. A9046), respectively. All other reagents and chemicals were purchased from Sigma-Aldrich.
Plasmid Constructs and DNA Cloning
The cDNA of mouse Shadoo protein (mSho) (Uniprot entry Q8BWU1) in a pSPORT1 plasmid was obtained from MRC Geneservice, and the cDNA of mouse PrP (mPrP) (Uniprot entry P04925) was from the Caughey laboratory (32). Plasmid vectors of the Sleeping Beauty transposon-based gene delivery system (SB CAGx100 (33) and pSB-CAG-Puro (34)) and the pRRL lentiviral vectors (pRRL-EF1-mCherry and pRRL-EF1-EGFP (35)) were kind gifts of Dr. Z. Izsvák and Dr. Z. Ivics and of Dr. K. Német, respectively. All plasmids were constructed with standard molecular biology techniques, briefly as follows.
The deletion of the central region (aa 105–125) of mPrP in the pcDNA3 vector and the introduction of a silent mutation for removing the XhoI restriction site of the mSho CDS in the pSPORT1 vector were carried out using the QuikChange site-directed mutagenesis protocol (Stratagene) with the following oligonucleotides: Delta105–125for and Delta105–125rev, XhoImutator5 and XhoImutator3, respectively. Subsequently, XhoI-mutated Shadoo was PCR-amplified, using mShoBamHI5 and XhoISho3 primers, and cloned to the pcDNA3 vector between the BamHI and XhoI restriction sites.
Sleeping Beauty Constructs (Fig. 1A)
FIGURE 1.
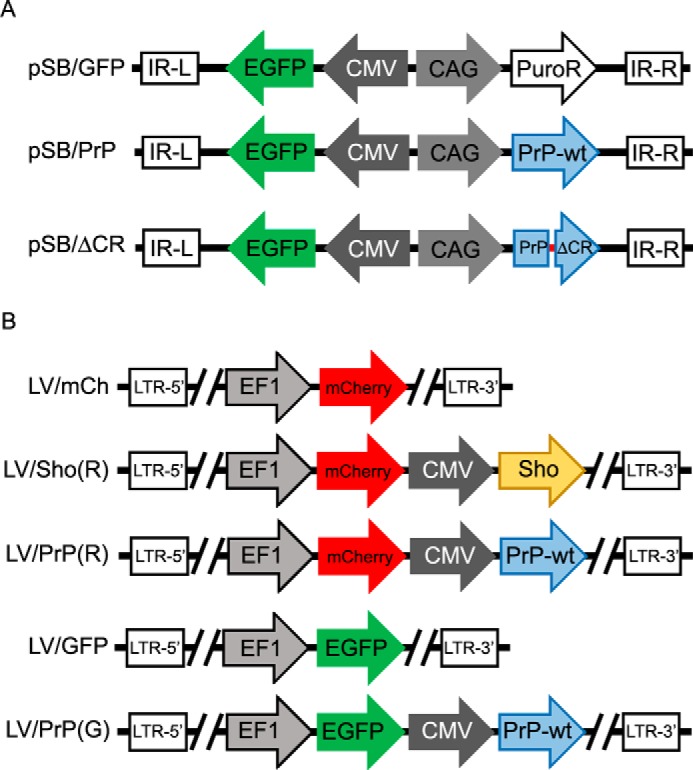
Topologies of the plasmid constructs used in these studies. A, Sleeping Beauty plasmids used for integration of the expression cassettes. B, pRRL plasmids used for lentivirus generation. IR-L and IR-R, left and right inverse repeats of SB. LTR-5′ and LTR-3′, 5′ and 3′ long terminal repeats. CMV, CMV intermediate early promoter. CAG, CMV early enhancer/chicken β-actin promoter. EF1, elongation factor-1 promoter. PuroR, puromycin resistance gene. PrP-wt, wild type mouse prion protein. PrPΔCR, mouse prion protein missing the central region (aa 105–125). Sho, mouse Shadoo protein.
The cDNA of the enhanced green fluorescent protein (EGFP) with a Kozak sequence was cloned into the polycloning site of the pcDNA3 eukaryotic expression vector, between the BamHI and NotI restriction sites. The whole expression cassette containing the CMV-IE promoter and EGFP followed by a BGH Poly(A) signal was amplified from the vector by PCR with the following primers: ApoICMV5 and ApoIBGH3. PCR fragments were purified and digested by ApoI enzyme and were cloned into the EcoRI site of a Sleeping Beauty plasmid containing a puromycin resistance gene driven by a CAG promoter (pSB-CAG-Puro). The resultant plasmid is named pSB/GFP.
The cDNAs of wild type mPrP and mPrPΔCR were PCR-amplified with the primers PrPNheI5 and PrPBamHI3 from the pcDNA3 vectors encoding the respective constructs. Subsequently, after being digested with NheI and BamHI enzymes, they were inserted downstream of the CAG promoter into the pSB/GFP vectors, between the restriction sites of NheI and BglII, which also removed the cDNA of the puromycin resistance gene. The resulted plasmids were named pSB/PrP and pSB/ΔCR.
Lentiviral Constructs (Fig. 1B)
A linker (linker2-3048) containing the unique sites of the AscI and BsiWI restriction endonucleases was cloned into the pRRL-EF1-mCherry or pRRL-EF1-EGFP lentiviral vector between the BsrGI and SalI sites. The modified pRRL vectors are called LV/mCh and LV/GFP. Expression cassettes containing the PrP or Sho coding sequences driven by CMV-IE promoters were amplified by PCR from the pcDNA3 vectors encoding the respective constructs with the following PCR primers: V-CMV-PrP fw and V-CMV-PrP rev (for PrP) and V-CMV-PrP fw and V-CMV-PA rev (for Sho). The PCR products were digested and inserted between the AscI and BsiWI sites of the modified pRRL-EF1-mCherry or pRRL-EF1-EGFP vectors. The vectors containing PrP or Shadoo expression cassettes are called LV/PrP(R), LV/Sho(R), and LV/PrP(G).
Recombinant Sho-PrP Fusion Polypeptide
The fusion polypeptide rSho-PrP was made by cloning a fragment that codes for the peptide from amino acids 81–116 of Sho (TGSGWRRTSG PGELGLEDDE NGAMGGNGTD RGVYSYS), corresponding to the Sho antibody epitope, into a fragment coding for PrP(23–230) in a pET41 expression vector, between positions corresponding to the 93rd and 94th amino acids of PrP using three DNA linkers with overlapping overhangs (Sho-epi1, -2, and -3) ligated into the unique Acc65 restriction enzyme site. For the sequences of the oligonucleotides used for PCR and linker ligation, see Table 1. The correct sequences for the expression cassettes in all plasmids generated in this study were confirmed by Sanger sequencing (Microsynth AG).
TABLE 1.
The DNA oligonucleotides used in the cloning processes
| Oligonucleotide name | DNA sequence |
|---|---|
| Delta105–125for | AACAAGCCCAGCAAACCAGGCTACATGCTGGGGAG |
| Delta105–125rev | CTCCCCAGCATGTAGCCTGGTTTGCTGGGCTTGTT |
| XhoImutator5 | CGGCCGCGGAGGCGCACGAGGCAGTGCCCGG |
| XhoImutator3 | CCGGGCACTGCCTCGTGCGCCTCCGCGGCCG |
| ApoICMV5 | CGCGCGAAATTTCTGCTTCGCGATGTACGGG |
| ApoIBGH3 | GGCCCGAAATTTCCACCGCATCCCCAGCATG |
| PrPNheI5 | GCCGGGCTAGCCACCATGGCGAACCTTGGCTACTG |
| PrPBamHI3 | TACCACGGATCCTCATCCCACGATCAGGAAGATG |
| mShoBamHI5 | CCCGAAGGATCCGCCACCATGAACTGGACTGCTGCCACG |
| XhoISho3 | GGCGCGCTCGAGCTAAGGCCGAAGCAGTTCTAG |
| Linker2-3048-fwd | GTACAAGTGAGGGCGCGCCAAACATATGAAACGTACGAA |
| Linker2-3048-rev | TCGATTCGTACGTTTCATATGTTTGGCGCGCCCTCACTT |
| V-CMV-PrP fw | TTTGCAGGCGCGCCCGATGTACGGGCCAGATATACG |
| V-CMV-PrP rev | CACTATTGTACAGGGCCCTCTAGATGCATGCTCGAGC |
| V-CMV-PA rev | ATAGAGTGTACAACATCCCCAGCATGCCTGC |
| Sho-epi1-fwd | GTACAGGCTCTGGCTGGAGGAGGACCTCAGGGCCTGGAGAGCTA |
| Sho-epi1-reva | AGGCCCTGAGGTCCTCCTCCAGCCAGAGCCT |
| Sho-epi2-fwda | GGCCTGGAGGACGATGAGAATGGGGCAATGGGAGGC |
| Sho-epi2-reva | TGCCCCATTCTCATCGTCCTCCAGGCCTAGCTCTCC |
| Sho-epi3-fwda | AACGGAACCGACCGAGGAGTCTACAGCTACA |
| Sho-epi3-rev | GTACTGTAGCTGTAGACTCCTCGGTCGGTTCCGTTGCCTCCCAT |
a These oligonucleotides are 5′-phosphorylated.
Expression, Purification, and Refolding of the Recombinant Sho-PrP Fusion Protein
Plasmid pET41a encoding the rSho-PrP fusion polypeptide encompassing both PrP and Sho epitopes was transformed into competent Escherichia coli BL21 (DE3) pLysS, and after induction by isopropyl-β-d-1-thiogalactopyranoside, it was expressed at 37 °C in inclusion bodies. Cells were harvested 8 h after induction. Proteins were purified as follows. Inclusion bodies were dissolved in buffer A (6 m guanidinium chloride (GdmCl), 100 mm Na2HPO4, 400 mm NaCl, 5 mm imidazole, and 5 mm β-mercaptoethanol, pH 8.0) and stirred overnight at 4 °C. After centrifugation, the soluble protein fraction was transferred to a nickel-nitrilotriacetic acid-agarose column. Before the oxidative refolding step, the column was washed with buffer B (10 mm Tris-HCl, 100 mm Na2HPO4, pH 8.0) containing at first 6 m GdmCl and then 1 m GdmCl. Oxidative refolding was performed in buffer B containing 1 m GdmCl, 10 mm glutathione (reduced), 5 mm glutathione (oxidized). To remove the nonspecifically bound protein impurities, the column was washed with buffer B containing 50 mm imidazole. Elution was carried out with 50 mm sodium acetate, pH 4.1. Proteins obtained were stored at −80 °C until use.
Cell Lines, Culturing, Transfection, and Transduction
SH-SY5Y human neuroblastoma and HEK293 cell lines were from ATCC (CRL-2266TM) and Gibco (11631-017), respectively. Each type of cell was cultured in high glucose DMEM complemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere with 5% CO2. Cells were passed at 90–95% confluence at 1:10 splitting ratios for each type of cells.
For transfection purposes, 1 × 105 SH-SY5Y cells were seeded on multiple wells of 6-well plates. The transfection was carried out at 50–70% confluence, using 1–4 μg of circular plasmid DNA with TurboFect transfection reagent, in accordance with the manufacturer's manual.
In the case of lentiviral transductions, the lentiviruses were generated in the Hungarian National Blood Transfusion Service's lentiviral facility. The transductions of SH-SY5Y and HEK293 cells were carried out on 24-well plates. 3 × 104 cells were seeded, and the transductions were carried out for 24 h at various multiplicities of infection from 1 to 5.
Fluorescence-activated cell sorting was used to separate cells with stable transgene expression in the transfected or transduced cell populations based on the expression of the fluorescent marker. In case of transfection with the Sleeping Beauty constructs, the EGFP-positive cells were sorted at 3 and 14 days post-transfection. In the case of transduced cells, mCherry-positive or mCherry and EGFP double-positive cells were sorted 7–10 days post-transduction.
All types of cells were regularly tested for mycoplasma contamination. EGFP and mCherry positivity were examined at every passage, and experiments were carried out on cultures in which at least 90% of the cells expressed the required fluorescent markers. In parallel with the execution of the experiments, the expression levels of the transgenes were determined by an immunoblotting technique.
Immunocytochemistry
Cells were seeded on Labtek-II 8-well slides (5 × 104 cell/well density) or on 96-well plates at 1.5 × 104 cells/well density for phosphorylated histone 2AX (γ-H2AX) detection. 24 h after seeding, cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature and washed three times in immunofluorescence washing (IF) solution (0.2% bovine serum albumin, 0.1% Triton X-100 in PBS) followed by blocking and permeabilization using 5% bovine serum albumin, 0.5% Triton X-100 in IF solution for 10 min at room temperature. Cells were washed again three times in IF solution before applying the primary antibody (SAF32 anti-PrP mouse IgG for PrP staining and purified anti-H2AX.phospho antibody for γ-H2AX staining) for 1 h at room temperature. The dilutions for the primary antibodies were as follows: 1:200 SAF32 and 1:250 anti-γ-H2AX in IF solution. The cells were then washed once by IF solution, and the secondary antibody (goat anti-mouse IgG (H+L) or Alexa Fluor 594 or 647) was applied for 30 min at room temperature. All secondary antibodies were used at 1:250 dilutions in IF solution. Cells were washed three more times for 10 min and once overnight, and then nuclei were stained with DAPI for 10 min at room temperature (1 μm DAPI in PBS).
The cells were observed using an Olympus FV500-IX confocal laser scanning microscope, with a PLAPO ×60 (1.4 numerical aperture) oil immersion objective (Olympus). DAPI, EGFP, mCherry, and Alexa Fluor 647 fluorophores were excited at 405, 488, 543, and 633 nm, respectively. Emission detection ranges were 430–460 nm (DAPI), 505–525 nm (EGFP), 560+ nm (mCherry), and 660+ nm (Alexa Fluor 647), respectively. Confocal images were recorded with Olympus Fluoview version 5.0 software. Image acquisition settings (laser intensity, photomultiplier settings, and confocal aperture size for each channel) were kept unchanged during confocal microscopy sessions. Images were captured in sequential scanning mode and using 1024 × 1024-pixel resolution.
Immunoblotting
Cells seeded on 100-mm cell culture dishes were harvested at 70–90% confluence after being washed once with PBS by scraping in 1 ml of PBS. Cells were pelleted by centrifugation (3 min, 200 × g) and resuspended in ice-cold lysis buffer (50 mm HEPES, pH 7.5, 0.2 mm EDTA, 10 mm NaF, 250 mm NaCl, 0.5% Nonidet P-40, with 1% proteinase inhibitor mixture, 1% calpain inhibitor, 1 mm DTT). The total protein concentration was measured by using a Bradford protein assay. Where needed, PNGase F treatment was carried out on samples of 50–100 μg of total protein, according to the manufacturer's protocol. Samples of 1–50 μg of total protein, depending on the necessities of the experiment, were run on 15% denaturing polyacrylamide gels and were blotted onto activated PVDF membrane, using a wet blotting system from Bio-Rad. The membrane was blocked for at least 1 h in Tris-buffered saline with Tween 20 (TBST), containing 5% nonfat milk powder, and primary antibodies were applied overnight at 4 °C at the following dilutions: SAF32, 1:5000; anti-Shadoo rabbit polyclonal antibody, 1:200; anti-β-actin chicken IgG, 1:1000. The next day, after several washing steps in TBST, HRP-conjugated secondary antibodies were applied to the membrane for 60 min in the following dilutions: anti-mouse (1:20,000) and anti-rabbit and anti-chicken antibodies (1:200,000) in blocking buffer. The proteins were visualized by adding chemiluminescent substrate (Millipore Immobilon ECL substrate).
Phosphatidylinositol-dependent Phospholipase C (PI-PLC) Treatment
Cells were seeded on 24-well plates. After reaching confluence, the PI-PLC treatment was carried out according to the manufacturer's protocol. Briefly, cells were washed twice in PBS, and the plate with cells having only PBS or PBS with PI-PLC (1 unit/ml PI-PLC) was rocked gently for 30 min at 4 °C. The supernatants were removed from the cells, centrifuged (5 min, 20,000 × g) to remove cells and debris, and processed for SDS-PAGE (noted as medium (M) samples in Fig. 4). The PI-PLC-treated and untreated cells were harvested from the plates by scraping and were processed for SDS-PAGE (noted as cell lysate (Cl) samples in Fig. 4).
FIGURE 4.

Overexpressed PrPΔCR and Sho have normal traffic and similar expression levels in SH-SY5Y cells. A, lentiviral transduction to express Shadoo of PrPΔCR cells does not affect its proliferation adversely. Fluorescence of SH/ΔCR, SH/ΔCR+Sho, and SH/ΔCR+PrP cells was measured at every 24th h after attachment for 8 days and in multiple wells, using PrestoBlue reagent. A representative plot is shown. 100% is the fluorescence value at day 0 after attachment for each type of cell. Values are means ± S.D. of replicas in the individual experiment. B, both PrP-WT and PrPΔCR overexpressed in SH-SY5Y cells are complex-glycosylated. PrPΔCR expression levels in SH/ΔCR, SH/ΔCR+mCh, and SH/ΔCR+PrP cells are comparable. Shown is Western blotting analysis of extracts from various cells as indicated above the lanes, untreated (−) or treated with PNGase F (+). Left and right panels are from the same x-ray film, corresponding to the left and right parts of the same membrane. C, Shadoo, overexpressed in SH/ΔCR+Sho cells, is complex-glycosylated. Shown is Western blotting analysis of extracts from SH/ΔCR+mCh and SH/ΔCR+Sho cells, incubated with (+) or without (−) PNGase F or without incubation (0). After PNGase F treatment, the C1 fragment (58) of Shadoo is more readily detectable. D, Shadoo protein is attached to the cell surface via a GPI anchor when expressed in SH-SY5Y cells. Shown is Western blotting analysis of cell lysates (Cl) and supernatant medium (M) from SH/GFP (left) and SH/Sho (right) cells, incubated with (+) or without (−) PI-PLC. Endogenous Shadoo expression is below the detection limit. The amount of Shadoo decreases below the detection limit in the cell samples of PI-PLC-treated cells (C− versus C+), whereas the amount of an intracellular protein, β-actin, remains unchanged. Shadoo remains below the detection limit in the medium samples. Left and right panels are from the same x-ray film, corresponding to the left and right parts of the same membrane. E, positive control of PI-PLC treatment for D; PrP-WT is attached to the cell surface via a GPI anchor. Shown is Western blotting analysis of cell lysate (Cl) and supernatant medium (M) from SH/PrP cells, untreated (−) or treated (+) with PI-PLC. A decrease in the PrP level is apparent in the lysates. Unlike Shadoo, PrP can be detected in the medium after PI-PLC treatment. F, PrPΔCR expression level in SH/ΔCR+PrP is similar to PrP-WT and is lower than in SH/ΔCR+Sho cells. Comparison was carried out by Western blotting analysis of serial dilutions of extracts from SH/ΔCR+PrP (12, 6, and 3 μg of total protein; left) and of extracts from SH/ΔCR+Sho (24, 12, 6, and 3 μg of total protein; right) treated with PNGase F. Left and right panels are from the same x-ray film, corresponding to the left and right parts of the same membrane. G, schematic design of rSho-PrP recombinant polypeptide. The mSho(81–116) fragment is inserted into mPrP(23–231) between the 93rd and 94th amino acids in order to produce a bacterially expressed polypeptide that contains epitopes for both Sho and PrP antibodies to compare relative Shadoo and PrP protein levels. H, PrPΔCR and Shadoo expression levels of SH/ΔCR+Sho cells are similar. Comparison was carried out by Western blotting analysis of serial dilutions of extracts from SH/ΔCR+Sho cells, treated with PNGase F, (24, 12, 6, or 3 μg of total protein; left) and of rSho-PrP recombinant polypeptide (200, 100, and 50 ng of purified recombinant protein; right). Left and right panels are from the same x-ray film, corresponding to the left and right parts of the same membrane. B–F and H, *, a nonspecific band. Numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa. As a loading control, β-actin was used.
Cell Viability Assays
Cells stably transfected or transduced were seeded onto 96-well plates at 3 × 104 cells/well density (SH-SY5Y cells) or 1 × 104 cells/well density (HEK293 cells). After the attachment of the cells, the medium was changed to fresh medium containing serial dilutions of various drugs; Zeocin and G418 treatments were administered for 48 h, and puromycin treatment was administered for 24 h. The measurement of PrPΔCR-induced Zeocin hypersensitivity (Fig. 3, A and B) was carried out using MTT; all other cell viability assays were carried out using PrestoBlue reagent according to the manufacturer's protocol, briefly as follows.
FIGURE 3.

Coexpression of PrP-WT but not Sho diminishes PrPΔCR caused Zeocin hypersensitivity. A and B, PrPΔCR makes SH-SY5Y cells hypersensitive to Zeocin. Shown are cytotoxicity assays, using MTT reagent. A, representative experiment carried out at Zeocin concentrations between 0 and 100 μg/ml on various cells, as indicated, for 48 h. Values are means ± S.D. (error bars) of replicas in the individual experiment. B, bars show the means ± S.D. of cell viabilities measured at 6.25 μg/ml Zeocin concentration in n = 4 independent experiments. C and D, coexpression of PrP-WT but not Sho diminishes PrPΔCR-caused Zeocin hypersensitivity. Shown is a cytotoxicity assay, using PrestoBlue reagent. C, representative experiment carried out at Zeocin concentrations between 0 and 100 μg/ml Zeocin on various cells as indicated for 48 h. Values are means ± S.D. of replicas. D, bars show the means ± S.D. of cell viabilities measured at 6.25 μg/ml Zeocin concentration in n = 3 independent experiments. A–D, 100% is the absorbance (A and B) or fluorescence (C and D) value of untreated cells of each cell type. B and D, samples were compared with the leftmost values on each diagram (B, SH-SY5Y; D, SH/ΔCR); *, p = 0.04; ***, p < 0.001.
MTT Assay
After drug treatment, the medium was changed to PBS containing 0.5 μg/ml MTT, and plates were placed back into the cell culture incubator. After 4 h of incubation, MTT solution was carefully removed, and the converted dye was solubilized in acidic isopropyl alcohol (isopropyl alcohol plus 1 n HCl, 9:1), and the absorbance of the solution was measured at 560 nm.
PrestoBlue Assay
After the drug treatment, the medium was changed to PBS containing 5% PrestoBlue, and cells were placed back for 60 min into the CO2 incubator before measuring fluorescence with a PerkinElmer Enspire multimode plate reader (excitation, 555 nm; emission, 585 nm).
Analysis of cell proliferation was carried out using 96-well plates and the PrestoBlue assay. About 4 h after seeding, when cell attachment was confirmed by microscopy analysis, the fluorescence in 4 wells of each cell type was measured in order to be used as initial values. After every 24 h for 8 days, 4 wells of each cell were measured, and the measured fluorescence values were normalized to the initial values to estimate the change in the number of cells.
Detection of Histone 2AX Phosphorylation
SH-SY5Y cells expressing either Sho, PrPΔCR, or their respective controls, mCherry or EGFP, were seeded on 96-well plates at 1.5 × 104 cells/well density. 24 h after seeding, the cells were subjected to 0, 20, or 100 μg/ml Zeocin or 50 μm etoposide in normal culture medium for 60 min at 37 °C and then washed once with PBS and immunostained as described above.
Image acquisition was performed with an ImageXpress® Micro XLS high content screening system (Molecular Devices), using a Nikon ×10 plan fluor objective (numerical aperture = 0.3). Blue and far-red fluorescence signals of DAPI and γ-H2AX for nuclei were detected using emission filters of 447/60 nm and 692/40 nm, respectively, with 377/50- and 635/18-nm excitation filters, and 4–6 fields of view were imaged per well. For analysis, the cells were segmented on the basis of DAPI staining, and the percentage of γ-H2AX-positive nuclei was determined using the MetaXpress software. At least 4000 cells were analyzed per condition.
Image Analysis
Microscopy images were analyzed using the ImageJ version 1.48 software with Bio-Formats plugin. During image processing, the lookup tables were always linear and covered the full range of the data.
Statistics
Cell viability assays were done with five parallel samples for every condition. The number of surviving cells in the case of each drug concentration was normalized to the number of cells receiving no drug treatments. For statistical analysis, one concentration was chosen from each drug treatment: 6.25 μg/ml for Zeocin, 250 μg/ml for G418, and 1.6 μg/ml for puromycin in the case of SH-SY5Y cells and 50 μg/ml for Zeocin and 250 μg/ml for G418 in the case of HEK293 cells.
Statistical analysis (normality tests, Student's t tests, and one-way analyses of variance with two-tailed Dunnett's or Tukey's HSD post hoc tests) was carried out on data from at least three independent experiments with SPSS Statistics version 20 software. On plots, mean ± S.D. values are shown. p values are as follows: *, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ***, p < 0.001.
Results
Establishment of the Cell Culture Model Systems
In order to examine the effect of Shadoo expression on the toxic effect of PrPΔCR, we established a vector system that allows the effective use of both transient and stable expressions in cell cultures. Because of the lack of an appropriate anti-Sho antibody for immunocytochemistry and to avoid the adverse effects of tagging Shadoo (i.e. loss of Shadoo function), we used individual fluorescent proteins (EGFP or mCherry) whose expressions are tightly coupled to the expression of the target proteins. Thus, the successfully transfected cells transiently expressing the target protein could be identified and examined separately from the untransfected cell population by fluorescence microscopy. Alternatively, the cells with stable transgene integration could be selected by using fluorescence-activated cell sorting (FACS). In order to average out the potential positional effects of individual integrations on the outcome of the experiments, the stably transfected cell population was used without establishing individual clones. The tight coupling between the expression of the target and marker protein is achieved through two approaches: the use of either 1) the Sleeping Beauty transposon-based gene delivery system (SB) (36) or 2) a lentiviral approach (37).
SB ensures the collective insertion of the two expression cassettes between the transposon arms, resulting in over 95% coupling (data not shown). When a second transgene expression was required, we used lentivirus for the collective integration of the second transgene with the second fluorescent marker. In order to find sufficiently high Shadoo expression, two virus vectors containing expression cassettes in different orientations in respect to each other were tested. Viruses with a “tandem” orientation were chosen, because this resulted in a higher mSho expression (data not shown). The cells generated are listed in Table 2, and the topologies of the plasmid constructs are shown in Fig. 1.
TABLE 2.
Cells with stable transgene expression used in these studies
The given abbreviated names and the specifics for each type of transformant cell are listed.
| Cell culture name | Introduced transgenes | Parental cell name | Vector used |
|---|---|---|---|
| SH/GFP | EGFP | SH-SY5Y | pSB/GFP |
| SH/PrP | mPrP and EGFP | SH-SY5Y | pSB/PrP |
| SH/ΔCR | mPrPΔCR and EGFP | SH-SY5Y | pSB/ΔCR |
| SH/mCh | mCherry | SH-SY5Y | LV/mCh |
| SH/Sho | mSho and mCherry | SH-SY5Y | LV/Sho(R) |
| SH/ΔCR+mCh | mCherry | SH/ΔCR | LV/mCh |
| SH/ΔCR+PrP | mPrP and mCherry | SH/ΔCR | LV/PrP(R) |
| SH/ΔCR+Sho | mSho and mCherry | SH/ΔCR | LV/Sho(R) |
| SH/Sho+GFP | EGFP | SH/Sho | LV/GFP |
| SH/Sho+PrP | mPrP and EGFP | SH/Sho | LV/PrP(G) |
| HEK/mCh | mCherry | HEK293 | LV/mCh |
| HEK/Sho | mSho and mCherry | HEK293 | LV/Sho(R) |
| HEK/Sho+GFP | EGFP | HEK/Sho | LV/GFP |
| HEK/Sho+PrP | mPrP and GFP | HEK/Sho | LV/PrP(G) |
First, using SB, we established stable expressions of PrPΔCR, PrP-WT, and EGFP in Zpl2-1 immortalized hippocampal Prnp-knock-out cells (38) and in SH-SY5Y human neuroblastoma cells that usually exhibit no detectable endogenous PrP expression (20, 39).
Their expression is corroborated by immunocytochemistry and immunoblotting (Fig. 2, A and B). PNGase F (Fig. 2B) and PI-PLC treatments (Fig. 2C) confirmed that both wild type and mutant PrPs are properly processed, N-glycosylated, acquired a GPI anchor, and are localized on the cell surface.
FIGURE 2.

Analysis of the glycosylation, localization, and expression levels of prion proteins in SH/PrP-WT and SH/PrPΔCR cells. A, both PrP-WT and PrPΔCR are present on the cell surface, and their expressions are coupled with the marker EGFP. Shown are laser scanning confocal microscopy images. Nuclei (blue) and PrPs (red) are stained by DAPI and SAF32 anti-PrP antibody, respectively. Scale bar, 40 μm. B, both PrP-WT and PrPΔCR are complex-glycosylated. Shown is Western blotting analysis of extracts from various cells as indicated above the lanes, untreated (−) or treated (+) with PNGase F and visualized using SAF32 anti-prion antibody. The higher mobility of PrPΔCR caused by the deletion of the central region (aa 105–125) is more apparent after a PNGase F treatment. Endogenous PrP levels are below the detection limit. β-Actin was used as loading control (bottom). C, both PrP-WT and PrPΔCR are attached to the cell surface via a GPI anchor. Shown is Western blotting analysis of extracts (cell lysate) and supernatant medium (medium) from various cells as indicated above the lanes, untreated (−) or treated (+) with PI-PLC (top). β-Actin was used as loading control (bottom) in the case of cell lysates and to detect cell contamination in supernatant medium samples (note that PrP coming from cell contamination remained below the detection limit in the case of PI-PLC-untreated samples). A decrease in the PrP level is apparent in the lysates with a concurrent increase in the medium of PI-PLC-treated samples. B and C, numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa.
Cell Protective and Toxic Effects of the PrP Constructs
We were not able to detect any significant spontaneous toxicity associated with the expression of PrPΔCR in Zpl2-1 as tested by annexin staining using FACS.3 Several reports suggested that PrPC exerts a cytoprotective activity that prevents the death of neurons and other cells caused by toxic stimulus (39–44). However, we were unable to detect a significant anti-apoptotic effect of PrP-WT expression under the condition of serum deprivation in Zpl2-1 cells.3 These results are in line with a former report where no robust cytoprotective effect of PrP was found in the various models tested (45). Because Zpl2-1 cells repeatedly lost the expression of the transgenes during our work, thwarting progress, we used the SH-SY5Y cells for further studies.
PrPΔCR is reported to cause hypersensitivity to Zeocin- and G418-related antibiotics, which is eliminated by PrP-WT coexpression (29). The expression of PrPΔCR caused Zeocin hypersensitivity in SH-SY5Y cells (Fig. 3, A and B) that was diminished by the coexpression of PrP-WT (Fig. 3, C and D). These data are in agreement with those obtained using different cells, such as HEK, CHO, and mouse neural stem cells (29, 46–48).
Shadoo Expression Causes Drug Hypersensitivity
To assess whether Sho expression exerts a PrP-WT-like effect on these phenotypes, we introduced Sho into SH/ΔCR cells by lentivirus. The transduction itself did not have an adverse effect on the proliferation of the resulting SH/ΔCR+Sho cells (Fig. 4A). The plasma membrane localization is a prerequisite for PrPΔCR neurotoxicity and probably for the neuroprotective activities of PrP-WT (46, 47). PNGase F and PI-PLC treatment indicated that Sho expressed here, like PrP, is both complex-glycosylated (Fig. 4, B and C) and attached to the cell surface via a GPI anchor (Fig. 4, D and E). An expression level of PrP-WT that is identical to that of PrPΔCR (Fig. 4F) is sufficiently high in SH/ΔCR+PrP cells to effectively eliminate the drug hypersensitivity phenotype of PrPΔCR (Fig. 3, C and D). The expression level of Sho, as determined by Western blotting using an rPrP-Sho fusion polypeptide (Fig. 4G), is comparable with that of PrPΔCR in SH/ΔCR+Sho cells (Fig. 4H). However, contrary to expectations, Sho expression does not diminish PrPΔCR-induced Zeocin hypersensitivity when expressed in SH/ΔCR cells (Fig. 3, C and D).
More surprisingly, the Sho-expressing control SH-SY5Y cells (SH/Sho; Table 2), like SH/ΔCR, showed hypersensitivity to Zeocin and G418 but not to puromycin (Fig. 5, A–F) in a dose-dependent manner (Fig. 5, G–I). These hypersensitivities are also eliminated by the coexpression of PrP-WT with Sho (Fig. 6), as has been demonstrated for PrPΔCR (Fig. 3, C and D). These phenomena were also observed in HEK293 cells (see Table 2 for cell lines). HEK/Sho cells, which overexpress Shadoo protein (Fig. 7, A and B), are more sensitive to Zeocin (Fig. 7, C and D) or G418 (Fig. 7, E and F) than HEK/mCh cells, and the hypersensitivity to both drugs is eliminated by the co-expression of wild type PrP (Fig. 7, C–E).
FIGURE 5.

The overexpression of Shadoo, like PrPΔCR, sensitizes cells to Zeocin and G418. A–H, cytotoxicity assays, using PrestoBlue reagent. A and B, SH-SY5Y cells expressing either Shadoo (SH/Sho) or PrPΔCR (SH/ΔCR) are hypersensitive to Zeocin (48-h treatment). Of note, PrPΔCR expression in SH/ΔCR used in this experiment was lower than in the cells used for the experiments presented on Fig. 3 (data not shown). A, representative experiment carried out at Zeocin concentrations between 0 and 100 μg/ml on various cells as indicated. B, bars show the means ± S.D. (error bars) of cell viabilities measured at 6.25 μg/ml Zeocin concentration in n = 4 independent experiments. C and D, SH-SY5Y cells expressing either Shadoo (SH/Sho) or PrPΔCR (SH/ΔCR) are hypersensitive to G418 (48-h treatment). C, representative experiment carried out at G418 concentrations between 0 and 1000 μg/ml on various cells as indicated. D, bars show the means ± S.D. of cell viabilities measured at 250 μg/ml G418 concentration in n = 3 independent experiments. E and F, neither SH/Sho nor SH/ΔCR cells are hypersensitive to puromycin (24-h treatment). E, representative experiment carried out at puromycin concentrations between 0 and 2 μg/ml on various cells as indicated. F, bars show the means ± S.D. of cell viabilities measured at 1.6 μg/ml puromycin concentration in n = 6 independent experiments. G and H, Shadoo induces G418 hypersensitivity in a dose-dependent manner (48-h treatment). G, representative experiment carried out at G418 concentrations between 0 and 1000 μg/ml on SH/Sho-low, SH/Sho-medium, and SH/Sho-high cells. H, bars show the means ± S.D. of cell viabilities measured at 250 μg/ml G418 concentration in n = 3 independent experiments. a–h, 100% is the fluorescence value of untreated cells for each cell type. A, C, E, and G, values are means ± S.D. of corresponding replicas within the experiment. B, D, F, and H, for testing significance, values were compared with the control (i.e. leftmost values on each diagram) (SH/GFP (B and D) and SH-SY5Y (F and H)), and bars connected with brackets (H) were also compared using Tukey's honest significant difference post hoc test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. I, relative Shadoo protein levels in SH/Sho cells. Shown is Western blotting analysis of extracts from SH/Sho-low, SH/Sho-medium, and SH/Sho-high as well as the parental SH/Sho and SH-SY5Y cells. *, a nonspecific band. Numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa. β-Actin was used as a loading control.
FIGURE 6.
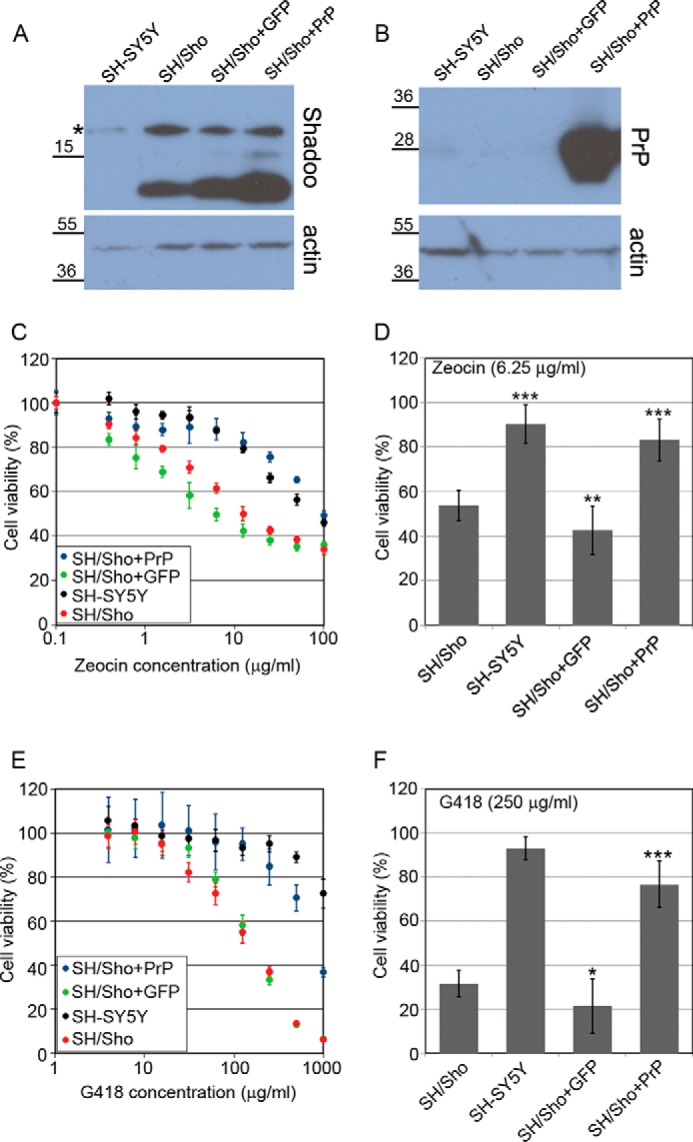
Coexpression of wild type PrP in SH/Sho cells eliminates both Zeocin and G418 hypersensitivity caused by Shadoo expression. A, SH/Sho+GFP or SH/Sho+PrP cells have slightly higher Shadoo expression than the parental SH/Sho cells. Shown is Western blotting analysis of extracts from various cells as indicated above the lanes. Samples were treated with PNGase F. B, PrP expression is detectable only in SH/Sho+PrP cells. Western blotting analysis of extracts from the same cells as in A. Endogenous PrP is below the detection limit. A and B, *, a nonspecific band. Numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa. β-Actin was used as a loading control. C–F, cytotoxicity assays, using PrestoBlue reagent. C and D, coexpression of PrP but not EGFP diminishes Zeocin hypersensitivity caused by Shadoo expression (48-h treatment). C, representative experiment carried out at Zeocin concentrations between 0 and 100 μg/ml on various cells as indicated. D, bars show the means ± S.D. (error bars) of cell viabilities measured at 6.25 μg/ml Zeocin concentration in n = 4 independent experiments. E and F, PrP coexpression diminishes G418 hypersensitivity caused by Shadoo expression (48-h treatment). E, representative experiment carried out at G418 concentrations between 0 and 1000 μg/ml on various cells, as indicated. F, bars show the means ± S.D. of cell viabilities measured at 250 μg/ml G418 concentration in n = 3 independent experiments. C–F, 100% is the fluorescence value of untreated controls of each cell line. C and E, values are means ± S.D. of corresponding replicas within the experiment. D and F, samples were compared with SH/Sho cells; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 7.
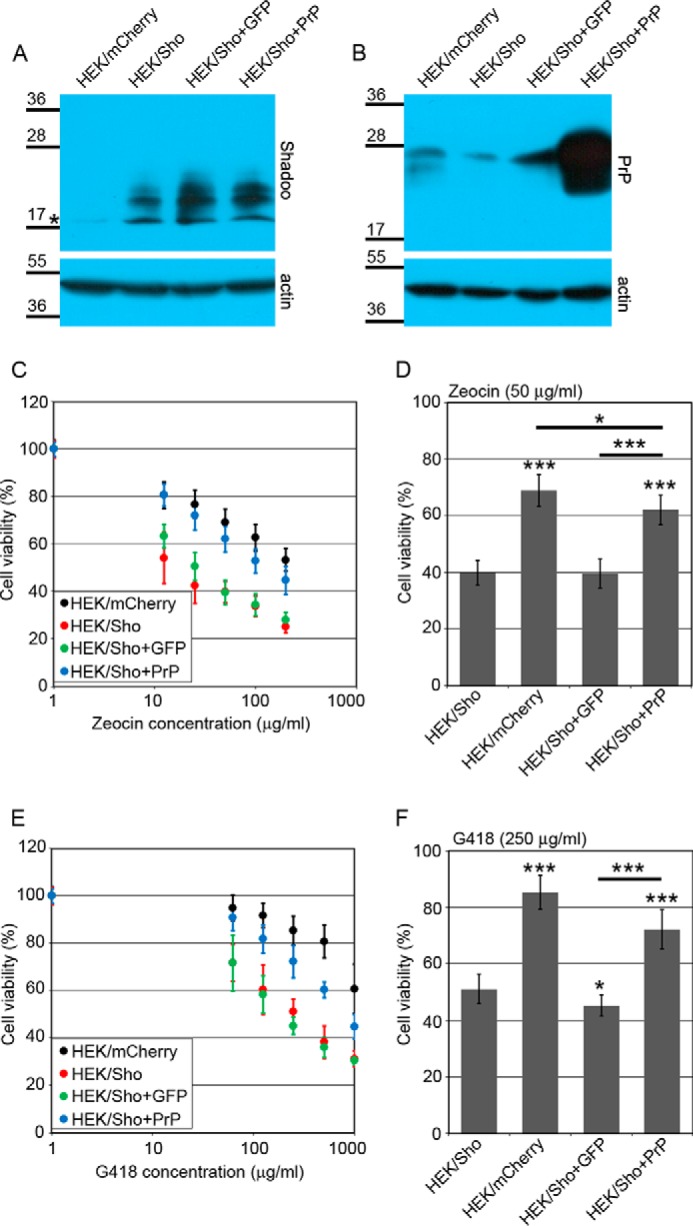
Shadoo expression causes Zeocin and G418 hypersensitivity in HEK293 cells that is eliminated by the coexpression of wild type PrP. A, HEK/Sho+GFP or HEK/Sho+PrP cells have slightly higher Shadoo expression than the parental HEK/Sho cells. Shown is Western blotting analysis of extracts from various cells as indicated above the lanes. *, a nonspecific band. B, PrP expression is detectable only in HEK/Sho+PrP cells. Shown is Western blotting analysis of extracts from the same cells as in A after PNGase F treatment. A and B, numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa. β-Actin was used as a loading control. C–F, cytotoxicity assays, using PrestoBlue reagent. C and D, Shadoo expression causes Zeocin hypersensitivity in HEK cells, which is diminished by the coexpression of PrP but not EGFP (48-h treatment). C, average of three independent experiments carried out at Zeocin concentrations between 0 and 200 μg/ml on various cells as indicated. D, bars show the means ± S.D. of cell viabilities measured at 50 μg/ml Zeocin concentration in n = 3 independent experiments. E and F, Shadoo expression causes G418 hypersensitivity in HEK cells that is diminished by the coexpression of PrP but not EGFP (48-h treatment). E, average of three independent experiments carried out at G418 concentrations between 0 and 1000 μg/ml on various cells as indicated. F, bars show the means ± S.D. (error bars) of cell viabilities measured at 250 μg/ml G418 concentration in n = 3 independent experiments. C–F, 100% is the fluorescence value of untreated controls of each cell line. C and E, values are means ± S.D. of three independent experiments. D and F, viabilities were compared using a Tukey post hoc test after one-way analysis of variance; asterisks above bars, difference when compared with HEK/Sho; brackets, significant difference between compared cells; *, p < 0.05; ***, p < 0.001.
Zeocin causes double strand breaks to DNA, triggering the recruitment and phosphorylation of histone 2AX at the break points (49). It has been shown that PrPΔCR causes an increased initial uptake of Zeocin, as assessed by histone 2AX immunocytochemistry (29, 49).
We observed an increased number of γ-H2AX-positive nuclei in SH/PrPΔCR cells compared with that seen in SH/mCh cells (Fig. 8, A and B), consistent with earlier reports (29). Similarly, a substantial increase was seen in the number of γ-H2AX-positive nuclei in Sho-expressing cells (Fig. 8, A and C); however, this effect was less pronounced despite the higher expression of Shadoo in SH/Sho cells as compared with that of PrPΔCR in SH/ΔCR cells (Fig. 8D). These results suggest that both Shadoo and PrPΔCR augment cellular uptake of Zeocin, although Shadoo might be less effective.
FIGURE 8.

Detection of histone 2AX phosphorylation caused by Zeocin treatment; Shadoo, like PrPΔCR, causes an elevated Zeocin-uptake in SH-SY5Y cells. A, representative high content screening images of SH/Sho, SH/ΔCR, and their respective control cells (SH/mCh and SH/GFP) stained with DAPI and an anti-γ-H2AX antibody after 60 min of 0, 20, or 100 μg/ml Zeocin treatment or SH/mCh cells that received either 50 μm etoposide treatment (positive control) or were left untreated and received only secondary antibody staining (staining control). Scale bar, 65 μm. B, the ratios of γ-H2AX-positive to total nuclei counted in SH/mCherry and SH/Sho cells, both untreated and treated with Zeocin (20 or 100 μg/ml for 60 min) in n = 3 independent experiments. C, the ratios of γ-H2AX-positive to total nuclei counted in SH/GFP and SH/ΔCR cells, both untreated and treated with Zeocin (20 or 100 μg/ml for 60 min) in n = 3 independent experiments. B and C, γ-H2AX positivity of cells that received the same treatment was compared with independent samples Student's t tests. **, p < 0.01. D, comparison of Shadoo and PrPΔCR expression in SH/Sho and SH/ΔCR cells using the rSho-PrP polypeptide. Shown is Western blotting analysis of serial dilutions made from PNGase F-treated samples of total cell lysates of SH/Sho and SH/ΔCR and from rSho-PrP polypeptide. *, a nonspecific band. β-Actin was used as loading control. Numbers and marks on the left indicate the positions of the corresponding molecular size markers in kDa. Error bars, S.D.
Discussion
One of the most challenging questions in prion biology is how the various forms of PrPs cause/contribute to the neurodegeneration seen in diseases such as Alzheimer disease and prion diseases in humans (familial, infectious, and sporadic forms), in ruminant, or even in model transgenic animals (7). Some transgenic mice expressing N-terminal deletion mutant forms of PrP on a PrP−/− background exhibit a lethal neurodegenerative phenotype that is dose-dependently suppressed by the coexpression of PrP-WT (14–16). This latter finding suggests that these deletion mutant PrPs subvert a normal functional activity of the prion protein and have long been studied with the expectations that these mechanisms might be the underlying cause of the neuronal loss apparent in some neurodegenerative disorders (11). Although these N-terminal deletion mutant PrPs do not cause any apparent toxic phenotype when expressed in immortalized cells, they cause hypersensitivity to a few cationic members of two classes of antibiotics, glycopeptides (Zeocin and bleomycin D1) and aminoglycosides (G418 and hygromycin) (29). Here, we found that the expression of Sho, like that of mutant PrPs, also makes SH-SY5Y and HEK293 cells hypersensitive to these two types of antibiotics and that the Sho-induced hypersensitivities are also diminished by the coexpression of PrP-WT.
These findings are surprising, because Sho is generally regarded as an analogue of PrP-WT, specifically an analogue of the N-terminal half of PrP, in paradigms where PrP exhibits a neuroprotective, cytoprotective activity (17, 20, 21). Here, not only does Sho not exhibit PrP-like cell protective activity; it actually mediates a toxic effect. This might reflect a role for the disordered N-terminal part of PrP in toxic phenotypes too (46, 48). Indeed, neurotoxic signals triggered by an interaction of PrP with certain anti-prion monoclonal antibodies were reported to critically depend on the flexible N-terminal part of PrP (50).
PrPΔCR and Sho Explore Identical Cellular Pathways to Induce Zeocin/Drug Hypersensitivity
Zeocin and G418 are unrelated in their chemical structure, cellular target, and mechanism of action. The specificity of how Sho renders cells hypersensitive to these two kinds of drugs but not to others (i.e. puromycin) (see Fig. 5, A–F) suggests that this phenotype caused by Sho involves identical pathways/initial interacting partners to the drug hypersensitivity induced by PrPΔCR (29, 46). This contention is further supported by the fact that PrP-WT coexpression non-variably eliminates these effects (29, 31, 51) (see Figs. 6 and 7) and by the increased initial Zeocin uptake observed with both Sho (see Fig. 8) and PrPΔCR (29) expression. This Sho-mediated hypersensitivity is especially intriguing if one considers that no extended sequence similarity exists between Shadoo and Doppel or between Shadoo and PrPΔCR. In the latter, the only short segment that would represent considerable sequence similarity between Sho and PrP is deleted.
Furthermore, whereas PrPΔCR and Doppel are not expressed physiologically in the CNS (16, 23), Shadoo is expressed (17). Thus, our results support a view that this drug hypersensitivity assay uncovers a pathway wherein the function of all three prion protein family members is preserved, suggesting that this pathway may also be responsible for the maintenance of a physiologically important process.
However, it is not clear whether the drug-sensitizing effects of Sho expression in immortalized cells demonstrated here operate in any of the prion-related pathological processes found in vivo, such as TSEs or Alzheimer disease.
Relation to Other Toxic Phenotypes Associated with Mutant or WT Prion Proteins
A handful of toxic phenotypes have been described involving wild type and mutant prion proteins, many of them being associated with the deletion of the hydrophobic domain or central region of PrP (7, 17, 20, 29, 31). One of the most interesting questions is how these toxic phenotypes seen in various systems relate to each other and to the neurotoxic effects seen in neurodegenerative diseases, such as TSE and Alzheimer disease. Our results offer a way to distinguish some of these pathways involved by the expression/coexpression of Sho.
Neurodegeneration in Mice Expressing Toxic Deletion Mutant Forms of PrP
The strongest toxicity that appears among mice expressing the various N-terminal deletion mutant PrPs is associated with PrPΔCR, conferring a neonatal lethal phenotype on a Prnp−/− background characterized by spontaneous degeneration of CGNs as well as white matter pathology in the brain and spinal cord (16).
Here, we found that Sho causes Zeocin/G418 hypersensitivity in the absence of PrP-WT. If the same cellular pathways are used by PrPΔCR to cause both the Zeocin/G418 hypersensitivity in cell model systems and CGN degeneration in mice, Sho-expressing Prnp-knock-out mice should also show some overt phenotypes. This could be tested by crossing Shadoo-overexpressing mice (52) with a Prnp-knock-out background. It is worth noting that Prnp-null mice that have endogenous Sho expression have no such apparent phenotype (53); however, the endogenous expression levels of Sho are thought to be much lower than those of PrP (see the AceView Web site), which might also contribute to the absence of a clear phenotype.
Inward Cationic Currents Induced by PrPΔCR Expression
Spontaneous ionic currents that may jeopardize the integrity of the plasma membrane are detected with whole cell patch clamping technique in a wide variety of cells of human, mouse, and insect origin (HEK; N2a; CGN, neuronal stem cells, and organotypic cerebellar slices from mice; Sf9 cells) that express PrP bearing ΔCR, Shmerling, or hydrophobic domain point mutations associated with familial TSEs (30, 31). This phenotype is also rescued by the coexpression of PrP-WT. It would be highly illuminating to learn whether Sho expression eliminates, induces, or is indifferent to this effect (31).
Without such data available for Sho, it is hard to envisage the possible molecular mechanism by which Sho exerts its sensitization effect observed here on SH-SY5Y and HEK293 cells. For PrPΔCR and related deletion mutants, the induction of inward currents in cultured cells (30) and the possible pore formation ability demonstrated on model membranes (54) point to one possible mechanism of sensitization. In this respect, the N-terminal region of PrP, especially the polybasic region 23–32, proved to be essential (46). Sho resembling the N-terminal domain of PrP also possesses a polybasic region, which precedes its hydrophobic domain. One could speculate that in a similar manner to the N-terminal tail of PrP, Sho could also in some circumstances exert sensitization through its basic region that may or may not involve pore formation and increased inward currents. The rescue effects of full-length PrP toward sensitization by both PrPΔCR and Sho indicate that the rescue process may involve similar molecular mechanisms. The N-terminal polybasic region of PrP proved to be critical for its neuroprotective effects as well, but a direct interaction of PrP with the deletion mutant PrPΔ32–134 in vivo did not take place in the process (55). Using a yeast two-hybrid system, Jiayu et al. (56) had shown that Sho can interact with the 108–126 region of PrP, involving its aa 61–67 region, and Ciric et al. (57) demonstrated that their interaction can interfere with the oligomerization of PrP. One can also speculate that this binding, although involving a different region of Sho, could silence the sensitizing effects of the basic region of Sho while not interfering with the N-terminal polybasic region of PrP, hence resulting in a protective effect of PrP. However, whether a direct binding of Sho and PrP takes place or not while rescuing the sensitization effects of Sho in our system is not known. The surprising and simultaneously intriguing findings presented here are remarkable for revealing a common cellular pathway where the function of all three members of the prion protein family seems to be preserved, suggesting that Sho might help discern some of the mysterious facets of prion biology.
Author Contributions
A. N. and E. W. designed the study, analyzed the results, and wrote the paper. A. N. designed pSB and pRRL plasmids encoding PrP or Shadoo protein constructs; constructed the pSB plasmids; generated stable cell lines; and performed MTT and PrestoBlue assays, immunocytochemistry, Western blotting analyses, and the H2AX detection experiments. P. B. constructed the plasmids encoding rSho-PrP peptide and plasmids for lentivirus generation and performed PrestoBlue assays. I. V. expressed and purified the rSho-PrP peptide and analyzed it with Western blotting. Z. H. set up and performed the H2AX detection experiments and analyzed the results. L. H. analyzed the results. E. F. contributed to stable cell line generation and immunoblotting, analyzed the results, and critically revised the paper. All authors critically read and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Katalin Német, Monika Bátkainé, and Áron Szepesi for the lentivirus generation and transductions and Dr. György Várady for the flow cytometry measurements.
This work was supported by Hungarian Scientific Research Fund Grant K-82090. The authors declare that they have no conflicts of interest with the content of this article.
A. Nyeste and E. Welker, unpublished data.
- PrP
- prion protein
- mPrP
- mouse PrP
- HD
- hydrophobic domain
- CR
- central region
- GPI
- glycosylphosphatidylinositol
- CGN
- cerebellar granular neuron
- TSE
- transmissible spongiform encephalopathy
- GdmCl
- guanidium chloride
- PNGase F
- protein:N-glycosidase F
- PI-PLC
- phosphatidylinositol-dependent phospholipase C
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SB
- Sleeping Beauty
- mSho
- mouse Sho
- rSho
- recombinant Sho
- aa
- amino acids
- EGFP
- enhanced GFP
- IF
- immunofluorescence washing
- H2AX
- histone 2AX.
References
- 1. Aguzzi A., and Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327 [DOI] [PubMed] [Google Scholar]
- 2. Ford M. J., Burton L. J., Morris R. J., and Hall S. M. (2002) Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 113, 177–192 [DOI] [PubMed] [Google Scholar]
- 3. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kellett K. A. B., and Hooper N. M. (2009) Prion protein and Alzheimer disease. Prion 3, 190–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nieznanski K., Choi J.-K., Chen S., Surewicz K., and Surewicz W. K. (2012) Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J. Biol. Chem. 287, 33104–33108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nieznanski K., Surewicz K., Chen S., Nieznanska H., and Surewicz W. K. (2014) Interaction between prion protein and Aβ amyloid fibrils revisited. ACS Chem. Neurosci. 5, 340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aguzzi A., Baumann F., and Bremer J. (2008) The prion's elusive reason for being. Annu. Rev. Neurosci. 31, 439–477 [DOI] [PubMed] [Google Scholar]
- 8. Le Pichon C. E., Valley M. T., Polymenidou M., Chesler A. T., Sagdullaev B. T., Aguzzi A., and Firestein S. (2009) Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat. Neurosci. 12, 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Criado J. R., Sánchez-Alavez M., Conti B., Giacchino J. L., Wills D. N., Henriksen S. J., Race R., Manson J. C., Chesebro B., and Oldstone M. B. (2005) Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol. Dis. 19, 255–265 [DOI] [PubMed] [Google Scholar]
- 10. Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A. D., Toyka K. V., Nave K.-A., Weis J., and Aguzzi A. (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318 [DOI] [PubMed] [Google Scholar]
- 11. Westergard L., Christensen H. M., and Harris D. A. (2007) The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim. Biophys. Acta 1772, 629–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Resenberger U. K., Winklhofer K. F., and Tatzelt J. (2011) Neuroprotective and neurotoxic signaling by the prion protein. Top. Curr. Chem. 305, 101–119 [DOI] [PubMed] [Google Scholar]
- 13. Barton K. A., and Caughey B. (2011) Is PrP the road to ruin? EMBO J. 30, 1882–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shmerling D., Hegyi I., Fischer M., Blättler T., Brandner S., Götz J., Rülicke T., Flechsig E., Cozzio A., von Mering C., Hangartner C., Aguzzi A., and Weissmann C. (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93, 203–214 [DOI] [PubMed] [Google Scholar]
- 15. Baumann F., Tolnay M., Brabeck C., Pahnke J., Kloz U., Niemann H. H., Heikenwalder M., Rülicke T., Bürkle A., and Aguzzi A. (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li A., Christensen H. M., Stewart L. R., Roth K. A., Chiesa R., and Harris D. A. (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 26, 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watts J. C., Drisaldi B., Ng V., Yang J., Strome B., Horne P., Sy M.-S., Yoong L., Young R., Mastrangelo P., Bergeron C., Fraser P. E., Carlson G. A., Mount H. T. J., Schmitt-Ulms G., and Westaway D. (2007) The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 26, 4038–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solomon I. H., Schepker J. A., and Harris D. A. (2010) Prion neurotoxicity: insights from prion protein mutants. Curr. Issues Mol. Biol. 12, 51–61 [PMC free article] [PubMed] [Google Scholar]
- 19. Radovanovic I., Braun N., Giger O. T., Mertz K., Miele G., Prinz M., Navarro B., and Aguzzi A. (2005) Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J. Neurosci. 25, 4879–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakthivelu V., Seidel R. P., Winklhofer K. F., and Tatzelt J. (2011) Conserved stress-protective activity between prion protein and Shadoo. J. Biol. Chem. 286, 8901–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watts J. C., and Westaway D. (2007) The prion protein family: diversity, rivalry, and dysfunction. Biochim. Biophys. Acta 1772, 654–672 [DOI] [PubMed] [Google Scholar]
- 22. Westaway D., Daude N., Wohlgemuth S., and Harrison P. (2011) The PrP-like proteins Shadoo and Doppel. Top. Curr. Chem. 305, 225–256 [DOI] [PubMed] [Google Scholar]
- 23. Moore R. C., Lee I. Y., Silverman G. L., Harrison P. M., Strome R., Heinrich C., Karunaratne A., Pasternak S. H., Chishti M. A., Liang Y., Mastrangelo P., Wang K., Smit A. F., Katamine S., Carlson G. A., Cohen F. E., Prusiner S. B., Melton D. W., Tremblay P., Hood L. E., and Westaway D. (1999) Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 292, 797–817 [DOI] [PubMed] [Google Scholar]
- 24. Rossi D., Cozzio A., Flechsig E., Klein M. A., Rülicke T., Aguzzi A., and Weissmann C. (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 20, 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sakaguchi S. (2007) Molecular biology of prion protein and its first homologous protein. J. Med. Invest. 54, 211–223 [DOI] [PubMed] [Google Scholar]
- 26. Didonna A., Sussman J., Benetti F., and Legname G. (2012) The role of Bax and caspase-3 in doppel-induced apoptosis of cerebellar granule cells. Prion 6, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Premzl M., and Gamulin V. (2007) Comparative genomic analysis of prion genes. BMC Genomics 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tóth E., Kulcsár P. I., Fodor E., Ayaydin F., Kalmár L., Borsy A. É., László L., and Welker E. (2013) The highly conserved, N-terminal (RXXX)8 motif of mouse Shadoo mediates nuclear accumulation. Biochim. Biophys. Acta 1833, 1199–1211 [DOI] [PubMed] [Google Scholar]
- 29. Massignan T., Stewart R. S., Biasini E., Solomon I. H., Bonetto V., Chiesa R., and Harris D. A. (2010) A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J. Biol. Chem. 285, 7752–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Solomon I. H., Huettner J. E., and Harris D. A. (2010) Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J. Biol. Chem. 285, 26719–26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Solomon I. H., Biasini E., and Harris D. A. (2012) Ion channels induced by the prion protein. Prion 6, 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Atarashi R., Sim V. L., Nishida N., Caughey B., and Katamine S. (2006) Prion strain-dependent differences in conversion of mutant prion proteins in cell culture. J. Virol. 80, 7854–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mátés L., Chuah M. K. L., Belay E., Jerchow B., Manoj N., Acosta-Sanchez A., Grzela D. P., Schmitt A., Becker K., Matrai J., Ma L., Samara-Kuko E., Gysemans C., Pryputniewicz D., Miskey C., Fletcher B., VandenDriessche T., Ivics Z., and Izsvák Z. (2009) Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 41, 753–761 [DOI] [PubMed] [Google Scholar]
- 34. Kolacsek O., Erdei Z., Apáti A., Sándor S., Izsvák Z., Ivics Z., Sarkadi B., and Orbán T. I. (2014) Excision efficiency is not strongly coupled to transgenic rate: cell type dependent transposition efficiency of Sleeping Beauty and piggyBac DNA transposons. Hum. Gene Ther. Methods 25, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tátrai P., Sági B., Szigeti A., Szepesi A., Szabó I., Bősze S., Kristóf Z., Markó K., Szakács G., Urbán I., Mező G., Uher F., and Német K. (2013) A novel cyclic RGD-containing peptide polymer improves serum-free adhesion of adipose tissue-derived mesenchymal stem cells to bone implant surfaces. J. Mater. Sci. Mater. Med. 24, 479–488 [DOI] [PubMed] [Google Scholar]
- 36. Izsvák Z., and Ivics Z. (2004) Sleeping beauty transposition: biology and applications for molecular therapy. Mol. Ther. 9, 147–156 [DOI] [PubMed] [Google Scholar]
- 37. Barde I., Salmon P., and Trono D. (2010) Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. 10.1002/0471142301.ns0421s37 [DOI] [PubMed] [Google Scholar]
- 38. Kim B.-H., Kim J.-I., Choi E.-K., Carp R. I., and Kim Y.-S. (2005) A neuronal cell line that does not express either prion or doppel proteins. Neuroreport 16, 425–429 [DOI] [PubMed] [Google Scholar]
- 39. Rambold A. S., Müller V., Ron U., Ben-Tal N., Winklhofer K. F., and Tatzelt J. (2008) Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 27, 1974–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. White A. R., Collins S. J., Maher F., Jobling M. F., Stewart L. R., Thyer J. M., Beyreuther K., Masters C. L., and Cappai R. (1999) Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 155, 1723–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fukuuchi T., Doh-ura K., Yoshihara S., and Ohta S. (2006) Metal complexes with superoxide dismutase-like activity as candidates for anti-prion drug. Bioorganic Med. Chem. Lett. 16, 5982–5987 [DOI] [PubMed] [Google Scholar]
- 42. Oh J.-M., Shin H.-Y., Park S.-J., Kim B.-H., Choi J.-K., Choi E.-K., Carp R. I., and Kim Y.-S. (2008) The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol. Cell. Neurosci. 39, 238–247 [DOI] [PubMed] [Google Scholar]
- 43. Roucou X., Gains M., and LeBlanc A. C. (2004) Neuroprotective functions of prion protein. J. Neurosci. Res. 75, 153–161 [DOI] [PubMed] [Google Scholar]
- 44. Kim B.-H., Lee H.-G., Choi J.-K., Kim J.-I., Choi E.-K., Carp R. I., and Kim Y.-S. (2004) The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res. Mol. Brain Res. 124, 40–50 [DOI] [PubMed] [Google Scholar]
- 45. Christensen H. M., and Harris D. A. (2008) Prion protein lacks robust cytoprotective activity in cultured cells. Mol. Neurodegener. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Solomon I. H., Khatri N., Biasini E., Massignan T., Huettner J. E., and Harris D. A. (2011) An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 286, 14724–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biasini E., Turnbaugh J. A., Massignan T., Veglianese P., Forloni G., Bonetto V., Chiesa R., and Harris D. A. (2012) The toxicity of a mutant prion protein is cell-autonomous, and can be suppressed by wild-type prion protein on adjacent cells. PLoS One 10.1371/journal.pone.0033472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Westergard L., Turnbaugh J. A., and Harris D. A. (2011) A nine amino acid domain is essential for mutant prion protein toxicity. J. Neurosci. 31, 14005–14017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Celeste A., Fernandez-Capetillo O., Kruhlak M. J., Pilch D. R., Staudt D. W., Lee A., Bonner R. F., Bonner W. M., and Nussenzweig A. (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5, 675–679 [DOI] [PubMed] [Google Scholar]
- 50. Sonati T., Reimann R. R., Falsig J., Baral P. K., O'Connor T., Hornemann S., Yaganoglu S., Li B., Herrmann U. S., Wieland B., Swayampakula M., Rahman M. H., Das D., Kav N., Riek R., Liberski P. P., James M. N. G., and Aguzzi A. (2013) The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106 [DOI] [PubMed] [Google Scholar]
- 51. Biasini E., Unterberger U., Solomon I. H., Massignan T., Senatore A., Bian H., Voigtlaender T., Bowman F. P., Bonetto V., Chiesa R., Luebke J., Toselli P., and Harris D. A. (2013) A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 33, 2408–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Watts J. C., Stöhr J., Bhardwaj S., Wille H., Oehler A., Dearmond S. J., Giles K., and Prusiner S. B. (2011) Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog. 7, e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steele A. D., Lindquist S., and Aguzzi A. (2007) The prion protein knockout mouse: a phenotype under challenge. Prion 1, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chu N. K., Shabbir W., Bove-Fenderson E., Araman C., Lemmens-Gruber R., Harris D. A., and Becker C. F. W. (2014) A C-terminal membrane anchor affects the interactions of prion proteins with lipid membranes. J. Biol. Chem. 289, 30144–30160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Turnbaugh J. A., Westergard L., Unterberger U., Biasini E., and Harris D. A. (2011) The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One 6, e25675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiayu W., Zhu H., Ming X., Xiong W., Songbo W., Bocui S., Wensen L., Jiping L., Keying M., Zhongyi L., and Hongwei G. (2010) Mapping the interaction site of prion protein and Sho. Mol. Biol. Rep. 37, 2295–2300 [DOI] [PubMed] [Google Scholar]
- 57. Ciric D., Richard C.-A., Moudjou M., Chapuis J., Sibille P., Daude N., Westaway D., Adrover M., Béringue V., Martin D., and Rezaei H. (2015) Interaction between Shadoo and PrP affects the PrP folding pathway. J. Virol. 89, 6287–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Daude N., Wohlgemuth S., Brown R., Pitstick R., Gapeshina H., Yang J., Carlson G. A., and Westaway D. (2012) Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrP(C)-deficiency. Proc. Natl. Acad. Sci. U.S.A. 109, 9035–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]