

Abstract
Mutations in the renal specific Na-K-2Cl co-transporter (NKCC2) lead to type I Bartter syndrome, a life-threatening kidney disease featuring arterial hypotension along with electrolyte abnormalities. We have previously shown that NKCC2 and its disease-causing mutants are subject to regulation by endoplasmic reticulum-associated degradation (ERAD). The aim of the present study was to identify the protein partners specifically involved in ERAD of NKCC2. To this end, we screened a kidney cDNA library through a yeast two-hybrid assay using NKCC2 C terminus as bait. We identified OS9 (amplified in osteosarcomas) as a novel and specific binding partner of NKCC2. Co-immunoprecipitation assays in renal cells revealed that OS9 association involves mainly the immature form of NKCC2. Accordingly, immunocytochemistry analysis showed that NKCC2 and OS9 co-localize at the endoplasmic reticulum. In cells overexpressing OS9, total cellular NKCC2 protein levels were markedly decreased, an effect blocked by the proteasome inhibitor MG132. Pulse-chase and cycloheximide-chase assays demonstrated that the marked reduction in the co-transporter protein levels was essentially due to increased protein degradation of the immature form of NKCC2. Conversely, knockdown of OS9 by small interfering RNA increased NKCC2 expression by increasing the co-transporter stability. Inactivation of the mannose 6-phosphate receptor homology domain of OS9 had no effect on its action on NKCC2. In contrast, mutations of NKCC2 N-glycosylation sites abolished the effects of OS9, indicating that OS9-induced protein degradation is N-glycan-dependent. In summary, our results demonstrate the presence of an OS9-mediated ERAD pathway in renal cells that degrades immature NKCC2 proteins. The identification and selective modulation of ERAD components specific to NKCC2 and its disease-causing mutants might provide novel therapeutic strategies for the treatment of type I Bartter syndrome.
Keywords: endoplasmic reticulum-associated protein degradation (ERAD), hypertension, intracellular trafficking, kidney, membrane trafficking, Na-K-Cl co-transporter (NKCC), sodium transport, hypertension
Introduction
The thick ascending limb of loop of Henle (TAL)3 of the kidney is responsible for absorbing 20–30% of the filtered load of NaCl (1, 2). Given that the reabsorptive capacity of downstream portions of the nephron is limited, inhibition of TAL transport capacity results in marked natriuresis and diuresis, making specific inhibitors of NaCl transport in TAL cells such as furosemide or bumetanide the most potent class of all diuretics (3). The apically located Na-K-2Cl co-transporter (NKCC2) is the pacemaker of TAL sodium chloride reabsorption (2). Hence, the activity of NKCC2 is a key determinant of final urinary salt excretion, consequently influencing long term blood pressure levels (2). This is of particular interest because changes in NKCC2 expression can be caused by several conditions such as high salt intake (4), diabetes mellitus (5), obesity (6), and aging (7). Inherited variation in the activity of NKCC2 or its regulators alters blood pressure in humans (8). Indeed, loss-of-function mutations in the NKCC2 gene, SLC12A1, cause type I Bartter syndrome (BS1), a life-threatening disease featuring arterial hypotension along with electrolyte abnormalities (2). Conversely, enhanced activity of NKCC2 has been linked to salt-sensitive hypertension (2, 8, 9). More recently, it was shown that carriers of rare NKCC2 mutations are protected against the development of arterial hypertension, further supporting the notion that factors governing the activity of NKCC2 are key determinants of essential hypertension in the general population (10–12). Intriguingly, despite the importance of NKCC2 in the regulation of blood pressure and the pathogenesis of Bartter syndrome, the molecular mechanisms underlying the trafficking, targeting, and turnover of NKCC2 remain largely unknown.
NKCC2 belongs to the superfamily of electroneutral cation-coupled chloride co-transporters, which also contains the Na-Cl and K-Cl co-transporters (13). Similar to nearly all membrane proteins, the preparation for appropriate trafficking starts as the co-transporter protein is inserted into the endoplasmic reticulum (ER) (14–16). The ER acts therefore as an important determinant of the amount of protein that reaches the plasma membrane (14, 15). The ER represents an important quality control mechanism for newly synthesized proteins given that it is the site where membrane and secretory proteins fold, and only properly folded proteins usually exit the ER (17). Incompletely folded proteins may be retained in the ER, and if folding cannot be achieved, they may form aggregates or be targeted for ER-associated protein degradation (ERAD) (18, 19). Based on their size and complex topologies, even the wild-type forms of integral membrane proteins such as cation-coupled chloride co-transporters are expected to encounter a significant number of hurdles during synthesis (19). The first integral membrane mammalian protein to be characterized as an ERAD substrate was the cystic fibrosis transmembrane conductance regulator (CFTR) (20, 21). CFTR folding and maturation in the ER is an inefficient, temperature-sensitive process as illustrated by the fact that 80% of wild-type CFTR is degraded via ERAD (20, 21). Likewise, the epithelial sodium channel (ENaC) assembles inefficiently after its insertion into the ER; a substantial portion of its subunits is targeted for ERAD (22, 23). The ERAD pathway is a multistep process that requires substrate recognition, retrotranslocation, ubiquitination, and proteolysis of aberrant proteins (18, 19). Generally, the selection of misfolded proteins requires substrate-specific interactions with molecular chaperones (18, 19). For instance, the ERAD of CFTR is a complex process requiring several chaperones such as members of the heat shock protein 70 (Hsp70) and 40 (Hsp40) protein family, Hsp70-interacting protein (CHIP), and derlin 1 (24). Similar to ENaC (22, 23) and CFTR (20, 21), we previously demonstrated that NKCC2 is a substrate for the ERAD quality control system (25, 26). Most importantly, we found evidence that the maturation of NKCC2 is also a relatively slow and inefficient process and suggested that export from the ER constitutes the limiting step in the maturation and cell surface expression of the co-transporter (26). Accordingly, the majority of newly synthesized NKCC2 proteins is trapped in the ER and destined for ERAD (25, 26). Despite this, our knowledge of the molecular mechanisms underlying the ER quality control of NKCC2 proteins is nil. More specifically, the identities of the protein partners of NKCC2 that are involved in the ERAD of the co-transporter are unknown. Accordingly, identifying proteins that specifically interact with NKCC2 at the ER should help to begin to define the pathway by which NKCC2 is subject to quality control. In this report, we describe a novel protein-protein interaction between the C-terminal tail of NKCC2 and OS9, a ubiquitous protein originally identified as being amplified in certain osteosarcomas (27). Previous studies have implicated a role for OS9 in ER quality control (28–31) and ER-to-Golgi transport (32–35). Here, we show that OS9 interacts with the ER-resident form of NKCC2 and promotes its degradation by the proteasome pathway, revealing therefore a new molecular pathway in the regulation of the co-transporter. Consequently, these findings may open up new avenues in studying the regulation of the ER-associated degradation of cation-coupled chloride co-transporters in general and in particular of renal Na-Cl co-transporters, proteins that are necessary for normal blood pressure homeostasis.
Experimental Procedures
Materials
All chemicals were obtained from Sigma unless otherwise noted. Penicillin and streptomycin were from Invitrogen. Subclonings were carried out with the following vectors: 1) pGKT7 (Clontech), 2) pCMV-Myc (Clontech), 3) pEGFP-C2 (Clontech), and 4) pcDNA3.1/V5-His-TOPO (Invitrogen).
Yeast Two-hybrid Assay
Yeast two-hybrid screening was performed as described previously in detail (29). Briefly, the cDNA fragment encoding the distal region of the NKCC2 C terminus (last 195 amino acids) was cloned in-frame with the GAL4 DNA-binding domain in pGBKT7-BD and transformed into the yeast strain AH109. AH109 expressing the bait was then mated with the Y187 yeast strain pretransformed with a human kidney cDNA library constructed in the pACT2-AD vector. Mated yeast cells were first grown on low stringency selection plates (−Leu, −Trp, −His) and then on high stringency selection plates (−Leu, −Trp, −His, −Ade). Colonies were tested for β-galactosidase activity, and DNA from positive clones encoding the putative interacting proteins was isolated from yeast cells using the RPM yeast plasmid isolation kit (BIO 101 Systems). Prey plasmids were rescued by transformation into DH5α bacteria (Invitrogen) and isolated using a Qiagen kit. Insert sizes were checked by BglII digestion. cDNA plasmids were then sequenced and assessed using the BLAST program.
Plasmid Construction and Site-Direct Mutagenesis
The cDNAs encoding mouse NKCC2 and Na-Cl co-transporter (NCC) were fused at the N-terminal end of Myc (Myc-NKCC2 and Myc-NCC) or EGFP (EGFP-NKCC2) using pCMV-Myc and pEGFP-C2 vectors, respectively. The mouse pcDNA3 plasmid encoding mouse OS9.1 was a kind gift from L. Litovchick (Dana-Farber Cancer Institute, Boston, MA). The mouse OS9 coding sequence was subcloned into the mammalian expression pcDNA3.1/V5 vector. Alanine, glutamine, or asparagine substitutions were introduced into the OS-V5 construct at Tyr-120 (Y120A), Gln-130 (Q130E), Arg-188 (R188A), Glu-212 (E212N), and Tyr-212 (Y212A) using the QuikChange mutagenesis system (Stratagene). All mutations were confirmed by sequencing.
Cell Culture
Opossum kidney (OKP) cells were grown in DMEM complemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin, and 100 units/ml streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. Human embryonic kidney (HEK) 293 cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For DNA transfection, cells were grown to 60–70% confluence on plastic culture dishes and then transiently transfected for 5 h with plasmids using a Lipofectamine Plus kit according to the manufacturer's instructions (Invitrogen). For protein degradation assays, transiently transfected cells were treated with MG132 (10 μm) or leupeptin (100 μm) overnight prior to cell lysis.
Measurement of Intracellular pH and Na-K-2Cl Co-transporter Activity
Measurement of cytoplasmic pH (pHi) was accomplished in cells grown to confluence on coverslips using the intracellularly trapped pH-sensitive dye 2′,7′-bis(2-carboxyethyl)-5-(and-6)-carboxyfluorescein as described earlier (25, 26, 36, 37). Likewise, Na-K-2Cl co-transport activity was measured as bumetanide-sensitive NH4 influx as described previously (25, 26, 36, 37). Briefly, cells were first bathed at 37 °C in a CO2-free Hepes/Tris-buffered medium to measure baseline pHi. 20 mm NH4Cl was then added to the medium, causing a very rapid initial cellular alkalinization followed by a pHi recovery. The initial rate of intracellular pH recovery (dpHi/dt) was measured over the first 20 s of recordings as reported earlier (36, 38). The dpHi caused by NH4Cl addition was used to calculate the cell buffer capacity, which was not different between the studied groups (data not shown). Hence, the Na-K-2Cl co-transporter transport activity is expressed as dpHi/dt.
Protein Preparation, Immunoblotting, and Immunoprecipitation
Forty-eight hours post-transfection, cells were washed with cold PBS and lysed in 0.2 or 0.5 ml of lysis buffer (120 mm Tris/Hepes, pH 7.4, 150 mm NaCl, 5 mm EDTA, 3 mm KCl, 1% (v/v) Triton X-100) containing protease inhibitors (Complete, Roche Diagnostics catalog number 1697498). Samples were harvested and centrifuged at 16,000 rpm for 15 min at 4 °C. Protein expression levels were assessed after normalizing and loading equal amounts of total protein for 7.5% SDS-PAGE separation and immunoblotting with the antibodies of interest. For immunoprecipitation, cells were solubilized with lysis buffer containing 0.4 m NaCl, 0.5 mm EGTA, 1.5 mm MgCl2, 10 mm Hepes, pH 7.9, 5% (v/v) glycerol, 0.5% (v/v) Nonidet P-40), and protease inhibitors (Complete). Immunoprecipitation was carried out using the primary antibody of interest and affinity purification using protein G-agarose beads (Dynal). After incubation with protein G-agarose beads for 1 h at room temperature, the immunocomplex was washed three times in PBS (Invitrogen). The protein samples were boiled in loading buffer, run on gradient 7.5% SDS-polyacrylamide gels, and probed with primary antibodies of interest and horseradish peroxidase-conjugated secondary antibody according to standard procedures. Proteins were visualized by enhanced chemiluminescence detection (PerkinElmer Life Sciences) according to the manufacturer's instructions.
Biotinylation
Cells were placed on ice and rinsed twice with a cold rinsing solution containing PBS+, pH 8.0, containing 1 mm MgCl2, and 0.1 mm CaCl2. Cells were then gently agitated at 4 °C for 1 h in PBS+ in containing 1 mg/ml NHS-biotin. Biotinylation was stopped by incubation with PBS supplemented with 100 mm glycine. Cells were then washed three times in PBS supplemented with 1 mm MgCl2 and 0.1 mm CaCl2. Washed cells were lysed for 1 h at 4 °C in solubilizing buffer (150 mm NaCl, 5 mm EDTA, 3 mm KCl, 120 mm Tris/Hepes, pH 7.4, 1% (v/v) Triton X-100) containing protease inhibitors (Complete). After quantification and normalization, an aliquot of the total cell extract was taken from each sample to provide a measure of total NKCC2 expression, and the rest of the cell lysates were incubated with NeutrAvidin-agarose beads (Thermo Fisher) overnight at 4 °C. After overnight incubation, samples were centrifuged at 16,000 rpm for 5 min, and the supernatant (the intracellular fraction) was removed. Avidin beads were then washed with solubilizing buffer and centrifuged for 7 min at 16,000 rpm seven times. Pellets were incubated in solubilizing buffer and denaturing buffer for 10 min at 95 °C and stored at −20 °C. Each fraction was subjected to SDS-PAGE and Western blotting analysis
Immunocytochemistry
48 h post-transfection, confluent cells were washed twice with PBS2+, pH 8, containing 1 mm MgCl2 and 0.1 mm CaCl2. Cells were then fixed with 2% paraformaldehyde in PBS for 20 min at room temperature, incubated with 50 mm NH4Cl, permeabilized with 0.1% Triton X-100 for 1 min, and incubated with Dako antibody diluent with background-reducing components for 30 min to block nonspecific antibody binding. Fixed cells were incubated for 1 h at room temperature with the primary antibodies at appropriate dilution in Dako antibody diluent. To assess protein expression at the cell surface, confluent cells were first incubated at 4 °C for 1 h in PBS2+ containing 1 mg/ml NHS-biotin. Cells were rinsed three times in rinsing solution with 100 mm glycine and reincubated at 4 °C in the same solution for 10 min. After washing with PBS2+, the monolayers were fixed and stained for cell surface biotin (avidin-Cy2; green). Mouse anti-Myc and anti-V5 antibodies were visualized with Texas Red-coupled secondary antibodies. Rabbit anti-calnexin was visualized with FITC-coupled IgG antibodies. Cells were then washed with PBS and mounted with Vectashield.
Pulse-Chase and Cycloheximide-Chase Assays
For pulse-chase assays, OKP and HEK 293 cells transiently transfected with NKCC2 plasmid cDNAs were incubated in cysteine- and methionine-free DMEM starvation medium for 1 h. Starvation medium was removed and replaced with DMEM labeling medium containing [35S]methionine/cysteine labeling mixture. After 1 h, cells were rinsed three times with PBS and another three times with normal growth medium and returned to normal growth medium for the duration of chase to the specified time points. Cells were washed twice with ice-cold PBS and incubated on ice for 1 h in lysis buffer with a mixture of protease inhibitors after which solubilized extracts were collected for immunoprecipitation. Proteins were immunoprecipitated with monoclonal anti-Myc antibody, resolved by SDS-PAGE, blotted onto nitrocellulose, and revealed by autoradiography. For cycloheximide-chase assays, cycloheximide was added at a concentration of 100 μm to OKP or HEK cells 12–14 h post-transfection with NKCC2 plasmids. Cell lysates were then collected at 0, 1, 2, and 4 h and analyzed by quantitative immunoblotting.
Small Interfering RNA (siRNA) Knockdown
The siRNAs for control and OS9 siRNAs were purchased from Dharmacon as ON-TARGETplus SMARTpools (L-010811-01-0005) and Santa Cruz Biotechnology siRNAs (sc-96230). HEK cells were first transfected with control or specific siRNA with Lipofectamine RNAiMAX (Invitrogen) using the manufacturer's specifications. 1 day after siRNA transfection, cells were transfected with NKCC2 plasmids. 24–48 h after NKCC2 transfection, cell lysates were analyzed for each protein using the indicated antibodies.
Statistical Analyses
Results are expressed as mean ± S.E. Differences between means were evaluated using paired or unpaired t test or analysis of variance as appropriate. p < 0.05 was considered statistically significant.
Results
Yeast Two-hybrid Screen Identifies OS9 as a Novel NKCC2-binding Protein
As described in previous reports, to identify novel NKCC2-interacting proteins, we used a yeast two-hybrid system to screen a human kidney cDNA expression library using a series of bait fragments spanning the predicted cytoplasmic C terminus (residues 661–1095) of murine NKCC2 named C1-term (residues 661–768), C2-term (residues 741–909), and C3-term (residues 898–1095) (36, 37). We previously identified two NKCC2 binding partners, aldolase B (36) and SCAMP2 (37), that specifically interact with the proximal region of NKCC2 C terminus (residues 661–768). In this report, we describe results obtained from the distal region of NKCC2 C terminus (residues 898–1095, C3-term). 29 positive clones were selected by activation of three reporter genes, ADE2, HIS3, and MEL1. Among these positives clones, five matched the sequence originating from the same cDNA sequence. The protein encoded by this cDNA was originally identified as amplified in osteosarcomas and termed OS9 (27). Three splicing isoforms of human OS9 were initially described (39). The longest isoform, isoform 1, contains 667 amino acids; isoform 2 lacks amino acids 535–589, whereas isoform 3 lacks amino acids 456–470 and 535–589. All five identified clones code for the C-terminal domain of OS9 isoform-1 starting at residue 358. The interaction between OS9 and NKCC2 C3-term could be easily confirmed by retransforming the cloned cDNA into yeast using constructs issued from the yeast two-hybrid screening as judged by growth of the AH109 reporter strain on selection medium (Fig. 1A). To check for the specificity of the interaction between OS9 and C3-term, AH109 yeast cells were co-transformed with OS9 and NKCC2 C1-term (residues 661–768). In contrast to aldolase B (36) and SCAMP2 (37), no growth was observed when AH109 yeast cells were transformed with OS9 and the proximal region of NKCC2 C terminus (Fig. 1A), indicating that the co-transporter interaction with OS9 is specific to the distal region of NKCC2 C terminus.
FIGURE 1.
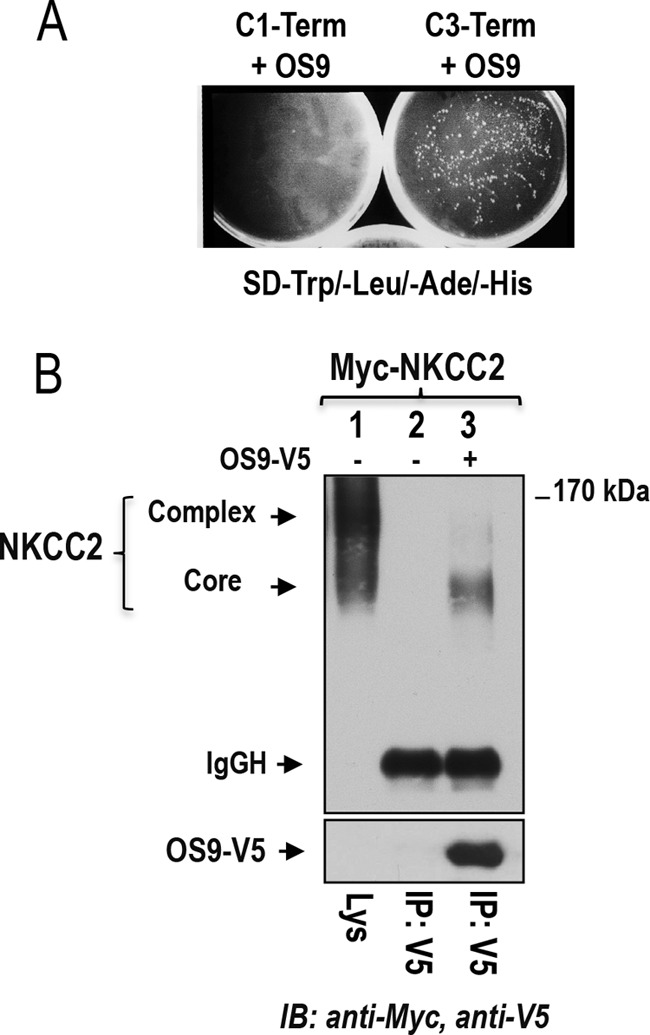
Identification of OS9 as a novel NKCC2-interacting protein. A, OS9 interacts specifically with the distal region of NKCC2 C terminus in yeast. A yeast two-hybrid assay was performed using the Matchmaker system as described under “Experimental Procedures.” The experiment confirmed the interaction of OS9 with the distal region of NKCC2 C terminus 5 (C3-term) as judged by growth of the AH109 reporter strain on selection medium (−Trp, −Leu, −His, −Ade). In contrast, no growth was observed when AH109 was transformed with OS9 and C1-term. B, NKCC2 interacts in vivo with OS9 in OKP cells. OKP cells transiently expressing Myc-NKCC2 singly or in combination with OS9 were immunoprecipitated (IP) with anti-V5 anti-body (lanes 2 and 3). 5% of total cell lysate (Lys) was resolved as a positive control. NKCC-2 co-immunoprecipitated with OS9 was detected by immunoblotting (IB) using anti-Myc (lane 3). The positions of the core glycosylated (immature) and complex glycosylated (mature) proteins of NKCC2 are indicated. The interaction of NKCC2 with OS9 involves mainly the core glycosylated, immature form of the co-transporter. SD, synthetic defined medium; IgGH, the heavy chain of IgG.
OS92 Interacts with NKCC2 in Renal Cells
The above data clearly indicate that OS9 interacts specifically with the distal region of NKCC2 C terminus in yeast. To show that the interaction between OS9 and NKCC2 also occurs in mammalian cells, Myc-NKCC2 construct was transiently expressed either singly or in combination with OS9-V5 in OKP cells. Cell lysates were incubated with anti-V5 or anti-Myc antibody, and the resultant immunoprecipitates were resolved by SDS-PAGE and immunoblotting. In agreement with our previous findings (25, 26, 36, 37), when NKCC2 is exogenously expressed in renal cultured cells, both the core (immature, ER-resident) and complex glycosylated (mature) forms are observed with the latter being more abundant. As shown in Fig. 1B, lane 3, immunoprecipitation of OS9 followed by immunoblotting for NKCC2 revealed robust co-immunoprecipitation of the two proteins as illustrated by the intensity of the bands corresponding to NKCC2 proteins. Interestingly, the interaction involves mainly the immature, ER-resident form of NKCC2 despite the predominant expression of mature, glycosylated NKCC2 in these cells. Moreover, the interaction appears to be specific because Myc-NKCC2 protein was not detected in control experiments in which OKP cells were not co-transfected with OS9-V5 (lane 2). In sum, these findings clearly indicate that the interaction of NKCC2 with OS9 is not an artifact of the yeast two-hybrid system and that the two proteins really associate in vivo. Most importantly, they demonstrate that this interaction involves mainly the core glycosylated form of the co-transporter.
OS9 Also Interacts with the NCC
The sequence of NKCC2 C terminus protein shares considerable homology with other members of the sodium-dependent chloride transporter family, namely the ubiquitous Na-K-2Cl co-transporter isoform NKCC1 and the related kidney-specific electroneutral NCC (2), suggesting that they may be have several common binding proteins. In support of this notion, we previously showed that SCAMP2 interacts with NKCC2 and with NCC (37). Hence, to determine whether OS9 also interacts with NCC, we performed a co-immunoprecipitation assay in OKP cells using Myc-NCC and OS9-V5 proteins. Moreover, to test for the specificity of the interaction, we also checked, under the same experimental conditions, whether OS9 could interact with the endothelin B (ETB) receptor. Like NKCC2, the ETB receptor is expressed in the TAL, but it is not structurally related to the Na-Cl co-transporter family. Similar to NKCC2 (Fig. 2A, lane 3), NCC protein could be recovered from OS9 immunoprecipitates (Fig. 2A, lanes 7 and 10), indicating interactions between the proteins. Moreover, like NKCC2, the interaction of OS9 with NCC involves mainly the immature form of the co-transporter (Fig. 2A, lanes 7 and 10). In contrast to the co-transporter proteins (Fig. 2, A and B), the ETB receptor did not co-immunoprecipitate with OS9 (Fig. 2B, lane 10), providing additional evidence for the specificity of the interaction of OS9 with NCC and NKCC2 in renal cells.
FIGURE 2.

OS9 interacts specifically with NKCC2 and the related NCC. A, co-immunoprecipitation of OS9 with NKCC2 and NCC in OKP cells. Cell lysates from cells transiently transfected with Myc-NKCC2 or Myc-NCC in the presence or absence of OS9-V5 were immunoprecipitated (IP) with anti-V5 antibody. Similar to NKCC2, NCC co-immunoprecipitated with OS9-V5 was detected by immunoblotting (IB) using anti-Myc (lanes 7 and 10). Again, the interaction with OS9 involves mainly the core glycosylated (immature) form of the co-transporters. 5% of total cell lysate (Lys) was resolved as a positive control. B, OS9 does not interact with ETB receptor. Cell lysates from OKP cells transiently transfected with the co-transporters proteins or ETB receptor in the presence or absence of OS9-V5 were immunoprecipitated with anti-V5 anti-body. Again, 5% of total cell lysate (Lys) was resolved as a positive control. In contrast to NKCC2 and NCC, ETB receptor protein was not recovered from OS9 immunoprecipitates (lane 10). IgGH, the heavy chain of IgG.
NKCC2 and OS9 Co-localize at the ER
The association of OS9 with the immature form of NKCC2 suggests that the two proteins interact in the ER. To support this hypothesis, we visualized the subcellular distribution of NKCC2 and OS9 proteins by immunofluorescence microscopy in renal cultured cells. To that end, OKP cells were co-transfected with EGFP-NKCC2 and OS9-V5 proteins. Of note, we have previously shown that, similar to Myc, N-terminal tagging of NKCC2 with EGFP does not influence the co-transporter intracellular trafficking (25, 36). As shown in Fig. 3A, NKCC2 (green) largely co-localized with OS9 (red), indicating that these two proteins share overlapping subcellular localization. Moreover, the two proteins displayed an immunofluorescence staining pattern that is more restricted to a perinuclear ER-like distribution, which is in agreement with an interaction of NKCC2 with OS9 at the ER. To confirm this, the subcellular localization of NKCC2 in the presence and absence of transfected OS9 was compared with the distribution of calnexin, an ER marker. As anticipated, when transfected alone, NKCC2 was found distributed primarily in the plasma membrane. Under these conditions, NKCC2 cell surface expression surrounded the calnexin signal (Fig. 3B, upper panels). In contrast, upon OS9 co-expression, a significant fraction of total NKCC2 appeared, most of the time, to be retained in the ER as judged by its excellent co-localization with the ER marker calnexin (Fig. 3B, lower panels). Accordingly, OS9 protein also displayed considerable co-localization with calnexin (Fig. 3C). Collectively, these findings provide additional evidence that NKCC2 and OS9 interact, an interaction that takes place mainly in the ER. Moreover, they suggest that OS9 regulates NKCC2 transit through the ER to alter its cell surface expression.
FIGURE 3.
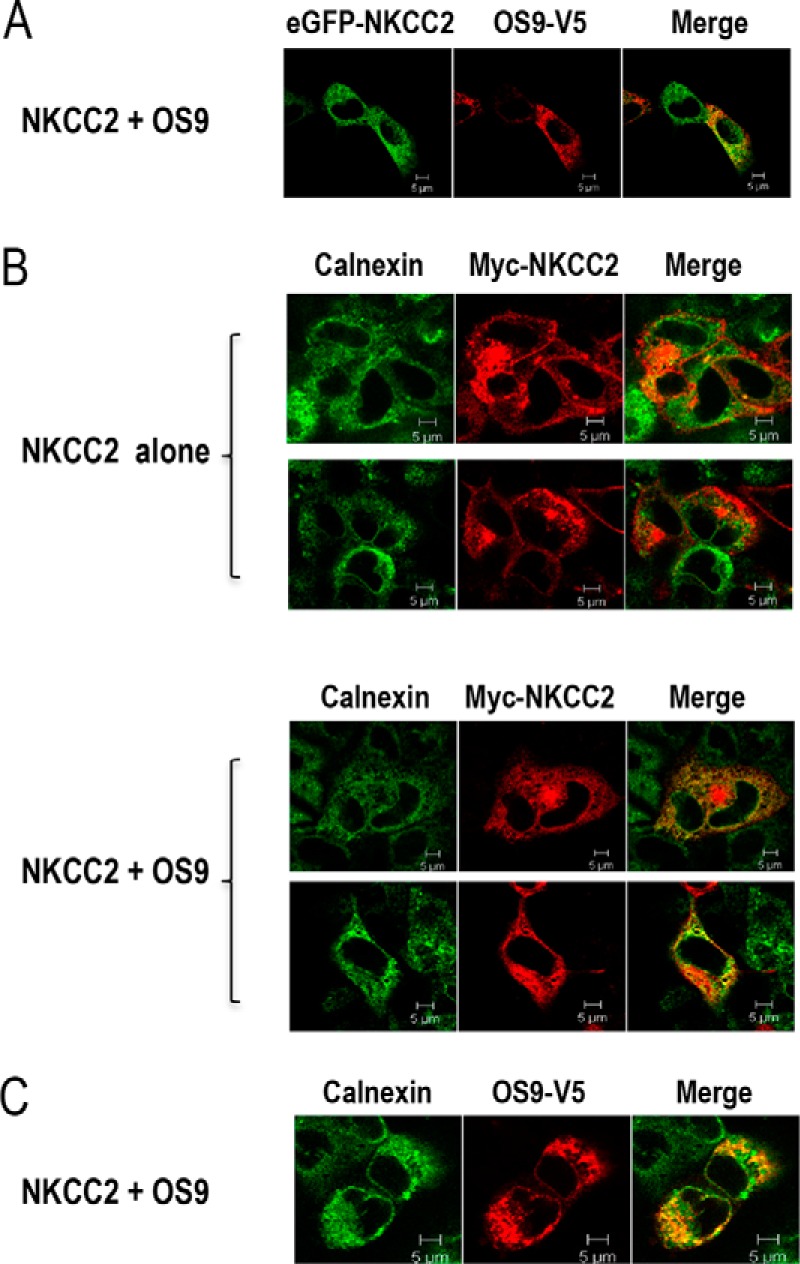
OS9 co-localizes with NKCC2 in the endoplasmic reticulum. A, immunofluorescence confocal microscopy showing distribution of NKCC2 and OS9 in OKP cells. OKP cells were transfected with NKCC2 N-terminally tagged with EGFP (green) and OS9-V5. Fixed and permeabilized cells were stained with mouse anti-V5 for OS9 (Texas Red). The yellow color (merged image) indicates co-localization of the proteins. B, effect of OS9 on subcellular distribution of NKCC2. OKP cells transfected with Myc-NKCC2 alone or with OS9 were stained with mouse anti-Myc (Texas Red; red) and rabbit anti-calnexin (fluorescein isothiocyanate; green). The yellow color indicates overlap between the Myc tag of NKCC2 protein (green) and the ER marker (red), representing co-localization of the proteins. C, subcellular distribution of OS9 in OKP cells. OKP cells overexpressing NKCC2 and OS9-V5 were stained with mouse anti-V5 (Texas Red; red) and rabbit anti-calnexin (FITC; green). The yellow color in the merged image indicates co-localization of OS9 (red) with the ER marker (green). The white scale bars represent 5 μm.
OS9 Regulates the Amount of NKCC2 Protein
Numerous previous reports documented that OS9 plays a pivotal role in the ER-associated regulation of several proteins (28–31). Hence, to assess the functional consequence of OS9-NKCC2 interactions, we first assessed the effects of OS9 on total NKCC2 expression. Toward this, Myc-NKCC2 cDNA was transfected into OKP cells in the absence (empty vector) or presence of OS9-V5 in OKP cells. The expression of NKCC2 and OS9 was assessed by immunoblotting lysates from OKP cells with either anti-Myc or anti-V5 antibody. As shown in Fig. 4A, OS9 co-expression strikingly decreased the amount of immature and mature forms of NKCC2 (Fig. 4 A, lower panel). Accordingly, the graph in Fig. 4A (lower panel), showing averaged results for eight independent experiments, demonstrates a significant decrease in total NKCC2 protein upon OS9 co-expression (−60%; p < 0.0001). Importantly, similar results were obtained when NKCC2 was co-transfected with untagged OS9 protein (−48%; p < 0.001). Moreover, it is worth noting that co-transfecting β-galactosidase, a non-related protein, had no effect on the expression of NKCC2 (Fig. 4B), suggesting that the OS9-induced down-regulation of OS9 is not an artifact of protein overexpression. Additionally, it is also of great interest to note that, under identical conditions, the co-expression of SCAMP2, another NKCC2 binding partner (37), had no effect on total NKCC2 protein abundance (Fig. 4B), further corroborating our conclusion that the effect of OS9 on total NKCC2 protein abundance is specific.
FIGURE 4.
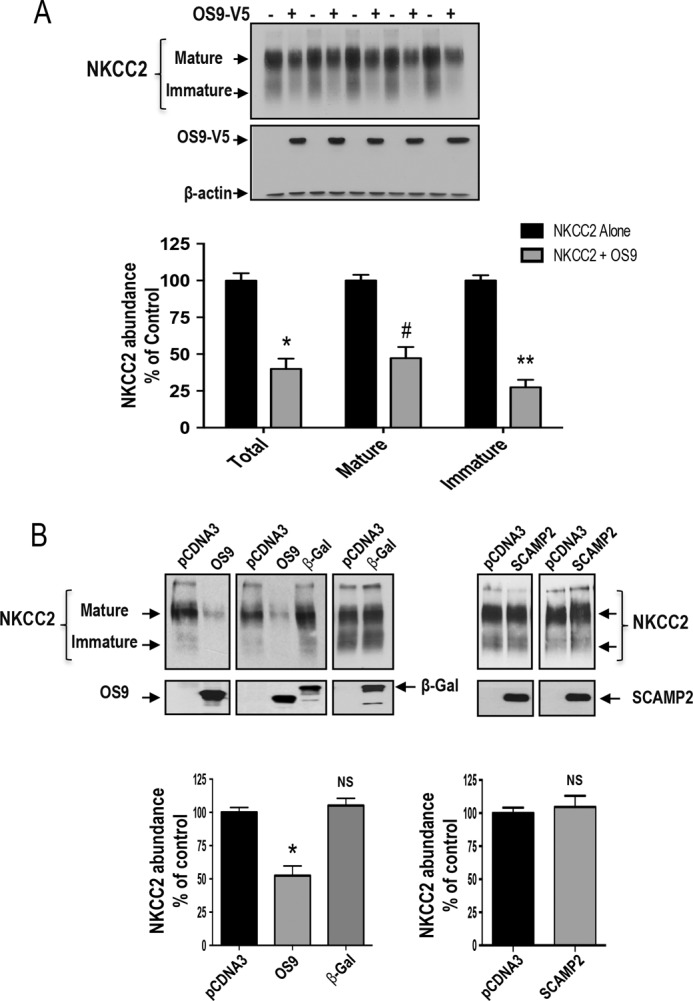
Regulation of steady-state protein level of NKCC2 by OS9. A, OS9 co-expression decreases the expression of NKCC2 proteins. Upper panel, representative immunoblotting analysis showing the effect of OS9 overexpression on NKCC2 protein abundance in OKP cells. Cells were transfected with Myc-NKCC2 alone or in the presence of OS9-V5. 48 h post-transfection, total cell lysates were subjected to immunoblotting analysis for Myc-NKCC2, OS9-V5, and actin. Actin was used as a loading control Lower panel, quantitation of steady-state mature, immature, and total NKCC2 expression levels with or without OS9 co-expression. NKCC2 expression is given as the percentage of that observed in controls co-expressing empty vector (n = 6; mean ± S.E.). Data are expressed as a percentage of control ±S.E. *, p < 0.0004; #, p < 0.0003; **, p < 0.0001 versus control. B, the effect of OS9 on NKCC2 is specific. Upper panel, representative immunoblotting analysis showing the effect of OS9, β-galactosidase (β-gal), and SCAMP2 on NKCC2 protein abundance in OKP cells. Cells were co-transfected with Myc-NKCC2 and the empty vector (pCDNA3) or with OS9.1, β-galactosidase-V5 (β-gal), or SCAMP2-V5 cDNAs. Bottom, summary of results. NS, not significant versus NKCC2 alone; *, p < 0.001 versus NKCC2 alone. NKCC2 alone, n = 8; NKCC2 with OS9.1, n = 8; NKCC2 with β-gal, n = 5. In contrast to OS9, SCAMP2-V5 overexpression had no effect NKCC2 protein abundance. OKP cells were co-transfected with Myc-NKCC2 (0.1 μg/well) alone or in combination with SCAMP2-V5 (0.3 μg/well). Bottom, summary of results. NS, not significant versus NKCC2 alone (NKCC2 alone, n = 5; NKCC2 with SCAMP2-V5, n = 5). Error bars, S.E.
OS9 Decreases NKCC2 Surface Expression and Function
To further elucidate the functional consequence of OS9-NKCC2 interactions, we checked the effect of OS9 on NKCC2 surface expression and activity. Toward that, we first performed cell surface biotinylation experiments. As expected, immunocytochemistry analysis showed that in the absence of OS9 transfection NKCC2 staining co-localized with biotinylated cell surface proteins, indicating appropriate trafficking of NKCC2 to the cell surface. Once again, in cells co-expressing both proteins, NKCC2 mostly disappeared from the cell membrane and clustered in an intracellular location, presumably the ER (Fig. 5A), suggesting that OS9 decreases the cell surface expression of the co-transporter. To study in a more quantitative fashion the effect of OS9 on NKCC2 surface expression, we opted for cell surface biotinylation assays. To this end, surface membrane proteins were biotinylated by reaction with sulfo-NHS-SS-biotin and isolated by precipitation with NeutrAvidin-bound agarose. Myc-NKCC2 protein was then identified by immunoblotting using anti-Myc antibody. We documented previously (25, 26, 36, 37) that only the complex glycosylated form of NKCC2 (160–170 kDa) is able to reach the cell membrane. Accordingly, only the fully glycosylated, mature form of NKCC2 was detected in biotinylated cell surface proteins. Most importantly, the decrease in total NKCC2 protein abundance in cells overexpressing OS9 was associated with a reduction (−55%; p < 0.004) in NKCC2 expression at the cell surface (Fig. 5B).
FIGURE 5.
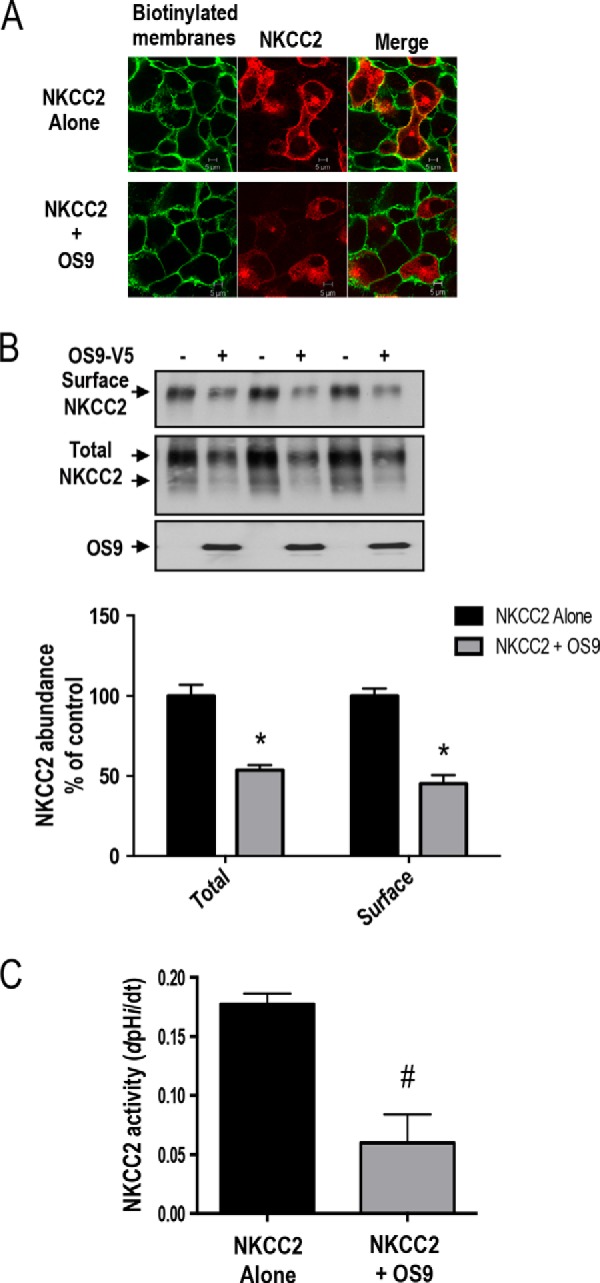
OS9 decreases NKCC2 cell surface expression and activity. A, effect of 0S9 on subcellular distribution of NKCC2. OKP cells were transfected with Myc-NKCC2 (red) alone or with SCAMP2-V5. Membrane proteins of confluent cells were biotinylated at 4 °C with the biotinylation reagent sulfo-NHS-SS-biotin. Then the monolayers were fixed and stained for cell surface biotin (avidin-Cy2; green). The stained specimens were evaluated by confocal microscopy. Optical sections (xy) at the cell surface are depicted for the Texas Red channel (red), Cy2 channel (red), and a merged channel. B, total and surface NKCC2 proteins are down-regulated by OS9. OKP cells were co-transfected with Myc-NKCC2 (0.1 μg/well) alone or in combination with OS9 (0.3 μg/well) as indicated. Biotinylated proteins were recovered from cell extracts by precipitation with NeutrAvidin-agarose. NKCC2 proteins on the cell surface were detected by immunoblotting with Myc antibody. An aliquot of the total cell extract from each sample was also run on a parallel SDS gel and Western blotted for total NKCC2 expression. Bottom, densitometric analysis of total and surface NKCC2 from cells expressing NKCC2 alone or with OS9. Data are expressed as a percentage of control. *, p < 0.004 versus NKCC2 alone (n = 3). C, measurement of Na-K-2Cl co-transport activity in OKP cells expressing NKCC2 alone or with OS9 proteins. Each bar represents the mean ± S.E. rates of cell pH recovery (dpHi/dt in pH units/min) from NH4+-induced alkaline load of three independent experiments. #, p < 0.03 versus NKCC2 alone (n = 3). Error bars represent S.E.
To elucidate the functional consequence of the observed reduction in the co-transporter cell surface level, we then checked the effect of OS9 co-expression on NKCC2 transport activity. As described previously (25, 26, 36, 37, 40), to assess Na-K-2Cl co-transporter activity, we took advantage of the fact that NH4+ can be carried by NKCC2 using its extracellular K+-binding site. OKP cells were exposed to NH4+, and the rate of cell acidification following the initial cell alkalinization was studied as an index of NH4+ transport into the cells. dpHi/dt, which is fully due to NH4+ entry, was measured over the first 20 s of recordings as described earlier (25, 36). As can be seen in Fig. 5C, Na-K-2Cl co-transporter activity decreased upon OS9 co-expression (−66%; p < 0.03). Taken together, these results clearly indicate that OS9 co-expression decreases the transport activity of NKCC2 by decreasing the cell surface level of the co-transporter.
OS9 Promotes NKCC2 ERAD in a Proteasome-dependent Manner
Based upon the above findings, we hypothesized that OS9 co-expression results in ER retention and/or degradation of NKCC2, leading to a decrease in total and cell surface expression of the co-transporter. This mechanism involves, in most cases, activation of the proteasome proteolysis pathway and/or the lysosomal machinery (18, 19, 41–43). Accordingly, we anticipated that treatment with proteasome and lysosome inhibitors might provide important insights into the possible mechanisms of NKCC2 degradation upon OS9 co-expression.
To investigate the possible involvement of proteasomal and/or lysosomal degradation pathways in the effect of OS9-induced down-regulation of NKCC2, cells were treated overnight with 10 μm MG132 or 100 μm leupeptin, and their lysates were subjected to Western blotting analysis. In agreement with our previous report (25), exposure to MG132 significantly increased the protein levels of the immature form of NKCC2 without an apparent increase in its mature form, an effect consistent with an ER-associated degradation (Fig. 6A). In support of this notion, OS9 was also up-regulated under these experimental conditions (Fig. 6A), which is in agreement with previous reports (44). Most importantly, when cells co-transfected with Myc-NKCC2 and OS9-V5 were treated with MG132, the OS9-dependent decrease of NKCC2 protein level was abrogated (Fig. 6A), suggesting that a proteasome-dependent mechanism underlies the degradation of NKCC2. In contrast to MG132, leupeptin was without action on the OS9-induced down-regulation of NKCC2 (Fig. 6A), indicating that the lysosome pathway is not involved in the action of OS9 on NKCC2 protein. Altogether, these results strongly suggest that OS9 decreases NKCC2 expression by promoting its ERAD in a proteasome-dependent manner.
FIGURE 6.
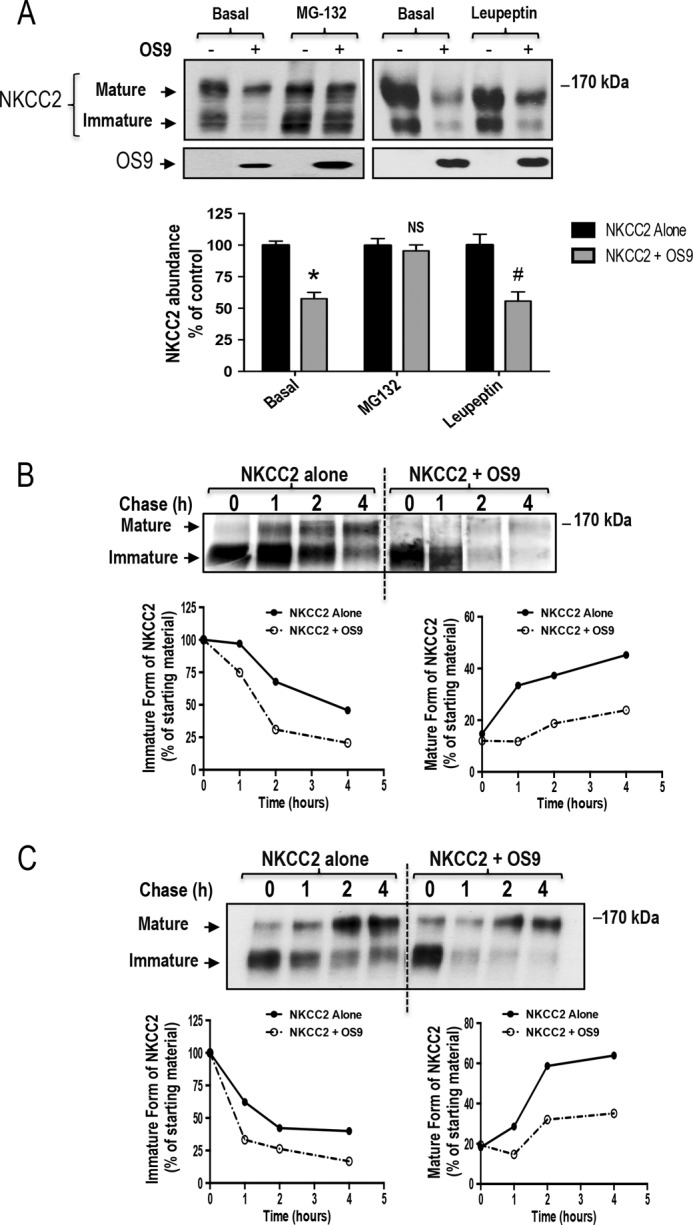
OS9 promotes NKCC2 degradation. A, OS9 decreases NKCC2 expression in a proteasome-dependent manner. OKP cells transiently transfected with Myc-NKCC2 alone or with OS9-V5 were treated with (+) or without (−) 10 μm MG132 or 100 μm leupeptin for 12–16 h prior to cell lysis. The cell lysates were subjected to SDS-PAGE and immunoblotted with anti-Myc and anti-V5 antibodies. Bottom, densitometric analysis of NKCC2 bands from untreated (−) cells and cells treated (+) with MG132 or leupeptin. Data are expressed as percentage of control ±S.E. *, p < 0.0001 versus control (n = 8); NS, not significant versus control (n = 9); #, p < 0.02 versus control (n = 3). B, pulse-chase experiments performed in OKP cells transfected with the indicated plasmids. Cells were labeled with [35S]methionine/cysteine and harvested at the indicated chase times for Myc-NKCC2 immunoprecipitation. Signals were detected by autoradiography. Lower left panel, quantitative analysis of immature NKCC2. The density of the immature form of NKCC2 protein was normalized to the density at time 0 (100%). Lower right panel, quantitative analysis of NKCC2 maturation. In these experiments, we compared the amount of the newly synthesized NKCC2 (immature form at time 0) and the conversion to the complex glycosylated form of the co-transporter (mature form) during the chase period. C, cycloheximide-chase analysis of NKCC2 in the presence or absence of OS9-V5. 14–16 h post-transfection, OKP cells transiently expressing WT NKCC2 alone or in combination with OS9 were chased for the indicated times after addition of cycloheximide. Total cell lysates were separated by SDS-PAGE and probed by anti-Myc antibody. The density of the mature and immature forms of NKCC2 proteins was normalized to the density at time 0. Error bars represent S.E.
To directly examine the effect of OS9 protein on the degradation and the maturation of NKCC2 protein, we traced the sorting delivery of newly synthesized proteins from the ER to Golgi compartments using pulse-chase analysis. In these experiments, cells transfected with Myc-tagged wild-type NKCC2 in the presence or absence of OS9 construct were labeled for 1 h with [35S]methionine/cysteine and chased with unlabeled methionine and cysteine during different intervals between 1 and 4 h. At t = 0, similar quantities of newly synthesized NKCC2 proteins were present in controls and in cells that overexpressed OS9. During the chase period, the core glycosylated (immature) form of NKCC2 is progressively converted to a more slowly migrating band, representing the mature and functional form of the co-transporter (25, 26, 36, 37). As shown in Fig. 6B, in cells transfected with OS9, immature NKCC2 protein showed a faster rate of decay during the chase period. In kinetic analysis, the estimated half-life of the immature form of NKCC2 protein was decreased by −55% upon OS9 co-expression. The decrease represents the conversion of the immature to the mature form as well as the degradation of the immature form of NKCC2 protein. Consequently, OS9 co-expression decreased the maturation efficiency of NKCC2 at different time points during the chase period. At t = 4 h, the effect of OS9 on NKCC2 maturation efficiency reached −49% (Fig. 6B). To further corroborate these observations, we used the cycloheximide decay assay as a second approach to assess the stability and maturation of NKCC2. To this end, 12–14 h post-transfection, cycloheximide was added to block protein synthesis, and at various times after addition of cycloheximide, NKCC2 levels were monitored by immunoblotting. As illustrated in Fig. 6, OS9 overexpression decreased the half-life of immature NKCC2 protein by 61%, which is in agreement with the pulse-chase analysis described above. Moreover, the decrease in immature NKCC2 stability in cells overexpressing OS9 was associated with a decrease in the conversion of the protein from the immature to the mature form. OS9-induced decrease in the maturation efficiency of NKCC2 was accentuated during the chase period to reach −45% at 4 h (Fig. 6C). Taken together, the results above indicate that OS9 targets the immature form of NKCC2 to the proteasome-dependent ER-associated degradation pathway.
N-Glycosylation of NKCC2 Is Critical for the OS9-dependent ERAD Pathway
The processing of N-linked oligosaccharides plays an important role in the ERAD of glycoproteins. NKCC2 is N-glycosylated at two sites, Asn-442 and Asn-452, located in the long extracellular loop (2, 36). Consequently, we next examined the role of N-glycan in the OS9-mediated ERAD of NKCC2. To this end, we mutated the two predicted glycosylation sites, Asn-442 and Asn-452, to glutamine (Gln) and assessed the effect of these mutations on OS9-induced down-regulation of NKCC2. Hence, N442Q/N452Q proteins were expressed in OKP cells in the presence or absence of OS9 construct. As expected, elimination of both N-glycosylated sites (N442Q/N452Q) resulted in the complete loss of the upper band with a lower band around 120 kDa remaining (Fig. 7A). Nevertheless, as can be seen in Fig. 7A, OS9 was still able to bind to the non-glycosylated form of the NKCC2. However, OS9 association with non-glycosylated NKCC2 was unproductive because, in contrast to its effect on WT NKCC2, it did not affect the protein abundance of the co-transporter under these conditions (Fig. 7B). To corroborate this observation, we performed a pulse analysis assay. In contrast to WT NKCC2, OS9 did not affect the kinetics of disappearance of the non-glycosylated form of the co-transporter (Fig. 7C). In sum, these data demonstrate that mutations of NKCC2 N-glycosylation sites (N442Q and N452Q) abolish the OS9 effects, indicating that OS9-induced protein degradation of the co-transporter is N-glycan-dependent.
FIGURE 7.
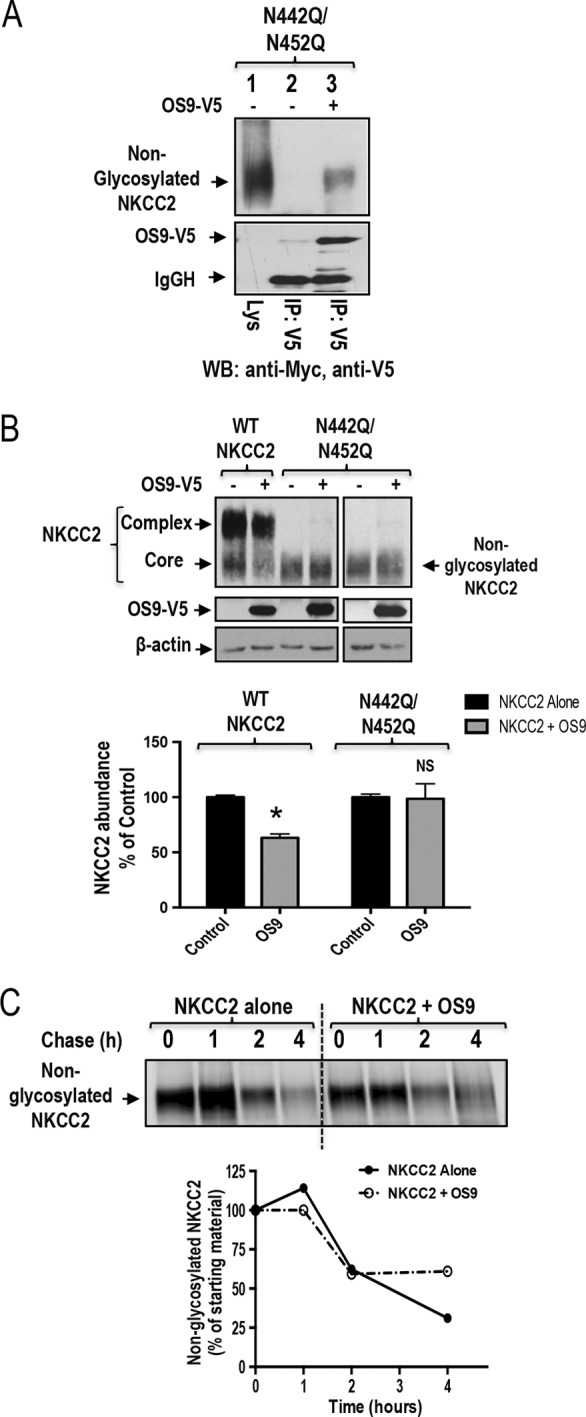
N-Glycosylation of NKCC2 is critical for the OS9-induced down-regulation of NKCC2. A, co-immunoprecipitation of OS9 with non-glycosylated NKCC2 (N442Q/N452Q). Cell lysates from cells transfected with N442Q/N452Q in the presence or absence of OS9-V5 were immunoprecipitated (IP) with anti-Myc or anti-V5 antibody. N442Q/N452Q co-immunoprecipitated with OS9 was detected by immunoblotting (WB) using anti-Myc (lane 3). B, OS9 association with non-glycosylated NKCC2 is unproductive. Cells were transfected with Myc-NKCC2 or N442Q/N452Q in the presence or absence of OS9-V5. 48 h post-transfection, total cell lysates were subjected to immunoblotting analysis for Myc-NKCC2, OS9-V5, and actin. Lower panel, quantitation of steady-state total NKCC2 expression levels with or without OS9 co-expression. *, p < 0.001 versus control (n = 4); NS, not significant versus control (n = 4). C, pulse-chase experiments performed in OKP cells transfected with non-glycosylated NKCC2 in the presence or absence of OS9 construct. Cells were labeled with [35S]methionine/cysteine and harvested at the indicated chase times for Myc-NKCC2 immunoprecipitation. Signals were detected by autoradiography. Lower panel, quantitative analysis of non-glycosylated NKCC2. The density of NKCC2 proteins was normalized to the density at time 0 (100%). IgGH, the heavy chain of IgG. Error bars represent S.E.
Mannose 6-Phosphate Receptor Homology (MRH) Domain of OS9 Is Dispensable for the Effect on NKCC2 Degradation
In yeast, it has been shown that the MRH domain of OS9 is required for its role in the ER quality control pathway (45). To examine the role of the lectin activity of the MRH domain in the OS9-induced down-regulation of NKCC2, we focused on five residues within this region (namely Tyr-120, Gln-130, Arg-188, Glu-212, and Tyr-212) that were found to be required for ERAD in yeast. The specific role of each of these amino acids was analyzed by site-directed mutagenesis converting each of them individually to alanine, glutamine, or asparagine (Fig. 8A). OKP cells were then transfected with NKCC2 singly or in combination with WT OS9 or mutated OS9 proteins. As can be seen in Fig. 8, inactivating the MRH domain of OS9 did not prevent the OS9-induced down-regulation of NKCC2 protein abundance. Indeed, similar to WT OS9, co-expression of mutated OS9 proteins decreased NKCC2 protein expression (Fig. 8, A and B), clearly indicating that the MRH domain of OS9 is dispensable for the ERAD of NKCC2.
FIGURE 8.
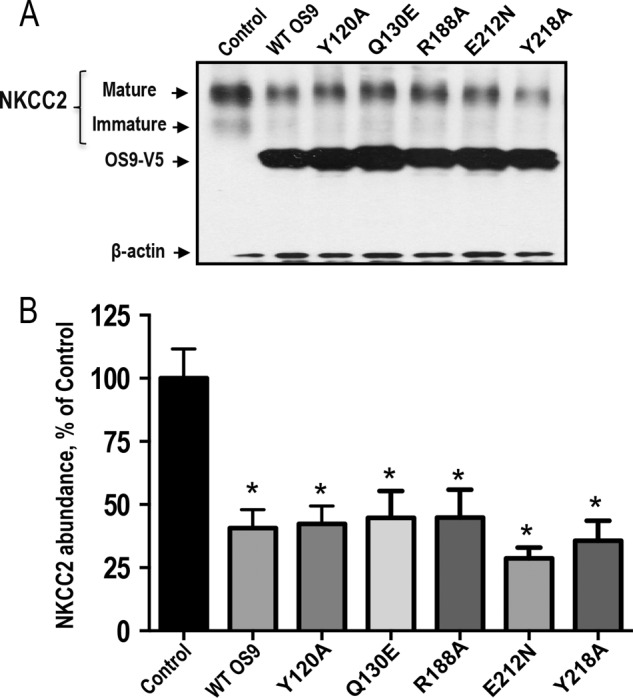
Effect of overexpression of OS9 with mutated MRH domain. A, representative immunoblotting analysis showing the effect of OS9 overexpression with mutated MRH domain on NKCC2 protein abundance. OKP cells were transfected with NKCC2 singly (Control) or in combination with WT OS9 or mutated OS9 proteins. 48 h later, total cell lysates were subjected to immunoblot analysis for Myc-NKCC2, OS9-V5, and actin. B, summary of results. *, p < 0.005 versus control (n = 5). Error bars represent S.E.
The Impact of OS9 on NKCC2 Expression Is Independent of the Expression System
Because the ER quality control mechanism and the ERAD system may be cell-dependent, we sought to corroborate our findings by conducting the experiments in a second renal cell line, HEK cells. As illustrated in Fig. 9A, similar to OKP cells, immunoprecipitation of OS9 followed by immunoblotting for NKCC2 also revealed robust interaction of the two proteins. Importantly, as in OKP cells, NKCC2 interaction with NKCC2 in HEK cells involves mainly the immature form of the co-transporter. Accordingly, NKCC2 and OS9 were also co-localized to the ER in HEK cells as indicated by co-localization with the ER marker calnexin (Fig. 9B). Most importantly, similar to OKP cells, OS9 co-expression in HEK cells decreased both immature and mature NKCC2 proteins (Fig. 9, C and D), clearly indicating that OS9 effects on the co-transporter expression are independent of the expression system.
FIGURE 9.

The effect of OS9 on NKCC2 ERAD is independent of the expression system. A, OS9 interacts with immature NKCC2 in HEK cells. Cell lysates from HEK cells transiently transfected with Myc-NKCC2 singly or in combination with OS9-V5 were immunoprecipitated (IP) with anti-Myc or anti-V5 antibody. NKCC2 protein was recovered from OS9 immunoprecipitates mainly in its immature form (lane 3). B, OS9 and NKCC2 co-localizes in the ER. Upper panel, immunofluorescence confocal microscopy showing the distribution of NKCC2 and OS9 in HEK cells. HEK cells were transfected with NKCC2 N-terminally tagged with EGFP (green) and OS9-V5. Fixed and permeabilized cells were stained with mouse anti-V5 for OS9 (Texas Red). The yellow color (merged image) indicates co-localization of the proteins. Lower panel, HEK cells transfected with NKCC2 and OS9-V5 were stained with mouse anti-V5 (Texas Red; red) and rabbit anti-calnexin (FITC; green). Yellow indicates overlap between the V5 tag of OS9 (red) and the ER marker (green), representing co-localization of the proteins. C, OS9 decreases the steady-state protein level of NKCC2. Cells were transfected with Myc-NKCC2 alone or in the presence of OS9-V5. 48 h post-transfection, total cell lysates were subjected to immunoblotting analysis for Myc-NKCC2, OS9-V5, and actin. D, summary of results. Data are expressed as a percentage of control ±S.E. *, p < 0.02 versus control (n = 3). Error bars represent S.E.
Endogenous OS9 Interacts with Immature NKCC2 and Regulates the Expression of the Co-transporter
The potential significance of our findings is that the interaction of OS9 with NKCC2 in the ER could be very important for sorting determinants for the ER quality control of the co-transporter. To further corroborate these findings, we next examined the role of endogenous OS9 in NKCC2 degradation. To this end, we first checked the ability of NKCC2 to interact with endogenously expressed OS9 in HEK cells. To address this, we used a rabbit OS9 antibody raised against native OS9 protein. Importantly, Western blotting analysis following immunoprecipitation with anti-OS9 antibody detected a protein band around 100 kDa, corresponding to the expected size band of isoform 1 of OS9. More importantly, immunoprecipitation of endogenous OS9 protein brought only the immature form NKCC2, thus again demonstrating physical interaction between OS9 and the ER-resident form of the co-transporter (Fig. 10A, lane 3). Of note, Myc-NKCC2 protein was not detected in control experiments in which immunoprecipitations were carried out using mouse anti-V5 antibodies (Fig. 10A, lane 2). Taken in concert, these findings clearly indicate that, similar to exogenously expressed OS9, endogenous OS9 interacts in vivo with NKCC2, an interaction that involves principally the immature form of the co-transporter.
FIGURE 10.

Knockdown of endogenous OS9 increases NKCC2 biogenesis. A, endogenous OS9 interacts with immature NKCC2. HEK cells overexpressing Myc-NKCC2 were immunoprecipitated (IP) with anti-Myc (positive control; lane 1), anti-V5 (negative control; lane 2), or anti-OS9 antibody (lane 3). Co-immunoprecipitated NKCC2 (mainly the immature form) was detected by immunoblotting (IB) using anti-Myc antibody (lane 3). IgGH, the heavy chain of IgG. B and C, knockdown of endogenous OS9 increases total and cell surface expression of NKCC2. Upper panels, representative immunoblotting analysis illustrating the effect of OS9 knockdown on total and surface NKCC2. HEK cells were transfected with NKCC2 in the absence (Control) or presence of specific OS9 siRNAs from Dharmacon (D) or Santa Cruz Biotechnology (S). 48 h post-transfection, biotinylated proteins were recovered from cell extracts by precipitation with NeutrAvidin-agarose. An aliquot of the total cell extract from each sample was also run on a parallel SDS gel and Western blotted for total NKCC2 expression. NKCC2 proteins were detected by immunoblotting with Myc antibody. Lower panels, summary of results. Data are expressed as a percentage of control. *, p < 0.004 versus control (n = 4); #, p < 0.007 versus control (n = 4); §, p < 0.0001 versus control (n = 5); **, p < 0.03 versus control (n = 5). D, knockdown of endogenous OS9 increases NKCC2 stability and maturation. HEK cells were first transfected in the absence (Control) or presence of OS9 siRNA followed by transfection with NKCC2 plasmids 24 h later. 12–14 h post-transfection of NKCC2, cells were chased for the indicated times after addition of cycloheximide. Total cell lysates were subjected to immunoblot analysis for NKCC2 and OS9. Lower left panel, quantitative analysis of immature NKCC2. The density of the immature form of NKCC2 proteins was normalized to the density at time 0 (100%). Lower right panel, quantitative analysis of NKCC2 maturation. The results are presented as relative intensity. Each point represents the mean ± S.E. of three independent experiments. *, p < 0.05 versus controls (n = 4). Error bars represent S.E.
To examine the role of endogenous OS9 in the regulation of NKCC2, we first checked the effect of OS9 knockdown on NKCC2 expression using siRNA. To this end, a sequential transfection in HEK cells was performed. Cells were first transfected with siRNAs for at least 24 h before transfection with Myc-NKCC2 plasmids for 48 h. As illustrated in Fig. 10B, relative to control, the endogenous expression of OS9 was reduced by 80% in cells transfected with OS9 specific siRNA (from Dharmacon). With this degree of OS9 knockdown, the steady-state level of NKCC2 was increased by 80% (p < 0.004). Importantly, this increase in total NKCC2 protein abundance was associated with an increase in the surface expression of the co-transporter protein (+76%; p < 0.007). To corroborate these findings, we also used a second set of OS9-specific siRNA (from Santa Cruz Biotechnology). As can be seen in Fig. 10C, using this second set of OS9 siRNA, OS9 knockdown again increased total and surface NKCC2 expression by 81 (p < 0.03) and 84% (p < 0.0001), respectively. Consequently, these data are fully in agreement with a role of endogenous OS9 in the ERAD of immature NKCC2 protein.
To further corroborate these observations, we used the cycloheximide decay assay to assess the effect of OS9 knockdown on the half-life of the immature form of NKCC2. To this end, a sequential transfection in HEK cells was performed as indicated above. However, in this series of experiments, 12–14 h after transfection of HEK cells with Myc-NKCC2 plasmids, cycloheximide was added to block protein synthesis, and at various times after addition of cycloheximide, NKCC2 levels were monitored by Western blotting. It is worth nothing that at the start of the chase period (t = 0) total NKCC2 protein levels were comparable between the studied groups (Fig. 10D), indicating that a modification of protein synthesis is not responsible for the increase in total NKCC2 protein levels observed above (Fig. 10, B and C). Most importantly, in cells transfected with OS9 siRNA, immature NKCC2 protein had a slower rate of decay at different time points during the chase period (Fig. 10D). Indeed, in kinetic analysis, the estimated half-life of immature form of NKCC2 protein was increased by +79% (p < 0.05) upon OS9 knockdown. Moreover, this increase in the stability of the immature form of NKCC2 was associated with an increase in the conversion of the protein from the immature to the mature form of the co-transporter (Fig. 10D). Altogether, these data strongly hint at a role of endogenous OS9 in the ERAD of immature NKCC2 protein.
Discussion
This study was carried out to gain insight into the molecular mechanisms underlying the regulation of NKCC2 transit in the ER. By means of yeast two-hybrid analysis and co-immunoprecipitation assays, we identified OS9 as a bona fide NKCC2-interacting protein. The interaction involves mainly the immature form of the co-transporter, therefore taking place in the ER. We have found that OS9 co-expression decreases the stability of core glycosylated NKCC2 by increasing its degradation by the proteasome pathway. Accordingly, OS9 depletion increases the stability and maturation of the co-transporter protein, indicating therefore that OS9 targets NKCC2 for the ERAD pathway.
OS9 was originally identified as a gene of unknown function amplified in osteosarcoma (27). OS9 is expressed ubiquitously and has alternatively splice versions (39). The function of OS9 remained mysterious until human OS9 and its yeast homolog Yos9p were implicated in ER-to-Golgi transport (35). Likewise, mammalian OS9 interacts with the C-terminal tails of meprin β (32) and DC-STAMP (33) to facilitate their transport from the ER. Besides its role in ER-to-Golgi transport, OS9 has been reported to interact with hypoxia-inducible factor 1-α in the cytosol to regulate its proteasome-mediated degradation in an O2-dependent manner (46). Moreover, more recent reports have also implicated OS9 in the ERAD of misfolded glycoproteins (28–31). It was first reported that removal of Yos9p from the yeast ER selectively inhibits degradation of glycosylated ERAD substrates (47). Subsequent studies in mammalian cells indicated that the degradation of misfolded glycoproteins is partially inhibited by OS9 knockdown, but this effect seems to depend on the ERAD substrates (45). In this regard, it is worth mentioning that it was initially thought that OS9 is required for disposal of substrates with luminal folding defects, whereas it is dispensable for disposal of proteins with defects in the transmembrane and cytosolic domains (28, 29, 48–51). However, a recent report showed evidence that the Arabidopsis homolog of the mammalian OS9 protein plays a key role in the ERAD of the receptor-like kinases, which are transmembrane proteins (52). Moreover, although Jansen et al. (33) have not explored in detail the role of OS9 in the degradation of DC-STAMP, they reported that preliminary biochemical data suggest that OS9 may also be involved in the ERAD of DC-STAMP (33). In the present study, we showed evidence for a specific interaction of mammalian OS9 with NKCC2 and NCC, two renal transmembrane proteins. Importantly, we demonstrated that OS9 binding in vivo involves mainly the immature form of NKCC2. Most importantly, we provided evidence that OS9 binding is involved in the regulation of NKCC2 ERAD.
In support of ERAD as a major regulator of NKCC2 trafficking and function, we previously demonstrated that the majority of newly synthesized NKCC2 proteins are targeted to proteasome degradation before export to the Golgi (26). In the present work, the function of OS9 in the ERAD of NKCC2 was first suggested by co-immunoprecipitation studies that demonstrated that the interaction in vivo involves mainly the immature, ER-resident form of NKCC2. Accordingly, the localization studies showed that OS9 co-localizes with NKCC2 at the endoplasmic reticulum. In contrast, OS9 did not co-immunoprecipitate with the ETB receptor, suggesting that OS9 is not simply interacting with all membrane proteins. In support of this notion, Wang et al. (34) demonstrated that OS9 specifically interacts with the TRP channels of the vanilloid group but not with other tested TRP channels or αNaC (34). Furthermore, we have shown that elevated OS9 levels, through the expression of exogenous OS9, prevent the cell surface localization of NKCC2 and cause the co-transporter to accumulate in a perinuclear location. The OS9-induced down-regulation of NKCC2 surface expression was associated with an increase in co-transporter degradation. Moreover, OS9 effects on the ER-resident form of NKCC2 were prevented in the presence of the proteasome inhibitor MG132 but not with leupeptin, an inhibitor of lysosomal function, indicating that OS9 targets the immature form of the co-transporter to the proteasome-dependent ERAD pathway. It is of interest to note that, under similar experimental conditions, Wang et al. (34) reported that OS9 overexpression appears to protect TRPV4 from the ER-associated degradation, indicating that the effect of OS9 is substrate-dependent (34). It is well documented that a single chaperone might be involved in either folding or degrading a given substrate that transits through the ER (53). OS9 is no exception, and therefore its actions also depend on the ERAD substrates (34). Consequently, it is conceivable that OS9 might play a protective role for TRPV4 but a destructive one for NKCC2 during ERAD. To further support the role of OS9 in the ERAD of NKCC2, we used the siRNA approach and demonstrated that OS9 knockdown delayed the degradation of the immature form of the co-transporter. Moreover, we showed that mutations of NKCC2 N-glycosylation sites also abolished OS9 effects, indicating that OS9-induced protein degradation is N-glycan-dependent. In contrast, the MRH domain of OS9 appears to be dispensable for the effect on NKCC2 degradation. In this regard, it is worth mentioning that the role of the MRH domain of OS9 remains unclear. Indeed, the significance of the lectin activity of the MRH domain of OS9 in vivo remains controversial and likely depends on the levels of overexpressed OS9, its sugar binding-defective mutants, and/or the yeast or mammalian expression system used (45, 54). In agreement with our findings, Bernasconi et al. (30) also demonstrated that the activity of OS9, in a mammalian cell background, does not require a functional MRH domain. In sum, our results demonstrate the presence of an OS9-mediated ERAD pathway in renal cells that degrades immature NKCC2 proteins.
ERAD starts with the recognition of a misfolded protein by molecular chaperones (19, 55). ERAD substrates can present a misfolded lesion either in the ER lumen, ER membrane, or cytoplasm, which will dictate the types of chaperones with which they interact (19, 55). Consequently, three different ERAD pathways have been proposed as ERAD-L, ERAD-M, and ERAD-C (19, 55). Soluble or membrane-bound proteins with a misfolded luminal domain are degraded via the ERAD-L (19, 55). Transmembrane substrates containing misfolded portions in the cytoplasmic and membrane domains are degraded via the ERAD-C and ERAD-M pathways, respectively (19, 55). For instance, to aid in folding and to select misfolded species for ERAD, nascent CFTR engages both cytoplasmic and ER luminal chaperones of CFTR (24). Likewise, NCC, another transmembrane protein, involves several cytoplasmic chaperones for its ERAD (56, 57). Similar to NCC, NKCC2 contains 12 transmembrane segments, two large cytoplasmic domains, and a large ER-exposed exofacial loop. Given the fact that the co-transporter contains domains in the ER and cytoplasm, NKCC2 could potentially interact with OS9 and/or other ER molecular chaperones on either side or on both sides of the ER membrane. In this regard, a remarkable difference between Yos9p, which is expressed only in the lumen of the ER, and its mammalian counterparts is the absence of a KDEL or other potential ER retention sequence from the mammalian proteins. However, mammalian OS9 contains a stretch of hydrophobic residues at the N terminus, suggesting at least transient localization in the ER (27). Accordingly, localization studies of OS9 in mammalian cells indicated that the protein might reside at both luminal and cytoplasmic surfaces of the ER (30, 32, 33). Hence, an interaction between OS9 and the C-terminal domain of NKCC2 at the cytoplasmic side of the ER is conceivable. However, one cannot exclude the possibility that OS9 may also interact with other domains of NKCC2 protein. In this respect, to further define the parts in the cytoplasmic tail of NKCC2 that are responsible for the interaction with OS9, we created a series of deletion mutants in the C-terminal tail of NKCC2. Subsequent co-immunoprecipitation experiments revealed that, even in the absence of the distal part of the NKCC2 C terminus, the co-transporter is still able to interact with OS9 (data not shown), opening the possibility for an interaction of OS9 with other parts of NKCC2 protein. Consequently, an involvement of OS9 in the ERAD-L, ERAD-M, and/or ERAD-C of NKCC2 remains possible. Undeniably, OS9 cannot work in isolation to mediate the ERAD of the co-transporter. Appropriately, one may reasonably postulate that OS9 works in concert, sequentially or simultaneously, with other NKCC2-binding proteins and ERAD components such as CHIP and the Hsp70/Hsp90 organizer protein that are involved in structurally related renal NCC ERAD (56, 57) to mediate the ER quality control and its associated protein degradation of the co-transporter. Obviously, further experiments are needed to uncover the precise mechanism behind this process.
Identification of proteins that interact specifically with the immature forms of NKCC2 is important to decipher the molecular mechanisms underlying the regulation of the ER-to-Golgi transport of the co-transporter. Although our study was conducted in renal cultured cells, it is likely that NKCC2 interaction with OS9 also takes place in native TAL cells to play a crucial role in the chronic adaptations of the co-transporter. Indeed, given that variation in the activities of Na-Cl co-transporters or their regulators alters blood pressure in humans (2, 9), it is conceivable that physiological or pathological changes in the expression level of their binding partners may affect their activities, therefore altering the sodium balance and blood pressure. In support of this notion, a recent report showed evidence that in vivo loss of Nedd4-2, a binding partner of NCC, leads to increased NCC abundance and function (58). Most importantly, renal tubular Nedd4-2 deficiency causes NCC-mediated salt-dependent hypertension (58). Similarly, it is reasonable to postulate that physiological or pathological changes in OS9 expression levels may also affect NKCC2 function. In support of this hypothesis, a previous report showed evidence that OS9 is up-regulated in response to ER stress and facilitates the ERAD of several glycosylated proteins under these conditions (44). The OS9 overexpression approach used in the present study may therefore actually mimic OS9 up-regulation under agents or conditions causing ER stress in TAL cells such as salt loading and aging (59, 60). This is of particular interest because NKCC2 protein expression is down-regulated by high salt intake (4) and in the aged kidney (7). Consequently, an increase in OS9 expression in response to ER stress could contribute to the down-regulation of NKCC2 expression via the ubiquitin-proteasome pathway under these conditions (4, 7).
Intensive research on the mechanisms underlying the ERAD machinery has provided new insights into how ERAD contributes to human health during both normal and diseases states (19, 60). The importance of ER quality control in general and the ERAD process in particular has been indicated in a large number of human pathologies called conformational diseases (19, 61, 62). One example is CFTR, the protein in which mutations give rise to cystic fibrosis (63). One emerging and promising strategy for the possible treatment of ERAD diseases is to overcome ER retention (61, 62, 64) because some mutant proteins such as the cystic fibrosis ΔF508-CFTR mutant are actually functional if they exit the ER and reach their normal cellular location (63, 65). In regard to NKCC2, previous reports demonstrated that, when expressed in a mammalian cell background, several NKCC2 mutants are retained in the ER (10, 25, 66). Moreover, although distinct cellular mechanisms may account for NKCC2 loss of function in BS1 disease, our preliminary data in the laboratory revealed that ER-associated protein degradation is the most common mechanism underpinning BS1.4 Consequently, understanding the involvement of this process in BS1 is essential to provide novel therapeutic strategies for the treatment of BS1. Indeed, the identification and selective modulation of ERAD components specific to NKCC2 and its disease causing mutants might help to define a strategy to release fractions of misfolded mutant proteins from the ER by preventing their interactions with the quality control components to rescue their surface expression and restore their functional activity. Hence, it will be of interest to determine whether alterations in NKCC2 protein levels in NKCC2-related diseases are also mediated by OS9.
In summary, we found that NKCC2 interacts specifically with OS9, a protein known to be involved in the regulation of the transit in the ER. OS9 interacts mainly with the ER-resident form of NKCC2 to accelerate its degradation by the ERAD pathway. To the best of our knowledge, this is the first study identifying a protein partner of NKCC2 that plays a role in proteasome-dependent ERAD of the co-transporter. Besides, this is also the first report providing evidence that mammalian OS9 can be also involved in the ERAD of transmembrane proteins. Moreover, OS9 also binds to the structurally related co-transporter NCC, suggesting that the interaction with OS9 is a common feature of members of the SLC12A family, a group of cation-chloride co-transporters that are targets of therapeutic drugs and mutated in several human diseases such as Bartter, Gitelman, and Andermann syndromes (2, 67, 68). Consequently, further studies of OS9-mediated NKCC2 degradation should be useful in defining the recognition principles associated with the disposition of NKCC2 and perhaps other members of the cation-coupled chloride co-transporter family, in particular NCC. The thorough characterization and identification of the molecular mechanisms underlying the ER quality control of the cation-chloride co-transporters may provide a foundation for the development of therapeutic strategies targeting co-transporter transport from the ER to the cell surface.
Author Contributions
E. S., N. D., S. D., and N. Z. performed the experiments and analyzed the data. K. L. designed the study, supervised the experiments, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Aurélie Edwards for helpful discussions and careful reading of the manuscript.
This work was supported in part by grants from INSERM, Paris, France. The authors declare that they have no conflicts of interest with the contents of this article.
E. Seaayfan, N. Defontaine, S. Demaretz, N. Zaarour, and K. Laghmani, unpublished data.
- TAL
- thick ascending limb
- OKP
- opossum kidney
- NCC
- Na-Cl co-transporter
- ER
- endoplasmic reticulum
- BS1
- type I Bartter syndrome
- pHi
- cytoplasmic pH
- NHS
- N-hydroxysuccinimide
- MRH
- mannose 6-phosphate receptor homology
- ERAD
- endoplasmic reticulum-associated degradation
- NKCC2
- Na-K-2Cl co-transporter
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ENaC
- epithelial sodium channel
- Hsp
- heat shock protein
- CHIP
- Hsp70-interacting protein
- EGFP
- enhanced GFP
- ETB
- endothelin B
- dpHi/dt
- initial rate of intracellular pH recovery
- TRP
- transient receptor potential
- DC-STAMP
- dendritic cell-specific transmembrane protein.
References
- 1. Greger R., and Velázquez H. (1987) The cortical thick ascending limb and early distal convoluted tubule in the urinary concentrating mechanism. Kidney Int. 31, 590–596 [DOI] [PubMed] [Google Scholar]
- 2. Gamba G. (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493 [DOI] [PubMed] [Google Scholar]
- 3. Gamba G. (1999) Molecular biology of distal nephron sodium transport mechanisms. Kidney Int. 56, 1606–1622 [DOI] [PubMed] [Google Scholar]
- 4. Wu J., Liu X., Lai G., Yang X., Wang L., and Zhao Y. (2013) Synergistical effect of 20-HETE and high salt on NKCC2 protein and blood pressure via ubiquitin-proteasome pathway. Hum. Genet. 132, 179–187 [DOI] [PubMed] [Google Scholar]
- 5. Kim D., Sands J. M., and Klein J. D. (2004) Role of vasopressin in diabetes mellitus-induced changes in medullary transport proteins involved in urine concentration in Brattleboro rats. Am. J. Physiol. Renal Physiol. 286, F760–F766 [DOI] [PubMed] [Google Scholar]
- 6. Riazi S., Khan O., Tiwari S., Hu X., and Ecelbarger C. A. (2006) Rosiglitazone regulates ENaC and Na-K-2Cl cotransporter (NKCC2) abundance in the obese Zucker rat. Am. J. Nephrol. 26, 245–257 [DOI] [PubMed] [Google Scholar]
- 7. Tian Y., Riazi S., Khan O., Klein J. D., Sugimura Y., Verbalis J. G., and Ecelbarger C. A. (2006) Renal ENaC subunit, Na-K-2Cl and Na-Cl cotransporter abundances in aged, water-restricted F344 × Brown Norway rats. Kidney Int. 69, 304–312 [DOI] [PubMed] [Google Scholar]
- 8. Lifton R. P., Gharavi A. G., and Geller D. S. (2001) Molecular mechanisms of human hypertension. Cell 104, 545–556 [DOI] [PubMed] [Google Scholar]
- 9. Ares G. R., Caceres P. S., and Ortiz P. A. (2011) Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Renal Physiol. 301, F1143–F1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monette M. Y., Rinehart J., Lifton R. P., and Forbush B. (2011) Rare mutations in the human Na-K-Cl cotransporter (NKCC2) associated with lower blood pressure exhibit impaired processing and transport function. Am. J. Physiol. Renal Physiol. 300, F840–F847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ji W., Foo J. N., O'Roak B. J., Zhao H., Larson M. G., Simon D. B., Newton-Cheh C., State M. W., Levy D., and Lifton R. P. (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 40, 592–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Acuña R., Martínez-de-la-Maza L., Ponce-Coria J., Vázquez N., Ortal-Vite P., Pacheco-Alvarez D., Bobadilla N. A., and Gamba G. (2011) Rare mutations in SLC12A1 and SLC12A3 protect against hypertension by reducing the activity of renal salt cotransporters. J. Hypertens. 29, 475–483 [DOI] [PubMed] [Google Scholar]
- 13. Russell J. (2000) Sodium-potassium-chloride cotransport. Physiol. Rev. 80, 211–276 [DOI] [PubMed] [Google Scholar]
- 14. Vitale A., and Denecke J. (1999) The endoplasmic reticulum-gateway of the secretory pathway. Plant Cell 11, 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yeaman C., Grindstaff K. K., and Nelson W. J. (1999) New perspectives on mechanisms involved in generating epithelial cell polarity. Physiol. Rev. 79, 73–98 [DOI] [PubMed] [Google Scholar]
- 16. Caplan M. J. (1997) Membrane polarity in epithelial cells: protein sorting and establishment of polarized domains. Am. J. Physiol. Renal Physiol. 272, F425–F429 [DOI] [PubMed] [Google Scholar]
- 17. Hebert D. N., and Molinari M. (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408 [DOI] [PubMed] [Google Scholar]
- 18. Stolz A., and Wolf D. H. (2010) Endoplasmic reticulum associated protein degradation: a chaperone assisted journey to hell. Biochim. Biophys. Acta 1803, 694–705 [DOI] [PubMed] [Google Scholar]
- 19. Guerriero C. J., and Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 21. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 22. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., and Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valentijn J. A., Fyfe G. K., and Canessa C. M. (1998) Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J. Biol. Chem. 273, 30344–30351 [DOI] [PubMed] [Google Scholar]
- 24. Ahner A., Gong X., and Frizzell R. A. (2013) Cystic fibrosis transmembrane conductance regulator degradation: cross-talk between the ubiquitylation and SUMOylation pathways. FEBS J. 280, 4430–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zaarour N., Demaretz S., Defontaine N., Mordasini D., and Laghmani K. (2009) A highly conserved motif at the COOH terminus dictates endoplasmic reticulum exit and cell surface expression of NKCC2. J. Biol. Chem. 284, 21752–21764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaarour N., Demaretz S., Defontaine N., Zhu Y., and Laghmani K. (2012) Multiple evolutionarily conserved di-leucine like motifs in the carboxyl terminus control the anterograde trafficking of NKCC2. J. Biol. Chem. 287, 42642–42653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su Y. A., Hutter C. M., Trent J. M., and Meltzer P. S. (1996) Complete sequence analysis of a gene (OS-9) ubiquitously expressed in human tissues and amplified in sarcomas. Mol. Carcinog. 15, 270–275 [DOI] [PubMed] [Google Scholar]
- 28. Bhamidipati A., Denic V., Quan E. M., and Weissman J. S. (2005) Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell 19, 741–751 [DOI] [PubMed] [Google Scholar]
- 29. Szathmary R., Bielmann R., Nita-Lazar M., Burda P., and Jakob C. A. (2005) Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol. Cell 19, 765–775 [DOI] [PubMed] [Google Scholar]
- 30. Bernasconi R., Pertel T., Luban J., and Molinari M. (2008) A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 283, 16446–16454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hüttner S., Veit C., Schoberer J., Grass J., and Strasser R. (2012) Unraveling the function of Arabidopsis thaliana OS9 in the endoplasmic reticulum-associated degradation of glycoproteins. Plant Mol. Biol. 79, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Litovchick L., Friedmann E., and Shaltiel S. (2002) A selective interaction between OS-9 and the carboxyl-terminal tail of meprin β. J. Biol. Chem. 277, 34413–34423 [DOI] [PubMed] [Google Scholar]
- 33. Jansen B. J., Eleveld-Trancikova D., Sanecka A., van Hout-Kuijer M., Hendriks I. A., Looman M. G., Leusen J. H., and Adema G. J. (2009) OS9 interacts with DC-STAMP and modulates its intracellular localization in response to TLR ligation. Mol. Immunol. 46, 505–515 [DOI] [PubMed] [Google Scholar]
- 34. Wang Y., Fu X., Gaiser S., Köttgen M., Kramer-Zucker A., Walz G., and Wegierski T. (2007) OS-9 regulates the transit and polyubiquitination of TRPV4 in the endoplasmic reticulum. J. Biol. Chem. 282, 36561–36570 [DOI] [PubMed] [Google Scholar]
- 35. Friedmann E., Salzberg Y., Weinberger A., Shaltiel S., and Gerst J. E. (2002) YOS9, the putative yeast homolog of a gene amplified in osteosarcomas, is involved in the endoplasmic reticulum (ER)-Golgi transport of GPI-anchored proteins. J. Biol. Chem. 277, 35274–35281 [DOI] [PubMed] [Google Scholar]
- 36. Benziane B., Demaretz S., Defontaine N., Zaarour N., Cheval L., Bourgeois S., Klein C., Froissart M., Blanchard A., Paillard M., Gamba G., Houillier P., and Laghmani K. (2007) NKCC2 surface expression in mammalian cells: down-regulation by novel interaction with aldolase B. J. Biol. Chem. 282, 33817–33830 [DOI] [PubMed] [Google Scholar]
- 37. Zaarour N., Defontaine N., Demaretz S., Azroyan A., Cheval L., and Laghmani K. (2011) Secretory carrier membrane protein 2 regulates exocytic insertion of NKCC2 into the cell membrane. J. Biol. Chem. 286, 9489–9502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amlal H., Paillard M., and Bichara M. (1994) NH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. NH4+ conductance and K+/NH4+(H+) antiport. J. Biol. Chem. 269, 21962–21971 [PubMed] [Google Scholar]
- 39. Kimura Y., Nakazawa M., and Yamada M. (1998) Cloning and characterization of three isoforms of OS-9 cDNA and expression of the OS-9 gene in various human tumor cell lines. J. Biochem. 123, 876–882 [DOI] [PubMed] [Google Scholar]
- 40. Amlal H., Legoff C., Vernimmen C., Paillard M., and Bichara M. (1996) Na+-K+(NH4+)-2Cl− cotransport in medullary thick ascending limb: control by PKA, PKC, and 20-HETE. Am. J. Physiol. Cell Physiol. 271, C455–C463 [DOI] [PubMed] [Google Scholar]
- 41. Fujita E., Kouroku Y., Isoai A., Kumagai H., Misutani A., Matsuda C., Hayashi Y. K., and Momoi T. (2007) Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16, 618–629 [DOI] [PubMed] [Google Scholar]
- 42. Bernasconi R., and Molinari M. (2011) ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 23, 176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arvan P., Zhao X., Ramos-Castaneda J., and Chang A. (2002) Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 3, 771–780 [DOI] [PubMed] [Google Scholar]
- 44. Alcock F., and Swanton E. (2009) Mammalian OS-9 is upregulated in response to endoplasmic reticulum stress and facilitates ubiquitination of misfolded glycoproteins. J. Mol. Biol. 385, 1032–1042 [DOI] [PubMed] [Google Scholar]
- 45. Hosokawa N., Kamiya Y., and Kato K. (2010) The role of MRH domain-containing lectins in ERAD. Glycobiology 20, 651–660 [DOI] [PubMed] [Google Scholar]
- 46. Baek J. H., Mahon P. C., Oh J., Kelly B., Krishnamachary B., Pearson M., Chan D. A., Giaccia A. J., and Semenza G. L. (2005) OS-9 interacts with hypoxia-inducible factor 1α and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1α. Mol. Cell 17, 503–512 [DOI] [PubMed] [Google Scholar]
- 47. Buschhorn B. A., Kostova Z., Medicherla B., and Wolf D. H. (2004) A genome-wide screen identifies Yos9p as essential for ER-associated degradation of glycoproteins. FEBS Lett. 577, 422–426 [DOI] [PubMed] [Google Scholar]
- 48. Carvalho P., Goder V., and Rapoport T. A. (2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 49. Denic V., Quan E. M., and Weissman J. S. (2006) A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359 [DOI] [PubMed] [Google Scholar]
- 50. Cormier J. H., Pearse B. R., and Hebert D. N. (2005) Yos9p: a sweet-toothed bouncer of the secretory pathway. Mol. Cell 19, 717–719 [DOI] [PubMed] [Google Scholar]
- 51. Kim W., Spear E. D., and Ng D. T. (2005) Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell 19, 753–764 [DOI] [PubMed] [Google Scholar]
- 52. Su W., Liu Y., Xia Y., Hong Z., and Li J. (2012) The Arabidopsis homolog of the mammalian OS-9 protein plays a key role in the endoplasmic reticulum-associated degradation of misfolded receptor-like kinases. Mol. Plant 5, 929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brodsky J. L. (2007) The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 404, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hosokawa N., Kato K., and Kamiya Y. (2010) Mannose 6-phosphate receptor homology domain-containing lectins in mammalian endoplasmic reticulum-associated degradation. Methods Enzymol. 480, 181–197 [DOI] [PubMed] [Google Scholar]
- 55. Ruggiano A., Foresti O., and Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Needham P. G., Mikoluk K., Dhakarwal P., Khadem S., Snyder A. C., Subramanya A. R., and Brodsky J. L. (2011) The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J. Biol. Chem. 286, 43611–43621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Donnelly B. F., Needham P. G., Snyder A. C., Roy A., Khadem S., Brodsky J. L., and Subramanya A. R. (2013) Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J. Biol. Chem. 288, 13124–13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rizzo F., and Staub O. (2015) NEDD4–2 and salt-sensitive hypertension. Curr. Opin. Nephrol. Hypertens. 24, 111–116 [DOI] [PubMed] [Google Scholar]
- 59. Mori T., and Cowley A. W. Jr. (2004) Renal oxidative stress in medullary thick ascending limbs produced by elevated NaCl and glucose. Hypertension 43, 341–346 [DOI] [PubMed] [Google Scholar]
- 60. Brown M. K., and Naidoo N. (2012) The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cohen F. E., and Kelly J. W. (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426, 905–909 [DOI] [PubMed] [Google Scholar]
- 62. Chaudhuri T. K., and Paul S. (2006) Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 273, 1331–1349 [DOI] [PubMed] [Google Scholar]
- 63. Amaral M. D. (2011) Targeting CFTR: how to treat cystic fibrosis by CFTR-repairing therapies. Curr. Drug Targets 12, 683–693 [DOI] [PubMed] [Google Scholar]
- 64. Chaari A., Hoarau-Véchot J., and Ladjimi M. (2013) Applying chaperones to protein-misfolding disorders: molecular chaperones against α-synuclein in Parkinson's disease. Int. J. Biol. Macromol. 60, 196–205 [DOI] [PubMed] [Google Scholar]
- 65. Brown C. R., Hong-Brown L. Q., Biwersi J., Verkman A. S., and Welch W. J. (1996) Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Welling P. A. (2014) Rare mutations in renal sodium and potassium transporter genes exhibit impaired transport function. Curr. Opin. Nephrol. Hypertens. 23, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Delpire E., and Mount D. B. (2002) Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 64, 803–843 [DOI] [PubMed] [Google Scholar]
- 68. Delpire E. (2000) Cation-chloride cotransporters in neuronal communication. News Physiol. Sci. 15, 309–312 [DOI] [PubMed] [Google Scholar]