

Abstract
Inhibiting class I histone deacetylases (HDACs) increases energy expenditure, reduces adiposity, and improves insulin sensitivity in obese mice. However, the precise mechanism is poorly understood. Here, we demonstrate that HDAC1 is a negative regulator of the brown adipocyte thermogenic program. The Hdac1 level is lower in mouse brown fat (BAT) than white fat, is suppressed in mouse BAT during cold exposure or β3-adrenergic stimulation, and is down-regulated during brown adipocyte differentiation. Remarkably, overexpressing Hdac1 profoundly blocks, whereas deleting Hdac1 significantly enhances, β-adrenergic activation-induced BAT-specific gene expression in brown adipocytes. β-Adrenergic activation in brown adipocytes results in a dissociation of HDAC1 from promoters of BAT-specific genes, including uncoupling protein 1 (Ucp1) and peroxisome proliferator-activated receptor γ co-activator 1α (Pgc1α), leading to increased acetylation of histone H3 lysine 27 (H3K27), an epigenetic mark of gene activation. This is followed by dissociation of the polycomb repressive complexes, including the H3K27 methyltransferase enhancer of zeste homologue (EZH2), suppressor of zeste 12 (SUZ12), and ring finger protein 2 (RNF2) from (and concomitant recruitment of H3K27 demethylase ubiquitously transcribed tetratricopeptide repeat on chromosome X (UTX) to) Ucp1 and Pgc1α promoters, leading to decreased H3K27 trimethylation, a histone transcriptional repression mark. Thus, HDAC1 negatively regulates the brown adipocyte thermogenic program, and inhibiting Hdac1 promotes BAT-specific gene expression through a coordinated control of increased acetylation and decreased methylation of H3K27, thereby switching the transcriptional repressive state to the active state at the promoters of Ucp1 and Pgc1α. Targeting HDAC1 may be beneficial in prevention and treatment of obesity by enhancing BAT thermogenesis.
Keywords: epigenetics, histone deacetylase 1 (HDAC1), histone methylation, obesity, uncoupling protein, H3K27, UCP1, brown adipocytes, histone deacetylation, thermogenesis
Introduction
Obesity develops when a persistent imbalance between energy intake and energy expenditure occurs (1). Whereas white adipose tissue (WAT)3 is involved in energy storage, the role of brown adipose tissue (BAT) is to dissipate energy as heat due to its unique expression of uncoupling protein 1 (Ucp1) (2–4). In rodents, there exist two types of brown adipocytes. Traditional brown adipocytes are located in discrete areas, whereas “inducible” beige adipocytes are dispersed in WAT (5–8) and can be induced by cold exposure or β3-adrenergic receptor activation (9–13). The ability of brown/beige adipocytes to produce adaptive thermogenesis depends on the unique expression of UCP1 in the inner mitochondrial membrane, which serves to uncouple oxidative phosphorylation from ATP synthesis, thereby profoundly increasing energy expenditure (2–4). Recent reports demonstrate that adult humans also possess metabolically active brown fat; the amount of brown fat is inversely correlated with body weight but positively correlated with energy expenditure (14–16). This important discovery provides new insight into the mechanisms regulating energy homeostasis in adult humans and suggests that increasing functional brown/beige adipocytes in humans is a novel and promising target in treating obesity.
Although the genome is fixed and identical in all cells, the epigenome, the combination of all genome-wide DNA and chromatin modifications, is continuously modified in response to developmental, environmental, physiological, and pathological cues (17–19). Epigenetic modifications, including DNA methylation, histone acetylation, and methylation, result in organization of the chromatin structure on different hierarchal levels, which regulate gene expression (17–19). Recent evidence suggests that epigenetic mechanisms have emerged as an important link between environmental factors and obesity (20–24). For example, Ucp1 promoter activity is regulated by changes in DNA methylation status (25). The histone H3 Lys-9 demethylase JHDM2a directly regulates Ucp1 expression, and genetic deletion of Jhdm2a in mice results in obesity (26). This is a newly emerging research area; however, much remains to be discovered regarding how epigenetic mechanisms regulate metabolism and energy homeostasis.
Histone acetylation and deacetylation are regulated by the balanced action of histone acetyltransferases and histone deacetylases (HDACs) (17–19). HDACs consist of four major classes: class I (HDAC1, -2, -3, and -8), class II (HDAC4, -5, -6, -7, -9, and -10), class III (SIRT1 to -7), and class IV (HDAC11). The class I, II, and IV HDACs act to remove acetyl groups from lysine residues in histone as well as cellular proteins, thereby regulating gene expression and cellular protein activity, whereas class III HDACs (Sirt1–Sirt7) form a structurally distinct class of NAD-dependent enzymes and can be inhibited by nicotinamide (17, 18). Recent data suggest that HDACs have emerged as important players in the regulation of energy and glucose homeostasis (27). For example, it is reported that class I HDAC inhibitor (HDACi) MS-275 ameliorates obesity and diabetes in animal models through stimulation of oxidative phosphorylation and mitochondrial function in muscle and fat (28). However, the precise mechanism by which class I HDACi exerts these effects is poorly understood.
In the current study, we have identified Hdac1 as a prominent epigenetic target in regulating the thermogenic program in brown adipocytes. Using loss- and gain-of-function approaches, we demonstrated that Hdac1 deficiency activated, whereas Hdac1 overexpression repressed, transcription of brown adipocyte-specific gene expression through regulating the acetylation and methylation status of histone H3 lysine 27 (H3K27) on promoter and enhancer regions of Ucp1 and peroxisome proliferator-activated receptor γ (Pparγ) co-activator 1-α (Pgc1α). Thus, our data suggest that epigenetics plays an important role in brown adipocyte thermogenesis, and Hdac1 may be an important regulator during this process.
Materials and Methods
Mice
C57BL/6J (B6) and A/J mice (Jackson Laboratories, Bar Harbor, ME) were housed with a 12/12-h light/dark cycle in temperature- and humidity-controlled rooms with free access to water and food. To study the role of Hdac1 in BAT thermogenic function, 7–8-week-old mice were exposed to cold conditions (4 °C) or intraperitoneally injected with β3-adrenergic agonist (CL316243 (Sigma-Aldrich), dose: 1 mg/kg) for up to 7 days. Mice were then sacrificed, and BAT was harvested for gene expression or chromatin immunoprecipitation (ChIP) analysis. All aspects of animal care were approved by Georgia State University's Animal Care and Use Committee.
Cell Culture
All cells were maintained at 37 °C with 5% CO2. Immortalized brown preadipocytes BAT1 (kindly provided by Dr. Patrick Seale, University of Pennsylvania) (29, 30) and HIB-1B (31, 32) were maintained in growth medium (DMEM/F-12 (BAT1) or DMEM (HIB-1B) containing 10% fetal bovine serum and 1% penicillin/streptomycin). Brown adipocyte differentiation was induced as described previously (29, 30). Briefly, cells were grown into 90% confluence in growth medium and were then induced to differentiate with a differentiation medium (growth medium plus 20 nm insulin, 1 nm triiodothyronine, 125 μm indomethacin, 500 μm isobutylmethylxanthine, and 0.5 μm dexamethasone). After 2 days, cells were cultured in maintenance medium (growth medium plus 20 nm insulin and 1 nm triiodothyronine). At day 6, all of the cells were differentiated as described previously (29, 30). To induce the thermogenic program, brown adipocytes were treated with 1 μm isoproterenol or norepinephrine (NE) for 3–4 h.
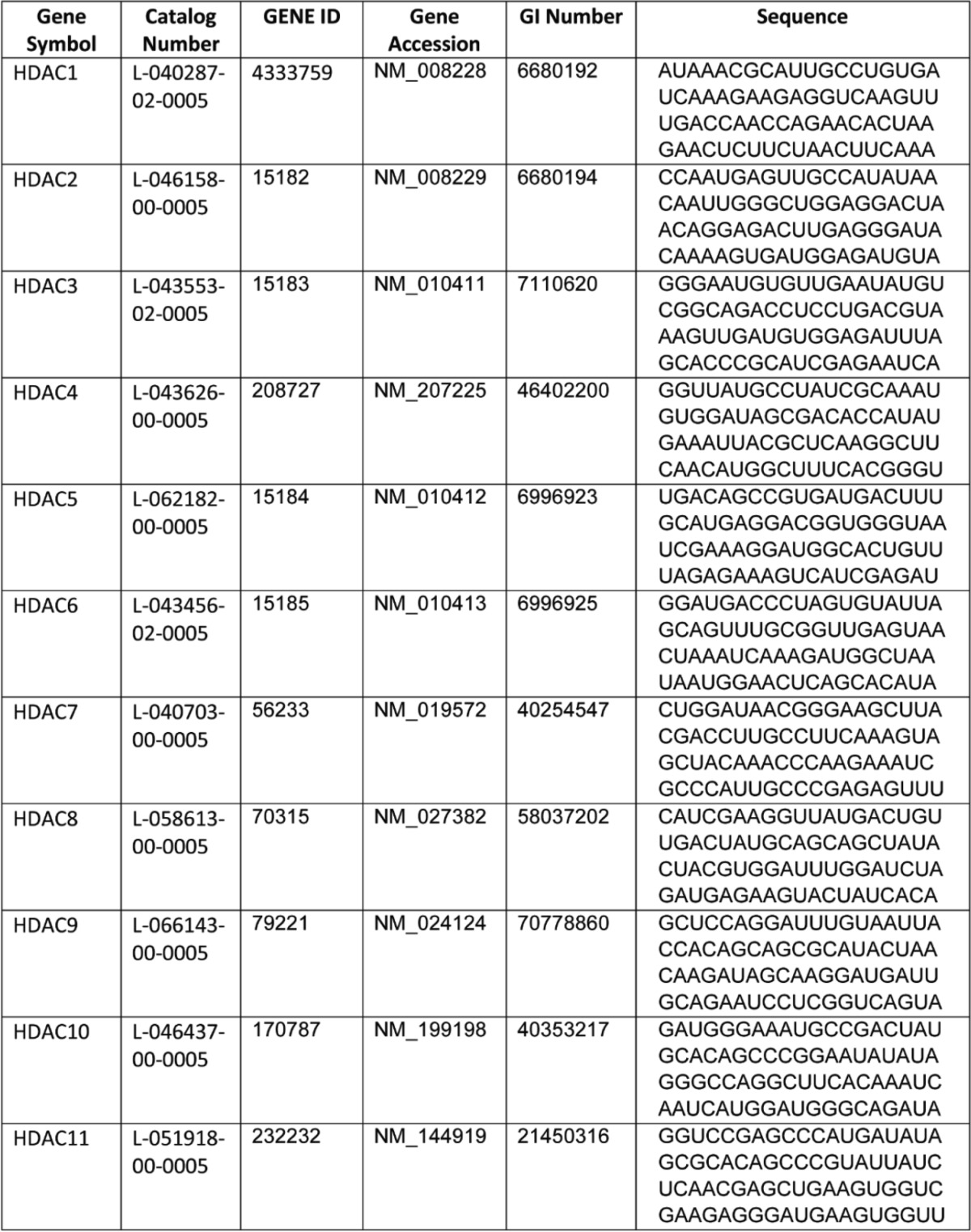
Small Interfering RNA (siRNA) and Plasmid DNA Transfection
The MGC fully sequenced mouse cDNA expression plasmids for Hdac1 (Clone ID 4976514), enhancer of zeste homologue (Ezh2) (Clone ID 3586689), suppressor of zeste 12 (Suz12) (Clone ID 6821922), ring finger protein 2 (Rnf2) (Clone ID 4021046), the ON-TARGET plus mouse Hdac1 siRNA-SMART pool (L-040287-02-0005), and Hdac1–Hdac11 siRNAs for our initial screening were purchased from GE Healthcare. The sequences for Hdac1–Hdac11 siRNAs are listed in Table 1. All other plasmids were in mammalian expression vector pSPORT6 except for Suz12, which was in a non-expressing vector, pXY-Asc. To clone Suz12 into pSPORT6 vector, PacI and SalI were used to release the Suz12 cDNA insert from pXY-Asc and subcloned into pSPORT6. The overexpressing plasmids or siRNAs were transfected into BAT1 or HIB-1B brown adipocytes by Amaxa Nucleofector II Electroporator (Lonza) using Amaxa cell line nucleofector kit L according to the manufacturer's instructions (Lonza). Briefly, at days 4–6 of differentiation, cells (2 × 106 cells/sample) were trypsinized and centrifuged at 90 × g for 5 min at room temperature. Cell pellet was resuspended in nucleofector solution (100 μl/sample) with 2 μg of plasmid DNA or 20 pmol of siRNA and seeded into 24-well plates. Cells were treated with NE or isoproterenol 2 days after transfection.
TABLE 1.
Sequences of siRNA smart pools for individual HDACs
For the co-immunoprecipitation (co-IP) experiment, HEK293T cells were electroporated with pSPORT6 vector or co-transfected with Hdac1, Ezh2, Suz12, and Rnf2 cDNA for 2 days. Cell lysates from transfected HEK293T cells or endogenous BAT1 brown adipocytes were collected to detect protein interactions by co-IP.
IP and Immunoblotting
IP and immunoblotting were performed as we described previously (33–36). Briefly, tissue or cell samples were homogenized in a modified radioimmune precipitation assay lysis buffer containing 50 mm Tris-HCl, 1 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 150 mm NaCl, 1 mm sodium fluoride, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, 1% protease inhibitor mixture (Sigma), and 1% phosphatase inhibitor mixture (Sigma). Cell homogenates were incubated on ice for 45 min to solubilize all proteins, and insoluble portions were removed by centrifugation at 14,000 × g at 4 °C for 15 min. Two mg of cell lysates was incubated overnight with the appropriate antibodies (Table 2) and protein A/G-agarose (Santa Cruz Biotechnology, Inc.) at 4 °C with constant gentle mixing. Agarose beads were collected by centrifugation, washed with ice-cold radioimmune precipitation assay lysis buffer and then with phosphate-buffered saline, and then boiled in Laemmli sample buffer. Proteins from IP or whole cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. The transferred membranes were blocked, washed, and incubated with various primary antibodies (Table 2), followed by incubation with Alexa Fluor 680-conjugated secondary antibodies (Life Technologies). The fluorescent signal was visualized with a LI-COR Imager System (LI-COR Biosciences, Lincoln, NE).
TABLE 2.
Antibodies used in immunoblotting and ChIP-quantitative PCR
| Antibody | Company | Catalog no. |
|---|---|---|
| UCP1 | Abcam | ab23841 |
| UTX | Abcam | ab36938 |
| H3K27ac | Abcam | ab4729 |
| HDAC1 | Cell Signaling Technology | 5356S |
| H3K27me3 | Cell Signaling Technology | 9733P |
| EZH2 | Cell Signaling Technology | 5246S |
| SUZ12 | Cell Signaling Technology | 3737S |
| RNF2 | Cell Signaling Technology | 5694S |
| H3K14ac | Millipore | 07-353 |
| H3K9ac | Millipore | 07-352 |
| H3K4ac | Millipore | 07-539 |
| Tubulin | Santa Cruz | sc-53646 |
Quantitative Real-time RT-PCR
Total RNA was extracted with TRI Reagent according to the manufacturer's instructions (Molecular Research Center, Cincinnati, OH). RNA expression was quantified by real-time RT-PCR with TaqMan one-step RT-PCR Master mix (Life Technologies, Inc.) using a Stratagene Mx3000p thermocycler (Stratagene, La Jolla, CA) and normalized to cyclophilin. The primer and probe pairs used in the assays were purchased from Applied Biosystems or designed using Primer Express software (Life Technologies) (Tables 3 and 4).
TABLE 3.
Primer/probe sequences for gene expression experiments
| Taqman | Primer |
Probe | |
|---|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | ||
| Cyclophilin | GGTGGAGAGCACCAAGACAGA | GCCGGAGTCGACAATGATG | ATCCTTCAGTGGCTTGTCCCGGCT |
| Ucp1 | CACCTTCCCGCTGGACACT | CCCTAGGACACCTTTATACCTAATGG | AGCCTGGCCTTCACCTTGGATCTGA |
| Pparα | GACAAGGCCTCAGGGTACCA | GCCGAATAGTTCGCCGAAA | AGCCCTTACAGCCTTCACATGCGTGA |
| Pgc1α | CATTTGATGCACTGACAGATGGA | CCGTCAGGCATGGAGGAA | CCGTGACCACTGACAACGAGGCC |
| Pgc1β | AGGAAGCGGCGGGAAA | CTACAATCTCACCGAACACCTCAA | AGAGATTTCGAATGTATACCACACGGCCTTCA |
| aP2 | GCGTGGAATTCGATGAAATCA | CCCGCCATCTAGGGTTATGA | GCTCTTCACCTTCCTGTCGTCTGCG |
| Srebp1c | GACCACGGAGCCATGGA | GGGAAGTCACTGTCTTGGTTGTT | TGCACATTTGAAGACATGCTCCAGCTCAT |
| C/ebpβ | CCAAGAAGACGGTGGACAAG | GTGCTGCGTCTCCAGGTT | CCGCATCTTGTACTCGTCGCTCAG |
| C/EBPα | TATAGACATCAGCGCCTACATCGA | GTCGGCTGTGCTGGAAGAG | CCGCCTTCAACGACGAGTTCC |
TABLE 4.
Primer/probe sets for gene expression experiments
| Gene symbol | Company | Catalog no. |
|---|---|---|
| Hdac1 | Applied Biosystems | Mm02391771-g1 |
| Prdm16 | Applied Biosystems | Mm00712556-m1 |
| Dio2 | Applied Biosystems | Mm00515664-m1 |
| Acox1 | Applied Biosystems | Mm01246834-m1 |
| Cox1 | Applied Biosystems | Mm04225243-g1 |
| Eva1 | Applied Biosystems | Mm00468397-m1 |
| Otop1 | Applied Biosystems | Mm00554705-m1 |
| Fgf21 | Applied Biosystems | Mm00840165-g1 |
| Cpt1b | Applied Biosystems | Mm00487191-g1 |
| Cidea | Applied Biosystems | Mm00432554-m1 |
| Pparγ | Applied Biosystems | Mm00440945-m1 |
ChIP Assay
The ChIP assay was performed using a ChIP assay kit (Upstate) as we described previously (37). For tissue ChIP assays, tissue samples were cut into small pieces and fixed with 1% of formaldehyde. The samples were then homogenized in cell lysis buffer (5 mm PIPES, 85 mm KCl, and 0.5% Nonidet P-40, supplemented with protease inhibitors, pH 8.0) using a Dounce homogenizer to isolate nuclei. The nuclei were resuspended in nuclei lysis buffer (50 mm Tris-HCl, 10 mm EDTA, and 1% SDS, supplemented with protease inhibitors, pH 8.1) and sonicated to shear genomic DNA to an average fragment length of 200–1,000 bp with a Diagenode Bioruptor (Diagenode, Denville, NJ). Lysates were centrifuged, and the supernatants were collected. Fifty μl of each sample was removed as the input control. The supernatants underwent overnight immunoprecipitation, elution, reverse cross-linking, and protease K digestion, according to the manufacturer's manual. A mock immunoprecipitation without antibody was also included for each sample. Eluted DNA was analyzed by real-time PCR using SYBR Green quantitative PCR (Life Technologies). Primer sequences used in this study were as follows: Ucp1 proximal promoter, 5′-CCCACTAGCAGCTCTTTGGA-3′ and 5′-CTGTGGAGCAGCTCAAAGGT; Ucp1 enhancer region, 5′-CTCCTCTACAGCGTCACAGAGG-3′ and 5′-AGTCTGAGGAAAGGGTTGA-3′; Pgc1α cAMP-response element (CRE) region, 5′-CAAAGCTGGCTTCAGTCACA-3′ and 5′-AAAAGTAGGCTGGGCTGTCA-3′.
Statistical Analysis
Data were expressed as mean ± S.E. Statistical tests were performed using SPSS software (version 16.0, SPSS Inc., Chicago, IL). One-way analysis of variance followed by the Student-Newman-Keuls test was used to determine multiple comparisons. Statistical significance was accepted at p < 0.05.
Results
Hdac1 Is Enriched in WAT Versus BAT and Is Down-regulated in BAT during β-Adrenergic Stimulation and during Brown Adipocyte Differentiation
Recent studies reported that class I but not class II HDACi enhanced whole body energy expenditure and attenuated high fat diet-induced insulin resistance through increased mitochondrial biogenesis in skeletal muscle and adipose tissues (28). However, little is known about whether these effects are exerted directly through activating brown fat thermogenesis and which HDAC is responsible for these beneficial effects. Thus, we first knocked down individual HDACs (Hdac1–Hdac11) in brown adipocyte cell line HIB-1B cells using an siRNA approach. Interestingly, although knockdown of other HDACs exerted minimal effects, reducing the expression of the class I HDAC family member Hdac1 by siRNA knockdown significantly enhanced NE-stimulated Ucp1 expression (Fig. 1A). This prompted us to investigate the role of HDAC1 in regulating the brown adipocyte thermogenic program.
FIGURE 1.

The expression pattern of HDAC1. A, reducing Hdac1 expression by siRNA knockdown in HIB-1B brown adipocytes up-regulates NE-stimulated Ucp1 expression. HIB-1B cells were transfected with scramble siRNA or siRNAs targeting individual HDACs. After 48 h, cells were treated with or without 1 μm norepinephrine for 4 h, and RNA was isolated for gene expression measurements. Class I HDACs are shown in blue. B, UCP1 and HDAC1 protein levels in BAT and epididymal (Epi) WAT from C57BL/6J (B6) mice. C and D, UCP1 and HDAC1 RNA (C) and protein (D) levels in BAT1 and HIB-1B brown adipocytes during differentiation. E and F, Hdac1 expression in A/J mouse BAT tissues exposed to 4 °C at the indicated time (E) or treated with β3-agonist CL-316,243 for 7 days. Data are expressed as mean ± S.E. (error bars). *, p < 0.05 versus undifferentiated samples (C), time 0 (E), or control (Ctrl) (F).
We first measured the HDAC1 expression pattern in brown and white adipose tissues. As expected, UCP1 protein was enriched in BAT but not detectable in epididymal WAT (Epi (WAT)) in adult mice housed at ambient temperature (Fig. 1B). Interestingly, in contrast to UCP1 expression, HDAC1 protein level was enriched in WAT but much lower in BAT (Fig. 1B). BAT1 and HIB-1B brown adipocyte differentiation was marked with significantly increased UCP1 mRNA and protein levels (Fig. 1, C and D), which were accompanied by decreased mRNA and protein levels of HDAC1 (Fig. 1, C and D).
It is well documented that cold temperature triggers sympathetic discharge, leading to the release of NE in BAT and WAT (3, 38). Thus, we tested whether Hdac1 expression in BAT was regulated by sympathetic stimulation. A/J mice were exposed to cold (4 °C) or intraperitoneally injected with β3-adrenergic agonist (CL-316,243) for up to 7 days. Hdac1 expression was profoundly reduced in BAT after 6 h of cold exposure and tended to stay reduced up to 7 days of cold exposure (Fig. 1E). A similar reduction of Hdac1 expression was also observed in BAT after CL-316,243 injection for 7 days (Fig. 1F). These data indicate that decreased Hdac1 may be a marker of mature brown adipocytes and that Hdac1 may negatively regulate the BAT thermogenic program.
Hdac1 Regulates Brown Adipocyte Thermogenic Gene Expression
To investigate the role of Hdac1 in the regulation of brown fat-specific gene expression, we performed gain- or loss-of-function experiments in differentiated BAT1 cells. It has been reported that Hdac1 regulates the early steps of adipocyte differentiation (39). Thus, to avoid the confounding effects of Hdac1 on differentiation, we have focused on the role of Hdac1 in mature brown adipocyte gene expression by knocking down or overexpressing Hdac1 in BAT1 cells after 4–6 days of differentiation. Hdac1 mRNA in knockdown cells was reduced by 80%, as measured by real-time RT-PCR, and HDAC1 protein expression was also significantly decreased, as assessed by immunoblotting (Fig. 2A). As expected, knocking down Hdac1 in mature BAT1 cells did not affect the mRNA levels of general adipocyte differentiation markers, including Pparγ, CCAAT/enhancer-binding protein α (C/ebpα), sterol regulatory element-binding protein 1C (Srebp1c), adipocyte protein 2 (aP2), and adiponectin (AdipoQ) (Fig. 2B). Remarkably, Hdac1 knockdown in BAT1 cells significantly increased basal and isoproterenol-stimulated expression of brown adipocyte-specific genes, including Ucp1, Pgc1α (Fig. 2C), Pgc1β, PR domain-containing protein 16 (Prdm16), Pparα, type 2 deiodinase (Dio2), acyl-CoA oxidase 1 (Acox1), cytochrome c oxidase 1 (Cox1), epithelial V-like antigen 1 (Eva1), and otopetrin 1 (Otop1) (Fig. 2D). In addition, isoproterenol significantly stimulated UCP1 protein expression in BAT1 cells, which was further enhanced by Hdac1 knockdown (Fig. 2E).
FIGURE 2.

Reducing HDAC expression promotes brown-specific gene expression in BAT1 brown adipocytes. BAT1 brown adipocytes were transfected with scramble or Hdac1 siRNA. Two days later, cells were treated with isoproterenol (Isop; 1 μm) for 3 h, and RNA and protein were isolated for gene expression and protein level measurements. A, HDAC1 RNA and protein levels in scramble or Hdac1 siRNA-transfected cells. B–D, the mRNA levels of various genes in scramble or Hdac1 siRNA-transfected cells. n = 6–8/group. E, UCP1 protein levels in scramble or Hdac1 siRNA-transfected cells. Left, UCP1 immunoblot; right, quantitation of UCP1 protein levels normalized to α-tubulin. n = 3/group. Data are expressed as mean ± S.E. (error bars). *, p < 0.05. n.s., not statistically significant.
We then overexpressed Hdac1 in BAT1 brown adipocytes, as shown by increased HDAC1 protein expression (Fig. 3A). Overexpression of Hdac1 in BAT1 brown adipocytes significantly suppressed basal and/or isoproterenol-stimulated brown adipocyte-specific gene expression, including Ucp1, Pgc1α, Pgc1β, Prdm16, C/ebpβ, fibroblast growth factor 21 (Fgf21), Pparα, carnitine palmitoyltransferase 1B (Cpt1b), Acox1, Cox1, Otop1, and cell death-inducing DNA fragmentation factor α subunit-like effector A (Cidea) (Fig. 3, B and C).
FIGURE 3.

Hdac1 overexpression attenuates mRNA expression of brown-specific genes. BAT1 brown adipocytes were transfected with pSPORT vector or Hdac1 cDNA. Two days later, cells were treated with isoproterenol (isop; 1 μm) for 3 h, and RNA and protein were isolated for gene expression and HDAC1 protein level measurements. A, HDAC1 protein levels in BAT1 cells transfected with pSPORT vector or Hdac1 cDNA. B and C, expression levels of BAT-specific genes in BAT1 cells transfected with pSPORT vector or Hdac1 cDNA. Data are expressed as mean ± S.E. (error bars) n = 6–8/group. *, p < 0.05. n.s., not statistically significant.
Similar results were observed in HIB-1B brown adipocyte cell line. In HIB-1B cells with Hdac1 siRNA knockdown, Hdac1 mRNA was reduced by more than 80% (Fig. 4A), and knocking down Hdac1 significantly increased basal and/or NE-stimulated brown adipocyte-specific gene expression, including Ucp1 (Fig. 4B), Pgc1α, Pgc1β, Cox1, Acox1, Cidea, Prdm16, Cpt1b, and Pparα (Fig. 4, C and D). Similarly, Hdac1 overexpression significantly reduced NE-stimulated Ucp1 expression in HIB-1B cells (Fig. 4E). These data demonstrate that Hdac1 is a negative regulator of the thermogenic program in mature brown adipocytes.
FIGURE 4.

HDAC1 negatively regulates brown-specific gene expression in HIB-1B brown adipocytes. A–D, reducing Hdac1 expression by siRNA knockdown in HIB-1B cells promotes brown-specific gene expression. HIB-1B brown adipocytes were transfected with scramble or Hdac1 siRNA. Two days later, cells were treated with 1 μm NE for 4 h, and RNA was isolated for gene expression analysis, including Hdac1 (A); Ucp1 (B); Pgc1α, Pgc1β, Cox1, and Acox1 (C); and Cidea, Prdm16, Cpt1b, and Pparα (D). E, Hdac1 overexpression attenuates Ucp1 mRNA levels. HIB-1B cells were transfected with pSPORT vector or Hdac1-overexpressing plasmids. Two days later, cells were treated with 1 μm NE for 4 h, and mRNA levels of Ucp1 were measured by RT-PCR. Data are expressed as mean ± S.E. (error bars). n = 6–8 in A–D and 4–6 in E. *, p < 0.05. n.s., not statistically significant.
Hdac1 Deficiency Mediates the Beneficial Effects of Class I HDACi on Improving Metabolic Phenotypes
It is reported that the pan-HDACi SAHA and class I HDACi MS-275, but not class II HDACi MC-1568, ameliorate obesity and diabetes in animal models, possibly through stimulation of oxidative phosphorylation and mitochondrial function in muscle and fat (28). To our surprise, treating BAT1 brown adipocytes with the pan-HDACi TSA and SAHA resulted in complex effects on brown fat-specific gene expression. Whereas TSA and SAHA enhanced isoproterenol-stimulated expression of genes involved in mitochondria oxidative activity, such as Pgc1α and Acox1, they significantly suppressed isoproterenol-stimulated expression of other BAT-specific genes, including Ucp1, Prdm16, Pparγ, and Otop1 (Fig. 5, A and B). In contrast, the class I HDACi MS-275 significantly enhanced isoproterenol-stimulated BAT-specific gene expression, including Ucp1, Elovl3, and Pgc1α, whereas it had minimal effects on Prdm16 and Pparγ expression (Fig. 5C).
FIGURE 5.

HDAC1 mediates the effects of class I HDACi MS-275 on BAT-1 brown adipocyte gene expression. A–C, isoproterenol (Isop)-stimulated gene expression in BAT1 cells treated with different concentration of pan-HDACi TSA (A), SAHA (B), and the class I HDACi MS-275 (C). D, isoproterenol-stimulated gene expression in BAT1 cells treated with MS-275 (5 μm) or Hdac1 siRNA individually or in combination. Data are expressed as mean ± S.E. (error bars). n = 6. *, p < 0.05.
We also tested whether Hdac1 might be the molecular target of class I HDACi MS-275 in regulating BAT-specific gene expression in BAT1 cells. As expected, MS275 or Hdac1 siRNA knockdown enhanced isoproterenol-stimulated gene expression of Ucp1 and Pgc1α in BAT1 cells to a similar extent when treated individually; however, they did not exert any additive effects on these gene expressions when treated in combination in BAT1 cells (Fig. 5D). Our data suggest that Hdac1 may be the molecular target of the class I HDACi MS-275 in stimulating brown fat function.
Sympathetic Activation in Brown Adipocytes Reduces HDAC1 Binding and Increases H3K27 Acetylation at Ucp1 and Pgc1α Promoters
HDACs repress gene expression by removing acetyl groups from lysine residues in histone proteins at specific gene promoters (18). We reasoned that if HDAC1 indeed negatively regulates brown fat functions, sympathetic activation in BAT may trigger HDAC1 dissociation from promoter regions of key genes regulating brown fat thermogenesis, which should allow transcriptional activation of these genes and subsequently the full activation of the brown adipocyte thermogenic program. UCP1 is responsible for mitochondrial uncoupling in BAT (2–4), whereas PGC1α is the central regulator of brown adipocyte thermogenesis (40, 41). A ∼220-bp upstream enhancer element along with the proximal promoter that is closer to the transcriptional start site have been identified at the Ucp1 promoter that mediates sympathetic stimulated and tissue-specific expression of Ucp1 (42–44). In addition, Pgc1α expression is highly induced via the β-adrenergic receptor-activated protein kinase A (PKA)/CRE-binding protein (CREB) pathway (45, 46) through the CRE cis-element on the Pgc1α promoter (28, 47). We therefore examined the binding of HDAC1 at the enhancer and proximal promoter regions of Ucp1 and the CRE cis-element on the Pgc1α promoter (Fig. 6A) upon sympathetic activation. As expected, in BAT of mice treated with β3-adrenergic agonist CL-243,316, binding of HDAC1 was significantly reduced at the enhancer and proximal promoter regions of Ucp1 (Fig. 6, B and C) and the CRE cis-element region at the Pgc1α promoter (Fig. 6D). In addition, isoproterenol treatment also significantly reduced HDAC1 binding to these promoter/enhancer regions in differentiated BAT1 brown adipocytes (Fig. 6E). These data indicate that β-adrenergic stimulation in brown adipocytes dissociates HDAC1 from promoters of key genes regulating brown adipocyte thermogenesis, including Ucp1 and Pgc1α. This may facilitate subsequent chromatin modifications and DNA unwinding, which can eventually lead to increased accessibility to transcription factor binding and gene activation (17–19).
FIGURE 6.

HDAC1 regulates H3K27ac at Pgc1α and Ucp1 promoters. A, schematic illustration of promoter/enhancer regions of Ucp1 and Pgc1α genes. B–D, β3-adrenergic agonist CL-316,243 treatment dissociates HDAC1 from Ucp1 and Pgc1α promoters in BAT of A/J mice. A/J mice were treated with CL-316.243 for the indicated time, and BAT was collected for a ChIP assay to measure HDAC1 binding to Ucp1 and Pgc1α promoters as described under “Materials and Methods.” E, isoproterenol treatment dissociates HDAC1 from Ucp1 and Pgc1α promoters in BAT1 brown adipocytes. BAT1 cells were treated with isoproterenol (Isop; 1 μm) for 3 h. Cells were collected, and ChIP assays were performed to measure HDAC1 binding to Ucp1 and Pgc1α promoters. F and G, H3K27ac levels in Ucp1 and Pgc1α promoters in BAT1 brown adipocytes. In F, BAT1 cells were transfected with scramble or Hdac1 siRNA for 2 days; in G, BAT1 cells were transfected with pSPORT6- or pSPORT6-HDAC1-overexpressing plasmids for 2 days. Cells were then treated with 1 μm isoproterenol for 3 h. A ChIP assay was performed as described under “Materials and Methods.” H, histone H3 lysine 14 (H3K14ac) and histone H3 lysine 9 (H3K9ac) levels in Ucp1 and Pgc1α promoters in BAT1 cells transfected with scramble or Hdac1 siRNA. Data are expressed as mean ± S.E. (error bars). n = 4–6. *, p < 0.05. n.s., not statistically significant.
Acetylation at histone H3 lysine 27 (H3K27ac) results in transcriptional activation (48). We found that isoproterenol treatment in BAT1 cells significantly increased H3K27ac levels at the enhancer and proximal promoter regions of Ucp1 and the CRE region at the Pgc1α promoter (Fig. 6, F and G). Importantly, reducing HDAC1 expression by siRNA knockdown induced a similar increase in H3K27ac levels and further enhanced isoproterenol-stimulated increase in H3K27ac levels (Fig. 6F), whereas Hdac1 overexpression completely blocked isoproterenol-induced increase in H3K27ac levels at these promoter/enhancer regions (Fig. 6G).
On the other hand, the acetylation levels at histone H3 lysine 14 (H3K14ac) and histone H3 lysine 9 (H3K9ac), two other lysine residues that can be modified by acetylation (18, 49), did not exhibit similar responsiveness to Hdac1 knockdown and/or isoproterenol stimulation in BAT1 cells (Fig. 6H). Thus, our data suggest that H3K27 may be the major target of HDAC1 in brown adipocytes regulating brown-specific gene expression.
Hdac1 Regulates Ucp1 and Pgc1α Expression by Further Modifying H3K27 Methylation
Except for acetylation, histone lysine residues can also be mono-, di-, and trimethylated and are associated with either gene repression or activation, depending on the lysine residues that are methylated and the degree of methylation. Trimethylation of H3K27 (H3K27me3) is a hallmark of gene repression, whereas trimethylation of histone H3 Lys-4 marks transcriptional activation (17–19). Importantly, histone acetylation and methylation mutually affect each other in the regulation of transcriptional process (49). We thus tested whether modulation of histone acetylation by HDAC1 led to further alterations in histone methylation. ChIP assay analysis demonstrated that isoproterenol treatment in BAT1 brown adipocytes resulted in a significant decrease in H3K27me3 levels at Ucp1 and Pgc1α promoters (Fig. 7, A and B). Reducing Hdac1 expression by siRNA knockdown mimicked isoproterenol's effect by reducing H3K27me3 levels at these promoter/enhancer regions to a similar extent as isoproterenol and did not further decrease isoproterenol-suppressed H3K27me3 levels (Fig. 7A). In contrast, Hdac1 overexpression significantly blocked isoproterenol-suppressed H3K27me3 levels at these promoter/enhancer regions (Fig. 7B).
FIGURE 7.

HDAC1 regulates trimethylation of H3K27me3 at Ucp1 and Pgc1α promoters through coordinated regulation of EZH2 and UTX binding to BAT-specific promoters. A–F, H3K27me3 levels (A and B), EZH2 (C and D), and UTX binding (E and F) at Ucp1 and Pgc1α promoters in BAR1 cells. BAT1 brown adipocytes were transfected with scramble or Hdac1 siRNA (A, C, and E) or pSPORT6- or pSPORT6-HDAC1-overexpressing plasmids (B, D, and F). Two days later, cells were treated with or without isoproterenol (Isop; 1 μm) for 3 h, and a ChIP assay was performed as described under “Materials and Methods.” Data are expressed as mean ± S.E. (error bars). n = 4–6. *, p < 0.05. n.s., not statistically significant.
H3K27 methylation is dynamically regulated by both histone methyltransferases and demethylases (18, 50, 51). EZH2 is a methyltransferase that specifically di- and trimethylates H3K27 (18, 50, 51), whereas lysine-specific demethylase 6A (KDM6A)/ubiquitously transcribed tetratricopeptide repeat on chromosome X (UTX) is a di- and trimethyl-H3K27 demethylase (52). A ChIP assay demonstrated that isoproterenol treatment in BAT1 brown adipocytes resulted in a significant decrease in EZH2 binding at Ucp1 and Pgc1α promoters (Fig. 7, C and D). Reducing Hdac1 expression by siRNA knockdown mimicked isoproterenol's effect by reducing EZH2 binding at these promoter/enhancer regions to a similar extent as isoproterenol and did not further decrease isoproterenol-suppressed EZH2 binding (Fig. 7C), whereas Hdac1 overexpression completely reversed isoproterenol-induced suppression of EZH2 binding (Fig. 7D).
In contrast, isoproterenol treatment in BAT1 brown adipocytes resulted in a significant increase in UTX binding at Ucp1 and Pgc1α promoters (Fig. 7, E and F). Reducing Hdac1 expression by siRNA knockdown also induced a similar increase in UTX binding to the Ucp1 enhancer region and further enhanced isoproterenol-induced binding of UTX to the Ucp1 proximal promoter and Pgc1α CRE region (Fig. 7E), whereas HDAC overexpression completely prevented the isoproterenol-induced increase in UTX binding to these promoter/enhancer regions (Fig. 7F). Overall, our data demonstrated that Hdac1 may coordinately regulate H3K27ac and H3K27me3 levels at Ucp1 and Pgc1α promoters through differential recruitment of the H3K27 methyltransferase EZH2 and the H3K27 demethylase UTX, thus regulating the expression of these genes.
EZH2 is a component of the polycomb group proteins, which are known to mediate gene silencing by regulating chromatin structure (51). Two major polycomb group proteins exist in mammals, namely polycomb repressive complex 1 and 2 (PRC1 and PRC2). PRC1 comprises three main components: ring finger protein 1 (RING1), RNF2, and Bmi1 polycomb ring finger oncogene (BMI1). PRC2 comprises three major components: EZH2, SUZ12, and EED (embryonic ectoderm development). PRC2 promotes H3K27 methylation through the methyltransferase activity of EZH2 and also facilitates the recruitment of PRC1 onto methylated H3K27, which in turn leads to further gene repression (51). It has been shown that HDAC1 may be associated with PRC2 (51). We thus investigated whether HDAC1 regulates the recruitment of components of PRC1/2 to Ucp1 and Pgc1α promoters. The ChIP assay demonstrated that isoproterenol treatment in BAT1 cells decreased binding of SUZ12 (Fig. 8, A and C) and RNF2 (Fig. 8, B and D) to Ucp1 and Pgc1α promoters. Importantly, reducing HDAC1 expression by siRNA knockdown mimicked isoproterenol's effect by inducing a similar decrease in SUZ12 and RNF2 binding and did not further reduce isoproterenol-suppressed SUZ12 and RNF2 binding at these promoters (Fig. 8, A and B), whereas Hdac1 overexpression completely prevented isoproterenol-suppressed SUZ12 and RNF2 binding to these promoters (Fig. 8, C and D).
FIGURE 8.

HDAC1 regulates the recruitment of polycomb repressor complexes to Ucp1 and Pgc1α promoters. A–D, SUZ12 and RNF2 binding at Ucp1 and Pgc1α promoters in BAT1 cells. BAT1 brown adipocytes were transfected with scramble or Hdac1 siRNA (A and B) or pSPORT6- or pSPORT-HDAC1-overexpressing plasmids (C and D). Two days later, cells were treated with or without isoproterenol (Isop; 1 μm) for 3 h, and a ChIP assay was performed as described under “Materials and Methods.” E and F, HDAC1 interacts with EZH2, SUZ12, and RNF2. In E, HEK293T cells were transfected with pSPORT vector or co-transfected with Hdac1, Ezh2, Suz12, and Rnf2 cDNA. Two days later, cell lysates were collected to detect protein interactions by co-IP. Left, overexpression of HDAC1, EZH2, SUZ12, and RNF2 was verified by immunoblotting using whole cell lysates. Right, IP of HDAC1 pulls down EZH2, SUZ12, and RNF2. In F, the interaction between HDAC1 and components of the polycomb repressor complexes in endogenous BAT1 cells was measured by co-IP. Data are expressed as mean ± S.E. (error bars) n = 4–6 in A–D. Blots in E and F are representative of two or three independent experiments. *, p < 0.05. n.s., not statistically significant. IB, immunoblotting.
We further performed co-IP experiments to test whether HDAC1 physically interacts with components of PRC1 and PRC2 complexes. Our data demonstrated that HDAC1 interacts with the PRC2 complex components EZH2 and SUZ12 and the PRC1 component RNF2 in HEK293T cells overexpressing Hdac1, Ezh2, Suz12, and Rnf2 (Fig. 8E) and, most importantly, in endogenous BAT1 brown adipocytes (Fig. 8F).
Discussion
Recent data suggest that HDACs have emerged as important players in the regulation of energy and glucose homeostasis (27). The pan-HDAC inhibitors sodium butyrate and trichostatin A increase energy expenditure, reduce adiposity, and improve insulin sensitivity in diet-induced obese (DIO) mice (53). This is possibly exerted through the inhibition of class I HDACs, because the specific class I HDACi exerts similar effects in DIO mice, whereas class II HDACi has no effects (28). However, the precise mechanism by which class I HDACi exerts these beneficial effects is poorly understood. Here, we demonstrate that the class I HDAC1 negatively regulates the brown adipocyte thermogenic program. This is based on the following observations. First, in contrast to Ucp1 expression, which is usually enriched in BAT, Hdac1 is enriched in WAT, but its expression is much lower in BAT. Second, brown adipocyte differentiation is associated with significantly up-regulated Ucp1 expression, which is concomitantly associated with reduction of Hdac1 RNA and protein levels. Third, sympathetic activation of BAT is associated with down-regulation of Hdac1 expression. These data suggest that Hdac1 may be a negative regulator of brown adipocyte thermogenic program. Indeed, knocking down Hdac1 in brown adipocytes significantly up-regulates BAT-specific gene expression, whereas overexpressing Hdac1 significantly suppresses BAT-specific gene expression, suggesting a causative link between Hdac1 inhibition and up-regulation of the brown adipocyte thermogenic program. Activating brown/beige adipocyte thermogenesis alleviates obesity and its associated metabolic diseases (9, 11, 54–56). Thus, our data suggest that HDAC1 may be important in regulating energy homeostasis in animal models, and inhibiting HDAC1 in brown adipocytes may contribute to the beneficial effects of class I HDACi in DIO mice (28). Further studies using genetic animal models with brown fat-specific deletion or overexpression of Hdac1 will be needed to study the role of HDAC1 in energy homeostasis and metabolism in whole animals.
Interestingly, we found that whereas both the pan-HDACi TSA and SAHA and the class I-specific HDACi MS-275 (57) stimulate the expression of genes involved in mitochondrial function and oxidative activity, such as Pgc1α, the effect of these different classes of HDACis on the expression of other genes that are important for brown adipocyte differentiation and determination is different. For example, the class I HDACi MS-275 potently stimulates, whereas the pan-HDACis TSA and SAHA significantly suppress, isoproterenol-stimulated Ucp1 expression in brown adipocytes. In addition, TSA also significantly inhibited Prdm16 and Pparγ expression, whereas MS-275 had minimal effects on these gene expressions. These TSA-suppressed genes are either key players in brown adipocyte differentiation and determination (Pparγ and Prdm16) or a marker of brown adipocyte (Ucp1). Because TSA and SAHA inhibit both class I and II HDACs (57), and because it has been shown that class II HDACs are mainly involved in the regulation of cellular differentiation (58), the difference in the gene regulation between the pan-HDACi TSA and the class I HDACi MS-275 may be mainly due to the different functions of class I and class II HDACs. Thus, further studies are warranted to elucidate the differential effects of class I and II HDACis on brown adipocyte function. Nonetheless, our data suggest that the class I HDACi MS-275 may contribute to its beneficial effect on improving metabolic phenotypes in DIO mice (as reported by Galmozzi et al. (28)) at least partly by directly stimulating brown adipocyte function, and HDAC1 may mediate this effect. Thus, class I HDACis, such as MS-275, and, more specifically, Hdac1, may serve as promising therapeutic targets in treating obesity and associated metabolic syndrome.
Histone acetylation and methylation are epigenetic mechanisms that regulate gene expression by remodeling chromatin structure (17–19). Whereas H3K27ac is a histone transcriptional activation mark, H3K27me3 serves as a histone transcriptional repression mark (17–19, 48). Histone acetylation is regulated by balanced action of histone acetyltransferases and HDACs, whereas histone methylation is regulated by histone methyltransferases and demethylases (17–19). Specifically, the di- and trimethylation of H3K27 are catalyzed by the PRC2 complex. Within PRC2, EZH2 is the catalytic subunit that possesses methyltransferase activity toward H3K27, and its activity also requires binding to the two other PRC2 protein components, SUZ12 and EED (51). The di- and trimethylated H3K27 further recruits PRC1 to the target genes, which, through the E3 ubiquitin ligase activity of the PRC1 components RING1 and RNF2, induces further chromatin compaction through histone 2A ubiquitination (51). On the other hand, UTX is a histone demethylase that specifically demethylates di- and trimethylated H3K27 (52). Interestingly, we find that HDAC1 physically interacts with the PRC2 components EZH2 and SUZ12 and the PRC1 catalytic subunit RNF2, suggesting a role of HDAC1 in connecting H3K27 deacetylation to methylation and possibly further chromatin compaction by histone ubiquitination. Indeed, we find that knocking down Hdac1 in brown adipocytes not only results in an increase in H3K27ac; it also leads to reduced H3K27me3 in promoters of BAT-specific genes, including Ucp1 and Pgc1α. This is exerted through the dissociation of the PRC2 components EZH2 and SUZ12 and PRC1 catalytic subunit RNF2 from these promoters and a reciprocal recruitment of the H3K27 demethylase UTX to these promoters. Thus, our data demonstrate that HDAC1 negatively regulates the brown adipocyte thermogenic program through interaction with PRC1/2 complexes, thereby promoting H3K27 deacetylation and methylation at promoters of BAT-specific genes, such as Ucp1 and Pgc1α. In addition, our data also demonstrate that β-adrenergic activation induces dissociation of HDAC1 along with PRC complexes from Ucp1 and Pgc1α promoters, which in turn leads to gene activation.
Sympathetic signaling is essential in brown/beige adipocyte activation (9–13). We find that β-adrenergic activation in BAT1 adipocytes is associated with increased H3K27ac and decreased H3K27me3 through dissociation of HDAC1 and PRC complexes and concomitant recruitment of UTX on BAT-specific gene promoters, including Ucp1 and Pgc1α. These data indicate that HDAC1 may be one of the epigenetic modulations triggered by sympathetic signaling in brown adipocytes, which eventually leads to thermogenic activation. It would be interesting to study how β-adrenergic stimulation triggers the dissociation of HDAC1 from BAT-specific gene promoters in brown adipocytes. HDAC1 is a part of the catalytic core of several multimeric corepressor complexes, including SIN3A, NuRD (nucleosome remodeling deacetylase), and CoREST (corepressor of RE1-silencing transcription factor) (59–61), and is also a part of the PRC2 complex (51). The recruitment of these multiprotein complexes is usually triggered by cell-specific transcriptional factors or the histone recognition motifs found within the complex components (59–61). In this context, pRB (retinoblastoma protein) and RIP140 (receptor-interacting protein 140) are potent negative regulators of BAT-specific gene expression (62). It has been reported that pRB and RIP140 silence promoter activity and gene expression through recruitment of HDACs, including HDAC1 (63–65). Thus, it is possible that HDAC1 may be recruited to BAT-specific promoters through association with negative transcriptional regulators of BAT, such as pRB and RIP140.
In addition, HDAC1 itself is subjected to various post-transcriptional modifications. For example, phosphorylation of HDAC1 by casein kinase II up-regulates its activity, whereas acetylation of HDAC1 by the acetyltransferase p300 suppresses its activity (61). Interestingly, recent studies using phosphoproteomics have identified casein kinase II as a negative regulator of BAT function through phosphorylating and regulating HDAC1 activity (66). Thus, our data fall in line with the results from Shinoda et al. (66) and further demonstrate the importance of HDAC1 in the regulation of BAT function.
Moreover, HDAC1 activity or protein levels can also be regulated by ubiquitination, SUMOylation, nitrosylation, and carbonylation (61). Further study is required to decipher the cellular signaling cascades and mechanisms that regulate HDAC1 activity and recruitment to BAT-specific promoters in response to sympathetic and other stimuli in brown adipocytes.
In the present study, we have focused on the epigenetic regulation of Pgc1α by Hdac1, because it is well established that PGC1α plays a central role in regulating important pathways involved in mitochondrial biogenesis and thermogenesis (40, 41). However, we have found that HDAC1 regulates H3K27 acetylation and methylation at the promoter and enhancer regions of Ucp1, a brown adipocyte terminal differentiation marker. Thus, it would be interesting to know whether HDAC1 also regulates H3K27 deacetylation and methylation at other genes' promoters to regulate their transcription. Unbiased approaches, such as ChIP sequencing, will be required to explore the gene profile that HDAC1 regulates through H3K27 deacetylation.
Although both express UCP1 and share striking similarities in morphological and biological properties, traditional brown fat and inducible beige adipocytes are derived from distinct cell origins during embryonic development (7, 8, 67). In rodents, traditional brown adipocytes are originated from the skeletal muscle lineage (7), whereas at least a subset of beige cells arise from the smooth muscle origin (8). A recent study shows that human brown adipocytes possess molecular features similar to those of rodent beige cells (68). We used the brown fat cell BAT1 in this study. Thus, it is not clear whether the role of Hdac1 in regulating brown adipocyte function can be extrapolated to beige cells. Additional studies, involving knockdown or overexpression of Hdac1 in beige lineage cells, will be warranted to determine the role of Hdac1 in the regulation of beige cell function.
In the present study, we investigated the role of HDAC1 in brown fat gene expression at the mature adipocyte stage. It has been reported that HDAC1 inhibits white adipocyte differentiation by deacetylating H4 at the promoter of C/ebpα, an important regulator of adipogenesis (39). In addition, we find that HDAC1 RNA and protein levels are down-regulated during brown adipocyte differentiation. Thus, it would be interesting to study whether HDAC1 also regulates brown adipogenesis and subsequently affects the brown fat thermogenic program.
In summary, we have identified Hdac1 as a negative regulator of brown adipocyte thermogenic program. Our data show that HDAC1 is down-regulated during brown adipocyte differentiation and is also suppressed by sympathetic activation in BAT. Overexpressing Hdac1 blocks, whereas knocking down Hdac1 further enhances, β-adrenergic agonist-stimulated BAT-specific gene expression in brown adipocytes. Remarkably, HDAC1 physically interacts with PRC1/2 complexes, and activation of β-adrenergic signaling dissociates HDAC1 along with PRC1/2 complexes from Ucp1 and Pgc1α promoters and concomitantly recruits UTX to these promoters, leading to increased H3K27 acetylation and decreased H3K27me3 levels in these promoters. These coordinated changes switch the transcriptional repressive state to the transcriptional active state at the promoters of Ucp1 and Pgc1α, which in turn activates the brown thermogenic program. Thus, our data demonstrate that HDAC1 negatively regulates brown adipocyte gene expression by coordinated regulation of H3K27 deacetylation and methylation, and inhibiting HDAC1 promotes the brown adipocyte thermogenic program. Targeting Hdac1 may be a novel therapeutic target in the treatment of obesity by promoting brown adipocyte thermogenesis and energy dissipation.
Author Contributions
F. L., R. W., X. C., and L. Z. performed most experiments. L. Y. contributed to discussion and reviewed/edited the manuscript. B. X., H. S., and F. L. conceived the hypothesis, designed the study, analyzed the data, and wrote the manuscript.
Acknowledgment
We thank Dr. Patrick Seale for providing the BAT1 cells.
This work was supported by National Institutes of Health Grants R01HL107500 (to B. X.), R01DK084172 (to H. S.), and R01DK085176 (to L. Y.); American Heart Association Grant 10SDG3900046 (to B. X.); and American Diabetes Association Grant 7-13-BS-159 (to H. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- WAT
- white adipose tissue
- BAT
- brown adipose tissue
- HDAC
- histone deacetylase
- HDACi
- HDAC inhibitor
- H3K27
- histone H3 Lys-27
- H3K27ac
- H3K27 acetylation
- H3K27me3
- H3K27 trimethylation
- PPARγ
- peroxisome proliferator-activated receptor γ
- PGC1α
- proliferator-activated receptor γ co-activator 1α
- NE
- norepinephrine
- IP
- immunoprecipitation
- C/EBPα
- CCAAT/enhancer-binding protein α
- CRE
- cAMP-response element
- CREB
- CRE-binding protein
- DIO
- diet-induced obese
- TSA
- trichostatin A
- SAHA
- suberoylanilidehydroxamic acid (vorinostat).
References
- 1. Hill J. O., Wyatt H. R., and Peters J. C. (2012) Energy balance and obesity. Circulation 126, 126–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cannon B., and Nedergaard J. (1985) The biochemistry of an inefficient tissue: brown adipose tissue. Essays Biochem. 20, 110–164 [PubMed] [Google Scholar]
- 3. Cannon B., and Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 4. Nicholls D. G., and Locke R. M. (1984) Thermogenic mechanisms in brown fat. Physiol. Rev. 64, 1–64 [DOI] [PubMed] [Google Scholar]
- 5. Ishibashi J., and Seale P. (2010) Medicine: beige can be slimming. Science 328, 1113–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petrovic N., Walden T. B., Shabalina I. G., Timmons J. A., Cannon B., and Nedergaard J. (2010) Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 285, 7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H. M., Erdjument-Bromage H., Tempst P., Rudnicki M. A., Beier D. R., and Spiegelman B. M. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu J., Boström P., Sparks L. M., Ye L., Choi J. H., Giang A. H., Khandekar M., Virtanen K. A., Nuutila P., Schaart G., Huang K., Tu H., van Marken Lichtenbelt W. D., Hoeks J., Enerbäck S., Schrauwen P., and Spiegelman B. M. (2012) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guerra C., Koza R. A., Yamashita H., Walsh K., and Kozak L. P. (1998) Emergence of brown adipocytes in white fat in mice is under genetic control: effects on body weight and adiposity. J. Clin. Invest. 102, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Himms-Hagen J. (1989) Brown adipose tissue thermogenesis and obesity. Prog. Lipid Res. 28, 67–115 [DOI] [PubMed] [Google Scholar]
- 11. Himms-Hagen J., Cui J., Danforth E. Jr., Taatjes D. J., Lang S. S., Waters B. L., and Claus T. H. (1994) Effect of CL-316,243, a thermogenic β3-agonist, on energy balance and brown and white adipose tissues in rats. Am. J. Physiol. 266, R1371–R1382 [DOI] [PubMed] [Google Scholar]
- 12. Xue B., Coulter A., Rim J. S., Koza R. A., and Kozak L. P. (2005) Transcriptional synergy and the regulation of Ucp1 during brown adipocyte induction in white fat depots. Mol. Cell Biol. 25, 8311–8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xue B., Rim J. S., Hogan J. C., Coulter A. A., Koza R. A., and Kozak L. P. (2007) Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. J. Lipid Res. 48, 41–51 [DOI] [PubMed] [Google Scholar]
- 14. Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., and Kahn C. R. (2009) Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J. (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 16. Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerbäck S., and Nuutila P. (2009) Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 17. Bäckdahl L., Bushell A., and Beck S. (2009) Inflammatory signalling as mediator of epigenetic modulation in tissue-specific chronic inflammation. Int. J. Biochem. Cell Biol. 41, 176–184 [DOI] [PubMed] [Google Scholar]
- 18. Bannister A. J., and Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maunakea A. K., Chepelev I., and Zhao K. (2010) Epigenome mapping in normal and disease States. Circ. Res. 107, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Campión J., Milagro F. I., and Martínez J. A. (2009) Individuality and epigenetics in obesity. Obes. Rev. 10, 383–392 [DOI] [PubMed] [Google Scholar]
- 21. Holness M. J., and Sugden M. C. (2006) Epigenetic regulation of metabolism in children born small for gestational age. Curr. Opin. Clin. Nutr. Metab. Care 9, 482–488 [DOI] [PubMed] [Google Scholar]
- 22. Ling C., and Groop L. (2009) Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 58, 2718–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maier S., and Olek A. (2002) Diabetes: a candidate disease for efficient DNA methylation profiling. J. Nutr. 132, 2440S–2443S [DOI] [PubMed] [Google Scholar]
- 24. Szarc vel Szic K., Ndlovu M. N., Haegeman G., and Vanden Berghe W. (2010) Nature or nurture: let food be your epigenetic medicine in chronic inflammatory disorders. Biochem. Pharmacol. 80, 1816–1832 [DOI] [PubMed] [Google Scholar]
- 25. Shore A., Karamitri A., Kemp P., Speakman J. R., and Lomax M. A. (2010) Role of Ucp1 enhancer methylation and chromatin remodelling in the control of Ucp1 expression in murine adipose tissue. Diabetologia 53, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tateishi K., Okada Y., Kallin E. M., and Zhang Y. (2009) Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ye J. (2013) Improving insulin sensitivity with HDAC inhibitor. Diabetes 62, 685–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galmozzi A., Mitro N., Ferrari A., Gers E., Gilardi F., Godio C., Cermenati G., Gualerzi A., Donetti E., Rotili D., Valente S., Guerrini U., Caruso D., Mai A., Saez E., De Fabiani E., and Crestani M. (2013) Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 62, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajakumari S., Wu J., Ishibashi J., Lim H. W., Giang A. H., Won K. J., Reed R. R., and Seale P. (2013) EBF2 determines and maintains brown adipocyte identity. Cell Metab. 17, 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seale P., Kajimura S., Yang W., Chin S., Rohas L. M., Uldry M., Tavernier G., Langin D., and Spiegelman B. M. (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab. 6, 38–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klaus S., Choy L., Champigny O., Cassard-Doulcier A. M., Ross S., Spiegelman B., and Ricquier D. (1994) Characterization of the novel brown adipocyte cell line HIB 1B. Adrenergic pathways involved in regulation of uncoupling protein gene expression. J. Cell Sci. 107, 313–319 [DOI] [PubMed] [Google Scholar]
- 32. Ross S. R., Choy L., Graves R. A., Fox N., Solevjeva V., Klaus S., Ricquier D., and Spiegelman B. M. (1992) Hibernoma formation in transgenic mice and isolation of a brown adipocyte cell line expressing the uncoupling protein gene. Proc. Natl. Acad. Sci. U.S.A. 89, 7561–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X., Yang Z., Xue B., and Shi H. (2011) Activation of the cholinergic antiinflammatory pathway ameliorates obesity-induced inflammation and insulin resistance. Endocrinology 152, 836–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xue B., Kim Y. B., Lee A., Toschi E., Bonner-Weir S., Kahn C. R., Neel B. G., and Kahn B. B. (2007) Protein-tyrosine phosphatase 1B deficiency reduces insulin resistance and the diabetic phenotype in mice with polygenic insulin resistance. J. Biol. Chem. 282, 23829–23840 [DOI] [PubMed] [Google Scholar]
- 35. Xue B., Pulinilkunnil T., Murano I., Bence K. K., He H., Minokoshi Y., Asakura K., Lee A., Haj F., Furukawa N., Catalano K. J., Delibegovic M., Balschi J. A., Cinti S., Neel B. G., and Kahn B. B. (2009) Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol. Cell Biol. 29, 4563–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang X., Wang X., Liu D., Yu L., Xue B., and Shi H. (2014) Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 28, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi H., Kokoeva M. V., Inouye K., Tzameli I., Yin H., and Flier J. S. (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lowell B. B., and Spiegelman B. M. (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660 [DOI] [PubMed] [Google Scholar]
- 39. Wiper-Bergeron N., Wu D., Pope L., Schild-Poulter C., and Haché R. J. (2003) Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. EMBO J. 22, 2135–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin J., Handschin C., and Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 41. Puigserver P., and Spiegelman B. M. (2003) Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1 α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24, 78–90 [DOI] [PubMed] [Google Scholar]
- 42. Cassard-Doulcier A. M., Gelly C., Bouillaud F., and Ricquier D. (1998) A 211-bp enhancer of the rat uncoupling protein-1 (UCP-1) gene controls specific and regulated expression in brown adipose tissue. Biochem. J. 333, 243–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kozak U. C., Kopecky J., Teisinger J., Enerbäck S., Boyer B., and Kozak L. P. (1994) An upstream enhancer regulating brown-fat-specific expression of the mitochondrial uncoupling protein gene. Mol. Cell Biol. 14, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rim J. S., and Kozak L. P. (2002) Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. J. Biol. Chem. 277, 34589–34600 [DOI] [PubMed] [Google Scholar]
- 45. Cao W., Daniel K. W., Robidoux J., Puigserver P., Medvedev A. V., Bai X., Floering L. M., Spiegelman B. M., and Collins S. (2004) p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 24, 3057–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herzig S., Long F., Jhala U. S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., and Montminy M. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183 [DOI] [PubMed] [Google Scholar]
- 47. Handschin C., Rhee J., Lin J., Tarr P. T., and Spiegelman B. M. (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 7111–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ge K. (2012) Epigenetic regulation of adipogenesis by histone methylation. Biochim. Biophys. Acta 1819, 727–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. An W. (2007) Histone acetylation and methylation: combinatorial players for transcriptional regulation. Subcell. Biochem. 41, 351–369 [PubMed] [Google Scholar]
- 50. Dambacher S., Hahn M., and Schotta G. (2010) Epigenetic regulation of development by histone lysine methylation. Heredity 105, 24–37 [DOI] [PubMed] [Google Scholar]
- 51. Morey L., and Helin K. (2010) Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 35, 323–332 [DOI] [PubMed] [Google Scholar]
- 52. Lee M. G., Villa R., Trojer P., Norman J., Yan K. P., Reinberg D., Di Croce L., and Shiekhattar R. (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450 [DOI] [PubMed] [Google Scholar]
- 53. Gao Z., Yin J., Zhang J., Ward R. E., Martin R. J., Lefevre M., Cefalu W. T., and Ye J. (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cederberg A., Grønning L. M., Ahrén B., Taskén K., Carlsson P., and Enerbäck S. (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 106, 563–573 [DOI] [PubMed] [Google Scholar]
- 55. Cohen P., Levy J. D., Zhang Y., Frontini A., Kolodin D. P., Svensson K. J., Lo J. C., Zeng X., Ye L., Khandekar M. J., Wu J., Gunawardana S. C., Banks A. S., Camporez J. P., Jurczak M. J., Kajimura S., Piston D. W., Mathis D., Cinti S., Shulman G. I., Seale P., and Spiegelman B. M. (2014) Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feldmann H. M., Golozoubova V., Cannon B., and Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 57. Chavan A. V., and Somani R. R. (2010) HDAC inhibitors: new generation of target specific treatment. Mini Rev Med. Chem. 10, 1263–1276 [DOI] [PubMed] [Google Scholar]
- 58. Nebbioso A., Dell'Aversana C., Bugge A., Sarno R., Valente S., Rotili D., Manzo F., Teti D., Mandrup S., Ciana P., Maggi A., Mai A., Gronemeyer H., and Altucci L. (2010) HDACs class II-selective inhibition alters nuclear receptor-dependent differentiation. J. Mol. Endocrinol. 45, 219–228 [DOI] [PubMed] [Google Scholar]
- 59. Kelly R. D., and Cowley S. M. (2013) The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41, 741–749 [DOI] [PubMed] [Google Scholar]
- 60. Laugesen A., and Helin K. (2014) Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 14, 735–751 [DOI] [PubMed] [Google Scholar]
- 61. Segré C. V., and Chiocca S. (2011) Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J. Biomed. Biotechnol. 2011, 690848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sharma B. K., Patil M., and Satyanarayana A. (2014) Negative regulators of brown adipose tissue (BAT)-mediated thermogenesis. J. Cell. Physiol. 229, 1901–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brehm A., Miska E. A., McCance D. J., Reid J. L., Bannister A. J., and Kouzarides T. (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391, 597–601 [DOI] [PubMed] [Google Scholar]
- 64. Magnaghi-Jaulin L., Groisman R., Naguibneva I., Robin P., Lorain S., Le Villain J. P., Troalen F., Trouche D., and Harel-Bellan A. (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391, 601–605 [DOI] [PubMed] [Google Scholar]
- 65. Wei L. N., Hu X., Chandra D., Seto E., and Farooqui M. (2000) Receptor-interacting protein 140 directly recruits histone deacetylases for gene silencing. J. Biol. Chem. 275, 40782–40787 [DOI] [PubMed] [Google Scholar]
- 66. Shinoda K., Ohyama K., Hasegawa Y., Chang H. Y., Ogura M., Sato A., Hong H., Hosono T., Sharp L. Z., Scheel D. W., Graham M., Ishihama Y., and Kajimura S. (2015) Phosphoproteomics identifies CK2 as a negative regulator of beige adipocyte thermogenesis and energy expenditure. Cell Metab. 22, 997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Long J. Z., Svensson K. J., Tsai L., Zeng X., Roh H. C., Kong X., Rao R. R., Lou J., Lokurkar I., Baur W., Castellot J. J. Jr., Rosen E. D., and Spiegelman B. M. (2014) A smooth muscle-like origin for beige adipocytes. Cell Metab. 19, 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shinoda K., Luijten I. H., Hasegawa Y., Hong H., Sonne S. B., Kim M., Xue R., Chondronikola M., Cypess A. M., Tseng Y. H., Nedergaard J., Sidossis L. S., and Kajimura S. (2015) Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 21, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]