

Abstract
The sodium (Na+)-calcium (Ca2+) exchanger 1 (NCX1) is an important regulator of intracellular Ca2+ homeostasis. Serine 68-phosphorylated phospholemman (pSer-68-PLM) inhibits NCX1 activity. In the context of Na+/K+-ATPase (NKA) regulation, pSer-68-PLM is dephosphorylated by protein phosphatase 1 (PP1). PP1 also associates with NCX1; however, the molecular basis of this association is unknown. In this study, we aimed to analyze the mechanisms of PP1 targeting to the NCX1-pSer-68-PLM complex and hypothesized that a direct and functional NCX1-PP1 interaction is a prerequisite for pSer-68-PLM dephosphorylation. Using a variety of molecular techniques, we show that PP1 catalytic subunit (PP1c) co-localized, co-fractionated, and co-immunoprecipitated with NCX1 in rat cardiomyocytes, left ventricle lysates, and HEK293 cells. Bioinformatic analysis, immunoprecipitations, mutagenesis, pulldown experiments, and peptide arrays constrained PP1c anchoring to the K(I/V)FF motif in the first Ca2+ binding domain (CBD) 1 in NCX1. This binding site is also partially in agreement with the extended PP1-binding motif K(V/I)FF-X5–8Φ1Φ2-X8–9-R. The cytosolic loop of NCX1, containing the K(I/V)FF motif, had no effect on PP1 activity in an in vitro assay. Dephosphorylation of pSer-68-PLM in HEK293 cells was not observed when NCX1 was absent, when the K(I/V)FF motif was mutated, or when the PLM- and PP1c-binding sites were separated (mimicking calpain cleavage of NCX1). Co-expression of PLM and NCX1 inhibited NCX1 current (both modes). Moreover, co-expression of PLM with NCX1(F407P) (mutated K(I/V)FF motif) resulted in the current being completely abolished. In conclusion, NCX1 is a substrate-specifying PP1c regulator protein, indirectly regulating NCX1 activity through pSer-68-PLM dephosphorylation.
Keywords: animal model, computer modeling, electrophysiology, heart failure, ion channel, peptide array, phosphoprotein phosphatase 1 (PP1), protein motif, protein-protein interaction, sodium-calcium exchange
Introduction
The sodium (Na+)-calcium (Ca2+) exchanger (NCX)3 is a bidirectional ion-transporting membrane protein, which exchanges three Na+ for one Ca2+ across the plasma membrane. Its mode of operation and activity are determined by the ion concentration gradients and membrane potential (1). In mammals, there are three distinct genes that control the expression of three NCX isoforms (NCX1, NCX2, and NCX3) that are expressed in a tissue-specific manner (2, 3). Among these, NCX1 is highly expressed in cardiomyocytes, where it modulates excitation-contraction coupling and mediates Ca2+ removal during diastole (4). Increased NCX1 mRNA and protein levels have been shown in human end-stage heart failure (HF) (5–7), and elevated activity of NCX1 has been linked to dysfunctional Ca2+ handling in chronic heart disease (8). Consequently, modulation of NCX1 activity constitutes a potential therapeutic target in the treatment of HF (9).
NCX1 cDNA encodes a protein of 973 amino acids in humans, which includes a 32-amino acid signaling peptide that is cleaved during processing (10). The eukaryotic exchanger is composed of 10 transmembrane domains (TM) (11, 12), with a large cytosolic loop between TM5 and TM6. Deletion of the cytosolic loop has revealed that it does not play a direct role in ion translocation, but rather it mediates regulation of the exchanger by associating with various cytosolic factors (13–16). Relevant to this study are the interaction sites for phospholemman (PLM), a membrane phosphoprotein (14, 16), and calpain, a non-lysosomal cysteine protease (15).
NCX1 has been shown to exist in a macromolecular complex comprising protein kinase A (PKA) and C (PKC), protein phosphatase 1 (PP1) and 2A (PP2A), as well as other anchoring and adaptor proteins (17). Direct regulation of NCX1 by kinases is controversial (18); in fact, we have shown that there is no phosphorylation of endogenous NCX1 following PKA activation, and we concluded that the identified phosphorylation site was not accessible in full-length NCX1 (19). It has been suggested that phosphorylation and dephosphorylation rather occur on accessory proteins in the NCX1 macromolecular complex, enabling fine-tuning of signals that converge on NCX1 (17). Accumulating data indicate that PLM is one of these regulatory players. This 72-amino acid transmembrane protein, belonging to the FXYD1 family of ion transporters (20), co-localizes and co-immunoprecipitates with NCX1 and has been shown to inhibit NCX1 activity when it is phosphorylated at serine 68 (pSer-68-PLM) (21–24). Interestingly, pSer-68-PLM relieves inhibition of the Na+/K+-ATPase (NKA), causing an increase in NKA activity (25, 26), suggesting that PLM may serve as a regulator of both NCX (24) and NKA (27) depending on its phosphorylation status. pSer-68-PLM is in turn regulated by PP1 (28). The latter is a ubiquitously expressed ∼38.5-kDa serine/threonine phosphatase that counters the effects of serine/threonine kinases and has little intrinsic specificity for its substrates (29). Mammalian genomes encode four distinct catalytic subunits of PP1 as follows: PP1α, PP1β/δ, and the splice variants PP1cγ1 and PP1cγ2 (30). The isoforms show 85% sequence identity, but the N- and C-terminal extremities show amino acid differences (30). The catalytic subunit (PP1c) achieves substrate specificity by forming holoenzymes with more than 200 targeting proteins (31). These targeting proteins localize PP1c to specific subcellular domains and fine-tune its activity, allowing for substrate-specific effects (31). More than 90% of targeting proteins interact with PP1c via a short degenerate RVXF-docking motif (32), which serves as an anchor for the initial recruitment of PP1c. Although dephosphorylation of pSer-68-PLM, by PP1c, was recently shown to modulate pSer-68-PLM regulation of NKA (28), it is unknown whether this mechanism is involved in NCX1 regulation.
In this study, we investigated whether a direct and functional NCX1-PP1c interaction is a prerequisite for pSer-68-PLM dephosphorylation. Bioinformatic and biomolecular approaches were used to investigate this hypothesis. We aimed to map the PP1c-targeting site on the NCX1 macromolecular complex and elucidate the biological effect of this interaction on NCX1 activity.
Experimental Procedures
Animal Model
Animal experiments were approved by the Norwegian Animal Research Committee (FOTS ID 3820) and conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23, revised 1996). Male Wistar rats (Møllergaard Breeding and Research Center, Skensved, Denmark) weighing ∼170 g were subjected to aortic banding, as described previously (33, 34). In short, anesthesia was induced in a chamber containing a mixture of 68% N2O, 28% O2, and 4% isoflurane. After endotracheal intubation, ventilation was performed with a respirator (Zoovent, Triumph Technical Services, Milton Keynes, UK), and anesthesia was maintained by administration of a mixture of 69% N2O, 29% O2, and 2% isoflurane. The chest was opened in the right second intercostal space, and the ascending aorta was carefully dissected. A significant stenosis was induced by a tight banding of the ascending aorta with a 3.0 silk suture. In sham-operated animals (SHAM), the silk suture around the ascending aorta was not tightened. Buprenorphine was administered for postoperative analgesia. After 6 weeks, echocardiography was performed with a Vevo 2100 (Fujifilm VisualSonics, Canada), and short and long axis images of the left ventricle (LV) and atrium were obtained. The flow through the mitral and aortic valve was measured. Aortic banding animals with congestive heart failure (ABHF) were selected based on echocardiographic, hemodynamic, and post-mortem analysis. The criteria for inclusion in the ABHF group were increased posterior wall diameter (>1.9 mm), increased LV weight (>0.75 g), increased lung weight (>2.5 g), and increased left atrial diameter (>5.0 mm) (34). The SHAM group served as control. All LV samples were snap-frozen in liquid nitrogen and stored at −70 °C until analysis.
Isolation of Neonatal Cardiomyocytes
Neonatal rat cardiomyocytes were isolated from 1- to 3-day-old Wistar rats. The LV was enzymatically digested in a collagenase solution. The cell mix was transferred onto uncoated culture flasks and incubated for 20 min. Unattached cardiomyocytes were seeded onto 6-well culture plates coated with 0.2% gelatin (G1890, Sigma) and 0.1% fibronectin (F1141, Sigma). The seeding density was 3.75 × 105 cells/ml in Dulbecco's modified Eagle's medium-high glucose (DMEM) (D1152, Sigma) supplemented with Medium 199 (M2520, Sigma), penicillin/streptomycin (P0781, Sigma), horse serum (14-403E, BioWhittaker, Walkersville, MD), and fetal bovine serum (FBS) (14-701F, BioWhittaker). The cultures were maintained in a humidified incubator with 5% CO2 at 37 °C for 24 h until used in protein fractionation experiments.
Adult Cardiomyocyte Isolation
Rats were anesthetized in a chamber filled with 95% room air and 5% isoflurane (Abbott Scandinavia Ab, Solna, Sweden) and were subsequently sacrificed by cervical dislocation. Hearts were then quickly excised and placed in ice-cold 0.15 m NaCl solution with heparin (Heparin LEO, 5000 IE/ml; Orifarm AS, Norway). The aorta was then cannulated and retrogradely perfused with a cell isolation buffer containing 130 mm NaCl, 25 mm Hepes, 22 mm d-glucose, 5.4 mm KCl, 0.5 mm MgCl2, 0.4 mm NaH2PO4, pH 7.4 (all chemicals from Sigma) to wash out the blood. The heart was thereafter perfused with cell isolation buffer containing 200 units/ml collagenase type II (Worthington) and 0.1 mm Ca2+. After 20 min of perfusion, the heart was cut down, and the atria and right ventricle were removed. The LV was minced and gently shaken at 37 °C for 3–4 min in the same solution used in the perfusion, but with addition of 1% bovine serum albumin (BSA) (A9647, Sigma) and 0.02 units/ml deoxyribonuclease I (LS002006, Worthington). The digested ventricular tissue was then filtered (200-μm nylon mesh), and cardiomyocytes were sedimented. The cardiomyocyte pellet was resuspended in cell isolation buffer with 1% BSA (A9647, Sigma) and 0.1 mm Ca2+ in the solution. Isolated cardiomyocytes were kept at room temperature until used.
Antibodies
The primary antibodies used are as follows: anti-NCX1 (custom-made, epitope: GQPVFRKVHARDHPIPST, Genscript Corp., Piscataway, NJ) (19); anti-rabbit IgG (sc-2027, Santa Cruz Biotechnology, Santa Cruz, CA); anti-PP1 (E-9) (sc-7482, Santa Cruz Biotechnology); anti-mouse IgG (sc-2025, Santa Cruz Biotechnology); anti-FXYD1 (total PLM) (ab76597, Abcam plc, Cambridge, UK); anti-vinculin (V9131, Sigma); anti-His6 (A00186, Genscript Corp.); anti-serine 68 phosphorylated phospholemman (pSer-68-PLM) (35); anti-GFP (632381, Clontech); anti-DYKDDDDK (FLAG tag) (A00170-40, Genscript Corp.); anti-FLAG® M2 (F3165, Sigma); anti-biotin horseradish peroxidase (HRP) (A0185, Sigma); anti-His6 HRP (against a polyhistidine (His6 tag) epitope) (R931-25, Life Technologies, Inc.); anti-GST HRP (RPN 1236V, GE Healthcare, Little Chalfont, Buckinghamshire, UK), and anti-calsequestrin (PA1-913, Thermo Fisher Scientific, Waltham, MA). The secondary antibodies used are as follows: anti-rabbit IgG HRP (NA934V, GE Healthcare); anti-mouse IgG HRP (NA931V, GE Healthcare); anti-goat IgG HRP (HAF109, R&D Systems, Bio-Techne, Minneapolis, MN); anti-sheep IgG HRP (6150-05, Southern Biotech, Birmingham, AL); and anti-mouse IgG HRP, light chain-specific (115-005-174, Jackson ImmunoResearch).
Bioinformatic Analyses
Human, mouse, and rat NCX1 protein sequences (P32418, O35157, and Q01728) as well as human and rat PLM sequences (O00168 and O08589) were screened in silico for putative PP1-binding motifs, (RK)X0–1(VI)X(FW), as defined by Wakula et al. (32). Using the Protein Pattern Find-Bioinformatics.org database, the consensus sequence input was as follows in the program, [RK].{0,1}[VI].[FW]. NCX1 sequences were also screened for the presence of the Φ1Φ2 and/or arginine motifs, as defined by Choy et al. (36). Lasergene (DNA Star, Madison, WI) was used for protein alignments.
Total Protein Extracts
Frozen rat LV lysates were pulverized in a mortar with liquid nitrogen before transfer to lysis buffer (20 mm Hepes, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% TritonTM X-100 (X100–500ML, Sigma)) supplemented with 1 mm PMSF (93482, Sigma) and a Complete Mini EDTA-free tablet (11836170001, Roche Diagnostics, Basel, Switzerland). Tissue samples were homogenized three times for 1 min on ice with a Polytron 1200 and centrifuged at 100,000 × g for 60 min at 4 °C. Supernatants were collected and stored at −70 °C until analysis.
Fractionation
LVs from rat heart and neonatal cardiomyocyte cultures were fractionated using the compartment protein extraction kit according to the manufacturer's protocol (2145, Merck Millipore Billerica, MA). The supernatants from cytosolic and/or membrane fractions were collected and stored at −70 °C. Protein concentrations, where applicable, was determined by the Micro BCA protein assay kit (23235, Thermo Fisher Scientific).
DNA Constructs
Cloning and mutations of NCX1, PP1, and PLM constructs were performed by Genscript Corp. The primary sequence numbering in all NCX variants includes the signal peptide sequence. The MGC mouse clone BC079673 was used for NCX1 constructs. WT NCX1 full-length (FL) was cloned into pEGFP-N1 (Clontech) or into the first reading frame of pAdTrack-cytomegalovirus (CMV) shuttle vector (plasmid 16405, Addgene, Cambridge, MA). Ala mutants (I406A and F408A or K405A and F407A) were mutated into NCX1/pEGFP-N1. A Pro substitution mutant (F407P) was cloned into NCX1/pEGFP-N1. Deletion mutants of the cytoplasmic loop of NCX1, NCX1(243–787), NCX1(243–705), NCX1(243–532), NCX1(243–402), and NCX1(Δ399–424), were cloned into pEGFP-C2 (Clontech). The rat catalytic subunit (P62138) was used for PP1 constructs, and an N-terminal FLAG and His6 tag were inserted. FLAG-His6-PP1c(1–330) and deletion mutants FLAG-His6-PP1c(1–212), FLAG-His6-PP1c(1–149), and FLAG-His6-PP1c(Δ232–263) were cloned into pCEP4 vector (Invitrogen). Single and double glycine mutants of FLAG-His6-PP1c (L243G,F257G) were mutated into pCEP4 (Invitrogen). Mouse PLM (AF089734) was cloned into pcDNA3.1/Myc-HisA (Invitrogen) by Genscript Corp. The fidelity of the cloning procedure and mutagenesis were verified by sequence analysis (Genscript Corp.). The empty vectors pcDNA3.1 and pEGFP-N1 were obtained from Invitrogen and Clontech, respectively. Tobacco etch virus (TEV) protease in pCS2MT was kindly provided by Prof. Pati (37).
Transient Transfection of HEK293 Cells
Human embryonic kidney 293 (HEK293) cells were cultured in DMEM (41965-039, Gibco, Life Technologies, Inc.) supplemented with 10% FBS (14-701F, BioWhittaker), 1% non-essential amino acids (10370-021, Gibco, Life Technologies, Inc.), 100 units/ml penicillin, and 0.1 mg/ml streptomycin (penicillin/streptomycin, P4333, Sigma) and maintained in a 37 °C, 5% CO2-humidified incubator. Cells were cultured in medium without antibiotics 24 h prior to transfection. Cells were transfected with DNA using Lipofectamine 2000, as instructed by the manufacturer (11668-019, Invitrogen), or a solution containing CaCl2. For this method, two solutions were prepared: solution A (124 mm CaCl2 and 8 μg of plasmid DNA diluted in 500 μl of purified water) and solution B (50 mm HEPES, 280 mm NaCl, and 1.5 mm Na2HPO4 in 500 μl of PBS, pH 7.0). The two solutions were then mixed together and incubated at room temperature for 30 min. After the 30 min, HEK293 cells were incubated with the mixed solution for 18–24 h (38). After 24 h, the cells were either transferred to coverslips for patch clamp/Ca2+-dependent fluorescence experiments or lysed in immunoprecipitation (IP)-buffer (20 mm Hepes, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% TritonTM X-100) supplemented with a Complete Mini EDTA-free tablet (11836170001, Roche Diagnostics) and used in molecular studies. In the patch clamp/Ca2+-dependent fluorescence experiments, the glass coverslips were pre-coated with poly-l-lysine (P4707, Sigma), and the cells on the coverslips were incubated for 24 h prior to analysis. GFP was used as a positive control for the transfection.
Peptide Synthesis
Peptides were purified to >80% purity by Genscript Corp. anti-NCX1-blocking peptide (amino acids 655–672), (C)-GQPVFRKVHARDHPIPST; biotin-NCX1 (KVFF) (amino acids 399–424, rat), ENDPVSKVFFEQGTYQCLENCGTVAL; biotin-NCX1 (KAFA) mutant, ENDPVSKAFAEQGTYQCLENCGTVAL; biotin-NCX1 (AVAF) mutant, ENDPVSAVAFEQGTYQCLENCGTVAL; biotin-NCX2 (amino acids 389–410, rat), EDDGASRIFFEPSLYHCLENCG; and biotin-NCX3 (amino acids 392–412, rat), EDFASKVFFDPCSYQCLENCG.
Recombinant Proteins
The recombinant proteins used were as follows: PP1α (14–595, Merck Millipore Billerica); custom-made His6-trigger factor (TF)-NCX1cyt and GST-PP1α (both Genscript Corp.); PP1α (P0754S, New England Biolabs, Ipswich, MA); and PP1 inhibitor 2 (P0755S, New England Biolabs).
Spot Membrane Synthesis
The cytosolic loop (amino acids 243–799) of rat NCX1 protein (EDM02743) and rat PP1 catalytic subunit (P62138) (PP1c) were synthesized as 20-mer peptides with a three-amino acid overlap on cellulose membranes using a Multipep automated peptide synthesizer (INTAVIS Bioanalytical Instruments AG, Cologne, Germany), as described previously (39).
Overlay Assay
Peptide array membranes, consisting of either the NCX1 or PP1c sequence, were activated by soaking in methanol for a few seconds and followed by three washes (three times for 10 min) with TBS-T (Tris-buffered saline with 1% Tween 20 (161-0781, Bio-Rad)). The membranes were then incubated with blocking solution (1% casein) (11921673001, Roche Diagnostics) at room temperature for 1 h. Next, the membranes were either overlaid with 1 μg/ml recombinant His6-TF-NCX1cyt or 1 μg/ml GST-PP1α recombinant protein (in 1% casein) overnight at 4 °C with gentle agitation. The membranes were then washed five times for 5 min with TBS-T and incubated with anti-His6 HRP or anti-GST HRP-conjugated antibody for 1 h at room temperature. The bound protein on the membranes was detected by immunodetection. Incubation of the membranes without recombinant protein was used as a negative control. For antibody epitope mapping, incubation of the membranes without primary antibody was used as a negative control.
Pulldown Experiments
Biotinylated peptides (10 mm) were incubated with 25 μl of anti-biotin-conjugated beads (A1559, Sigma) and 100 μl of PBS for 2 h at 4 °C under gentle rotation. The beads with bound peptides were then washed three times with PBS followed by incubation with 1 μg of GST-PP1α recombinant protein in 100 ml of IP-buffer supplemented with 1% BSA (805095, Bio-Rad) followed by gentle rotation for 2 h at 4 °C. The peptide complex was washed three times with 1 ml of IP-buffer. To elute the complex, it was boiled in 2× SDS loading buffer and analyzed by immunoblotting.
Immunoprecipitation (IP)
IP was performed using 2 μg of the appropriate antibody. The immunocomplexes were collected by 50 μl of protein A/G PLUS-agarose beads (sc-2003, Santa Cruz Biotechnology) overnight at 4 °C. They were then washed three times with 1 ml of IP-buffer followed by the boiling of the complexes in 2× SDS loading buffer. Analysis was done by immunoblotting. Equal amounts of rabbit IgG (sc-2027, Santa Cruz Biotechnology) and mouse IgG (sc-2025, Santa Cruz Biotechnology) were used as negative controls. Blocking peptide (antigen, cardiac NCX1; sequence, (C)-GQPVFRKVHARDHPIPST) (Genscript Corp.) was incubated with anti-NCX1 prior to immunoprecipitation (negative control).
Proximity Ligation Assay (Duolink)
Isolated adult rat cardiomyocytes were plated on laminin (23017-015, Life Technologies, Inc.)-coated glass coverslips and left to adhere for 1 h. The cells were fixed in 4% paraformaldehyde (158127, Sigma), permeabilized with 0.03% Triton X-100 (X100–500ML, Sigma), and incubated with the two primary antibodies anti-NCX1 (rabbit polyclonal antibody) and anti-PP1c (mouse monoclonal antibody) overnight at 4 °C. Staining without primary antibodies and use of an anti-NCX1 blocking peptide was used as a negative control. The proximity ligation assay was performed using the Duolink kit (DUO92014, Sigma) according to the manufacturer's protocol, as described previously (15). The cells were then incubated with 600 nm SYTOX Orange (S-11368, Life Technologies, Inc.), a nucleic acid stain, for 10 min at room temperature and rinsed three times for 5 min with PBS. Imaging experiments were performed at 25 °C. The water-based Duolink® In Situ Mounting Medium (refractive index 1.44), provided in the kit, was used to mount the glass coverslip to the glass slide. The fluorochromes used were the Duolink® In Situ Detection Reagents Green (excitation 488 nm and emission 510 nm) and SYTOX Orange (excitation 543 nm and emission 650 nm). The cells were visualized with an inverted LSM 710 confocal microscope (Zeiss GmbH, Jena, Germany) equipped with an LD C-Apochromat ×40 objective (numerical aperture 1.1). Sequential optical scans were acquired using the Zeiss ZEN imaging software.
Immunoblotting
LV lysates and immunoprecipitates were resolved on either 4–15% or 15% CriterionTM Tris-HCl precast gels (Bio-Rad) and blotted onto PVDF membranes (RPN 303F, GE Healthcare). The PVDF membranes and peptide arrays were blocked in 5% nonfat dry milk or 1% casein in TBS-T for 60 min at room temperature, followed by incubation with primary antibodies either for 1 h at room temperature or overnight at 4 °C. The membranes were washed three to five times for 5 min in TBS-T and incubated with a horseradish peroxidase-conjugated secondary antibody. Blots were developed using ECL Plus or Prime (RPN 2132 or RPN 2232, GE Healthcare). The chemiluminescence signals were detected by Las-4000 (Fujifilm, Tokyo, Japan). Membranes were re-probed after stripping using the Restore Western blot Stripping buffer (21059, Thermo Scientific).
Protein Phosphatase 1 Activity Assay
Phosphatase activity was measured in vitro by a nonradioactive assay using a malachite green-based phosphatase assay kit (40) (17-128, Merck Millipore), according to the manufacturer's protocol. Briefly, 1 unit of PP1α recombinant protein (P0754S, New England Biolabs) was incubated with 200 μm (RRApSVA) peptide substrate together with either a range of His6-TF-NCX1cyt recombinant protein concentrations or biotinylated NCX1 peptides in a phosphatase buffer (50 mm HEPES, 10 mm NaCl, 2 mm DTT, 0.01% Brij 35, pH 7.5) supplemented with 1 mm MnCl2 for 20 min at 37 °C. Next, 100 ml of malachite green dye was added to the solution and incubated for 15 min at room temperature. A spectrophotometer, absorbance set at 650 nm, was used to measure the colored complex formed upon the release of free phosphate. The amount of released phosphate was calculated using a standard curve created with various concentrations of KH2PO4. PP1 inhibitor 2 was used as a control for the assay.
Protein Kinase A (PKA) Phosphorylation Assay
PKA assays were performed using cAMP-dependent protein kinase, catalytic subunit (PKA-C) (P6000S, New England Biolabs) as instructed by the manufacturer. PKA-C and 1× NEBuffer for protein kinases (P6000S, New England Biolabs), supplemented with 200 μm ATP, were added to the HEK293 cell lysates, followed by incubation for 30 min at 30 °C. PKA-C treatment was omitted for control samples. Kinase reaction was stopped by adding sample buffer. The samples were subsequently subjected to SDS-PAGE and immunoblotting.
Patch Clamp Studies
Whole cell patch clamp experiments were conducted on HEK293 cells transfected with WT NCX1(FL), WT NCX1(FL) co-transfected with PLM or Pro substitution mutant NCX1(F407P) co-transfected with PLM using an Axoclamp 200B amplifier (Axon Instruments) and low resistance pipettes (2–4 megaohms). We also determined the current of the NCX1(F407P) mutant alone, as a control for the experiment. The recordings were performed at 37 °C in an extracellular solution containing (in mm) the following: 140 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 CaCl2, 10 HEPES, 10 glucose, pH 7.4 (CsOH), and osmolality at 290 mOsm. To block K+, Ca2+, Cl−, and NKA currents, we used cesium, nifedipine (20 μm), niflumic acid (30 μm), and ouabain (1 mm), respectively. The patch pipettes were filled with a solution composed of (in mm) the following: 100 cesium glutamate, 4 NaCl, 1 MgCl2, 10 HEPES, 2 Na2-ATP, 10 EGTA, 6 CaCl2, pH 7.2, and osmolality 270 mOsm. Free intracellular Ca2+ was 369 nm. Liquid junction potential was calculated using pCLAMP 10 software (Molecular Devices) and corrected by 15 mV (Vmembrane = Vpipette − (15 millivolts (mV))). The NCX1 reversal potential under these conditions was −40 mV at 37 °C. Cells were voltage-clamped at −40 mV for 4–5 min to allow sufficient intracellular dialysis. NCX1 current was elicited by a descending voltage ramp from 120 to −100 mV (0.05 V/s) and isolated using 5 μm Ni2+. The currents were normalized to cell capacitance and the current (I)-voltage (V) relations were plotted from −80 to 80 mV.
Measurement of Intracellular Ca2+
Cytosolic Ca2+ concentrations were determined from HEK293 cells transfected with WT NCX1(FL), WT NCX1(FL) co-transfected with PLM, or Pro substitution mutant NCX1(F407P) co-transfected with PLM. Cells were seeded and transfected on poly-l-lysine-coated coverslips 48 h prior to the experiment. On the day of the experiment, transfected HEK293 cells were preincubated in Tyrode's solution (140 mm NaCl, 5 mm HEPES, 5.4 mm KCl, 0.5 mm MgCl2, 5.5 mm d(+)-glucose monohydrate, 0.4 mm NaH2PO4, 1.8 mm CaCl2, 1 μm ouabain, and 1 μm thapsigargin, 37 °C). The cells were then loaded with 5 μm fluo-4 acetoxymethyl ester (F14202, Molecular Probes®-Thermo Fisher) for 10 min at room temperature, mounted in a superfusion chamber on a Zeiss LSM 510 confocal microscope (Zeiss Gmbh, Jena Germany), and visualized with a W Plan-Apochromat ×40 objective (numerical aperture = 1.0, Zeiss). Fluorescence was excited by a 480 nm light-emitting diode (Colibri lamp, Zeiss), and emission was collected at >515 nm with a photomultiplier tube (R1527P, Horiba Scientific, Edison, NJ). Ca2+-dependent fluorescence was monitored as the superfusing solution was changed from the initial Tyrode's solution ([Na+] = 140 mm) to a Na+-free solution (140 mm choline chloride, 5 mm HEPES, 5.4 mm KCl, 0.5 mm MgCl2, 5.5 mm d(+)-glucose monohydrate, 1.8 mm CaCl2, 1 μm ouabain, and 1 μm thapsigargin, 37 °C), to induce Ca2+ influx via reverse-mode NCX1. Data acquisition and analysis were performed with Clampex 10.4 and Clampfit 10.4 software, respectively (Molecular Devices LLC, Sunnyvale, CA). Ca2+ recordings were background-subtracted and are presented normalized to baseline fluorescence (F/F0).
SPR Analysis
SPR analysis was performed using BIAcore X100. A desorb step in IP-buffer supplemented with 1% BSA was performed before each analysis. Recombinant His6-TF-NCX1cyt (ligand) in acetate, pH 4.0, was immobilized on CM5 chips using N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide. His6-TF-NCX1cyt was immobilized in three independent experiments to 189, 419, and 443 resonance units. Recombinant GST-PP1α (analyte) was diluted over a range of concentrations (31.3–500, 6.25–100, and 9.8–50 nm) in the buffer above and injected over the sensor surface at a flow rate of 30 μl/min for 180 s. The dissociation time was 600 s at the same flow rate. Obtained sensorgrams were analyzed by BIAcore X100 evaluation software.
Structure Modeling
Atomic coordinates for human PP1 in complex with the PP1 and PDZ domains of rat spinophilin (PDB code 3EGG (41)) were used to build a model of the NCX1 tetrapeptide 405KVFF408 binding to PP1. The peptide was built by homology modeling using the 448KIHF451 segment of the spinophilin as a template. The side chain conformations of the middle Val and Phe residues were selected based on the orientation of the corresponding Ile and His residues in spinophilin, respectively. The structural models of the complexes were analyzed and visualized by PyMOL (Schrodinger LLC).
Densitometric Analysis
Densitometric analysis was performed using ImageJ (National Institutes of Health), and blots were processed in Adobe Photoshop CS2 (Adobe Systems Inc., San Jose, CA).
Statistics
All data were expressed as mean ± S.E. relative to control. Comparisons between two groups were analyzed using the unpaired t test or Mann-Whitney test (GraphPad software, Prism 5.04). A p value of <0.05 was considered statistically significant.
Results
Bioinformatic Analysis to Identify PP1-binding Sites in NCX1
The Protein Pattern Find-Bioinformatics.org database was used to search for putative PP1-binding motifs in NCX1 and PLM, with the consensus sequence (R/K)(V/I)X(F/W) (see under “Experimental Procedures” for details). No PP1-binding sites were found in PLM; however, three putative binding sites were identified in human, mouse, and rat NCX1. The first site, RVFF, is localized within the cytosolic loop connecting TM3 and -4. The second site, KIFF/KVFF (human and mouse/rat), is localized in the large cytosolic loop in the first Ca2+ binding domain (CBD1), the primary sensor of calcium (42). The KIFF/KVFF site does not overlap with the sites in CBD1 that coordinate the four Ca2+ ions (43). The third site, KVLF, is localized at the end of the intracellular loop (Fig. 1).
FIGURE 1.
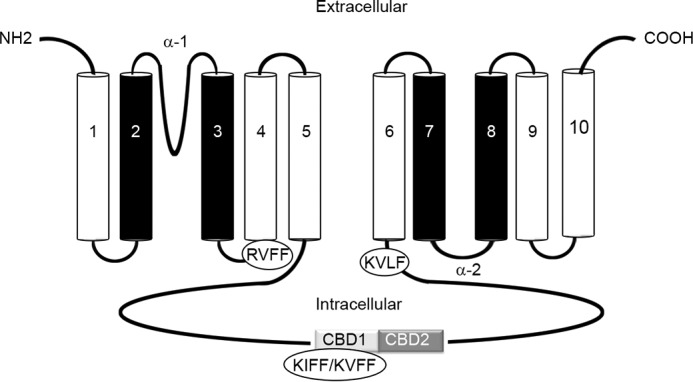
Putative PP1-binding motifs within NCX1. Topology model of Na+/Ca2+ exchanger 1 (NCX1) consisting of 10 transmembrane domains (TMs) and a large cytosolic loop between TM5 and TM6 (11, 12) is shown. Within the TMs are two α repeats (α1 and α2) that face opposite sides of the membrane and catalyze ion translocation (36) (TMs involved in ion translocation are shown in black). The cytosolic loop mediates regulation of the exchanger and contains the two Ca2+-binding domains (CBD1 and CBD2). In silico screening of human, rat, and mouse NCX1 primary sequences identified three putative PP1-binding motifs. The first site, RVFF, is localized within the membrane loop connecting TM3 and -4. The second site, KIFF/KVFF (human and mouse/rat), is localized in CBD1, and the third site, KVLF, is localized at the end of the intracellular loop.
PP1 Binds Directly to NCX1
Several experiments were performed to show that PP1 interacts with NCX1. First, immunodetection with specific antibodies for NCX1 (as published previously (19)) and PP1 (Fig. 2A, antibody epitope mapping) showed that a pool of PP1 catalytic subunit (PP1c) is localized with NCX1 in membrane fractions isolated from rat LV (Fig. 2B, left panels) and neonatal cardiomyocytes (Fig. 2B, right panels). Second, immunoprecipitation of NCX1 in LV lysate and immunoblotting with antibodies against PP1c and PLM identified a NCX1-PLM-PP1c macromolecular complex (Fig. 2C). Third, using the proximity ligation assay, we showed that NCX1 and PP1c co-localize in isolated adult rat cardiomyocytes, strongly indicating that NCX1 and PP1c are in the same molecular complex (as indicated by the green fluorescent spots in Fig. 2D, 1st panel). No signal was observed when primary antibodies were omitted (Fig. 2D, 2nd panel) or when the NCX1 antibody was pre-incubated with a specific anti-NCX1-blocking peptide (Fig. 2D, 3rd panel). Fourth, immunoprecipitation of recombinant His6-TF-NCX1cyt (Fig. 2E, schematic figure) using His6 or NCX1 antibodies showed that recombinant PP1α precipitated with His6-TF-NCX1cyt (Fig. 2F), suggesting NCX1 and PP1α to be direct binding partners. Fifth, the binding kinetics of the GST-PP1α-NCX1 interaction was analyzed by SPR. A range of concentrations of recombinant GST-PP1α was injected over immobilized recombinant His6-TF-NCX1cyt on a CM5 chip and analyzed with the fit of a 1:1 interaction model (Langmuir). The dissociation equilibrium constant (KD) for the interaction was 3.02 ± 1.48 nm, with an association rate constant (ka) = (6.10 ± 3.35) × 104 m−1 s−1 and a dissociation rate constant (kd) = (1.71 ± 0.95) × 10−4 s−1 (Fig. 2G). Conclusively, SPR data indicated that the GST-PP1α-NCX1 interaction was strong and stable.
FIGURE 2.

PP1c binds directly to NCX1. A, epitope mapping was performed by overlaying an array of immobilized overlapping 20-mer rat PP1-α (P62138) peptides with anti-PP1 (E-9) (sc-7482, top panel). Amino acids in bold constitute the core epitope, localized in the PP1 catalytic subunit (PP1c), and are relevant for anti-PP1c binding (n = 2). Immunodetection without anti-PP1c was used as a negative control (bottom panel). B, NCX1 and PP1c were analyzed in cytoplasmic and membrane fractions isolated from rat LV and neonatal cardiomyocytes using anti-NCX1 and anti-PP1c (in triplicate). Vinculin and epidermal growth factor receptor (EGFR) were used as controls for cytoplasmic and membrane fractions, respectively. C, rat LV lysates were subjected to immunoprecipitation (IP) using anti-NCX1 (in triplicate). Immunoprecipitates (right panels) and lysate (input; positive control for the immunoblot, left panels) were immunoblotted with NCX1, PP1c, and PLM antibodies. As a negative control, NCX1 antibody pre-incubated with a specific anti-NCX1 blocking peptide or non-relevant rabbit IgG was used. D, confocal images of in situ proximity ligation assay. NCX1-PP1c co-localization was analyzed in adult cardiomyocytes using anti-NCX1 and anti-PP1c (left panel, interaction indicated by green spots, see “Experimental Procedures” for details). Incubation without primary antibodies (middle panel) and pre-incubating anti-NCX1 with blocking peptide (right panel) were used as negative controls. Nuclei were stained with SYTOX orange. E, schematic figure of recombinant proteins used in this study. The cytosolic loop of NCX1 was purified, and His6 and trigger factor (TF) tags were added to the N terminus (His6-TF-NCX1cyt, top panel). Because of the size of the tags, His6-TF-NCX1cyt migrates as a 150-kDa protein. Commercially available PP1α was used in the initial analysis of the interaction (in F) after which rat PP1-α (P62138) with a GST tag (GST-PP1α) was generated (bottom panel) and used in the remainder of the study. F, recombinant proteins were subjected to immunoprecipitation using anti-NCX1 and anti-His6 (in triplicates). Immunoprecipitates (right panels) and proteins (input panels; positive control for the immunoblot) were immunoblotted with anti-NCX1 and anti-PP1c. As a negative control, non-relevant rabbit and mouse IgG were used. G, SPR analysis was performed by immobilizing recombinant His6-TF-NCX1cyt (ligand) on a CM5 chip and measuring the response when injecting a range of concentrations of GST-PP1α (analyte) (n = 3).
PP1 Anchors to the KIFF/KVFF Motif in NCX1-CBD1
To identify PP1 binding in NCX1, mouse NCX1(FL)-GFP and a series of GFP-NCX1 deletion mutants were expressed in HEK293 cells (Fig. 3A, schematic figure). Fishing with anti-PP1c and recombinant GST-PP1α in the HEK293 lysates and immunoblotting with anti-GFP revealed that all NCX1 variants precipitated, except for GFP-NCX1(243–402) (Fig. 3B), which was lacking CBD1 containing the KIFF/KVFF motif. The bottom panel shows expression of the NCX1 variants and GFP (negative control) in HEK293 lysates (Fig. 3B). To map the PP1-binding site more precisely, the cytosolic loop of rat NCX1 was synthesized as 20-mer overlapping peptides on a membrane and incubated with recombinant GST-PP1α, followed by anti-GST HRP incubation and immunodetection. Interestingly, GST-PP1α bound to 399ENDPVSKVFFEQGTYQCLENCGTVALTII427 containing the KVFF motif (Fig. 3C, boxed sequence). Notably, some GST-PP1α binding was also observed to amino acids C-terminal to the KVFF motif (408FEQGTYQCLENCGTVALTII427) (Fig. 3C, underlined amino acids). Introducing Gly sequentially to the C terminus showed that GST-PP1α binding was lost when the asparagine (Asn-418) was deleted (Fig. 3D), suggesting additional amino acid-binding sites C-terminal to the KVFF motif to be a requisite for PP1 binding. Noteworthy, no GST-PP1α binding was observed for the KVLF-containing sequence, which has previously been suggested to be a putative PP1-binding site (Fig. 3C, upper panel, lower box) (17).
FIGURE 3.

Mapping of the PP1c interaction site in NCX1. A, schematic figure showing full-length NCX1-GFP and a series of GFP-NCX1 deletion mutants that were generated to map the PP1c interaction site in NCX1. The numbering of NCX1 includes the N-terminal amino acid signal peptide sequence. B, GST-PP1α together with NCX1 deletion variants were subjected to immunoprecipitation (IP) using anti-PP1c (in triplicates). Precipitates were analyzed with immunoblotting. The asterisks in the top panel indicate the NCX1 dimer (64) and missing GFP-NCX1(243–402) fragment. Noteworthy, the GFP-NCX1(243–402) fragment was not visible when we re-ran the samples on a second gel with good separation around 50 kDa and used a light chain-specific antibody (data not shown). The middle panel shows anti-PP1c-precipitated GST-PP1α. In the bottom panel the input lysates were immunoblotted to show the migration and corresponding molecular weight of NCX1-GFP and the deletion mutants. The asterisks in this panel indicate the NCX1 dimer, monomer, and migration of the GFP-NCX1(243–402) deletion fragment. C, identification of PP1 binding by overlaying GST-PP1α on 20-mer overlapping NCX1 peptides synthesized on membrane. Rat NCX1 protein (EDM02743) was used spanning amino acids 243–799. Binding was analyzed using anti-GST HRP (n = 2) top panel. Boxed areas show KVFF and KVLF respectively. Underlined amino acids in the first boxed area represent the RVXF -PP1-binding motif and a putative Φ1Φ2 motif, whereas amino acids in bold indicate the common sequence in the four peptide sequences. Incubation without GST-PP1α was used as a negative control (lower panel). D, one of the peptide sequences identified in C; 402PVSKVFFEQGTYQCLENCGT421, was synthesized with a glycine spacer and was overlaid with GST-PP1α (right panel) to determine the effect on PP1 binding when EN, putative Φ1Φ2 motif, was deleted (n = 2). The peptide sequence without GST-PP1α served as negative control (left panel).
Several mutation strategies were employed to test the effect of PP1 binding to the KIFF/KVFF mouse/rat, motif. Ala substitution at the second and fourth position in the KIFF/KVFF motif (KAFA mutant) has been shown to be an effective mutation strategy, as it disrupts PP1 binding (44–46). However, upon examination of the Ca2+-bound CBD1 crystal structure of dog NCX1 (PDB code 2FWS), we noted that the Ile and Phe in the second and fourth position, localized in the α-1 strand in β-sheet 1, seem to be buried in the CBD1 structure. Thus, it seemed more sensible to mutate the Lys, at the first position, and Phe, at the third position (AIAF mutant), as these are the accessible residues. We also generated a Pro substitution mutant (F407P), and in a fourth construct the KIFF motif was deleted (GFP-NCX1(Δ399–424)). All variants were expressed in HEK293 cells and immunoprecipitated with anti-NCX1. PP1c co-precipitated with NCX1(FL)-GFP but not with GFP-NCX1(I406A,F408A) (Fig. 4A), GFP-NCX1(K405A,F407A) (Fig. 4B), GFP-NCX1(F407P) or the deletion, GFP-NCX1(Δ399–424) (Fig. 4C). Interestingly, more PP1c co-precipitated with NCX1 in the presence of 3 mm CaCl2 (Fig. 4D). In a final experiment, the rat peptide sequence 402PVSKVFFEQGTYQCLENCGT421 (identified in Fig. 3C) and corresponding sequences with KVFF substituted for KAFA or AVAF were overlaid with GST-PP1α. Strong binding was observed for the native peptide, but not to the KAFA or AVAF mutations (Fig. 4E). Altogether, the data supports that PP1α anchors to the KIFF/KVFF motif in NCX1.
FIGURE 4.

Confirmation of the PP1c interaction site in NCX1 by mutagenesis. NCX1(FL)-GFP, containing the KIFF motif (mouse sequence), together with either GFP-NCX1(I406A, F408A) representing KAFA mutant (A), GFP-NCX1(K405A, F407A) representing the AIAF mutant (B), or GFP-NCX1(F407P) representing the proline substitution KIPF and GFP-NCX1(Δ399–424) representing KIFF deletion (C) were generated and expressed in HEK293 cells after which they were used in immunoprecipitation (IP) experiments. The NCX1 antibody was used to precipitate NCX1 followed by immunoblotting with anti-PP1c for determination of PP1 association. D, rat LV lysates were subjected to immunoprecipitation using anti-NCX1, with and without the addition of 3 mm CaCl2. Immunoprecipitates (right panels) and lysate (input panels, positive controls for the immunoblot) were immunoblotted using relevant antibodies. E, peptide sequence 402PVSKVFFEQGTYQCLENCGT421 (identified in Fig. 3C) with KVFF motif was mutated to KAFA and AVAF and overlaid with GST-PP1α to determine the effect of mutations (n = 2). GST-PP1α binding was analyzed by immunodetection with anti-GST HRP. All experiments were run in triplicates (A–D).
NCX1-KIFF/KVFF Is an Independent, Non-regulatory PP1c-anchoring Site
To confirm NCX1-KIFF/KVFF as an independent PP1c-binding site, we generated biotinylated NCX1-KVFF, NCX1-KAFA, and NCX1-AVAF peptides (Fig. 5A, schematic figure). Pulldown experiments using anti-biotin-coupled agarose beads showed that GST-PP1α precipitated with biotin-NCX1-KVFF but not biotin-NCX1-KAFA (Fig. 5B) or biotin-NCX1-AVAF (data not shown). When the NCX1-KVFF peptide or His6-TF-NCX1cyt recombinant protein was tested in an in vitro PP1 activity assay, no change in recombinant PP1 activity was observed (Fig. 5, C and D, respectively), indicating that the NCX1-KIFF/KVFF motif is a non-regulatory PP1c-anchoring site. Sequence alignments show that the NCX1-KIFF/KVFF PP1c-anchoring site is highly conserved across human, rat, and mouse NCX1 (Fig. 5E) and that NCX2 and NCX3 harbor RIFF and KVFF motifs, respectively (Fig. 5F). Therefore, biotinylated NCX isoform peptides (schematically illustrated in Fig. 5A) were also generated and used in pulldown experiments. Using equal amounts of peptides, we found strong GST-PP1c binding to biotin-NCX1(399–424), some binding to biotin-NCX3(392–412), and weak binding to biotin-NCX2(389–410) (Fig. 5G).
FIGURE 5.

Independence and isoform specificity of the NCX1-KIFF/KVFF-anchoring site. A, schematic figure of NCX1-biotinylated peptides used in pulldown and PP1 activity assays. B, pulldown assay with the biotinylated NCX1 peptides and GST-PP1α recombinant protein (in triplicates). PP1 binding was analyzed by immunoblotting with anti-PP1c (upper panel). Anti-biotin HRP was used to show the presence of biotinylated peptides. Incubation of the GST-PP1α recombinant protein with only the beads was used as negative control. C, effect of NCX1-PP1 interaction on PP1 activity was assessed. In each experiment, 1 unit of active PP1c was incubated with a range of recombinant His6-TF-NCX1cyt protein concentrations or (D) biotinylated NCX1 peptides for 20 min, after which the activity was determined. Inhibitor 2 (PP1 inhibitor) was used as control for the assay. The KIFF/KVFF-anchoring motif, a putative Φ1Φ2 motif (EN) and a conserved arginine motif (R), are shown in the alignment of human, rat, mouse NCX1 (E) and NCX1–3 isoforms (F) (black boxes indicate similar functional amino acids (DNA Star). G, pulldown assay with the biotinylated NCX1–3 peptides and GST-PP1α recombinant protein (in triplicates). PP1 binding was analyzed by immunoblotting with anti-PP1c (upper panel). Incubation of GST-PP1α recombinant protein with only the beads was used a negative control. GST-PP1α recombinant protein and biotinylated NCX peptides were used as positive controls for the immunoblot in B and G (input panels on left).
PP1c Dephosphorylates pSer-68-PLM and Regulates NCX1 Activity through NCX1-KIFF/KVFF Anchoring
First, existence of the NCX1-PLM-PP1c macromolecular complex was confirmed in lysates from HEK293 cells co-transfected with NCX1(FL)-GFP and PLM and immunoblotting with antibodies against NCX1, PP1c, and PLM (Fig. 6A). Second, to determine whether NCX1 anchoring is required for dephosphorylation of pSer-68-PLM by PP1c, FLAG-His6-PP1c(1–330) and PLM were expressed with and without NCX1(FL)-GFP in HEK293. PLM has been reported to be 30–40% phosphorylated by endogenous kinases in HEK293 (24, 47), and immunodetection with a specific anti-pSer-68-PLM (Fig. 6B, antibody epitope mapping) showed that PLM was highly phosphorylated in HEK293 (Fig. 6C, lanes 4–6, anti-pSer-68-PLM level). Importantly, the pSer-68-PLM/total PLM level was significantly decreased in PLM + FLAG-His6-PP1c + NCX1(FL)-GFP compared with PLM + FLAG-His6-PP1c (0.4 ± 0.03 versus 1.0 ± 0.05). This suggests that NCX1 anchoring is a prerequisite for dephosphorylation of pSer-68-PLM by PP1c (Fig. 6C, lanes 7–9). Consistently, when the KIFF/KVFF motif was mutated (GFP-NCX1(F407P)) or deleted (GFP-NCX1(Δ399–424)), PP1c was not able to dephosphorylate pSer-68-PLM, as shown by a significant increase of pSer-68-PLM/total PLM level in PLM + FLAG-His6-PP1c + GFP-NCX1(F407P) (2.4 ± 0.04 versus 1.0 ± 0.09) and a significant increase of pSer-68-PLM/total PLM level in PLM + FLAG-His6-PP1c + GFP-NCX1(Δ399–424) (2.0 ± 0.10 versus 1.0 ± 0.09) compared with PLM + FLAG-His6-PP1c + NCX1(FL)-GFP (Fig. 6D, lanes 4–9 versus lane 1–3).
FIGURE 6.

Analysis of the NCX1, PP1c, and PLM macromolecular complex in HEK293 cells. A, HEK293 cells were co-transfected with PLM and NCX1(FL)-GFP. The lysates were subjected to immunoprecipitation (IP) using anti-NCX1. Immunoprecipitates (right panels) and lysate (input; positive control for the immunoblot, left panel) were immunoblotted with NCX1, PP1c, and PLM antibodies. As a negative control NCX1 antibody pre-incubated with a specific anti-NCX1 blocking peptide or non-relevant rabbit IgG was used. B, epitope mapping was performed by overlaying an array of immobilized overlapping 20-mer PLM peptides with anti-pSer-68-PLM (right panel) (n = 2) (35). Immunodetection without anti-pSer-68-PLM was used as a negative control (left panel). FLAG-His6-PP1c(1–330) and PLM were expressed with and without either NCX(FL)-GFP (C) or GFP-NCX1(F407P) (D), representing the proline substitution KIPF, or GFP-NCX1(Δ399–424), representing KIFF deletion, in HEK293 followed by immunoblotting with anti-NCX1, FLAG, pSer-68-PLM, and total PLM. Non-transfected HEK293 cells were used as control in C. Bar graph shows relative pSer-68-PLM/total PLM level quantified by densitometry analysis. Differences were tested using unpaired t test (**, p < 0.01; ***, p < 0.001). E, lysate from FLAG-His6-PP1c(1–330) and PLM co-expressed with or without NCX1(FL)-GFP in HEK293 were treated with or without active PKA-C, followed by immunoblotting with anti-pSer-68-PLM and total PLM. Bar graph shows the relative pSer-68-PLM/total PLM level quantified by densitometry analysis. Differences were tested using unpaired t test (ns.; not significant). All experiments were performed in triplicates (A–E).
To exclude that the presence of NCX1 prevents PLM phosphorylation, lysate from FLAG-His6-PP1c(1–330) and PLM co-expressed with or without NCX1(FL)-GFP was treated with or without active PKA-C. Our results indicated that NCX1 did not block the pSer-68-PLM phosphorylation, as the increase in the pSer-68-PLM/total PLM level was similar between the two groups (Fig. 6E).
Finally, the biological role of endogenous PP1c on NCX1 activity through dephosphorylation of PLM was analyzed in HEK293 cells. First, the whole cell patch clamp technique was employed. NCX1 current was isolated by measuring Ni2+-sensitive current elicited during a slow voltage ramp from 120 to −100 mV. Co-expression of PLM with WT NCX1(FL) resulted in NCX1 inhibition (both forward and reverse mode), consistent with what was reported previously (Fig. 7F) (PLM is endogenously phosphorylated 30–40% by kinases in HEK293) (22). Moreover, mutation of the PP1c-anchoring site in NCX1 (NCX1(F407P)) resulted in more NCX1 inhibition (Fig. 7F), indicating that endogenous PP1c was not able to dephosphorylate the pool of phosphorylated PLM. Noteworthy, the current of NCX1(F407P) alone was similar to that of WT NCX1(FL) (Fig. 7E). Current traces of the various transfections are given in Fig. 7, A–D.
FIGURE 7.

Whole cell patch clamp analysis of NCX1 regulation by PLM and PP1c. HEK293 cells were transfected with WT NCX1(FL), WT NCX1(FL), and PLM or Pro substitution mutant NCX1(F407P) and PLM. NCX1 current was elicited by a descending voltage ramp from 120 to −100 mV. Current traces of WT NCX1(FL) (n = 9) (A), NCX1(F407P) (n = 7) (B), WT NCX1(FL) and PLM (n = 6) (C), and NCX1(F407P) and PLM (n = 5) (D) are shown. E and F, relationship between current (normalized to cell capacitance) and membrane voltage (Vm), presented for potentials between +80 and −80 mV. *, p < 0.05 (WT NCX1(FL) + PLM versus WT NCX1(FL) and NCX1(F407P) + PLM versus WT NCX1(FL)). #, p < 0.05 (NCX1(F407P) + PLM versus WT NCX1(FL) + PLM (n = 5)).
NCX1 function was further examined by measuring Ca2+-dependent fluorescence in transfected HEK293 cells. Extracellular Na+ concentration was rapidly changed from 140 to 0 mm to induce Ca2+ entry via reverse-mode NCX1 activity. In accordance with observations in the patch clamp experiments, NCX1-mediated Ca2+ influx observed in WT NCX1-transfected cells was markedly reduced when PLM was co-expressed with WT NCX1(FL) (F/F0 = 1.6 ± 0.08 versus 1.9 ± 0.18). Mutation of the PP1c-anchoring site in NCX1 (NCX1(F407P)) resulted in a further decrease in the intracellular calcium level when co-expressed with PLM (F/F0 = 1.2 ± 0.05) (Fig. 8). Altogether, our data strongly suggest that NCX1-associated PP1c dephosphorylates pSer-68-PLM.
FIGURE 8.

NCX regulation by PLM and PP1c analyzed by Ca2+-dependent fluorescence. Ca2+ entry into HEK293 cells via reverse-mode NCX1 function was induced by rapidly decreasing extracellular [Na+]. A, representative recordings of Ca2+ fluorescence from HEK293 cells transfected with WT NCX1(FL) (n = 11), or NCX1(FL) and PLM (n = 11), or NCX1(F407P) and PLM (n = 6). B, bar graph shows Ca2+ recordings that were background-subtracted and are presented normalized to baseline fluorescence (F/F0) level quantified by Clampex 10.4 and Clampfit 10.4 software, respectively. Differences were tested using unpaired t test: *, p < 0.05 (WT NCX1(FL) + PLM versus WT NCX1-(FL)). **, p < 0.005 (NCX1(F407P) + PLM versus WT NCX1(FL)). #, p < 0.05 (NCX1(F407P) + PLM versus WT NCX1(FL) + PLM)).
Mapping of the NCX1 Interaction Site in PP1c
To determine the reciprocal NCX1-binding site in PP1, PP1c with an N-terminal FLAG and His6 tags, along with two deletion mutants, were generated and expressed in HEK293 cells (Fig. 9A, schematic figure). Immunoprecipitation experiments using His6-TF-NCX1cyt together with the PP1c deletion variants revealed that His6-TF-NCX1cyt precipitated only with FLAG-His-PP1c(1–330) but not with the deletions (Fig. 9B). To map NCX1 binding in PP1c more precisely, 20-mer overlapping peptides of rat PP1c were synthesized on membranes that were overlaid with recombinant His6-TF-NCX1cyt. The residues forming the RVXF binding pocket are dispersed in the PP1 primary sequence (Fig. 9E, denoted with stars). Nevertheless, weak NCX1 binding was observed for the peptide sequence 235FLHKHDLDLICRAHQVVEDGYEFFAK260, containing Asp-242, Leu-243, and Phe-257 (Fig. 9C, boxed sequences). These amino acids form part of the RVXF binding pocket (30). Interestingly, Phe-257 is reported as an important anchoring site in PP1c (48). To validate this interaction site, a deletion mutant, FLAG-His6-PP1c(Δ232–263), without the putative interaction site, was generated and expressed in HEK293 cells. Lysates were incubated with recombinant His6-TF-NCX1cyt. Immunoprecipitation with anti-FLAG showed loss of interaction with FLAG-His6-PP1c(232–263) deletion, suggesting that the constrained binding region is important for NCX1 binding (Fig. 9D).
FIGURE 9.

Mapping of the NCX1 interaction site in PP1. A, schematic figure showing PP1c and two deletion mutants that were generated to map the NCX1 interaction site in PP1. A FLAG and His6 tags were inserted on the N terminus. The arrow indicates NCX1 binding region. B, His6-TF-NCX1cyt together with the PP1 deletion variants were subjected to immunoprecipitation using anti-FLAG (in triplicates). Precipitates were analyzed with immunoblotting. C, identification of NCX1 binding by overlaying His6-TF-NCX1cyt on 20-mer overlapping rat PP1 (P62138) peptides synthesized on membrane. Binding was analyzed using anti-His6 HRP (n = 2). Boxed area shows His6-TF-NCX1cyt binding to PP1c; amino acids in bold indicate the common sequence in the three peptide sequences, and underlined amino acids form part of the RVXF binding pocket. D, FLAG-His6-PP1c(1–330) together with a variant, where the NCX1 binding region was deleted; FLAG-His6-PP1c(Δ232–263) was used in immunoprecipitation experiments. The anti-FLAG antibody was used to precipitate FLAG-His6-PP1c variants expressed in HEK293 cells followed by immunoblotting with anti-NCX1 for determination of NCX1 association with PP1c (in triplicates). His6-TF-NCX1cyt in lysate from non-transfected HEK293 cells was used as a negative control. E, residues that are important for PP1-binding of target proteins (30) is shown by the stars in the alignment of human, mouse, and rat PP1. The boxed area show His6-TF-NCX1cyt binding to PP1 (identified in C).
Structural Modeling of NCX1-KVFF into PP1
A homology model was generated of the rat NCX1 tetrapeptide 405KVFF408, binding to rat/human PP1c using the 448KIHF451 PP1c binding segment of rat spinophilin (PDB code 3EGG) as template (Fig. 10A). It shows that Val-406 may fit into a hydrophobic pocket formed by Ile-169, Leu-243, and Leu-289 in PP1c and that Phe-407 may interact with Met-290. The peptide backbone forms hydrogen bonds with Asp-242, Leu-289, and Cys-291. Phe-257 and Arg-261 also play an important role in peptide binding. The conformation of the KVFF motif in this model is essentially similar to the conformation of the homologous KIFF motif as found in the NMR and x-ray structures of the Canis Lupus NCX1 ((PDB code 2FWS) (42) and (PDB code 2DPK (49)), the only difference being the orientation of the Lys side chain. However, large rearrangements in the NCX1 must take place upon binding to PP1c in order for the motif to be accessible for binding. To further test the model, Leu-243 and Phe-257 were single and double-mutated to Gly as follows: FLAG-His6-PP1c (L243G), FLAG-His6-PP1c (F275G), and FLAG-His6-PP1c (L243G,F275G). Immunoprecipitation showed that FLAG-His6-PP1c(1–330) co-precipitated His6-TF-NCX1cyt, but no binding was observed for the PP1c mutants (Fig. 10B). This lends support to Leu-243 and Phe-257 in PP1c being important residues for NCX1 anchoring.
FIGURE 10.

Model of NCX1-KVFF peptide binding to PP1. A, homology model of rat NCX1 peptide motif KVFF binding to human/rat PP1. The KVFF peptide (green stick model) interacts with PP1 (light blue surface and stick model of selected residues) via polar hydrogen bonds (black dashed lines) and hydrophobic contacts (red dashed lines). The directions of extension of the peptide are indicated by two black arrows. B, PP1c (FLAG-His6-PP1c(1–330)) together with FLAG-His6-PP1c single and double mutants (L243G, F257G) were immunoprecipitated with anti-FLAG, followed by immunoblotting with anti-NCX1 to determine co-precipitation of recombinant His6-TF-NCX1cyt (in triplicates).
pSer-68-PLM-NCX1-PP1c Complex Is Increased in HF
To analyze the pSer-68-PLM-NCX1-PP1c levels in failing hearts, membrane fractions were isolated from SHAM and ABHF rats. Animal characteristics are listed in Table 1. Immunoblotting showed that the PP1c/calsequestrin level was significantly increased in ABHF hearts compared with SHAM (1.9 ± 0.10 versus 1.0 ± 0.03). The pSer-68-PLM/total PLM level was also significantly increased in ABHF hearts compared with SHAM (1.3 ± 0.10 versus 1.0 ± 0.03) (Fig. 11A). We have previously shown that the NCX1 level is up-regulated in response to pressure overload in our animal model (15), similar to what others have shown (5, 50). Anti-calsequestrin and anti-total PLM were used as controls. Recently, we have identified a calpain cleavage site at Met-369 in NCX1, which resides between the PLM-anchoring site (16) and PP1c-anchoring site (this study). Wanichawan et al. (15) showed that calpain activity was greatly increased in ABHF hearts and that this correlated with increased cleavage of NCX1. To test whether calpain cleavage is responsible for dissociating PP1c away from PLM, HEK293 cells were co-transfected with PLM, FLAG-His6-PP1c(1–330), and NCX1(TEV). In NCX1(TEV), the calpain cleavage site is engineered into a TEV protease site. Interestingly, the pSer-68-PLM/total PLM level was significantly increased in PLM + FLAG-His6-PP1c + NCX1 (TEV) + TEV/pCS2MT (HEK293 cells co-transfected with the enzyme that cleaves the TEV sequence) compared with PLM + FLAG-His6-PP1c + NCX1(TEV) (HEK293 cells are not co-transfected with the enzyme) (1.3 ± 0.08 versus 1.0 ± 0.05) (Fig. 11B), indicating that calpain cleavage of NCX1 at Met-369 separates the PLM- and PP1c-anchoring sites. A model of PP1c anchored onto NCX1-KIFF/KVFF leading to dephosphorylation of pSer-68-PLM is summarized in Fig. 12, A and B (effect of calpain cleavage).
TABLE 1.
Animal characteristics of rats showing SHAM controls and ABHF
The following abbreviations are used: LV, left ventricle; BW, body weight; RV, right ventricle; IVSd, interventricular septum in diastole; PWd, posterior wall thickness in diastole; LVDd/s, left ventricular diameter in diastole or systole, respectively; FS, fractional shortening; LAD, left atrial diameter; HR, heart rate; CO, cardiac output; LVOT, left ventricular outlet tract.
| SHAM | ABHF | |
|---|---|---|
| Organ weights | ||
| Number (n) | 9 | 5 |
| BW (g) | 364 ± 6 | 353 ± 12 |
| Heart weight (g) | 1.01 ± 0.05 | 2.31 ± 0.19a |
| LV weight (g) | 0.66 ± 0.03 | 1.23 ± 0.04a |
| LV/BW ratio (mg/g) | 1.8 ± 0.1 | 3.5 ± 0.2a |
| RV weight (g) | 0.17 ± 0.01 | 0.29 ± 0.02a |
| Lung weight (g) | 1.33 ± 0.05 | 3.28 ± 0.34a |
| Echocardiography | ||
| M-mode | ||
| IVSd (mm) | 1.6 ± 0.0 | 2.2 ± 0.1a |
| PWd (mm) | 1.6 ± 0.1 | 2.0 ± 0.1a |
| LVDd (mm) | 6.3 ± 0.2 | 7.3 ± 0.3a |
| LVDs (mm) | 3.4 ± 0.2 | 4.3 ± 0.5 |
| FS (%) | 55 ± 5 | 42 ± 5a |
| LAD (mm) | 3.5 ± 0.1 | 5.5 ± 0.4a |
| Doppler | ||
| Peak mitral flow (mm/s) | 908 ± 42 | 1039 ± 20 |
| Mitral deceleration (mm/s2) | 2974 ± 190 | 4243 ± 750 |
| HR (beats/min) | 438 ± 5 | 390 ± 19a |
| CO in LVOT (ml/min) | 130 ± 11 | 105 ± 12 |
| Tissue Doppler | ||
| Maximal systolic velocity (mm/s) | 70 ± 4 | 49 ± 8a |
| Minimal diastolic velocity (mm/s) | 72 ± 3 | 54 ± 5a |
a p < 0.05 SHAM versus ABHF.
FIGURE 11.

Analysis of NCX1, PP1c, and PLM in SHAM and ABHF rats. A, PP1c, pSer-68-PLM and PLM levels were analyzed in isolated cardiac membrane fractions by immunoblotting using relevant antibodies (n = 5). Anti-calsequestrin and total PLM were used as loading controls. Bar graph show relative PP1c/calsequestrin level and pSer-68-PLM/total PLM level quantified by densitometry analysis. Differences were tested using unpaired t test (*, p < 0.05; ***, p < 0.001). B, FLAG-His6-PP1c(1–330), PLM, and NCX1 (TEV) were expressed with or without TEV protease followed by immunoblotting using relevant antibodies (in triplicates). Bar graph show relative pSer-68-PLM/total PLM level quantified by densitometry analysis. Differences were tested using unpaired t test (*, p < 0.05).
FIGURE 12.

Proposed model of indirect regulation of NCX1 by PP1c. A, under normal physiological conditions, pSer-68-PLM (phosphorylation denoted by a circled P) interacts with the PASKT- and QKHPD-containing sequences in the intracellular loop of NCX1 and inhibits NCX1 activity (14, 16, 23, 24). PP1c anchoring onto NCX1-KIFF/KVFF in CBD1 dephosphorylates pSer-68-PLM, relieving inhibition of the exchanger. B, calpain activity is increased in ABHF hearts (simulating heart failure), and this correlates with increased cleavage of NCX1 at Met-369 (15). This cleavage hinders dephosphorylation of pSer-68-PLM by PP1c because it dissociates PP1c away from pSer-68-PLM. Cleavage of NCX1 at Met-369 down-regulates NCX1 current (both forward and reverse mode) (15).
Discussion
In an ongoing effort to understand regulation of cardiac NCX1 activity, we have investigated the molecular basis of PP1c targeting to the NCX1 macromolecular complex, and we determined the functional consequences of this interaction.
PP1c Anchors to the NCX1-KIFF/KVFF Motif in CBD1
PP1 targeting is predominantly mediated by a short RVXF-docking motif, which is present in 90% of targeting proteins (31, 51). Therefore, bioinformatic analysis was used to search for RVXF motifs on PLM and NCX1. No RVXF motifs were identified in PLM, but three putative RVXF motifs were identified in NCX1, two of which were localized to the cytosolic loop, which mediates NCX1 regulation.
Several experiments were conducted, which suggest that the KVFF motif in the rat NCX1 cytosolic loop (KIFF in human and mouse NCX1) is the PP1c-targeting site responsible for pSer-68-PLM dephosphorylation. 1) PP1c bound directly to the cytosolic loop of recombinant NCX1. 2) Peptide array data localized PP1c binding to NCX1-KVFF. Binding to other putative PP1c-binding motifs was not observed. 3) PP1c binding was abolished when the KVFF motif was deleted or mutated to KAFA or AVAF, both in the NCX1 full-length protein and in 20-mer NCX1 peptides. 4) Presence of NCX1 was a pre-requisite for dephosphorylation of pSer-68-PLM by PP1c. No dephosphorylation was observed when the NCX1 full-length protein was absent or when the KVFF motif was mutated. 5) The NCX1 cytosolic loop, containing KVFF, had no effect on PP1 activity, confirming an anchoring role of NCX1. 6) Finally, the electrophysiological experiments demonstrated that mutation of the PP1c KVFF site suppresses NCX1 activity and that this suppression is mediated through an increased pSer-68-PLM level.
As mentioned above, loss of the NCX1-PP1c interaction was observed with both the NCX1-KAFA and NCX1-AVAF mutations. We also generated a proline substitution mutant, NCX1-(F407P), and the obtained results are in agreement with studies reporting that proline is not tolerated at position X in RVXF because it prevents β-strand formation (31). Other proteins that adopt a more rigid structure upon PP1-binding are spinophilin and MYPT1. Spinophilin forms a β-strand upon binding to PP1, which extends a β-strand structure in PP1 (52). Although regulator proteins adopt a distinct conformation when bound to PP1, they do not significantly change the PP1 structure.
It has been suggested that most PP1-targeting proteins combine multiple docking motifs to form a stable complex with PP1 (53). Thus, in addition to looking for the RVXF motifs, we scanned the NCX1 primary sequence for additional PP1-docking motifs. NCX1 was found to partially fulfill the extended PP1-binding motif, RVXF-X5–8Φ1Φ2-X8–9-R (36). By introducing glycine mutations to the C terminus of KVFF in the array, loss of binding was observed with N418G, suggesting Asn-418 to be part of the Φ1Φ2 motif. Using the JackHMMER (54), Peti et al. (30) constrained Φ2 to amino acids FIYRHNQSC and Φ1 to amino acids VIYFH. Thus, Glu-417 does not fit into the pure Φ1 definition; however, the Φ1Φ2 motif is considered to be variable. Additionally, there is a conserved Arg-428 in NCX1. Therefore, in addition to the RVXF PP1-binding motif, the partial Φ1Φ2 motif and the conserved Arg-428 on NCX1 may form part of a high affinity PP1-binding site. The Arg motif in the PP1 nuclear targeting subunit (PNUTS)-PP1 holoenzyme has been shown to work as a substrate selectivity filter, by blocking access to the C-terminal binding groove (36). Therefore, dephosphorylation of pSer-68-PLM may not require binding to the C-terminal groove, and PP1 interaction sites such as SILK (55) and MyPhoNE (56) were not found in NCX1. Several PP1 holoenzyme complexes have been reported to be regulated by phosphorylation of amino acids flanking the RVXF motifs (31). NCX1 possesses both a serine and threonine close to the KIFF/KVFF motif, suggesting that PP1 targeting to NCX1 might be dynamic. Scansite.mit.edu software identified the flanking phosphorylation sites in NCX-KIFF/KVFF as putative casein kinase or Src kinase phosphorylation sites.
NCX1 Binds to the Hydrophobic Pocket in PP1c
PP1c deletion mutants constrained NCX1 binding to amino acids 212–330 in the N terminus of PP1c. Consistently, the peptide array assay showed very weak binding to the sequence 235FLHKHDLDLICRAHQVVEDGYEFFAK260. The Asp-242, Leu-243, and Phe-257 residues are localized in β-sheets 10 and 11 and form part of the RVXF binding pocket, where Asp-242 exhibits a high excess of negative charges, and Leu-243 and Phe-257 form part of the hydrophobic pocket (30, 57). These different properties of the PP1 surface determine the amino acid types tolerated at the different ligand positions. To confirm that NCX1 anchoring to PP1 is mediated by the residues in the hydrophobic pocket, we generated glycine single and double mutations of FLAG-His6-PP1c (L243G and F257G). Our peptide docking model, with the NCX-KVFF motif in human NCX1 based on the PP1-spinophilin complex, shows that NCX1-Val-406 may fit onto a hydrophobic pocket formed by Ile-169, Leu-243, and Leu-289 in PP1. Furthermore, the NCX1-Phe-407 may interact with PP1-Met-290. The conformation of the KVFF motif in this model is similar to the conformation of the homologous KIFF motif, as found in the NMR and x-ray structures of the Canis Lupus NCX1 (Protein Data Bank code 2FWS (42) and Protein Data Bank code 2DPK (49)).
The PP1α-NCX1 interaction was strong (Kd of 3.0 nm), in a similar range to that of PNUTS-PP1α/β/γ (9.3 nm) (36) and spinophilin-PP1α (8.7 nm) (41). Two-thirds of all PP1-targeting proteins have a PP1 interaction domain that is predicted to be intrinsically disordered. These interaction domains adopt a defined fold upon binding of PP1. X-ray crystallography and NMR studies have shown a substantial loss of structural integrity in the Ca2+-binding sites of NCX1-CBD1 in the absence of Ca2+, but upon Ca2+ binding NCX1-CBD1 is arranged into antiparallel β-strands (58). Interestingly, Ile-406 and Phe-408 in canine NCX1, which are part of the identified NCX1-KIFF/KVFF motif, have been suggested to be important for stability of the protein structure (58). We speculate that the PP1-Asp-242 might form a salt bridge with the NCX1-Lys-408, facilitating high affinity binding, as shown in our SPR analysis results. Importantly, NCX1 was able to bind PP1-Phe-257 on the peptide array, which has been shown to be an important anchoring site in PP1, and mutation of this residue to alanine resulted in loss of PP1-binding (48).
PP1c Anchored to NCX1-KIFF/KVFF, Dephosphorylates pSer-68-PLM, and Regulates NCX1 Activity in HEK293
We confirmed that PLM co-precipitates with NCX1 in lysates from rat cardiomyocytes and HEK293, as reported previously (22). Immunoblotting with a specific anti-pSer-68-PLM antibody showed that PLM was highly phosphorylated in HEK293, consistent with reports that PLM is endogenously phosphorylated (30–40%) by kinases in HEK293 (24, 47). Noteworthy, both PLM and non-phosphorylatable PLM bind to NCX1 (22); however, it is pSer-68-PLM that exerts an inhibitory effect on NCX1 activity (24). The pSer-68-PLM level was significantly reduced in the presence of NCX1(FL)-GFP, suggesting that NCX1 anchoring is a prerequisite for dephosphorylation of pSer-68-PLM by PP1c. Co-expression of PLM with WT NCX1(FL) reduced the current in both forward and reverse mode. Moreover, NCX1 function was almost completely suppressed when HEK293 cells were co-transfected with PLM and NCX1(F407P) (mutated PP1-anchoring site). These results suggest that PP1 plays a significant role in relieving NCX1 inhibition, by dephosphorylating pSer-68-PLM. Reverse mode NCX1 was also observed to be inhibited in Ca2+ fluorescence measurements. This result is in agreement with Ahlers et al. (22), who used a radioactive tracer uptake method in HEK293 cells. They found that co-expressing NCX1 and PLM resulted in 15% reduction of 45Ca2+ uptake after cells were exposed to a solution containing zero Na+.
PLM-NCX1-PP1c Macro-molecular Complex in HF
Consistent with previous findings (15), we found an up-regulation of NCX1 expression in our pressure overload model. However, evaluation of NCX1 function is complex, as an increase in NCX1 expression does not imply an increase in NCX1 activity. Indeed, there have been some models showing a decreased or unchanged NCX1 activity (9, 59). Regulator proteins of NCX1 might also be altered in HF, which also affects NCX1 activity. The PLM level may vary in different models of hypertrophy or HF, as indicated in the study by El-Armouche et al. (28), where they showed a down-regulation of PLM phosphorylation in human heart failure. Bossuyt et al. (60) found a reduction in total PLM expression in HF rabbit myocytes, LV homogenates, and in human HF but an up-regulation in the pSer-68-PLM. The existence of different pools of PLM in the heart could account for the different phosphorylation outcomes for the NKA-associated and NCX1-associated PLM. Such pools are known to exist in myocytes (61). Global PP1c activity and expression is increased in human and experimental HF and is suggested to be a unique characteristic of HF (62). Mice overexpressing the catalytic subunit of PP1α to similar levels as observed in HF had depressed cardiac function, dilated cardiomyopathy, and premature mortality (63). We found up-regulation of both PP1c/calsequestrin levels and pSer-68-PLM/total PLM levels in our pressure overload model, suggesting that PP1 is not able to dephosphorylate pSer-68-PLM. PLM interacts with two distinct regions in the intracellular loop of NCX1 (amino acids 238–270 and 300–328) (16) and inhibits the activity of exchanger. Recently, we showed that NCX1 was cleaved by calpain in HF and mapped a calpain cleavage site to Met-369 in NCX1, which separates the PLM- and PP1c (this study)-anchoring sites. Cleavage at this site led to a reduction in NCX1 activity (both forward and reverse mode) compared with WT NCX1(FL) (15). Interestingly, cleavage at this site in HEK293 up-regulated pSer-68-PLM/total PLM levels (Fig. 10B; this study), suggesting that dephosphorylation of pSer-68-PLM by PP1c is inhibited. This finding could account for why it is reported that pSer-68-PLM and PP1c levels are up-regulated in HF.
In conclusion, NCX1 is a substrate-specifying PP1c-targeting protein. NCX1 also facilitates regulation of its own activity by mediating dephosphorylation of pSer-68-PLM.
Author Contributions
T. L. H. conceived and coordinated the study, performed and analyzed the experiments shown in Figs. 1, 2, A and C–F, 3, A–C, 4, A–C, 5, A–D, 5G, 6, A–D, 8, A and B, 9, A–D, 10B, 11A, and 12, wrote the paper, reviewed and approved the final version of the manuscript. K. H. performed and analyzed the experiments shown in Fig. 7, reviewed the results, and approved the final version of the manuscript. P. W. performed experiments in Figs. 2B and 6E, reviewed the results, and approved the final version of the manuscript. J. M. A. performed the surgery on the animals and did the echocardiographic, hemodynamic, and post-mortem analysis in Table 1, reviewed the results, and approved the final version of the manuscript. B. D. made the structure model shown in Fig. 10A, was involved in the planning of experiments shown in Figs. 3D and 10B, reviewed the results, and approved the final version of the manuscript. P. K. L. was involved in the planning of the experiment shown in Fig. 8, reviewed the results, and approved the final version of the manuscript. M. L. performed the experiments shown in Figs. 3D, 4, D–E, and 11B, reviewed the results, and approved the final version of the manuscript. M. M. performed the transfections used in Figs. 6, C–E, 7, and 11B, reviewed the results, and approved the final version of the manuscript. U. H. E. performed the experiments in Fig. 2G, reviewed the results, and approved the final version of the manuscript. W. F. provided the antibody shown in Fig. 6B and optimization conditions, reviewed the results, and approved the final version of the manuscript. I. S. performed the surgery on the animals and did the echocardiographic, hemodynamic, and post-mortem analysis in Table 1, reviewed the results, and approved the final version of the manuscript. W. E. L. conceived, coordinated, and analyzed the experiment shown in Fig. 8 and reviewed the results, and approved the final version of the manuscript. O. M. S. conceived the study, reviewed the results, and approved the final version of the manuscript. C. R. C. conceived and coordinated the study, performed the experiments in Figs. 2G, 5, E and F, and 9E, analyzed the experiments, supervised the writing of the paper, reviewed and approved the final version of the manuscript.
This work was supported in part by the South-Eastern Norway Regional Health Authority, the Norwegian National Health Association, Research Council of Norway, Stiftelsen Kristian Gerhard Jebsen, the Simon Fougner Hartmann's Family Fund, Denmark, and Anders Jahre's Fund for the promotion of Science. The authors declare that they have no conflicts of interest with the contents of this article.
- NCX
- Na+-Ca2+ exchanger
- PLM
- phospholemman
- pSer-68-PLM
- serine 68-phosphorylated phospholemman
- PP1
- protein phosphatase 1
- NKA
- Na+/K+-ATPase
- TM
- transmembrane domains
- CBD
- Ca2+ binding domain
- HF
- heart failure
- LV
- left ventricle
- ABHF
- aortic banding animals with congestive heart failure
- SHAM
- sham-operated animals
- SPR
- surface plasmon resonance
- FL
- full-length
- IP
- immunoprecipitation
- TEV
- tobacco etch virus
- PDB
- Protein Data Bank
- TF
- trigger factor
- His
- His6.
References
- 1. Blaustein M. P., and Lederer W. J. (1999) Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854 [DOI] [PubMed] [Google Scholar]
- 2. Lee S. L., Yu A. S., and Lytton J. (1994) Tissue-specific expression of Na+-Ca2+ exchanger isoforms. J. Biol. Chem. 269, 14849–14852 [PubMed] [Google Scholar]
- 3. Linck B., Qiu Z., He Z., Tong Q., Hilgemann D. W., and Philipson K. D. (1998) Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3). Am. J. Physiol. 274, C415–C423 [DOI] [PubMed] [Google Scholar]
- 4. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 5. Hasenfuss G., Schillinger W., Lehnart S. E., Preuss M., Pieske B., Maier L. S., Prestle J., Minami K., and Just H. (1999) Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 99, 641–648 [DOI] [PubMed] [Google Scholar]
- 6. Studer R., Reinecke H., Bilger J., Eschenhagen T., Böhm M., Hasenfuss G., Just H., Holtz J., and Drexler H. (1994) Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ. Res. 75, 443–453 [DOI] [PubMed] [Google Scholar]
- 7. Pogwizd S. M., Qi M., Yuan W., Samarel A. M., and Bers D. M. (1999) Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 85, 1009–1019 [DOI] [PubMed] [Google Scholar]
- 8. Khananshvili D. (2014) Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch. 466, 43–60 [DOI] [PubMed] [Google Scholar]
- 9. Sipido K. R., Volders P. G., Vos M. A., and Verdonck F. (2002) Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc. Res. 53, 782–805 [DOI] [PubMed] [Google Scholar]
- 10. Philipson K. D., and Nicoll D. A. (2000) Sodium-calcium exchange: a molecular perspective. Annu. Rev. Physiol. 62, 111–133 [DOI] [PubMed] [Google Scholar]
- 11. Ren X., and Philipson K. D. (2013) The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J. Mol. Cell. Cardiol. 57, 68–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shattock M. J., Ottolia M., Bers D. M., Blaustein M. P., Boguslavskyi A., Bossuyt J., Bridge J. H., Chen-Izu Y., Clancy C. E., Edwards A., Goldhaber J., Kaplan J., Lingrel J. B., Pavlovic D., Philipson K., et al. (2015) Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 593, 1361–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsuoka S., Nicoll D. A., Reilly R. F., Hilgemann D. W., and Philipson K. D. (1993) Initial localization of regulatory regions of the cardiac sarcolemmal Na+-Ca2+ exchanger. Proc. Natl. Acad. Sci. U.S.A. 90, 3870–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X. Q., Wang J., Carl L. L., Song J., Ahlers B. A., and Cheung J. Y. (2009) Phospholemman regulates cardiac Na+/Ca2+ exchanger by interacting with the exchanger's proximal linker domain. Am. J. Physiol. Cell Physiol. 296, C911–C921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wanichawan P., Hafver T. L., Hodne K., Aronsen J. M., Lunde I. G., Dalhus B., Lunde M., Kvaløy H., Louch W. E., Tønnessen T., Sjaastad I., Sejersted O. M., and Carlson C. R. (2014) Molecular basis of calpain cleavage and inactivation of the sodium-calcium exchanger 1 in heart failure. J. Biol. Chem. 289, 33984–33998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang X. Q., Wang J., Song J., Ji A. M., Chan T. O., and Cheung J. Y. (2011) Residues 248–252 and 300–304 of the cardiac Na+/Ca2+ exchanger are involved in its regulation by phospholemman. Am. J. Physiol. Cell Physiol. 301, C833–C840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schulze D. H., Muqhal M., Lederer W. J., and Ruknudin A. M. (2003) Sodium/calcium exchanger (NCX1) macromolecular complex. J. Biol. Chem. 278, 28849–28855 [DOI] [PubMed] [Google Scholar]
- 18. Ruknudin A. M., Wei S. K., Haigney M. C., Lederer W. J., and Schulze D. H. (2007) Phosphorylation and other conundrums of Na/Ca exchanger, NCX1. Ann. N.Y. Acad. Sci. 1099, 103–118 [DOI] [PubMed] [Google Scholar]
- 19. Wanichawan P., Louch W. E., Hortemo K. H., Austbø B., Lunde P. K., Scott J. D., Sejersted O. M., and Carlson C. R. (2011) Full-length cardiac Na+/Ca2+ exchanger 1 protein is not phosphorylated by protein kinase A. Am. J. Physiol. Cell Physiol. 300, C989–C997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sweadner K. J., and Rael E. (2000) The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 68, 41–56 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X. Q., Qureshi A., Song J., Carl L. L., Tian Q., Stahl R. C., Carey D. J., Rothblum L. I., and Cheung J. Y. (2003) Phospholemman modulates Na+/Ca2+ exchange in adult rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 284, H225–H233 [DOI] [PubMed] [Google Scholar]
- 22. Ahlers B. A., Zhang X. Q., Moorman J. R., Rothblum L. I., Carl L. L., Song J., Wang J., Geddis L. M., Tucker A. L., Mounsey J. P., and Cheung J. Y. (2005) Identification of an endogenous inhibitor of the cardiac Na+/Ca2+ exchanger, phospholemman. J. Biol. Chem. 280, 19875–19882 [DOI] [PubMed] [Google Scholar]
- 23. Song J., Zhang X. Q., Ahlers B. A., Carl L. L., Wang J., Rothblum L. I., Stahl R. C., Mounsey J. P., Tucker A. L., Moorman J. R., and Cheung J. Y. (2005) Serine 68 of phospholemman is critical in modulation of contractility, [Ca2+]i transients, and Na+/Ca2+ exchange in adult rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 288, H2342–H2354 [DOI] [PubMed] [Google Scholar]
- 24. Zhang X. Q., Ahlers B. A., Tucker A. L., Song J., Wang J., Moorman J. R., Mounsey J. P., Carl L. L., Rothblum L. I., and Cheung J. Y. (2006) Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger. Role of phosphorylation. J. Biol. Chem. 281, 7784–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crambert G., Fuzesi M., Garty H., Karlish S., and Geering K. (2002) Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc. Natl. Acad. Sci. U.S.A. 99, 11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bossuyt J., Despa S., Martin J. L., and Bers D. M. (2006) Phospholemman phosphorylation alters its fluorescence resonance energy transfer with the Na/K-ATPase pump. J. Biol. Chem. 281, 32765–32773 [DOI] [PubMed] [Google Scholar]
- 27. Han F., Bossuyt J., Martin J. L., Despa S., and Bers D. M. (2010) Role of phospholemman phosphorylation sites in mediating kinase-dependent regulation of the Na+-K+-ATPase. Am. J. Physiol. Cell Physiol. 299, C1363–C1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El-Armouche A., Wittköpper K., Fuller W., Howie J., Shattock M. J., and Pavlovic D. (2011) Phospholemman-dependent regulation of the cardiac Na/K-ATPase activity is modulated by inhibitor-1 sensitive type-1 phosphatase. FASEB J. 25, 4467–4475 [DOI] [PubMed] [Google Scholar]
- 29. O'Connell N., Nichols S. R., Heroes E., Beullens M., Bollen M., Peti W., and Page R. (2012) The molecular basis for substrate specificity of the nuclear NIPP1:PP1 holoenzyme. Structure 20, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peti W., Nairn A. C., and Page R. (2013) Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 280, 596–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bollen M., Peti W., Ragusa M. J., and Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wakula P., Beullens M., Ceulemans H., Stalmans W., and Bollen M. (2003) Degeneracy and function of the ubiquitous RVXF motif that mediates binding to protein phosphatase-1. J. Biol. Chem. 278, 18817–18823 [DOI] [PubMed] [Google Scholar]
- 33. Brattelid T., Qvigstad E., Birkeland J. A., Swift F., Bekkevold S. V., Krobert K. A., Sejersted O. M., Skomedal T., Osnes J. B., Levy F. O., and Sjaastad I. (2007) Serotonin responsiveness through 5-HT2A and 5-HT4 receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J. Mol. Cell. Cardiol. 43, 767–779 [DOI] [PubMed] [Google Scholar]
- 34. Lunde I. G., Aronsen J. M., Kvaløy H., Qvigstad E., Sjaastad I., Tønnessen T., Christensen G., Grønning-Wang L. M., and Carlson C. R. (2012) Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genomics 44, 162–172 [DOI] [PubMed] [Google Scholar]
- 35. Fuller W., Howie J., McLatchie L. M., Weber R. J., Hastie C. J., Burness K., Pavlovic D., and Shattock M. J. (2009) FXYD1 phosphorylation in vitro and in adult rat cardiac myocytes: threonine 69 is a novel substrate for protein kinase C. Am. J. Physiol. Cell Physiol. 296, C1346–C1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choy M. S., Hieke M., Kumar G. S., Lewis G. R., Gonzalez-DeWhitt K. R., Kessler R. P., Stein B. J., Hessenberger M., Nairn A. C., Peti W., and Page R. (2014) Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc. Natl. Acad. Sci. U.S.A. 111, 4097–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panigrahi A. K., Zhang N., Mao Q., and Pati D. (2011) Calpain-1 cleaves Rad21 to promote sister chromatid separation. Mol. Cell. Biol. 31, 4335–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jordan M., Schallhorn A., and Wurm F. M. (1996) Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24, 596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frank R., and Overwin H. (1996) SPOT synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol. Biol. 66, 149–169 [DOI] [PubMed] [Google Scholar]
- 40. McAvoy T., and Nairn A. C. (2010) Serine/threonine protein phosphatase assays. Curr. Protoc. Mol. Biol. Chapter 18, Unit 18.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ragusa M. J., Dancheck B., Critton D. A., Nairn A. C., Page R., and Peti W. (2010) Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol. 17, 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hilge M., Aelen J., and Vuister G. W. (2006) Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 22, 15–25 [DOI] [PubMed] [Google Scholar]
- 43. Ottolia M., Nicoll D. A., and Philipson K. D. (2009) Roles of two Ca2+-binding domains in regulation of the cardiac Na+-Ca2+ exchanger. J. Biol. Chem. 284, 32735–32741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beullens M., Van Eynde A., Vulsteke V., Connor J., Shenolikar S., Stalmans W., and Bollen M. (1999) Molecular determinants of nuclear protein phosphatase-1 regulation by NIPP-1. J. Biol. Chem. 274, 14053–14061 [DOI] [PubMed] [Google Scholar]
- 45. Bollen M. (2001) Combinatorial control of protein phosphatase-1. Trends Biochem. Sci. 26, 426–431 [DOI] [PubMed] [Google Scholar]
- 46. Egloff M. P., Johnson D. F., Moorhead G., Cohen P. T., Cohen P., and Barford D. (1997) Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 16, 1876–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang X. Q., Moorman J. R., Ahlers B. A., Carl L. L., Lake D. E., Song J., Mounsey J. P., Tucker A. L., Chan Y. M., Rothblum L. I., Stahl R. C., Carey D. J., and Cheung J. Y. (2006) Phospholemman overexpression inhibits Na+-K+-ATPase in adult rat cardiac myocytes: relevance to decreased Na+ pump activity in postinfarction myocytes. J. Appl. Physiol. 100, 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lesage B., Beullens M., Nuytten M., Van Eynde A., Keppens S., Himpens B., and Bollen M. (2004) Interactor-mediated nuclear translocation and retention of protein phosphatase-1. J. Biol. Chem. 279, 55978–55984 [DOI] [PubMed] [Google Scholar]
- 49. Nicoll D. A., Sawaya M. R., Kwon S., Cascio D., Philipson K. D., and Abramson J. (2006) The crystal structure of the primary Ca2+ sensor of the Na+/Ca2+ exchanger reveals a novel Ca2+ binding motif. J. Biol. Chem. 281, 21577–21581 [DOI] [PubMed] [Google Scholar]
- 50. Lu Y. M., Huang J., Shioda N., Fukunaga K., Shirasaki Y., Li X. M., and Han F. (2011) CaMKIIδB mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic constriction-induced failing heart. PLoS ONE 6, e24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heroes E., Lesage B., Görnemann J., Beullens M., Van Meervelt L., and Bollen M. (2013) The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J. 280, 584–595 [DOI] [PubMed] [Google Scholar]
- 52. Choy M. S., Page R., and Peti W. (2012) Regulation of protein phosphatase 1 by intrinsically disordered proteins. Biochem. Soc. Trans. 40, 969–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boens S., Szekér K., Van Eynde A., and Bollen M. (2013) Interactor-guided dephosphorylation by protein phosphatase-1. Methods Mol. Biol. 1053, 271–281 [DOI] [PubMed] [Google Scholar]
- 54. Johnson L. S., Eddy S. R., and Portugaly E. (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hurley T. D., Yang J., Zhang L., Goodwin K. D., Zou Q., Cortese M., Dunker A. K., and DePaoli-Roach A. A. (2007) Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J. Biol. Chem. 282, 28874–28883 [DOI] [PubMed] [Google Scholar]
- 56. Terrak M., Kerff F., Langsetmo K., Tao T., and Dominguez R. (2004) Structural basis of protein phosphatase 1 regulation. Nature 429, 780–784 [DOI] [PubMed] [Google Scholar]
- 57. Meiselbach H., Sticht H., and Enz R. (2006) Structural analysis of the protein phosphatase 1 docking motif: molecular description of binding specificities identifies interacting proteins. Chem. Biol. 13, 49–59 [DOI] [PubMed] [Google Scholar]
- 58. Hilge M. (2012) Ca2+ regulation of ion transport in the Na+/Ca2+ exchanger. J. Biol. Chem. 287, 31641–31649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hasenfuss G. (1998) Alterations of calcium-regulatory proteins in heart failure. Cardiovasc. Res. 37, 279–289 [DOI] [PubMed] [Google Scholar]
- 60. Bossuyt J., Ai X., Moorman J. R., Pogwizd S. M., and Bers D. M. (2005) Expression and phosphorylation of the Na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 97, 558–565 [DOI] [PubMed] [Google Scholar]
- 61. Wypijewski K. J., Howie J., Reilly L., Tulloch L. B., Aughton K. L., McLatchie L. M., Shattock M. J., Calaghan S. C., and Fuller W. (2013) A separate pool of cardiac phospholemman that does not regulate or associate with the sodium pump: multimers of phospholemman in ventricular muscle. J. Biol. Chem. 288, 13808–13820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nicolaou P., Hajjar R. J., and Kranias E. G. (2009) Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J. Mol. Cell. Cardiol. 47, 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Carr A. N., Schmidt A. G., Suzuki Y., del Monte F., Sato Y., Lanner C., Breeden K., Jing S. L., Allen P. B., Greengard P., Yatani A., Hoit B. D., Grupp I. L., Hajjar R. J., DePaoli-Roach A. A., and Kranias E. G. (2002) Type 1 phosphatase, a negative regulator of cardiac function. Mol. Cell. Biol. 22, 4124–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ren X., Nicoll D. A., Galang G., and Philipson K. D. (2008) Intermolecular cross-linking of Na+-Ca2+ exchanger proteins: evidence for dimer formation. Biochemistry 47, 6081–6087 [DOI] [PubMed] [Google Scholar]