

Abstract
Cdk5 is a versatile protein kinase that is involved in various neuronal activities, such as the migration of newborn neurons, neurite outgrowth, synaptic regulation, and neurodegenerative diseases. Cdk5 requires the p35 regulatory subunit for activation. Because Cdk5 is more abundantly expressed in neurons compared with p35, the p35 protein levels determine the kinase activity of Cdk5. p35 is a protein with a short half-life that is degraded by proteasomes. Although ubiquitination of p35 has been previously reported, the degradation mechanism of p35 is not yet known. Here, we intended to identify the ubiquitination site(s) in p35. Because p35 is myristoylated at the N-terminal glycine, the possible ubiquitination sites are the lysine residues in p35. We mutated all 23 Lys residues to Arg (p35 23R), but p35 23R was still rapidly degraded by proteasomes at a rate similar to wild-type p35. The degradation of p35 23R in primary neurons and the Cdk5 activation ability of p35 23R suggested the occurrence of ubiquitin-independent degradation of p35 in physiological conditions. We found that p35 has the amino acid sequence similar to the ubiquitin-independent degron in the NKX3.1 homeodomain transcription factor. An Ala mutation at Pro-247 in the degron-like sequence made p35 stable. These results suggest that p35 can be degraded by two degradation pathways: ubiquitin-dependent and ubiquitin-independent. The rapid degradation of p35 by two different methods would be a mechanism to suppress the production of p25, which overactivates Cdk5 to induce neuronal cell death.
Keywords: cyclin-dependent kinase (CDK), neuron, proteasome, protein degradation, serine/threonine protein kinase, ubiquitin, cyclin-dependent kinase 5, degron
Introduction
Cyclin-dependent kinases (Cdks)2 are a family of Ser/Thr kinases that are activated by binding a regulatory subunit called cyclin. Most members of Cdks are expressed in proliferating cells to promote cell cycle progression (1). In contrast, Cdk5 is activated by p35 or p39 non-cyclin proteins, which are mainly expressed in post-mitotic neurons (2). Cdk5 is a versatile kinase that is involved in many neuronal activities, including neuronal cell layer formation, synaptic transmission, membrane trafficking, and neuron cell death (3). p35 and p39 appear to share common and/or distinct functions for Cdk5, with p35 being the predominant activator. This is shown by the phenotypes of knock-out (KO) mice; p35 KO mice display abnormal neural layers in the cerebral cortex (4), and p39 KO mice do not show apparent abnormalities, whereas p35 and p39 double KO mice are perinatal lethal with abnormal neural layers, as are the Cdk5 KO mice (5–7). To understand the precise function of Cdk5-p35 in various neuronal activities, it is important to reveal the regulation mechanism of Cdk5 activity.
As well as being cell cycle Cdks, Cdk5 is a stable protein and is expressed more abundantly than p35 in neurons (8, 9). Therefore, Cdk5 activity is determined primarily by the available amount of activator protein p35, and the protein amounts of p35 are regulated by the balance between synthesis and degradation (2). Although the synthesis of p35 is stimulated by NGF or BDNF (10, 11), the degradation of p35 is carried out by proteasomes (12, 13). The degradation is a major determinant of the p35 level, which is reduced by treating neurons with excitatory neurotransmitter glutamate (14). p35 associates with membranes via myristoylation at the N-terminal glycine (15, 16), and this association enhances the degradation of p35 (17). On the other hand, p35 is cleaved by a calcium-dependent protease calpain to produce the C-terminal stable fragment p25 (15, 18, 19). Although the physiological function of Cdk5-p25 has been recently reported (20, 21), its abundance induces neuronal cell death in neurodegenerative diseases (22). Rapid turnover of p35 is suggested to be a mechanism to prevent p25 production (2). Therefore, it is particularly important to determine the degradation mechanism of p35. Interestingly, the addition of the N-terminal hepta-peptide containing the myristoylation site of p35 facilitates p35 lability (17), indicating that the degradation of p35 occurs selectively on membranes. Although p35 has previously been demonstrated to be post-translationally modified by ubiquitination (12), the E3 ligase responsible has not been identified yet in the neuron, and its degradation pathway is not completely understood.
The ubiquitin-proteasome system is a major component of the proteolytic machinery that performs the degradation of proteins in cells (23, 24). Ubiquitin is a small protein that is tagged to substrate proteins to be degraded. The proteasome is a large complex of multicatalytic proteases that degrades proteins to small peptides. The 26S proteasome is a complex of 20S proteasome and 19S particles. The 20S proteasome is the core of the proteasome, and 19S is a regulatory particle (PA700) that recognizes and unfolds ubiquitinated proteins. The unfolded proteins are proteolyzed by being inserted into the 20S chamber. Ubiquitination is a critical step in the ubiquitin-proteasome system for selective degradation, which is catalyzed by E3 ubiquitin ligases. There are large numbers of E3 ligases with a specific substrate (25). The E3 ligase for p35 has not been found in the brain, although in pancreatic β-cells, Pja2 has been recently reported to have E3 ligase activity to p35 (26).
However, polyubiquitination is not an absolute requirement for proteasomal degradation. Ornithine decarboxylase is a well known example of ubiquitin-independent proteasomal degradation (27, 28). The number of proteins susceptible to ubiquitin-independent proteasomal degradation had recently been increasing. They include thymidylate synthase, Rpn4, p21 Cdk inhibitor, p53 tumor suppressor, c-Fos, Nkx3.1, and so on (29–35). Degron sequences recognized by proteasomes have been investigated in these proteins, but their degradation mechanisms and physiological meanings are largely unknown. Herein, we intended to identify the ubiquitination sites on p35 for a better understanding of its ubiquitin-proteasome-dependent degradation mechanism. Unexpectedly, however, we found that non-ubiquitinated p35 was degraded at a comparable rate to wild-type p35. Our results indicate that p35 is degraded by proteasomes by two pathways: ubiquitin-dependent and ubiquitin-independent.
Experimental Procedures
Antibodies and Chemicals
Anti-HA (Y11, SC805), anti-p35 (C19), and anti-Cdk5 (DC17) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin (A2066) anti-Myc (9E10) antibodies and cycloheximide (CHX) were purchased from Sigma. Benzyloxycarbonyl-leucyl-leucyl-leucinal (MG132) and epoxomicin were obtained from Calbiochem. Protein A-Sepharose was obtained from GE Healthcare.
Construction of Mammalian Cell Expression Vectors
Expression vectors of p35, N7-p25-myc, and Cdk5 were previously described (17). Lys-to-Arg mutants were constructed by polymerase chain reaction (PCR) with Pfu Ultra DNA polymerase (Agilent Technologies, Santa Clara, CA) using the primers listed in Table 1. Mutant p35 P247A and mutant p35 23R P247A were constructed by PCR using pCMV5-p35 and pCMV5-p35 23R, respectively, as a template using primers included in Table 1. pCAG-p35 and pCAG-p35 23R were constructed by insetting p35 and p35 23R, respectively, into the pCAG-MCS2 vector (36) at the BamHI/HindIII sites.
TABLE 1.
List of primers using for mutations
| Name of primers | Forward sequence | Reverse sequence |
|---|---|---|
| K13R | CCCCAGCTATCGGAGGGCCACACTGTTTG | CAAACAGTGTGGCCCTCCGATAGCTGGGG |
| K34R | CGTGCAGAACAGCAGGAACGCCAAGGACA | TGTCCTTGGCGTTCCTGCTGTTCTGCACG |
| K34R/K37R | CAGCAGGAACGCCAGGGACAAGAACCTGA | TCAGGTTCTTGTCCCTGGCGTTCCTGCTG |
| K37R/K39R | GAACGCCAGGGACAGGAACCTGAAGCGGC | GCCGCTTCAGGTTCCTGTCCCTGGCGTTC |
| K39R/K42R | GGACAGGAACCTGAGGCGGCACTCCATCA | TGATGGAGTGCCGCCTCAGGTTCCTGTCC |
| K53R | GGTGCTGCCTTGGAGGAGGATCGTGGCGG | CCGCCACGATCCTCCTCCAAGGCAGCACC |
| K61R/K62R/K63R | GGTGTCAGCGAGGAGGAGGAACTCCAGGA | TCCTGGAGTTCCTCCTCCTCGCTGACACC |
| K66R/K67R | GAAGAAGAACTCCAGGAGGGCGCAGCCCA | TGGGCTGCGCCCTCCTGGAGTTCTTCTTC |
| K87R/K88R | CAATGAGAACCTGAGGAGGTCGCTGTCCT | AGGACAGCGACCTCCTCAGGTTCTCATTG |
| K126R/K127R | CTCCTCTTCTGTCAGGAGGGCCCCGCACC | GGTGCGGGGCCCTCCTGACAGAAGAGGAG |
| K140R | TGCAGGGACACCCAGACGGGTCATCGTCC | GGACGATGACCCGTCTGGGTGTCCCTGCA |
| K167R | GTGCTACCGCCTGAGGCACTTGTCCCCAA | TTGGGGACAAGTGCCTCAGGCGGTAGCAC |
| K246R | CTCCTACCCGCTCAGGCCCTTCCTGGTAG | CTACCAGGAAGGGCCTGAGCGGGTAGGAG |
| K254R | GGTAGAGAGCTGTAGGGAAGCCTTTTGGG | CCCAAAAGGCTTCCCTACAGCTCTCTACC |
| K271R | CCTCATGAGCTCCAGGATGCTGCAGATCA | TGATCTGCAGCATCCTGGAGCTCATGAGG |
| K290R | GTTCTCTGACTTGAGGAATGAGAGCGGTC | GACCGCTCTCATTCCTCAAGTCAGAGAAC |
| K298R/K299R | CGGTCAGGAGGACAGGAGGCGACTCCTCCTGG | CCAGGAGGAGTCGCCTCCTGTCCTCCTGACCG |
| MycKtoR | GGATCGGGAACAAAGACTCATCTCAGAAG | CTTCTGAGATGAGTCTTTGTTCCCGATCC |
| R126K/R127K | CTCCTCTTCTGTCAAGAAGGCCCCGCACC | GGTGCGGGGCCTTCTTGACAGAAGAGGAG |
| R167K | GTGCTACCGCCTGAAGCACTTGTCCCCAA | TTGGGGACAAGTGCTTCAGGCGGTAGCAC |
| R298K/R299K | CGGTCAGGAGGACAAGAAGCGACTCCTCC | GGAGGAGTCGCTTCTTGTCCTCCTGACCG |
| P247A | TACCCGCTCAAGGCCTTCCTGGTAGAGA | TCTCTACCAGGAAGGCCTTGAGCGGGTA |
| 23R-P247A | TACCCGCTCAGGGCCTTCCTGGTAGAGA | TCTCTACCAGGAAGGCCCTGAGCGGGTA |
Cell Culture and Transfection
Neuro2a and HEK293T cells were obtained from the JCRB Cell Bank (Osaka, Japan) and cultured in Dulbecco's modified Eagle's Medium (DMEM, Sigma) containing 10% fetal bovine serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Neuro2a and COS-7 cells were transfected with expression plasmids using Hilly Max transfection regent (Dojindo, Kumamoto, Japan), Lipofectamine 2000 (ThermoFisher, Waltham, MA), or PolyFect transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's protocol.
ICR mice were obtained from Sankyo Laboratory Service (Tokyo, Japan). Animal experiments were performed according to the guidelines for Animal Experiments of Tokyo Metropolitan University (approval numbers: 23-13, 24-15, and 25-12). The mice were housed in a temperature-controlled room under a 12-h light/12-h dark cycle with free access to food and water. Primary neurons were prepared from mouse brain cortex at embryonic day 16 (E16) and plated on polyethyleneimine-coated dishes in DMEM and Ham's F-12 (1:1) supplemented with 5% fetal bovine serum, 5% horse serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin at a density of 2.5 × 105 cells/cm2 (37). The medium was then changed to neurobasal medium supplemented with 1% B-27 (Invitrogen), 1 mm l-glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Primary cortical neurons were transfected at 3 days in vitro by the calcium phosphate method using the ProFection Mammalian Transfection System (Promega, Madison, WI).
Immunoprecipitation and Kinase Assay
Neuro2a cells expressing Cdk5 and N7-p25, N7-p25 11R, p35, or p35 23R were collected and lysed in 20 mm MOPS at pH 6.8, 1 mm EGTA, 0.1 mm EDTA, 0.15 m NaCl, 1 mm MgCl2, 10 μg/ml leupeptin, 0.4 mm Pefabloc, and 0.3% (v/v) Nonidet P-40. After centrifugation, the supernatants were immunoprecipitated using anti-Cdk5 (C8) as previously described (17). The kinase activity of the immunoprecipitates was measured with histone H1 and [γ-32P]ATP as substrates.
For the polyubiquitination assay, Neuro2a cells expressing N7-p25 or its mutants were treated with 20 μm MG132 for 5 h. At the end of the treatment the cells were collected, and the cell extracts were incubated with anti-p35 for p35 or anti-myc for N7-p25, which was followed by coprecipitation with protein A-Sepharose beads (17).
SDS-PAGE, Immunoblotting, and Statistical Analysis
Sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed with 12.5% polyacrylamide gels. The phosphorylation levels of p35 were assessed by Phos-tag SDS-PAGE with 10% polyacrylamide gels containing 50 μm Phos-tag and 100 μm MnCl2 as previously described (38). Immunoblotting was performed as previously described (17). All of the statistical treatments were performed by one-way analysis of variance with Tukey's post hoc test.
Results
Effect of Arginine Mutation of the Lysine Residues in the C-terminal p25 Region on the Stability of p35 or N7-p25
p35 has been shown to be degraded by proteasomes through its polyubiquitination (15, 17, 39). To better understand the regulation of p35 degradation, a good goal would be to identify the E3 ligase that catalyzes its ubiquitination. An approach toward identifying the E3 ligase is to determine the ubiquitination sites in p35. Ubiquitin conjugation occurs on the amino group of lysine residue(s) or the N-terminal residue of the protein. The α-NH2 group of the N-terminal glycine of p35 is blocked by myristoylation (15, 16, 17). Therefore, as candidates for possible ubiquitination sites, we first focused on the lysines at amino acids 126/127, 140, and 167 because they are relatively close to Thr-138, the phosphorylation of which is involved in the degradation of p35 (37, 40). Also, amino acids 298/299 in the C-terminal region share homology with double Lys residues that serve as ubiquitination sites in p53, a tumor suppressor protein with a short half-life (41) (Fig. 1A). We mutated these Lys to Arg and cotransfected the mutant with Cdk5 in Neuro2a cells to observe the degradation in the presence of cycloheximide, a protein synthesis inhibitor, for 1 and 3 h. However, none of the mutations stabilized p35 (Fig. 1B).
FIGURE 1.
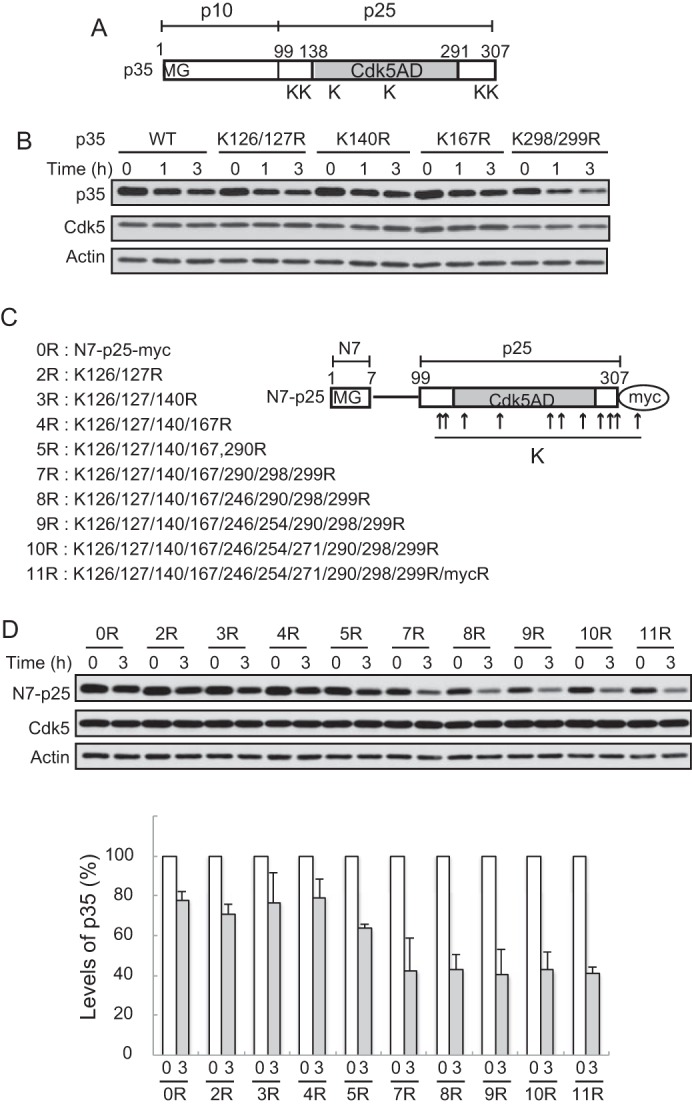
Effect of Lys-to-Arg mutations in the C-terminal p25 region of p35 on degradation. A, schematic representation of the p35 molecule. p35 consists of the N-terminal p10 and the C-terminal p25 Cdk5 activation domain (Cdk5AD). Lysine residues that we thought possible ubiquitination sites are indicated by K. B, effect of Arg mutations at these possible ubiquitination sites on the degradation of p35. Neuro2a cells expressing p35 (WT) or its Arg mutants K126R/K127R, K140R, K167R, or K298R/K299R were treated with CHX for 1 or 3 h. The amounts of p35 protein were assessed by immunoblotting with anti-p35 (top). Cdk5 is shown in the middle. Actin is the loading control. C, N7-p25 molecule and its Arg mutants at Lys residues. N7-p25 is p25 fused with the seven N-terminal amino acids (N7), where there is a myristoylation consensus motif. The positions of the Lys residues are indicated by arrows. The sequential mutation of all of the Lys to Arg in p25 is shown on the left. 11R contains an Arg mutation at the Lys in the myc-tag sequence. D, degradation of Arg mutants of N7-p25 in Neuro2a cells. N7-p25 and its Arg mutants coexpressed with Cdk5 into Neuro2a cells were detected by Immunoblotting with anti-myc after treatment with CHX. Quantification is shown in the lower panel (mean ± S.E., n = 3).
Although p25 is a stable C-terminal fragment of p35 (15, 18, 19), the addition of the seven N-terminal amino acids (N7), including the myristoylation motif, confers p25 with susceptibility to proteasomal degradation (17). The results suggest that the ubiquitination site(s) is present in the p25 region because there is no lysine in the N7 sequence. Accordingly, we mutated the Lys residues in N7-p25 in the order described in Fig. 1C. We expected that N7-p25 would become stable when the critical Lys is replaced with Arg. In this experiment we used N7-p25 with a myc tag at the C terminus because we wondered if multiple Lys-to-Arg mutations might affect the reactivity of anti-p35 antibodies. Mutations at the N-terminal Lys residues at 126, 127, 140, and 167 did not change the stability (Fig. 1D, 1R-4R). Arg mutants of N7-p25 became more labile when Lys residues in the C-terminal region were further changed (Fig. 1D, 5R-7R). Additional mutations at Lys at 246, 254, and 271 in the core of the Cdk5 activation domain did not further alter the stability (Fig. 1D, 8R-10R). Although we mutated all of the Lys residues in p25, N7-p25 was still unstable. There was a Lys residue in the myc-tag EQKLISEEDL. The Lys null version of N7-p25 11R was constructed, and its stability was examined. Surprisingly, however, N7-p25 11R was still unstable (Fig. 1D, 11R).
We confirmed that N7-p25 11R was not ubiquitinated. N7-p25 11R as well as 10R was cotransfected with HA-ubiquitin into Neuro2a cells. After immunoprecipitation with anti-myc, ubiquitination was assessed by immunoblotting with anti-HA or anti-polyubiquitin antibodies. In contrast to the strong ubiquitination of N7-p25, it was greatly reduced in N7-p25 11R. Some signal of N7-p25 10R suggested ubiquitination at Lys in the myc-tag sequence. We also tested whether there are preferential ubiquitination sites among the Lys residues in the p25 region using several add-back mutants. Although Lys at amino acid 167 did not induce ubiquitination at all, Lys add-back mutants at residues 126/127 or 298/299 showed intermediate strength ubiquitination signals (Fig. 2A). These results indicate that N7-p25 can be ubiquitinated at multiple Lys residues if they are available.
FIGURE 2.
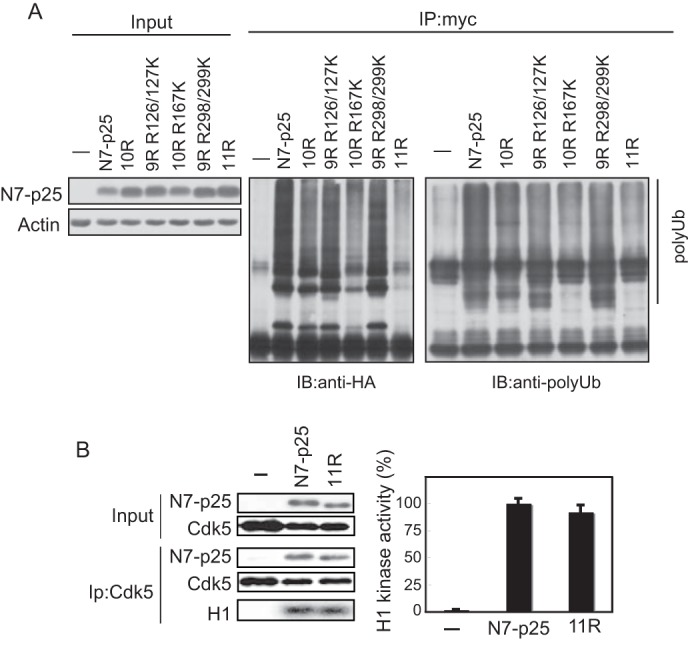
Ubiquitination and Cdk5 activation ability of N7-p25 Arg mutants. A, ubiquitination of N7-p25 and its Arg mutants. N7-p25 and its Arg mutants expressed with HA-ubiquitin into Neuro2a cells were immunoprecipitated (IP) by anti-p35, and ubiquitination was assessed by immunoblotting (IB) with anti-HA antibody (middle) and anti-polyubiquitin (polyUb) antibody (right). B, activation of Cdk5 by N7-p25 11R. N7-p25 or N7-p25 11R was cotransfected with Cdk5 into Neuro2a cells. The binding of N7-p25 or N7-p25 11R to Cdk5 was assayed by the co-immunoprecipitation with anti-Cdk5. The kinase activity was measured by the phosphorylation of histone H1. The autoradiograph is shown at the bottom, and quantification is shown on the right (mean ± S.E., n = 3).
We investigated whether N7-p25 11R would lose its conformation and, therefore, be degraded by proteasomes as a misfolded protein. To test this possibility, the N7-p25 11R ability to bind or activate Cdk5 was compared with that of N7-p25. N7-p25 11R was immunoprecipitated with Cdk5 as much as N7-p25 (Fig. 2B, left). The kinase activity of Cdk5 bound to N7-p25 11R was measured with histone H1 phosphorylation. No significant difference was observed between Cdk5-N7-p25 and Cdk5-N7-p25 11R (Fig. 2B, right). These results suggest that N7-p25 11R retains the same capacity for activating Cdk5 as does N7-p25.
Lys Null Mutant of p35, p35 23R, Is Also Degraded by Proteasomes
The above results with N7-p25 raised the possibility that p35 would be degraded without ubiquitination. However, there are still 13 additional Lys residues in the N-terminal p10 region. To test for the possibility that the Lys residues in the N-terminal p10 region provide ubiquitination sites, we constructed a lysine-null mutant of full-length p35 (p35 23R) in which all 23 lysine residues of p35 were change to arginine (Fig. 3A). Here, we used p35 WT and p35 23R without any tag because the Arg mutations in the C-terminal region of p35 did not affect the immunoreactivity to anti-p35 antibodies (C19). Because the N terminus of p35 is blocked as described above, p35 23R has no ubiquitination sites, and in fact, polyubiquitination was not detected in p35 23R (Fig. 3B). We observed the degradation of p35 23R in Neuro2a cells. p35 23R was decreased in the presence of CHX, and this decrease was suppressed with the proteasomal inhibitor MG132 as well as the more specific proteasome inhibitor Epoxomicin (Fig. 3C). These results indicate that p35 23R is degraded by proteasomes without ubiquitination. We confirmed the proper folding of p35 23R by its binding and activation of Cdk5. p35 23R was co-immunoprecipitated with anti-Cdk5, and Cdk5-p35 23R displayed histone H1 phosphorylation activity as much as Cdk5-p35 WT (Fig. 4A).
FIGURE 3.
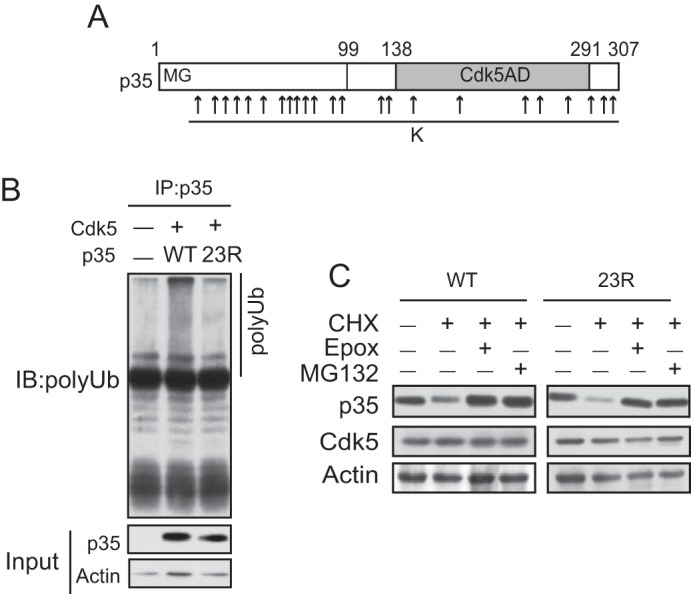
p35 is degraded by proteasomes without ubiquitination. A, Arg mutations of all of the Lys residues in p35. There are 23 lysine residues at amino acid numbers 13, 34, 37, 39, 42, 53, 61, 62, 63, 66, 67, 87, 88, 126, 127, 140, 167, 246, 254, 271, 290, 298, and 299. All of these Lys residues were replaced with Arg to make lysine-null mutants of p35 (p35 23R). B, ubiquitination of p35 or p35 23R. Neuro2a cells expressing p35 (WT) or p35 23R were treated with MG132 for 5 h. p35 were immunoprecipitated (IP) with anti-p35 and immunoblotted (IB) with anti-polyubiquitin (polyUb). C, p35 23R is degraded by proteasomes. Neuro2a cells expressing Cdk5 and p35 (WT) or p35 23R were treated with CHX in the presence or absence of the proteasome inhibitor epoxomicin or MG132 for 5 h. p35 (WT) (left) or p35 23R (right) was detected by immunoblotting with anti-p35.
FIGURE 4.

p35 23R activates Cdk5 as does p35. A, binding to and activation of Cdk5 by p35 23R. p35 (WT) or p35 23R expressed in Neuro2a cells was immunoprecipitated with anti-Cdk5, and their binding of p35 23R was examined by the immunoblotting of p35 in the Cdk5 immunoprecipitates (IP). The kinase activity was measured by the phosphorylation of histone H1 (H1). Histone H1 phosphorylation is quantified in the right panel (means ± S.E., n = 3). B, phosphorylation of p35. Phosphorylation of p35 expressed in Neuro2a cells was examined in the presence or absence of Cdk5-p35 using Phos-tag SDS-PAGE (lower panel). The phosphorylation states of the respective bands are indicated at the left side of the blot, based on the results by Hosokawa et al. (38). The upper two panels are immunoblotted with anti-p35 and anti-Cdk5 after Laemmli SDS-PAGE.
Degradation of p35 is affected by phosphorylation at Thr-138 (37, 40). Phosphorylation of p35 was assessed by the electrophoretic mobility shift in Phos-tag SDS-PAGE. p35 was separated into three bands; the intense lowest band is unphosphorylated p35, the slightly higher band is Thr-138-phosphorylated p35, and the upper band is p35 phosphorylated at Thr-138 and Ser-91 according to our previous results (38). Co-expression with Cdk5 reduced the mobility of p35 mainly by phosphorylation at Ser-8 (Fig. 4B). p35 23R showed a similar but not identical banding pattern to p35 in the presence or absence of Cdk5, which was probably because of the presence of Arg mutations at as many as 23 sites. However, we believe that Arg mutations did not affect the phosphorylation at Thr-138 because bands containing Thr-138 phosphorylation were detected. Furthermore, we observed that p35 23R showed the same cellular localization of p35 by immunofluorescent staining (data not shown). These results suggest that p35 23R retains a conformation that is capable of binding and activating Cdk5 as well as that wild-type p35 is degraded by proteasomes ubiquitin-independently.
Effect of Cdk5 Binding on the Degradation of p35 and p35 23R
When p35 or p35 23R alone was transfected into Neuro2a cells, the expression levels were higher with p35 23R than with p35. In contrast, p35 and p35 23R showed similar expression levels when Cdk5 was co-expressed (Fig. 5A). The results suggest that Cdk5 affects the stability of p35 23R and p35 differently, and we examined the effect of exogenous co-expression of Cdk5 on the degradation of p35.
FIGURE 5.

Degradation rate of p35 or p35 23R in the presence or absence of Cdk5. A, expression levels of p35 or p35 23R in the absence of Cdk5 in Neuro2a cells. Immunoblotting with anti-p35 shown in the top, Cdk5 is in the middle, and actin is at the bottom. B, p35 (WT) and p35 23R have the same degradation rate when co-expressed with Cdk5. Neuro2a cells expressing Cdk5 and p35 (WT or 23R) were treated with CHX for the indicated times. The quantification is shown in the lower panel (mean ± S.E., n = 3). C, p35 23R is stable more than p35 in the absence of exogenous Cdk5. Neuro2a cells expressing p35 (WT) or p35 23R were treated with CHX for the indicated times. p35 was detected by immunoblotting with anti-p35. Actin is the loading control. Quantification is shown in the lower panel (mean ± S.E. n = 3. *, p < 0.05, Tukey's post hoc test). D, degradation of p35 23R in primary neurons. p35 and p35 23R are degraded at the same speed in primary cortical neurons. Cerebral cortical neurons expressing p35-HA (WT) or p35-HA 23R were treated with CHX for the indicated times. p35 (WT) and p35 23R were detected by immunoblotting with anti-HA antibody. Cdk5 and actin are shown in middle and lower panels, respectively. Quantification is shown below (mean ± S.E., n = 3).
We measured the degradation rates of p35 and p35 23R in the presence of Cdk5 by CHX chase assay. p35 23R showed the same degradation rate as p35, and the p35 and p35 23R levels were reduced to ∼60% in 1 h after CHX addition (Fig. 5B). In contrast, in the absence of Cdk5, p35 23R was degraded slower than p35 (Fig. 5C). Although p35 was decreased to ∼60% in 1 h, which was as fast as in the presence of Cdk5, >70% of the p35 23R remained at 3 h after the addition of CHX (Fig. 5C). These results suggest that the degradation of p35 23R is slowed down in the absence of Cdk5, whereas the degradation rate of p35 is not affected by the expression of Cdk5.
Degradation of p35 23R in Primary Cortical Neurons
We used Neuro2a cells in the above experiments. Neuro2a is a neuronal cell line that is derived from the central nervous system, but these cells are not neurons themselves. To validate the physiological relevance of ubiquitin-independent degradation, we examined whether p35 23R was degraded as p35 was in neurons. p35 or p35 23R tagged with HA, which contains no Lys residues, were constructed and transfected into primary cortical neurons at 3 days in vitro. Their degradation rates were assessed by the CHX chase assay. The time course of degradation was quite similar between p35 and p35 23R (Fig. 5D). Their half-life was ∼1 h in primary neurons, which is consistent with our previous reports (17). This result indicated that p35 can be degraded independently of ubiquitin by proteasomes in neurons.
Ubiquitin-independent Degron Sequence in p35
We investigated how p35 is recognized by proteasomes without ubiquitination. The number of proteins, which are known to be degraded by proteasomes independent of ubiquitin, have increased recently. Among them, we were interested in a ubiquitin-independent degron that is found in the C-terminal region of NKX3.1, a tumor repressor (42). The ubiquitin-independent degron sequence is composed of 21 amino acids with a PXL motif in the middle (35). p35 has a similar, but not identical, sequence in residues 240–258 (Fig. 6A). To determine if the sequence is involved in the ubiquitin-independent degradation of p35, we mutated Pro-247 to Ala in p35 WT or p35 23R and examined the degradation of the P247A mutants. Pro-247 corresponds to Pro-221 in the degron of NKX3.1, a critical amino acid for the degron activity. p35 P247A was degraded as fast as p35 WT in the presence of exogenous Cdk5; however, the P247A mutation made p35 23R stable (Fig. 6B). This was also found in the absence of exogenous Cdk5. When the activators were expressed alone, the P247A mutation did not affect the half-life of wild-type p35, but the P247A mutation of p35 23R became more stable than p35 23R itself, which had a longer half-life than wild-type p35 (Fig. 6C). These results suggested that the degron-like sequence is involved in degradation of p35 23R whether p35 23R binds to Cdk5 or not.
FIGURE 6.

Ubiquitin-independent degron sequence in p35. A, the degron-like amino acid sequence in p35. Amino acids 240–258 of p35 are similar to the ubiquitin-independent degron of NKX3.1. Pro-221 in NKX3.1 is a critical amino acid for degron activity (42). p35 has Pro-247 in a similar position to Pro-221 in NKX3.1 (underlined). B, effect of the Ala mutation at Pro-247 of p35 on its degradation when co-expressed with Cdk5. Neuro2a cells expressing Cdk5 and p35 (WT) or a mutant (P247A, 23R, or 23R P247A) were treated by CHX for 0, 3, or 6 h. C, degradation of p35 or its mutants, P247A, 23R, or 23R P247A in the absence of exogenous Cdk5. p35 (WT) or one of its mutants was expressed in HEK293T cells, and the degradation rate was measured by immunoblotting with anti-p35. Cdk5 and actin are also shown. The lower panels are for quantification (the mean ± S.E. n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001, Tukey's post hoc test). D, the position of Pro-247 in p25. A ribbon structure of p25 is depicted using a Waals software (Altif Laboratories, Inc., Tokyo, Japan) based on the crystal structure of p25 (53). Pro-247 and the α3 and α5 helices are indicated.
Discussion
p35 is an unstable protein with a half-life of 30–60 min, and it is degraded by proteasomes after ubiquitination (12, 13, 17). Because the p35 protein amount is a critical determinant of Cdk5 activity, elucidating the p35 degradation mechanism is central for understanding Cdk5 functions. To this end we searched for the polyubiquitination site(s) in p35, but unexpectedly, we found that the degradation of p35 can occur without ubiquitination. We also showed that ubiquitin-independent degradation was mediated by an α-helical degron-like sequence in the C-terminal region of p35. Thus, p35 is subjected to two different degradation mechanisms: ubiquitin-dependent and ubiquitin-independent.
In contrast to cell cycle Cdks, whose activation is regulated by its phosphorylation/dephosphorylation upon cyclin binding (43), Cdk5 is activated only by binding to its activation subunit p35 (2). On the other hand, similar to cell cycle Cdks, whose inactivation is induced by the degradation of cyclin, Cdk5 is inactivated by the degradation of p35 by proteasomes. Because Cdk5 is expressed more than p35 in neurons (8, 9), the protein level of p35 is a limiting factor determining the total Cdk5 activity. Cyclins are typical well studied proteins to be targeted by proteasomes via ubiquitination in a cell cycle-dependent manner (44). Therefore, it is natural to expect that p35 is also targeted by proteasomes when it is ubiquitinated. In fact, ubiquitination of p35 has been demonstrated by several previous studies by groups including ours (12, 17, 45). Therefore, it was surprising for us to find that the lysine-less mutant of p35 underwent degradation at a rate similar to wild-type p35 in cultured cell lines and primary neurons.
Polyubiquitin works as a degradation signal for proteins targeted by proteasomes (46, 47). Thus, a question arises as to how proteasomes recognize and degrade p35 without the polyubiquitin tag. Some misfolded or impaired proteins are degraded without ubiquitination by default. There are several examples of proteins that display default degradation, although they are degraded physiologically in a ubiquitin-dependent manner. p53 is a tumor suppressor protein that is degraded by proteasomes via polyubiquitination by E3 ubiquitin ligases, such as Mdm2 (33), but it is also degraded by the 20S proteasome by default if its N-terminal unstructured region is not protected by other proteins or modification. c-Fos proto-oncoprotein is an unstructured protein and degraded independently of ubiquitin by proteasomes when it does not form a transcriptional heterodimer with a partner protein (34). p35 functions exclusively as the activator of Cdk5. Only a few p35 molecules exist as free p35 in vivo because Cdk5 is significantly more abundant than p35. If p35 fails to bind Cdk5, however, p35 would be recognized as a misfolded protein and degraded without ubiquitination by default. However, p35 23R appeared to maintain the proper conformation to fully bind and activate Cdk5, and it was degraded at a similar rate to that of wild-type p35. In this study we carried out most of the experiments under excess amounts of Cdk5 by co-expression. Thus, we think that it is unlikely that p35 23R is degraded through the default pathway of degradation.
There are at least three types of substrate protein recognition by proteasomes in the ubiquitin-independent degradation systems, which are as follows: by the 19S regulatory particle of the 26S proteasome as an example of ornithine decarboxylase (48, 49); by REGγ, also known as 11S or PA28, complexed with the 20S proteasome that is known for p21 Cdk inhibitor (50); by a core subunit of the 20S proteasome as exemplified by the F protein of hepatitis C virus (51). On the other hand, the amino acid sequence(s) required for degradation has also been investigated with several substrate proteins. In the case of the Rpn4 transcription factor that activates the expression of proteasome genes in yeast, the N-terminal unstructured segment and the following folded domain are essential for ubiquitin-independent degradation (31). A similar requirement of two elements, including an unstructured region and a following α-helical sequence, is shown for thymidylate synthase (30). The two elements of thymidylate synthase function as a degradation signal if they are tagged at the C terminus of a reporter protein and called a ubiquitin-independent degron (52). According to the two-step model of degradation, the α-helical degron region is recognized by the proteasome, and then the disordered region enters into the proteasomal cavity. p35 may be degraded similarly because p35 has an unstructured ∼13-amino acids extension at the C terminus downstream of an α-helix-rich domain called the cyclin fold (Fig. 6D).
NKX3.1 is a homeodomain transcription factor that regulates prostate cancer initiation and progression (42). NKX3.1 turnover is regulated by ubiquitination, but it is also proteolyzed by proteasomes independent of ubiquitination. This ubiquitin-independent degradation is mediated by a 21-amino acid sequence in its C-terminal region (35). The proline residue in the sequence is essential for its ubiquitin-independent degron activity. p35 has a homologous (∼53% identity) sequence at amino acids 240–258 with Pro-247 in a similar position (Fig. 6A). The mutation of Pro-247, which is in the ordered structure of the cyclin fold (53), to Ala slowed the turnover rate of p35 down remarkably. Considering that Pro-247 is positioned in the shallow concave (Fig. 6D), the structure, but not the amino acid sequence, around Pro-247 may provide the proteasome recognition site. Thus, p35 has two elements of an unstructured and a structured region next to each other that conform to the two-step degradation as shown by other proteins displaying ubiquitin-independent degradation.
The physiological role of ubiquitin-independent degradation and its regulation are largely unknown for most proteins (46, 47). In the case of p21, however, it is indicated that the cell cycle-regulated degradation is ubiquitin-dependent (54, 55), and its degradation during resting conditions is ubiquitin-independent (50, 56). Similar differential usage may be in operation for p35. p35 is unstable endogenously in neurons or when expressed heterogeneously in cultured cell lines, and p35 P247A showed a similar degradation rate to wild-type p35, suggesting that the degradation of p35 in resting neurons could be ubiquitin-independent. p35 is acutely degraded in neurons when treated with glutamate (14, 57). This stimulated degradation of p35 may be dependent on ubiquitination. In any case, rapid p35 turnover would be crucial for neurons to serve for their long life. Overactivation of Cdk5, which is induced by the p25 C-terminal stable fragment, is toxic for neurons. p25 is produced by the cleavage of p35 with calpain, and Cdk5 activated by p25 acquires a long lasting activity with free accessibility to proteins (15, 18, 19), which Cdk5-p35 cannot access. The longer half-life of p35 may enhance the probability of the overproduction of p25. Two degradation pathways for p35 would be a mechanism to secure the long life of neurons.
Author Contributions
T. T. conceived the study and wrote the paper. S. M. designed, performed, and analyzed the experiments shown in Figs. 1 and 2. A. A. and T. S. provided technical assistance and performed a portion of the experiments shown in Figs. 3 and 4, respectively. T. S. and H. K. provided technical assistance and contributed to the preparation of the figures. S. H. coordinated the study and wrote the paper. All of the authors reviewed the results and approved the final version of the manuscript.
This work was supported in part by Grants-in-aid For Scientific Research on Priority Area from the Ministry of Education, Culture, Sports, Science, and Technology in Japan 25290024 and 26117004 (to S. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- Cdk5
- cyclin-dependent kinase 5
- CHX
- cycloheximide
- 23R
- 23 Lys residues to Arg
- N7
- the 7 N-terminal amino acid.
References
- 1. Malumbres M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hisanaga S., and Endo R. (2010) Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J. Neurochem. 115, 1309–1321 [DOI] [PubMed] [Google Scholar]
- 3. Shah K., and Lahiri D. K. (2014) Cdk5 activity in the brain: multiple paths of regulation. J. Cell Sci. 127, 2391–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chae T., Kwon Y. T., Bronson R., Dikkes P., Li E., and Tsai L. H. (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 18, 29–42 [DOI] [PubMed] [Google Scholar]
- 5. Ohshima T., Ward J. M., Huh C. G., Longenecker G., Veeranna, Pant H. C., Brady R. O., Martin L. J., and Kulkarni A. B. (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology, and perinatal death. Proc. Natl. Acad. Sci. U.S.A. 93, 11173–11178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilmore E. C., Ohshima T., Goffinet A. M., Kulkarni A. B., and Herrup K. (1998) Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J. Neurosci. 18, 6370–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ko J., Humbert S., Bronson R. T., Takahashi S., Kulkarni A. B., Li E., and Tsai L. H. (2001) p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci. 21, 6758–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee K. Y., Rosales J. L., Tang D., and Wang J. H. (1996) Interaction of cyclin-dependent kinase 5 (Cdk5) and neuronal Cdk5 activator in bovine brain. J. Biol. Chem. 271, 1538–1543 [DOI] [PubMed] [Google Scholar]
- 9. Zhu Y. S., Saito T., Asada A., Maekawa S., and Hisanaga S. (2005) Activation of latent cyclin-dependent kinase 5 (Cdk5)-p35 complexes by membrane dissociation. J. Neurochem. 94, 1535–1545 [DOI] [PubMed] [Google Scholar]
- 10. Harada T., Morooka T., Ogawa S., and Nishida E. (2001) ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 3, 453–459 [DOI] [PubMed] [Google Scholar]
- 11. Bogush A., Pedrini S., Pelta-Heller J., Chan T., Yang Q., Mao Z., Sluzas E., Gieringer T., and Ehrlich M. E. (2007) AKT and CDK5/p35 mediate brain derived neurotrophic factor induction of DRPP-32 in medium size spiny neuron in vitro. J. Biol. Chem. 282, 7352–7359 [DOI] [PubMed] [Google Scholar]
- 12. Patrick G. N., Zhou P., Kwon Y. T., Howley P. M., and Tsai L. H. (1998) p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5), is degraded by the ubiquitin-proteasome pathway. J. Biol. Chem. 273, 24057–24064 [DOI] [PubMed] [Google Scholar]
- 13. Saito T., Ishiguro K., Onuki R., Nagai Y., Kishimoto T., and Hisanaga S. (1998) Okadaic acid-stimulated degradation of p35, an activator of Cdk5, by proteasome in cultured neurons. Biochem. Biophys. Res. Commun. 252, 775–778 [DOI] [PubMed] [Google Scholar]
- 14. Wei F. Y., Tomizawa K., Ohshima T., Asada A., Saito T., Nguyen C., Bibb J. A., Ishiguro K., Kulkarni A. B., Pant H. C., Mikoshiba K., Matsui H., and Hisanaga S. (2005) Control of cyclin-dependent kinase 5 (Cdk5) activity by glutamatergic regulation of p35 stability. J. Neurochem. 93, 502–512 [DOI] [PubMed] [Google Scholar]
- 15. Patrick G. N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., and Tsai L. H. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622 [DOI] [PubMed] [Google Scholar]
- 16. Asada A., Yamamoto N., Gohda M., Saito T., Hayashi N., and Hisanaga S. (2008) Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J. Neurochem. 106, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 17. Minegishi S., Asada A., Miyauchi S., Fuchigami T., Saito T., and Hisanaga S. (2010) Membrane association facilitates degradation and cleavage of the cyclin-dependent kinase 5 activators p35 and p39. Biochemistry 49, 5482–5493 [DOI] [PubMed] [Google Scholar]
- 18. Kusakawa G., Saito T., Onuki R., Ishiguro K., Kishimoto T., and Hisanaga S. (2000) Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 275, 17166–17172 [DOI] [PubMed] [Google Scholar]
- 19. Lee M. S., Kwon Y. T., Li M., Peng J., Friedlander R. M., and Tsai L. H. (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364 [DOI] [PubMed] [Google Scholar]
- 20. Engmann O., Hortobágyi T., Thompson A. J., Guadagno J., Troakes C., Soriano S., Al-Sarraj S., Kim Y., and Giese K. P. (2011) Cyclin-dependent kinase 5 activator p25 is generated during memory formation and is reduced at an early stage in Altzheiner's disease. Biol. Psychiatry 70, 159–168 [DOI] [PubMed] [Google Scholar]
- 21. Rei D., Mason X., Seo J., Gräff J., Rudenko A., Wang J., Rueda R., Siegert S., Cho S., Canter R. G., Mungenast A. E., Deisseroth K., and Tsai L. H. (2015) Basolateral amygdala bidirectionally modulates stress-induced hippocampal learning and memory deficits through a p25/Cdk5-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 7291–7296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cruz J. C., and Tsai L. H. (2004) Cdk5 deregulation in pathogenesis of Alzheimer disease. Trends Mol. Med. 10, 452–458 [DOI] [PubMed] [Google Scholar]
- 23. Hershko A., and Ciechanover A. (1992) The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61, 761–807 [DOI] [PubMed] [Google Scholar]
- 24. Ravid T., and Hochstrasser M. (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanaka K., Suzuki T., Hattori N., and Mizuno Y. (2004) Ubiquitin, proteasome and parkin. Biochim. Biophys. Acta 1695, 235–247 [DOI] [PubMed] [Google Scholar]
- 26. Sakamaki J., Fu A., Reeks C., Baird S., Depatie C., Al Azzabi M., Bardeesy N., Gingras A. C., Yee S. P., and Screaton R. A. (2014) Role of the SIK-p35 PJA2 complex in pancreatic β-cell functional compensation. Nat. Cell Biol. 16, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murakami Y., Matsufuji S., Hayashi S., Tanahashi N., and Tanaka K. (2000) Degradation of ornithine decarboxylase by the 26S proteasome. Biochem. Biophys. Res. Commun. 267, 1–6 [DOI] [PubMed] [Google Scholar]
- 28. Hoyt M. A., Zhang M., and Coffino P. (2003) Ubiquitin-independent mechanisms of mouse ornithine decarboxylase are conserved between mammalian and fugal cells. J. Biol. Chem. 278, 12135–12143 [DOI] [PubMed] [Google Scholar]
- 29. Peña M. M., Xing Y. Y., Koli S., and Berger F. G. (2006) Role of N-terminal residues in the ubiquitin-independent degradation of human thymidylate synthase. Biochem. J. 394, 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peña M. M., Melo S. P., Xing Y. Y., White K., Barbour K. W., and Berger F. G. (2009) The intrinsically disordered N-terminal domain of thymidylate synthase targets the enzyme to the ubiquitin-independent proteasomal degradation pathway. J. Biol. Chem. 284, 31597–31607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ha S. W., Ju D., and Xie Y. (2012) The N-terminal domain of Rpn4 serves as a portable ubiquitin-independent degron and is recognized by specific 19S RP subunits. Biochem. Biophys. Res. Commun. 419, 226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen X., Chi Y., Bloecher A., Aebersold R., Clurman B. E., and Roberts J. M. (2004) N-Acetylation and ubiquitin-independent proteasomal degradation of p21 (Cip 1). Mol. Cell 16, 839–847 [DOI] [PubMed] [Google Scholar]
- 33. Tsvetkov P., Reuven N., and Shaul Y. (2010) Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 17, 103–108 [DOI] [PubMed] [Google Scholar]
- 34. Basbous J., Jariel-Encontre I., Gomard T., Bossis G., and Piechaczyk M. (2008) Ubiquitin-independent versus ubiquitin-dependent proteasomal degradation of the c-Fos and Fra-1 trunscription factors: is there a unique answer? Biochimie 90, 296–305 [DOI] [PubMed] [Google Scholar]
- 35. Rao V., Guan B., Mutton L. N., and Bieberich C. J. (2012) Proline-mediate proteasomal degradation of the prostate-specific tumor suppressor NKX3.1. J. Biol. Chem. 287, 36331–36340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawauchi T., Chihama K., Nishimura Y. V., Nabeshima Y., and Hoshino M. (2005) MAP1B phosphorylation is differentially regulated by Cdk5/p35, Cdk5/p25, and JNK. Biochem. Biophys. Res. Commun. 331, 50–55 [DOI] [PubMed] [Google Scholar]
- 37. Saito T., Onuki R., Fujita Y., Kusakawa G., Ishiguro K., Bibb J. A., Kishimoto T., and Hisanaga S. (2003) Developmental regulation of the proteolysis of the p35 cyclin-dependent kinase 5 activator by phosphorylation. J. Neurosci. 23, 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hosokawa T., Saito T., Asada A., Fukunaga K., and Hisanaga S. (2010) Quantitative measurement of in vivo phosphorylation states of Cdk5 activator p35 by Phos-tag SDS-PAGE. Mol. Cell Proteomics 9, 1133–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sato K., Minegishi S., Takano J., Plattner F., Saito T., Asada A., Kawahara H., Iwata N., Saido T. C., and Hisanaga S. (2011) Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J. Neurochem. 117, 504–515 [DOI] [PubMed] [Google Scholar]
- 40. Kamei H., Saito T., Ozawa M., Fujita Y., Asada A., Bibb J. A., Saido T. C., Sorimachi H., and Hisanaga S. (2007) Suppression of calpain-dependent cleavage of the CDK5 activator p35 to p25 by site-specific phosphorylation. J. Biol. Chem. 282, 1687–1694 [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., and Hay R. T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 20, 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guan B., Pungaliya P., Li X., Uquillas C., Mutton L. N., Rubin E. H., and Bieberich C. J. (2008) Ubiquitination by TOPORS regulate the prostate tumor suppressor NKX3.1. J. Biol. Chem. 283, 4834–4840 [DOI] [PubMed] [Google Scholar]
- 43. Morgan D. O. (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291 [DOI] [PubMed] [Google Scholar]
- 44. Benanti J. A. (2012) Codination of cell growth and divition by the ubiquitin-proteasome system. Semin. Cell Dev. Biol. 23, 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Endo R., Saito T., Asada A., Kawahara H., Ohshima T., and Hisanaga S. (2009) Commitment of 1-methyl-4-phenylpyrinidinium ion-induced neuronal cell death by proteasome-mediated degradation of p35 cyclin-dependent kinase 5 activator. J. Biol. Chem. 284, 26029–26039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kravtsova-Ivantsiv Y., and Ciechanover A. (2012) Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 125, 539–548 [DOI] [PubMed] [Google Scholar]
- 47. Erales J., and Coffino P. (2014) Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 1843, 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coffino P. (2001) Antizyme, a mediator of ubiquitin-independent proteasomal degradation. Biochimie 83, 319–323 [DOI] [PubMed] [Google Scholar]
- 49. Takeuchi J., Chen H., Hoyt M. A., and Coffino P. (2008) Structural element of the ubiquitin-independent proteasomal degron of ornithine decarboxylase. Biochem. J. 410, 401–407 [DOI] [PubMed] [Google Scholar]
- 50. Chen X., Barton L. F., Chi Y., Clurman B. E., and Roberts J. M. (2007) Ubiquitin-independent degradation of cell-cycle inhibitor by the REGgammma proteasome. Mol. Cell 26, 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yuksek K., Chen W. L., Chien D., and Ou J. H. (2009) Ubiquitin-independent degradation of hepatitis C virus F protein. J. Virol. 83, 612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Melo S. P., Barbour K. W., and Berger F. G. (2011) Cooperation between an intrinsically disordered region and a helical segment is required for ubiuquitin-independent degradation by the proteasome. J. Biol. Chem. 286, 36559–36567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tarricone C., Dhavan R., Peng J., Areces L. B., Tsai L. H., and Musacchio A. (2001) Structure and regulation of the CDK5-p25(nck5a) complex. Mol. Cell 8, 657–669 [DOI] [PubMed] [Google Scholar]
- 54. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., and Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Gene Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bornstein G., Bloom J., Sitry-Shevah D., Nakayama K., Pagano M., and Hershko A. (2003) Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 278, 25752–25757 [DOI] [PubMed] [Google Scholar]
- 56. Zhang H., and Cohen S. N. (2004) Smurf 2 up-regulation activates telomere-dependent senescence. Genes Dev. 18, 3028–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hosokawa T., Saito T., Asada A., Ohshima T., Itakura M., Takahashi M., Fukunaga K., and Hisanaga S. (2006) Enhanced activation of Ca2+/calmodulin-dependent protein kinase II upon down-regulation of cyclin-dependent kinase 5-p35. J. Neurosci. Res. 84, 747–754 [DOI] [PubMed] [Google Scholar]