

Summary
Bacteria encode heat shock proteins that aid in survival during stressful growth conditions. In addition, the major heat shock proteins of the intracellular bacterium Chlamydia trachomatis have been associated with immune pathology and disease. We developed a ChIP-qPCR method to study the regulation of chlamydial heat shock gene regulation during an intracellular infection. This approach allowed us to show that chlamydial heat shock genes are regulated by the transcription factor HrcA within an infected cell, providing validation for previous in vitro findings. Induction of chlamydial heat shock gene expression by elevated temperature was due to loss of HrcA binding to heat shock promoters, supporting a mechanism of derepression. This heat shock response was rapid, while recovery of HrcA binding and return to non-stress transcript levels occurred more slowly. We also found that control of heat shock gene expression was differentially regulated over the course of the intracellular Chlamydia infection. There was evidence of HrcA-mediated regulation of heat shock genes throughout the chlamydial developmental cycle but the level of repression was lower at early times. This is the first study of Chlamydia-infected cells showing the effect of an environmental signal on transcription factor-DNA binding and target gene expression in the bacterium.
Keywords: HrcA, CIRCE, stress response, heat shock, Chlamydia
Introduction
Chlamydia trachomatis is an obligate intracellular bacterium and a major cause of human infection (Batteiger & Tan, 2014). C. trachomatis genital infections are the most common reportable infectious disease in the United States (CDC, 2014), and C. trachomatis ocular infections are the leading cause of infectious blindness in the world (Burton & Mabey, 2009). C. trachomatis replicates by means of an unusual developmental cycle in which there is conversion between two bacterial forms with specialized functions. The elementary body (EB) is the infectious, but non-replicating, form that binds and enters a host cell. At early times in infection, prior to 8 hours post infection (hpi), the EB converts to a reticulate body (RB) within a parasitophorous vacuole called the inclusion. RBs replicate by binary fission during the mid-stage of the developmental cycle to produce hundreds to several thousand progeny per infected cell. At late times in the developmental cycle (24 hpi to approximately 48 hpi) RBs asynchronously convert into EBs, prior to exiting the host cell and initiating a new round of infection.
The pathology of chlamydial infections is due in large part to chronic inflammation, which results in tissue damage and scarring (Darville & Hiltke, 2010). The major chlamydial stress response proteins, GroEL1 (commonly referred to as GroEL or Hsp60), GroES (Hsp10), and DnaK (Hsp70) have been proposed to play an important role in this deleterious host immune response (Brunham & Peeling, 1994, LaVerda et al., 2000). For example, serological responses to chlamydial GroEL1 have been associated with immunopathology in trachoma (Peeling et al., 1998, Morrison et al., 1989), pelvic inflammatory disease (Peeling et al., 1997), female infertility (Ault et al., 1998), and ectopic pregnancy (Brunham et al., 1992). An antibody response to GroES has also been associated with development of infertility in women (LaVerda et al., 2000). Additionally, expression of groEL1 is increased during infection of monocytes (Klos et al., 2009) and is associated with reactive arthritis (Gaston et al., 1996, Gerard et al., 2004).
Stress response proteins are highly conserved molecular chaperones and proteases. They assist in protein folding, stabilization, and prevention of protein aggregation (Georgopoulos & Welch, 1993), and their levels are transiently induced by elevated temperature and other stress conditions (Narberhaus, 1999). In Escherichia coli, this heat shock response is positively regulated by alternative sigma factors, which direct RNA polymerase to increase transcription of stress response genes (Narberhaus, 1999). In many other bacteria, stress response genes are negatively regulated by a transcriptional repressor HrcA and its cognate operator called CIRCE (Controlling Inverted Repeat of Chaperone Expression) (Narberhaus, 1999, Schulz & Schumann, 1996, Zuber & Schumann, 1994, Minder et al., 2000). Under non-stress conditions, HrcA binds to the CIRCE operator, which is located near promoters for stress response genes, and represses transcription. However, with heat shock or other cellular stress, HrcA binding to CIRCE is transiently disrupted, leading to upregulated transcription of the stress response genes.
The major chlamydial stress proteins are heat shock proteins that are induced by higher temperature. For example, increasing the temperature of Chlamydia-infected cells from 37 to 42 or 45°C transiently upregulates the transcription of groEL1, groES and dnaK (Engel et al., 1990). These chlamydial stress response genes are regulated by an HrcA ortholog (Wilson & Tan, 2002, Wilson & Tan, 2004), rather than by a heat shock sigma factor. In in vitro studies, recombinant C. trachomatis HrcA bound and repressed the dnaK P1 and groESL promoters, which are its only known targets (Fig. 1) (Wilson & Tan, 2002, Wilson & Tan, 2004). These findings support a model in which the dnaK P1 and groESL promoters are repressed by HrcA under non-stress conditions, but can be upregulated through a mechanism of derepression in response to stress. However, elevated temperature by itself was not sufficient to relieve binding and repression by HrcA in vitro (Wilson & Tan, 2004), and the role of HrcA during the intracellular infection has not been explored. HrcA does not appear to control the expression of a second C. trachomatis dnaK promoter, dnaK P2, nor the putative promoters for two groEL1 paralogs, groEL2 and groEL3, which all lack an identifiable CIRCE operator (McNally & Fares, 2007, Yu et al., 2006a).
Figure 1.

Transcriptional organization of C. trachomatis stress response genes and paralogs. Transcripts originating from each promoter are shown as an arrow above the genes. The CIRCE operator recognized by HrcA is boxed and shown adjacent to its target promoters.
In this report, we used a cell culture infection model to study the regulation of the chlamydial heat shock response by HrcA within a Chlamydia-infected cell. We developed a chromatin immunoprecipitation assay (ChIP) to detect HrcA binding to its target promoters in chlamydiae. This approach allowed us to measure changes in HrcA binding and target gene expression in response to stress conditions and to analyze HrcA-mediated regulation over the course of the chlamydial developmental cycle. Our results provide experimental support for the HrcA repression model of chlamydial heat shock regulation in the context of the intracellular infection. This approach has general applicability because it can be used to investigate the association of other chlamydial transcription factors and their target promoters within an infected cell.
Results
We established a protein-DNA co-immunoprecipitation assay, more commonly referred to as chromatin immunoprecipitation (ChIP), to quantitatively measure interactions between a Chlamydia transcription factor and its cognate DNA sequence within the bacterium. This approach has been used to investigate transcription factors and their target DNA sequences in prokaryotic organisms, but has been under-utilized for obligate intracellular bacteria, such as Chlamydia. We optimized the conditions to cross-link protein-DNA complexes in chlamydiae residing in the chlamydial inclusion within an infected host cell. After isolating specific protein-DNA complexes by immunoprecipitation, we used quantitative real-time PCR (qPCR), to compare the enrichment of target and non-target DNA sequences relative to total input DNA.
We used this ChIP assay to analyze binding of the Chlamydia stress response regulator HrcA to its target promoters in Chlamydia-infected cells grown in cell culture. L929 cells were infected with C. trachomatis, and at 24 hours post infection (hpi) infected cells were collected, cross-linked with formaldehyde, and analyzed by ChIP-qPCR. We measured binding to two known HrcA target promoters, dnaK P1 and the groESL promoter, which each contain a CIRCE sequence that is the HrcA operator (Narberhaus, 1999, Schulz & Schumann, 1996, Zuber & Schumann, 1994). As negative controls, we assayed binding to the dnaK P2, groEL2, and groEL3 promoters, which lack the CIRCE operator. We measured a 10-fold enrichment of the dnaK P1 promoter (Fig. 2A) when we co-immunoprecipitated DNA with anti-HrcA antibody compared to control antibody from pre-immune rabbit serum (0.041% vs. 0.0041% recovery of input DNA, respectively, Fig. 2B). There was an even greater 50.7-fold enrichment of the groESL promoter (Fig. 2A) (0.076% recovery with anti-HrcA antibody vs. 0.0015% with control antibody, Fig. 2B). In contrast, there was no enrichment of dnaK P2 and the groEL2 and groEL3 promoters (0.9-fold, 1.3-fold and 1.1-fold enrichment, respectively, Fig. 2A and Fig. 2B). The results provide validation of in vitro observations that HrcA binds in a CIRCE-dependent manner to its target promoters, dnaK P1 and the groESL promoter. In addition, this study demonstrates that ChIP can be used to measure the binding of a transcription factor to a target promoter in chlamydiae within an infected host cell.
Figure 2.
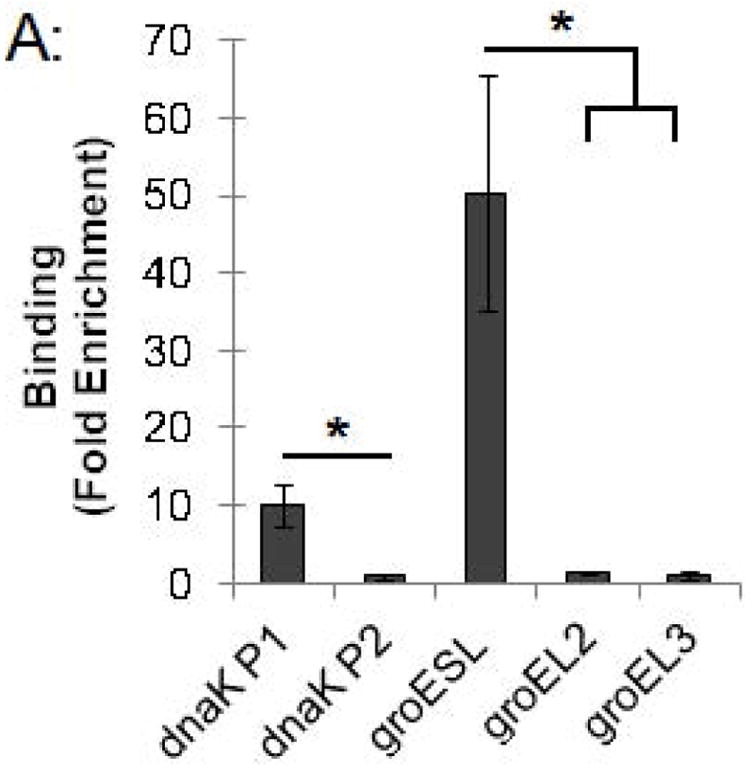
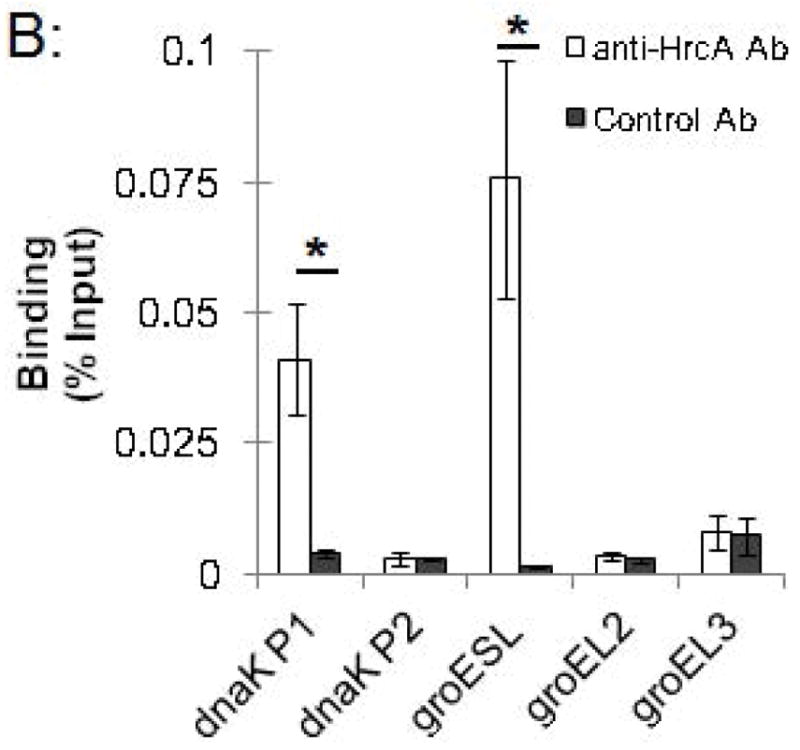
ChIP-qPCR assay measuring binding of C. trachomatis HrcA to promoters of stress response genes and paralogs. Recovery of promoter DNA reported as (A) fold enrichment with anti-HrcA antibody compared to a control antibody (pre-immune rabbit serum); and (B) the percentage of input DNA immunprecipitated with each antibody. Fold enrichment and % input were calculated as described in the Materials and Methods. Results are the mean of three independent ChIP experiments with the standard deviation indicated by error bars. The asterisk (*) indicates p ≤ 0.01.
This ChIP assay allowed us to measure changes in HrcA binding to its target promoters in response to stress conditions such as increased temperature. Chlamydia-infected cells grown at 37°C were collected at 24 hpi and transferred to media at 37°C or 45°C for 20 min, prior to ChIP analysis. The dnaK P1 promoter was enriched by 10-fold at 37°C but only by 2.8-fold at 45°C (Fig. 3A and Fig. S1). Similarly, the groESL promoter was enriched 54.2-fold at 37°C, but only 3.6-fold at 45°C (Fig. 3A and Fig. S1). There was minimal enrichment (<2.6-fold) of the control dnaK P2 and groEL2 and groEL3 promoters with the anti-HrcA antibody at both 37°C and 45°C (Fig. 3A and Fig. S1). These results demonstrate that HrcA binding to target promoters containing the CIRCE operator was abrogated when Chlamydia-infected cells were exposed to higher temperature. In contrast, a previous study reported that HrcA binding and repression in vitro was not disrupted at 42°C (Wilson & Tan, 2004), however when we repeated the ChIP analysis with heat shock at 42°C instead of 45°C we saw a similar loss of HrcA binding to its target promoters (Fig. S4).
Figure 3.
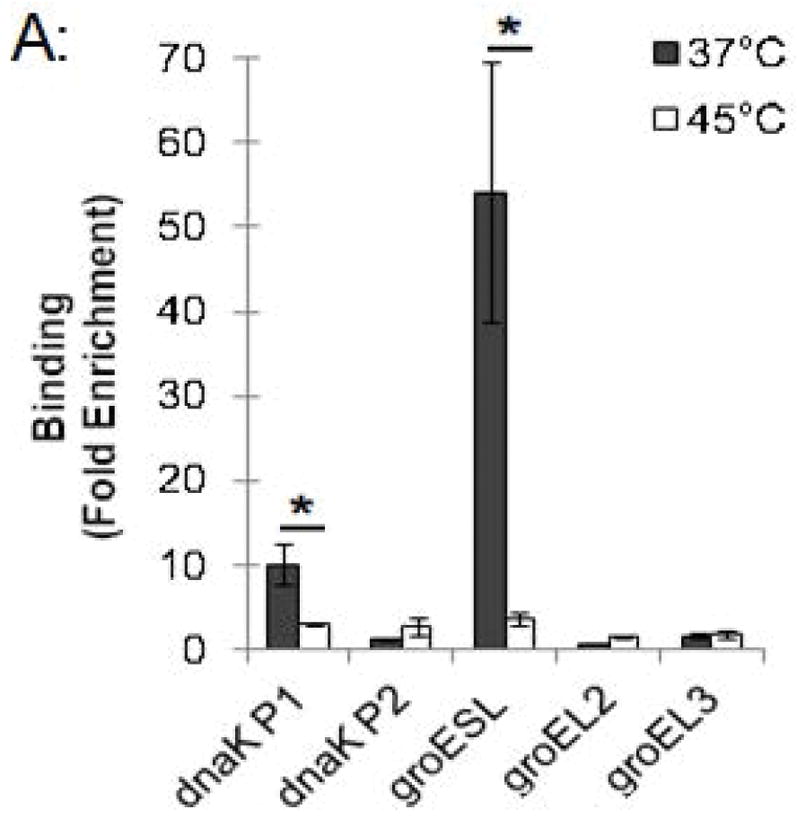
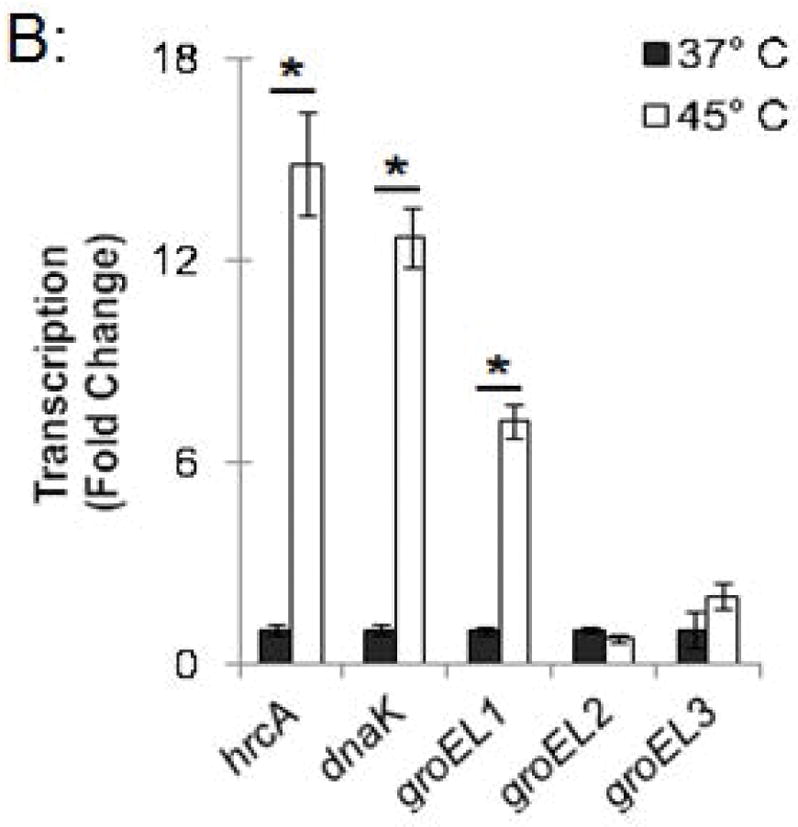
Effect of heat shock on HrcA binding and repression of stress response genes in Chlamydia-infected cells (A) ChIP-qPCR assay measuring recovery of promoter DNA with anti-HrcA antibody relative to control antibody in infected cells at 37°C or 45°C. Results are from three independent ChIP experiments with the standard deviation indicated with error bars. Asterisk (*) indicates p < 0.01 and NS indicates no statistical significance. (B) Fold change in transcript levels of stress response genes after incubating infected cells at 37°C or 45°C. Transcripts were first normalized to genome copy and then to the number of transcripts per genome present at 37°C. Results are the mean from three independent heat-shock experiments done in parallel with samples used for ChIP, with standard deviation indicated by error bars. The asterisk (*) indicates p < 0.01.
In a parallel experiment, RNA was isolated from these heat-shocked cells, and transcription of stress response genes was analyzed by qRT-PCR. hrcA and dnaK, which are both in the dnaK operon controlled by dnaK P1 (Fig. 1), were transcribed at 14.9-fold and 12.7-fold higher levels, respectively, at 45°C compared to 37°C (Fig. 3B). Similarly, groEL1, which is in the groESL operon (Fig. 1), was transcribed at a 7.2-fold higher level at 45°C compared to 37°C (Fig. 3B). In contrast, heat shock from 37°C to 45°C had no substantial effect on transcription of groEL2 or groEL3 (0.7-fold and 2.0-fold higher, respectively), which are stress response genes that are not regulated by HrcA. These ChIP and transcription studies demonstrate that heat shock from 37°C to 45°C decreases HrcA binding to CIRCE-containing promoters while also increasing transcription from these promoters. This correlation is consistent with the proposed role of HrcA as a repressor of specific stress response genes in Chlamydia.
To investigate the kinetics of the chlamydial heat shock response, we repeated these ChIP and qRT-PCR analyses with infected cells incubated at 45°C for shorter time periods. For dnaK P1, a 34.0-fold enrichment at 37°C was reduced to 1.7-fold and 1.4-fold after incubation at 45°C for 5 min and 10 min, respectively (Fig. 4A and Fig. S2). Similarly, a 66.8-fold enrichment of the groESL promoter at 37°C was reduced to 2.9-fold and 1.0-fold after heat shock for 5 min and 10 min, respectively (Fig. 4A and Fig. S2). This loss of HrcA binding corresponded with increased transcription at 45°C by 6.4 fold after 5 min and 11.1-fold after 10 min for dnaK, and by 4.1-fold after 5 min and 6.0-fold after 10 min for groEL1 (Fig. 4B).
Figure 4.
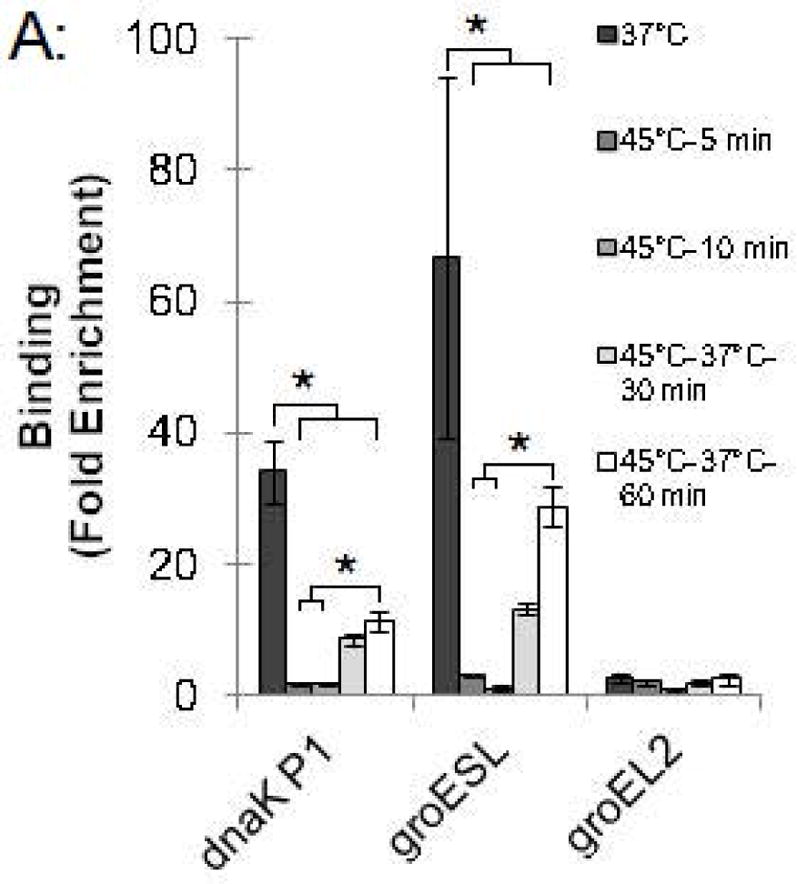
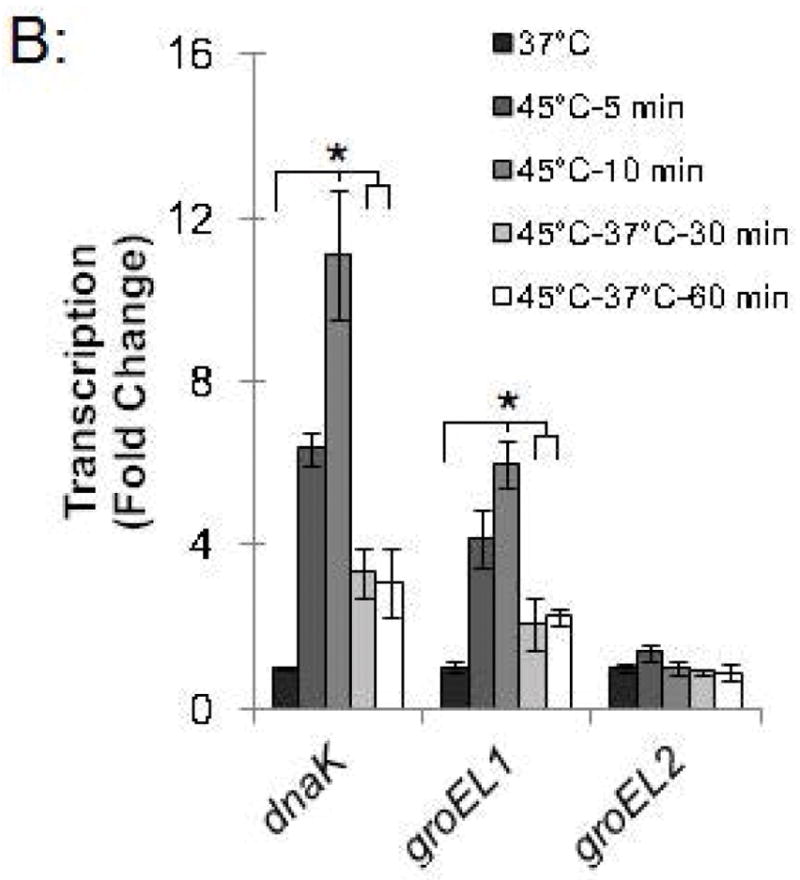
Time course of chlamydial heat shock response. (A) ChIP-qPCR assay with anti-HrcA antibody measuring fold enrichment of promoters in Chlamydia-infected cells prior to heat shock (37°C), after heat shock for 5 min or 10 min (45°C-5 min and 45°C-10 min), or after heat shock for 20 min and recovery at 37°C for 30 min or 60 min (45°C-37°C-30 min and 45°C-37°C-60 min). (B) Effect of these manipulations on transcription, reported as fold change relative to transcription at 37°C. These results are the mean from assays performed on infected cells collected at 24 hpi from three independent experiments with standard deviation indicated by error bars. Within each promoter or gene set, the asterisk (*) indicates p < 0.05.
We also examined HrcA binding and target gene transcription when infected cells were allowed to recover at 37°C for 30 or 60 min after a 20 min heat shock at 45°C. For dnaK P1, there was 8.4-fold enrichment after a 30 min recovery and 11.1-fold, after a 60 min recovery (Fig. 4A and Fig. S2), indicating that HrcA binding had been partially restored. Similarly, the groESL promoter was enriched by 13.0-fold and 28.7-fold after 30 and 60 min of recovery, respectively (Fig. 4A and Fig. S2). Transcription levels for dnaK were 3.3-fold higher than pre-heat shock levels after 30 min of recovery, and 3.1-fold higher after 60 min of recovery. groEL1 transcription levels also remained slightly above baseline during this period (2.1- and 2.2-fold higher than pre-heat shock levels after 30 and 60 min of recovery, respectively) (Fig. 4B). In contrast, there was minimal enrichment for the HrcA-independent groEL2 promoter regardless of the heat shock and recovery conditions (<2.5-fold, Fig. 4A and Fig. S2), and transcription of groEL2 was not altered (Fig. 4B). Together these results document the rapid onset of the heat shock response in Chlamydia, with loss of HrcA binding at target promoters within 5 min of temperature upshift to 45°C, and induction of target gene transcription by 5–10 min. Recovery from heat shock was slower and incomplete over the 60 min period examined, with partial restoration of HrcA binding at target promoters, and transcript levels that were decreased from their peak heat shock values but still slightly above baseline.
Chlamydial gene regulation is temporally regulated during the Chlamydia intracellular infection, and so we examined whether stress response genes are regulated by HrcA throughout the developmental cycle. Chlamydia-infected cells grown at 37°C were collected at 8, 16, 24, 32, and 40 hpi, and transferred to media at either 37°C or 45°C for 20 min. At 16 hpi and later, we detected HrcA binding to its target promoters at 37°C, together with an intact heat shock response. For instance, at these time points dnaK P1 was enriched by at least 11.8-fold (Fig. 5A and S3), and temperature upshift to 45°C caused a large and consistent loss of HrcA binding (Fig. 5A and Fig. S3) as well as > 4.0-fold induction of transcription (Fig. 5B). In a similar fashion, there was at least 39.9-fold enrichment of the groESL promoter at 37°C (Fig. 5A, Fig. S3A, Fig. S3B), and heat shock caused loss of HrcA binding and > 3.7-fold induction of transcription (Fig. 5B). In contrast, there was minimal enrichment of the HrcA-independent groEL2 promoter throughout the developmental cycle with less than 3.1-fold enrichment at 37°C and 45°C, at all times tested ((Fig. 5B, Fig. S3A, Fig. S3B). Additionally, groEL2 transcription was not responsive to a temperature upshift from 37°C to 45°C (< 1.9-fold) (Fig. 5B), indicating that this paralog of groEL1 is not regulated by heat shock.
Figure 5.


Chlamydial heat shock response over the course of the intracellular infection. Chlamydia-infected cells at selected times during the infection were incubated at 37°C or 45°C. and analyzed by (A) ChIP-qPCR assay, measuring fold enrichment of promoters with anti-HrcA antibody compared to a control antibody; and (B) qRT-PCR, measuring fold change in transcription of stress response genes at 37°C compared to 45°C. Results are the mean of three independent experiments with standard deviation indicated by error bars. The asterisk (*) indicates p < 0.05, and NS indicates no statistically significant difference between conditions.
There appeared to be an incomplete heat shock response at 8 hpi. At this early time in the intracellular infection, there was a minimal 1.9-fold HrcA enrichment for dnaK P1 at 37°C (Fig. 5A and S3) and only a 2.0-fold induction of transcription at 45°C. The groESL promoter was enriched at 8 hpi, but there was less binding than later times (14.4-fold enrichment at 8 hpi vs. > 39.9-fold enrichment at other times tested) (Fig. 5A, Fig. S3A, Fig. S3B). Induction of groEL1 transcription at 45°C was also slightly lower at 8 hpi (3.7-fold increase vs. > 5.0-fold increase at other times) (Fig. 5B). Together these results provide evidence that stress response genes can be regulated by HrcA throughout the chlamydial developmental cycle. However there appears to be a reduced heat shock response at early times, which was more evident for the dnaK operon.
Discussion
This report demonstrates for the first time that a chlamydial transcription factor within the bacterium is able to regulate its target genes in response to an environmental signal. Previously chlamydial transcription factors have been shown to bind DNA and modulate transcription in vitro (Tan, 2006, Tan, 2012), but their biological functions had not been validated in intracellular chlamydiae. Using a ChIP and qPCR approach, we quantitatively measured binding of the stress response regulator HrcA to its target heat shock gene promoters within chlamydiae. We also quantified transcription of heat shock genes under the same conditions so that we could correlate HrcA binding and target gene expression. This approach allowed us to monitor HrcA-mediated repression and derepression in Chlamydia-infected cells over the course of the developmental cycle and in response to cellular stress. We demonstrated rapid relief of HrcA binding and repression with heat shock, supporting a model of negative transcriptional regulation of the chlamydial heat shock response. We also showed that this stress response appears to be present throughout the chlamydial developmental cycle, although the magnitude of the response differed during the intracellular infection, particularly at early and late times.
ChIP has been widely used to study bacterial transcription (Grainger et al., 2009) but there have been few studies in the context of an intracellular infection (Rolando et al., 2013, Hickey et al., 2011). We have optimized the ChIP procedure so that we could cross-link chlamydial DNA-binding proteins to their cognate DNA sequences within intracellular chlamydiae, without having to first separate bacteria from the host cell. Important optimization steps included extensive blocking and wash steps to minimize isolation of non-specific DNA, and the specificity of the anti-HrcA antibody, which allowed us to selectively immunoprecipitate HrcA-CIRCE operator complexes. This method can be applied to study other chlamydial transcription factors and DNA-binding proteins, such as RNA polymerase. More broadly, this approach and methodology can be used to study transcriptional regulation in other obligate or facultative intracellular bacteria.
Prior to this study, C. trachomatis HrcA had been shown to bind and repress heat shock promoters in vitro (Wilson & Tan, 2004), but there was no direct evidence that it regulated the chlamydial heat shock response. In fact, HrcA-mediated binding to the CIRCE operator and repression of the groESL and dnaK P1 promoters had been shown to be unresponsive to elevated temperature in vitro (Wilson & Tan, 2004). The current study demonstrates that heat shock of Chlamydia-infected cells induces loss of HrcA binding to the groESL and dnaK P1 promoters with a concomitant increase in transcription from these promoters. The discrepancy between the in vitro and in vivo results suggests that elevated temperature is not sufficient to disrupt HrcA binding and that additional factors are involved in the in vivo regulation of the HrcA-mediated heat shock response. A likely cofactor is GroEL1 because it binds the HrcA-CIRCE complex in Chlamydia (Wilson et al., 2005), and other bacteria (Mogk et al., 1997), and enhances the ability of HrcA to bind and repress target promoters in vitro (Wilson et al., 2005, Chen et al., 2011). Another candidate is the GroESL chaperone complex, which helped to restore Helicobacter pylori HrcA binding activity after heat shock (Roncarati et al., 2014).
Our results provide strong evidence that the two paralogs of GroEL1 are not involved in the HrcA-regulated heat shock response in Chlamydia. We confirmed published findings that transcription of groEL2 and groEL3 in Chlamydia-infected cells is not induced by elevated temperature (Karunakaran et al., 2003). We also showed that HrcA did not bind in the vicinity of the putative groEL2 and groEL3 promoters at any of the times examined in the developmental cycle. Previously we showed that GroEL2 and GroEL3 did not promote HrcA binding to its operator nor repression of the groESL and dnaK P1 promoters (Wilson et al., 2005). Chlamydia GroEL2 and GroEL3 have also been shown to be functionally divergent from GroEL1 in an in silico analysis (McNally & Fares, 2007). Thus the two chlamydial GroEL paralogs have different functions from GroEL1, but their precise roles in the chlamydial infection remain unclear. In other bacteria, GroEL paralogs have been found to have non-heat-shock related functions such as predation and macromolecular feeding in Myxococcus xanthus (Li et al., 2010), and mycolic acid synthesis and biofilm formation in Mycobacterium smegmatis (Ojha et al., 2005).
Our study indicates that the heat shock response is operational throughout the chlamydial developmental cycle but is blunted at early times. There was less induction of transcription from the heat shock genes at 8 hpi, which indicates that these genes are in a relatively unrepressed state at early times. This reduced repression does not appear to be due to a complete absence of HrcA, because we detected HrcA binding to the groESL promoter in our ChIP studies at 8 hpi, as well as hrcA transcripts by qRT-PCR (data not shown). However, there may be limiting amounts of HrcA at early times, because there was a greater loss of the heat shock response at dnaK P1 (Fig. 5B), which has a lower binding affinity for HrcA than the groESL promoter (Fig. 5A). Another possibility is that the binding affinity of HrcA for its operator is lower at early times.
We also noted a selective decrease in heat shock induction of dnaK at late times (Fig. 5B), even though there was no corresponding decrease in HrcA binding to dnaK P1 (Fig. 5A). However, dnaK is also transcribed at late times from a HrcA-independent promoter, dnaK P2, which is controlled by the late regulator σ28 (Yu et al., 2006b). We propose that the chlamydial heat shock response is intact at late times, but overall dnaK expression is less responsive to heat shock because only one of its two promoters is regulated by HrcA. There is precedent in Bacillus subtilus (Mogk et al., 1997), Corynebacterium glutamicum (Barreiro et al., 2004), and Caulobacter crescentus (Avedissian et al., 1995) for dnaK expression to be regulated by tandem heat shock-inducible and heat shock-independent promoters.
In summary, this is the first study in Chlamydia measuring changes in the binding of a transcription factor to its target promoters in response to an environmental signal and concomitant changes in gene expression. We found that HrcA controls the expression of the major chlamydial heat shock proteins throughout the developmental cycle. However, the heat shock response appears to be temporally regulated so that there is a greater reserve of heat shock proteins that can be quickly produced in response to stress during the mid-stage of the developmental cycle. We used increased temperature as a convenient experimental method to study HrcA-mediated repression and the heat shock response in Chlamydia-infected cells. However, this transcriptional response is induced by additional cellular stressors in other bacteria. For example HrcA-regulated genes are induced by oxidative stress in Fusobacterium nucleatum (Steeves et al., 2011), and the HrcA-regulated DnaK protein is important for survival during nutrient starvation in E. coli, (Spence et al., 1990). In our study, the chlamydial heat shock genes were in a repressed, non-stressed state over the course of the developmental cycle, indicating that chlamydiae do not appear to encounter stress conditions in the cell culture infection model. However chlamydiae are likely to encounter multiple cellular stressors within an infected host animal, although the specific signals that regulate chlamydial HrcA remain to be identified.
Experimental Procedures
Cell culture and Chlamydia infections
L929 mouse fibroblast cells were grown in suspension culture at 37°C with 5% CO2 in RPMI 1640 medium (Cellgro) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Omega Scientific). L929 cells were infected with C. trachomatis serovar L2 (strain L2/434/Bu) at a multiplicity of infection (MOI) of three for all experiments. Unless specified otherwise, infected cells were collected at 24 hours post infection (hpi).
Heat shock
For each heat shock condition, 6 × 107 Chlamydia-infected L929 cells were collected with centrifugation and resuspended in 30 ml pre-heated RPMI 1640 (without FBS). Cells subject to heat shock were incubated at 45°C for 20 minutes (min), while control cells were incubated at 37°C. In experiments investigating the kinetics of the heat shock response, infected cells were incubated at 45°C for 5 or 10 min. Additional samples examining the recovery from heat shock were incubated at 45°C for 20 min, collected with centrifugation, and then resuspended and incubated in 30 ml RPMI 1640 (without FBS) at 37°C for 30 or 60 min. To examine the heat shock response during the developmental cycle, infected cells were collected at 8, 16, 24, 32, or 40 hpi and treated at either 37°C or 45°C for 20 min as described above.
Chromatin immunoprecipitation (ChIP)
1. Cross-linking
Cells were treated with formaldehyde (EMD Chemicals), which was added to the media at a final concentration of 1%, and rocked for 30 min at room temperature. To quench the cross-linking reaction, 3.3 ml of 2.5 M glycine pH 7.5 was added, and the infected cells were incubated at room temperature with rocking for an additional 15 min. Cross-linked infected cells were then collected with centrifugation and stored at −80°C.
2. Lysate preparation
To prepare lysates for chromatin immunoprecipitation the cross-linked infected cells were thawed and resuspended in 4 ml lysis buffer (25 mM Tris pH 7.5, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS) with a protease inhibitor cocktail (Sigma). The cells were then sonicated on ice using a Branson Digital Sonifier with an output setting of 22% for a total of 2 min in 15 second bursts. These sonication conditions resulted in fragmentation of DNA to a size range between 300–1200 bp. The lysate was centrifuged to remove debris and pre-cleared by incubating with 50 μl (bed volume) of Protein-G sepharose (GE Healthcare) at 4°C with rocking for 2 hours. Protein-G beads were removed from the lysate with centrifugation, and 250 μl aliquots were stored at −80°C. Each 250 μl aliquot represented lysate from approximately 3.75 × 106 infected cells.
3. Immunoprecipitation
Protein-G beads (25 μl bed volume) were washed with PBS (Cellgro) and incubated with 5 μl of pre-immune rabbit antibody (control antibody, Harlan Laboratories) or with 5 μl of anti-HrcA antibody (Harlan Laboratories) in 100 μl PBS at 4°C for 60 min with rotation. Antibody beads were then blocked with 500 μl of 5% BSA (Fisher Scientific) and 200 μg/ml of sheared salmon sperm DNA (Invitrogen) at 4°C for another 60 min with rotation. Blocked antibody beads were washed twice with PBS and added to 250 μl of pre-cleared cross-linked lysate (from 3.75 × 106 infected cells = 100% input), prepared as described above and incubated at 4°C overnight with rotation. The lysate was then removed and beads washed four times with 250 μl of ice-cold wash buffer (40 mM HEPES, 4 mM MgCl2, 70 mM KCl, 7.5% glycerol). Beads were then resuspended in 100 μl of 0.1 M glycine pH 2.5 to dissociate DNA-protein complexes from the antibody. The supernatant was transferred to a new tube and 20 μl of 1 M Tris pH 8.5 added to neutralize the pH. The eluate was then incubated at 95°C for 20 min to reverse the cross-linking, and the DNA isolated with a Nucleospin PCR clean up kit (Machery-Nagel), and stored at −20°C. Immunoprecipitations were performed at least three times.
DNA isolation
For each heat shock experimental condition examined by ChIP, a parallel sample of 6 × 106 infected cells (equivalent to 160% of the ChIP input sample) was collected to isolate and quantify genomic DNA. Infected cells were processed using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions. DNA quality and quantity was assessed using a NanoDrop ND-1000 (Thermo Scientific). The genomic DNA was diluted to a concentration equivalent to 0.01% ChIP input/μl and stored at −20°C. For normalization of qRT-PCR results the genomic DNA was diluted to 0.01% of total DNA/μl.
RNA isolation
An additional aliquot of 6 × 106 infected cells was collected in parallel for each experimental condition for isolation of RNA using the RNeasy Plus mini kit (Qiagen) according to the manufacturer’s instructions. RNA was eluted into 50 μl of DEPC-treated ddH2O. 2 μl (4%) of this RNA solution was incubated with 10 units of RQ1 RNase-free DNase (Promega) at 37°C for 60 min, after which an additional 10 units of DNase was added for another 60 min incubation. Following DNase treatment, the RNA was re-purified using the RNeasy Plus mini kit and eluted into 200 μl of DEPC-treated ddH2O, giving a final concentration of 0.02% of total RNA/μl.
Primer design
Primer3plus software (Untergasser et al., 2012) was used to design primers based on the C. trachomatis L2/434/Bu genomic sequence (NC_010287.1). Primer pairs were designed to amplify a 60–100 bp region for each promoter and gene sequence.
Quantitative PCR
For analysis of DNA immunoprecipitated in the ChIP experiments, quantitative PCR (qPCR) using the iQ SYBR Green kit (BioRad) was performed in triplicate with primers diluted to a final concentration of 250 nM and a total reaction volume of 20 μl. As a template, 2 μl (1.67%) of ChIP eluate or 2 μl (0.02%) of genomic DNA input was added to each reaction. Reactions were carried out on a BioRad iCycler with an initial denaturation step at 95°C for 5 min followed by 40 cycles of 30 seconds at 95°C and 30 seconds at 60°C. Fluorescent detection occurred during the annealing phase and subsequently during a dissocation curve analysis to confirm amplification of a single product. The threshold cycles (Ct) were determined using the BioRad iCycler software. Primer pairs used to amplify promoter DNA obtained in the ChIP experiments are listed in Table S1.
Quantitative reverse-transcriptase PCR
For analysis of RNA transcript levels, qRT-PCR using the iTaq Universal SYBR Green One-Step Kit was performed in triplicate with primers diluted to a final concentration of 250 nM and a total reaction volume of 20 μl. cDNA was synthesized from 2 μl (0.04%) of DNase-treated RNA with a 10 min step at 50°C, followed by a 5 min denaturation step at 95°C, and 40 cycles of 30 seconds at 95°C and 30 seconds at 60°C. Fluorescent detection and dissociation curve analysis were performed as described for qPCR. Reactions lacking reverse-transcriptase were performed in parallel for each RNA sample to confirm the absence of contaminating DNA. Primer pairs used to amplify specific genes are listed in Table S1.
ChIP data analysis
For normalization of ChIP Ct values to % input, the Ct for 100% input (= lysate from 3.75 × 106 infected cells) was calculated as Ct100% = Ct0.02% - log2(DF), where Ct0.02% is equal to the threshold cycle for 0.02% input and DF is the dilution factor (100%/0.02% = 5000). The same process was used to adjust the ChIP Ct value from 1.67% of the ChIP reaction to 100%, with CtCHIP_100% = CtCHIP_1.67% – log2(DF), where the DF = 100%/1.67% = 59.88. The % input ChIP recovery was then calculated as 100% × 2(Ct100% - CtCHIP_100%). Fold enrichment of each promoter was calculated as the % input obtained with the anti-HrcA antibody (HrcA%input) divided by the % input obtained with the control antibody (Control%input), or calculated as (HrcA%input/Control%input).
qRT-PCR data analysis
To normalize transcripts to genome copy number, standard curves were generated for each gene-specific primer pair by performing qPCR on C. trachomatis genomic DNA over a range of 3 × 102 – 3 × 106 copies per reaction. The Ct value obtained by adding 0.02% of total DNA for each sample was fit to the standard curve to determine the number of corresponding gene copies, and then multiplied by a dilution factor of 5000 (100%/0.02%) to yield total chlamydial genome copies in each sample of 6 × 106 infected cells. The Ct value obtained by adding 0.04% of total RNA for each sample using qRT-PCR was also fit to the gene-specific standard curve to determine the number of transcript copies, and then multiplied by a dilution factor of 2500 to yield total transcript copies in each sample of 6 × 106 infected cells. To calculate relative transcripts per genome copy, the number of transcripts was divided by the number of genome copies for each paired set of samples. Fold change was then calculated as the relative transcripts per genome copy of treated samples divided by the relative transcripts per genome copy of untreated samples, ((transcripts/genome copy at 45°C)/(transcripts/genome copy at 37°C)).
Statistics
The mean and standard deviation were calculated and experimental conditions were compared using the unpaired Student’s t-test. Where appropriate, conditions were compared using a one-way ANOVA followed by Levene’s test to assess homogeneity of variance. Post hoc analysis was subsequently performed using Tukey’s range test or the Games-Howell test.
Supplementary Material
Acknowledgments
We would like to thank Jennifer Lee, Chris Rosario and Emilie Orillard for critical reading of the manuscript. This work was supported by a grant from the NIH (AI44198). B.R.H. was supported by NRSA postdoctoral fellowship F32-AI108097.
Footnotes
The authors have no conflict of interest to report.
References
- Ault KA, Statland BD, King MM, Dozier DI, Joachims ML, Gunter J. Antibodies to the chlamydial 60 kilodalton heat shock protein in women with tubal factor infertility. Infect Dis Obstet Gynecol. 1998;6:163–167. doi: 10.1002/(SICI)1098-0997(1998)6:4<163::AID-IDOG5>3.0.CO;2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avedissian M, Lessing D, Gober JW, Shapiro L, Gomes SL. Regulation of the Caulobacter crescentus dnaKJ operon. J Bacteriol. 1995;177:3479–3484. doi: 10.1128/jb.177.12.3479-3484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro C, González-Lavado E, Pátek M, Martín JF. Transcriptional Analysis of the groES-groEL1, groEL2, and dnaK genes in Corynebacterium glutamicum: Characterization of Heat Shock-Induced Promoters. Journal of Bacteriology. 2004;186:4813–4817. doi: 10.1128/JB.186.14.4813-4817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Batteiger BE, Tan M. Chlamydia trachomatis (trachoma, genital infections, perinatal infections, and lymphogranuloma venereum) In: Bennett JE, Dolin R, Mandell GL, editors. Mandell, Douglas, and Bennett’s: Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier Inc; 2014. pp. 2154–2170. [Google Scholar]
- Brunham RC, Peeling R, Maclean I, Kosseim ML, Paraskevas M. Chlamydia trachomatis-associated ectopic pregnancy: serologic and histologic correlates. J Infect Dis. 1992;165:1076–1081. doi: 10.1093/infdis/165.6.1076. [DOI] [PubMed] [Google Scholar]
- Brunham RC, Peeling RW. Chlamydia trachomatis antigens: role in immunity and pathogenesis. Infect Agents Dis. 1994;3:218–233. [PubMed] [Google Scholar]
- Burton MJ, Mabey DC. The global burden of trachoma: a review. PLoS Negl Trop Dis. 2009;3:e460. doi: 10.1371/journal.pntd.0000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. Summary of Notifiable Diseases - United States, 2012. MMWR. 2014;61:1–124. [PubMed] [Google Scholar]
- Chen AL, Wilson AC, Tan M. A Chlamydia-specific C-terminal region of the stress response regulator HrcA modulates its repressor activity. J Bacteriol. 2011;193:6733–6741. doi: 10.1128/JB.05792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darville T, Hiltke TJ. Pathogenesis of Genital Tract Disease Due to Chlamydia trachomatis. Journal of Infectious Diseases. 2010;201:S114–S125. doi: 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JN, Pollack J, Perara E, Ganem D. Heat shock response of murine Chlamydia trachomatis. Journal of Bacteriology. 1990;172:6959–6972. doi: 10.1128/jb.172.12.6959-6972.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston JSH, Deane KHO, Jecock RM, Pearce JH. Identification of 2 Chlamydia trachomatis antigens recognized by synovial fluid T Cells from patients with Chlamydia induced reactive arthritis. Journal of Rheumatology. 1996;23:130–136. [PubMed] [Google Scholar]
- Georgopoulos C, Welch W. Role of the major heat shock proteins as molecular chaperones. Annual review of cell biology. 1993;9:601–634. doi: 10.1146/annurev.cb.09.110193.003125. [DOI] [PubMed] [Google Scholar]
- Gerard HC, Whittum-Hudson JA, Schumacher HR, Hudson AP. Differential expression of three Chlamydia trachomatis hsp60-encoding genes in active vs. persistent infections. Microb Pathog. 2004;36:35–39. doi: 10.1016/j.micpath.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Grainger DC, Lee DJ, Busby SJW. Direct methods for studying transcription regulatory proteins and RNA polymerase in bacteria. Current Opinion in Microbiology. 2009;12:531–535. doi: 10.1016/j.mib.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Hickey JM, Weldon L, Hefty PS. The atypical OmpR/PhoB response regulator ChxR from Chlamydia trachomatis forms homodimers in vivo and binds a direct repeat of nucleotide sequences. J Bacteriol. 2011;193:389–398. doi: 10.1128/JB.00833-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran KP, Noguchi Y, Read TD, Cherkasov A, Kwee J, Shen C, Nelson CC, Brunham RC. Molecular analysis of the multiple GroEL proteins of Chlamydiae. J Bacteriol. 2003;185:1958–1966. doi: 10.1128/JB.185.6.1958-1966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klos A, Thalmann J, Peters J, Gérard HC, Hudson AP. The transcript profile of persistent Chlamydophila (Chlamydia) pneumoniae in vitro depends on the means by which persistence is induced. FEMS Microbiology Letters. 2009;291:120–126. doi: 10.1111/j.1574-6968.2008.01446.x. [DOI] [PubMed] [Google Scholar]
- LaVerda D, Albanese LN, Ruther PE, Morrison SG, Morrison RP, Ault KA, Byrne GI. Seroreactivity to Chlamydia trachomatis Hsp10 correlates with severity of human genital tract disease. Infect Immun. 2000;68:303–309. doi: 10.1128/iai.68.1.303-309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang Y, Zhang C-y, Zhang W-y, Jiang D-m, Wu Z-h, Liu H, Li Y-z. Myxococcus xanthus Viability Depends on GroEL Supplied by Either of Two Genes, but the Paralogs Have Different Functions during Heat Shock, Predation, and Development. Journal of Bacteriology. 2010;192:1875–1881. doi: 10.1128/JB.01458-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally D, Fares MA. In silico identification of functional divergence between the multiple groEL gene paralogs in Chlamydiae. BMC Evol Biol. 2007;7:81. doi: 10.1186/1471-2148-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minder AC, Fischer HM, Hennecke H, Narberhaus F. Role of HrcA and CIRCE in the heat shock regulatory network of Bradyrhizobium japonicum. J Bacteriol. 2000;182:14–22. doi: 10.1128/jb.182.1.14-22.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Homuth G, Scholz C, Kim L, Schmid FX, Schumann W. The GroE chaperonin machine is a major modulator of the CIRCE heat shock regulon of Bacillus subtilis. EMBO J. 1997;16:4579–4590. doi: 10.1093/emboj/16.15.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison RP, Lyng K, Caldwell HD. Chlamydial disease pathogenesis. Ocular hypersensitivity elicited by a genus-specific 57-kD protein. J Exp Med. 1989;169:663–675. doi: 10.1084/jem.169.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narberhaus F. Negative regulation of bacterial heat shock genes. Mol Microbiol. 1999;31:1–8. doi: 10.1046/j.1365-2958.1999.01166.x. [DOI] [PubMed] [Google Scholar]
- Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR, Jr, Hatfull GF. GroEL1: A Dedicated Chaperone Involved in Mycolic Acid Biosynthesis during Biofilm Formation in Mycobacteria. Cell. 2005;123:861–873. doi: 10.1016/j.cell.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Peeling RW, Bailey RL, Conway DJ, Holland MJ, Campbell AE, Jallow O, Whittle HC, Mabey DC. Antibody response to the 60-kDa chlamydial heat-shock protein is associated with scarring trachoma. J Infect Dis. 1998;177:256–259. doi: 10.1086/517367. [DOI] [PubMed] [Google Scholar]
- Peeling RW, Kimani J, Plummer F, Maclean I, Cheang M, Bwayo J, Brunham RC. Antibody to chlamydial hsp60 predicts an increased risk for chlamydial pelvic inflammatory disease. J Infect Dis. 1997;175:1153–1158. doi: 10.1086/516454. [DOI] [PubMed] [Google Scholar]
- Rolando M, Sanulli S, Rusniok C, Gomez-Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C. Legionella pneumophila Effector RomA Uniquely Modifies Host Chromatin to Repress Gene Expression and Promote Intracellular Bacterial Replication. Cell Host & Microbe. 2013;13:395–405. doi: 10.1016/j.chom.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Roncarati D, Danielli A, Scarlato V. The HrcA repressor is the thermosensor of the heat-shock regulatory circuit in the human pathogen Helicobacter pylori. Molecular Microbiology. 2014;92:910–920. doi: 10.1111/mmi.12600. [DOI] [PubMed] [Google Scholar]
- Schulz A, Schumann W. hrcA, the first gene of the Bacillus subtilis dnaK operon encodes a negative regulator of class I heat shock genes. Journal of Bacteriology. 1996;178:1088–1093. doi: 10.1128/jb.178.4.1088-1093.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J, Cegielska A, Georgopoulos C. Role of Escherichia coli heat shock proteins DnaK and HtpG (C62.5) in response to nutritional deprivation. J Bacteriol. 1990;172:7157–7166. doi: 10.1128/jb.172.12.7157-7166.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeves CH, Potrykus J, Barnett DA, Bearne SL. Oxidative stress response in the opportunistic oral pathogen Fusobacterium nucleatum. Proteomics. 2011;11:2027–2037. doi: 10.1002/pmic.201000631. [DOI] [PubMed] [Google Scholar]
- Tan M. Regulation of gene expression. In: Bavoil PM, Wyrick PB, editors. Chlamydia: Genomics and Pathogenesis. Wymondham, U.K: Horizon Bioscience; 2006. pp. 103–131. [Google Scholar]
- Tan M. Temporal gene regulation during the chlamydial development cycle. In: Tan M, Bavoil PM, editors. Intracellular Pathogens I: Chlamydiales. Washington, D.C: ASM Press; 2012. pp. 149–169. [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Tan M. Functional Analysis of the Heat Shock Regulator HrcA of Chlamydia trachomatis. Journal of Bacteriology. 2002;184:6566–6571. doi: 10.1128/JB.184.23.6566-6571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Tan M. Stress Response Gene Regulation in Chlamydia Is Dependent on HrcA-CIRCE Interactions. Journal of Bacteriology. 2004;186:3384–3391. doi: 10.1128/JB.186.11.3384-3391.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Wu CC, Yates JR, 3rd, Tan M. Chlamydial GroEL autoregulates its own expression through direct interactions with the HrcA repressor protein. J Bacteriol. 2005;187:7535–7542. doi: 10.1128/JB.187.21.7535-7542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HH, Kibler D, Tan M. In silico prediction and functional validation of sigma28-regulated genes in Chlamydia and Escherichia coli. J Bacteriol. 2006a;188:8206–8212. doi: 10.1128/JB.01082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HHY, Kibler D, Tan M. In Silico Prediction and Functional Validation of σ28-Regulated Genes in Chlamydia and Escherichia coli. Journal of Bacteriology. 2006b;188:8206–8212. doi: 10.1128/JB.01082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber U, Schumann W. CIRCE, a novel heat shock element involved in regulation of heat shock operon dnaK of Bacillus subtilis. J Bacteriol. 1994;176:1359–1363. doi: 10.1128/jb.176.5.1359-1363.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.