

Abstract
Homozygous recessive mutations in the PRICKLE1 gene were originally reported in three consanguineous families with myoclonic epilepsy. Subsequently, several studies have identified neurological abnormalities in animal models with both heterozygous and homozygous mutations in PRICKLE1 orthologues, including epilepsy in flies and in mice with heterozygous PRICKLE1 mutations. We describe a fetus with a novel de novo mutation in PRICKLE1 associated with agenesis of the corpus callosum.
INTRODUCTION
Homozygous recessive mutations in the PRICKLE1 gene were originally reported in three consanguineous families with myoclonic epilepsy(Bassuk, Wallace et al. 2008). Subsequently, several studies have identified neurological abnormalities in animal models with both heterozygous and homozygous mutations in PRICKLE1 orthologues, including epilepsy in flies and in mice with heterozygous PRICKLE1 mutations(Bosoi, Capra et al. 2011; Tao, Manak et al. 2011; Paemka, Mahajan et al. 2013; Ehaideb, Iyengar et al. 2014). Neuronal migration abnormalities have also been reported in several animal models of PRICKLE1 mutations(Tao, Manak et al. 2011; Yang, Bassuk et al. 2014). While heterozygous PRICKLE1 mutations have been reported in several human conditions including epilepsy(Tao, Manak et al. 2011), autism(Paemka, Mahajan et al. 2013) and spina bifida(Bosoi, Capra et al. 2011), these mutations have all been inherited (or inheritance was not determined). We describe a fetus with a novel de novo mutation in PRICKLE1 associated with agenesis of the corpus callosum.
MATERIALS AND METHODS
The family was consented by an IRB approved protocol at UCSF, after informed consent was obtained from an IRB approved protocol from USCF, clinical whole exome sequencing was performed on DNA isolated from the fetus and from the parents. Clinical exome sequencing was perfomed by GeneDx (Gaithersburg, MD) using standard techniques (as in Tao, Manak et al. 2011). Briefly, the clinical exome sequencing pipeline begins with massive parallel sequencing using the Illumina sequencing system with 2x100 bp paired-end reads. Bidirectional sequence were assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool (Xome Analyzer). 95% of the targeted region was covered at >10X coverage.
Sanger sequencing to confirm the gene variant was performed using PRICKLE1 specific primers (Bassuk, Wallace et al. 2008). Fetal MRI was performed using standard A1-Coronal, A2-Axial and A3-Parasagittal images. Peptide sequences with high sequence similarity were downloaded from NCBI BLAST and the alignment was performed using the software ClustalW.
RESULTS
Case report
A 37 year-old G1P0 female presented at 19 weeks for fetal ultrasound which demonstrated agenesis of the corpus callosum. A subsequent brain MRI was performed at 22 weeks 2 days by last menstrual period (LMP), confirming complete callosal agenesis and demonstrating mild ventriculomegaly (11 mm in both left and right lateral ventricles; Figure 1(A–C) and possible polymicrogyria). The fetus was delivered at 23 weeks. Anatomic pathology did not demonstrate any craniofacial or other organ dysmorphism. After informed consent was obtained from an IRB approved protocol from USCF, clinical whole exome sequencing was performed on DNA isolated from the fetus and from the parents, revealing a de novo mutation in the PRICKLE1 gene C.427T>G, S143A. No other de novo mutations were identified, nor were any other mutations in known disease-causing genes uncovered (including known polymicrogyria or lissencepahly genes including DCX, LIS1, LAMB1, LARGE, POMT1, POMT2, POMGNT1, POMGNT2, PHGDNDE1, FKTN, FKRP, ARX, ADGRG1, FIG4, NHEJ1, PI4KA, RTTN, SRPX2, AND TUBA1A, and TUBB2B, nor any other known neurodevelopmental genes). Parentage was validated using standard microsatellite analysis, and the de novo mutation was validated by Sanger sequencing (Figure 2). This variant was absent from over 6500 alleles in the Exome Variant Server (http://evs.gs.washington.edu/EVS/) nor was it present in the 1000 Genomes Project (http://www.1000genomes.org/). Sequence alignment demonstrates that the S142A encoding PRICKLE1 mutation alters an amino acid conserved throughout vertebrate evolution (Figure 3).
Figure 1.
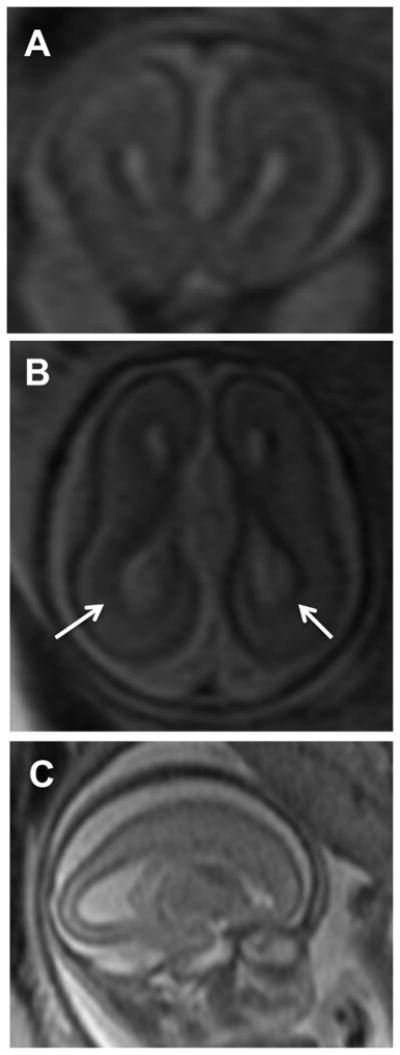
IMAGES. A-Coronal, B-Axial and C-Parasagittal images from a fetal MRI conducted at 22 wk and 2 days gestation showing enlarged lateral ventricles (more posteriorly (white arrows); colpocephaly) and agenesis of the corpus callosum
Figure 2.

Chromatogram. The top two chromatograms are from the mother, the middle two are from the father, and the bottom two (showing the heterozygous mutation) is from the fetus.
Figure 3.

Protein Feature View for PRICKLE1 from the RCSB Protein Data Bank (PDB) website and a multiple species protein alignment of the region surrounding the amino acid change caused by the de novo mutation in PRICKLE1. An arrow indicates the reference amino acid (S) in humans and other vertebrates. The mutation is in the LIM zinc-binding 1-domain of PRICKLE1. The amino acid is completely conserved in the vertebrates used in the alignment, and is within a highly conserved domain across all species. Peptide sequences with high sequence similarity were downloaded from NCBI BLAST and the alignment was performed using the software ClustalW.
DISCUSSION
PRICKLE1 mutations in humans were originally described as recessive mutations in families with syndromic myoclonic epilepsy(Bassuk, Wallace et al. 2008) and variation in PRICKLE1 was subsequently described in patients with non-syndromic epilepsy(Tao, Manak et al. 2011), autism(Paemka, Mahajan et al. 2013), and spina bifida(Bosoi, Capra et al. 2011). Neurological phenotypes including epilepsy(Tao, Manak et al. 2011) and learning abnormalities(Paemka, Mahajan et al. 2013) and neuronal migration abnormalities have been described in multiple animal models of PRICKLE1 mutations(Bosoi, Capra et al. 2011; Tao, Manak et al. 2011; Mei, Wu et al. 2013; Paemka, Mahajan et al. 2013; Ehaideb, Iyengar et al. 2014; Yang, Bassuk et al. 2014) yet to our knowledge, a de novo mutation in human PRICKLE1 had yet to be reported. Our description of a de novo PRICKLE1 mutation associated with agenesis of the corpus calllosum and polymicrogyria in the absence of any other de novo mutation or known disease causing mutation strongly implicates the PRICKLE1 gene as causative of the severe cerebral malformation in this fetus. These findings likely expand the clinical spectrum of abnormalities associated with PRICKLE1 mutations in humans. Our own previous studies of PRICKLE1 mutations have focused on patients with normal neuroimaging finding and epilepsy, which may in part explain why PRICKLE1 mutations have not been observed with human brain malformations. While we cannot conclude from this single case that some PRICKLE1 mutations might be embryonic lethal, the extreme phenotype in this report suggests that PRICKLE1 de novo heterozygous mutations may be underreported, and that many of the cases may involve fetal demise. With the expansion of massive parallel sequencing techniques to the clinical realm, including fetal testing, we expect that mutations in PRICKLE1 and related genes will be increasingly recognized as contributors to human neurological conditions.
Acknowledgments
Supported by NIH 1R01 NS064159 (AGB)
Footnotes
Drs. Bassuk and Sherr report no disclosures.
Author contributions: Drs. Bassuk and Sherr co-wrote the manuscript and performed all genotype-phenotype correlations.
References
- Bassuk AG, Wallace RH, et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet. 2008;83(5):572–581. doi: 10.1016/j.ajhg.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosoi CM, Capra V, et al. Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum Mutat. 2011;32(12):1371–1375. doi: 10.1002/humu.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehaideb SN, Iyengar A, et al. prickle modulates microtubule polarity and axonal transport to ameliorate seizures in flies. Proc Natl Acad Sci U S A. 2014;111(30):11187–11192. doi: 10.1073/pnas.1403357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei X, Wu S, et al. Mechanisms of prickle1a function in zebrafish epilepsy and retinal neurogenesis. Dis Model Mech. 2013;6(3):679–688. doi: 10.1242/dmm.010793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paemka L, V, Mahajan B, et al. PRICKLE1 interaction with SYNAPSIN I reveals a role in autism spectrum disorders. PLoS One. 2013;8(12):e80737. doi: 10.1371/journal.pone.0080737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Manak JR, et al. Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet. 2011;88(2):138–149. doi: 10.1016/j.ajhg.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Bassuk AG, et al. Prickle1 is necessary for the caudal migration of murine facial branchiomotor neurons. Cell Tissue Res. 2014;357(3):549–561. doi: 10.1007/s00441-014-1925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]