

Abstract
The d or l form of 2-hydroxyglutarate (2HG) accumulates in certain rare neurometabolic disorders, and high d-2-hydroxyglutarate (d-2HG) levels are also found in several types of cancer. Although 2HG has been detected in Saccharomyces cerevisiae, its metabolism in yeast has remained largely unexplored. Here, we show that S. cerevisiae actively forms the d enantiomer of 2HG. Accordingly, the S. cerevisiae genome encodes two homologs of the human d-2HG dehydrogenase: Dld2, which, as its human homolog, is a mitochondrial protein, and the cytosolic protein Dld3. Intriguingly, we found that a dld3Δ knock-out strain accumulates millimolar levels of d-2HG, whereas a dld2Δ knock-out strain displayed only very moderate increases in d-2HG. Recombinant Dld2 and Dld3, both currently annotated as d-lactate dehydrogenases, efficiently oxidized d-2HG to α-ketoglutarate. Depletion of d-lactate levels in the dld3Δ, but not in the dld2Δ mutant, led to the discovery of a new type of enzymatic activity, carried by Dld3, to convert d-2HG to α-ketoglutarate, namely an FAD-dependent transhydrogenase activity using pyruvate as a hydrogen acceptor. We also provide evidence that Ser3 and Ser33, which are primarily known for oxidizing 3-phosphoglycerate in the main serine biosynthesis pathway, in addition reduce α-ketoglutarate to d-2HG using NADH and represent major intracellular sources of d-2HG in yeast. Based on our observations, we propose that d-2HG is mainly formed and degraded in the cytosol of S. cerevisiae cells in a process that couples d-2HG metabolism to the shuttling of reducing equivalents from cytosolic NADH to the mitochondrial respiratory chain via the d-lactate dehydrogenase Dld1.
Keywords: dehydrogenase, enzyme kinetics, flavoprotein, inborn error of metabolism, yeast metabolism, 2-hydroxyglutarate, transhydrogenase
Introduction
2-Hydroxyglutarate (2HG)3 is a 5-carbon dicarboxylic acid that was first detected in human urine in the late 1970s (1). Because of the hydroxyl group on the second carbon, 2HG exists under two enantiomeric configurations (l or d) that can be separated by gas or liquid chromatography after derivatization with another chiral compound and that can therefore be differentially assayed in biological samples using GC-MS or LC-MS methods (2, 3). The interest in 2HG increased when it was found to accumulate in urine of patients with suspected inborn errors of metabolism (4, 5). Most 2-hydroxyglutaric aciduria patients present elevations of either l-2HG or d-2HG in their extracellular fluids, and the clinical phenotype depends on the configuration of the accumulated organic acid. More recently, cases of “combined d,l-hydroxyglutaric aciduria” have been reported (6).
2-Hydroxyglutaric acidurias remained enigmatic diseases because neither l-2HG nor d-2HG are intermediates of any known metabolic pathway, and the causal gene deficiencies were only discovered many years after the first patient case reports. It is now established that l-2-hydroxyglutaric aciduria is caused by loss-of-function mutations in the L2HGDH gene, encoding a specific l-2HG dehydrogenase, whereas in many cases d-2-hydroxyglutaric aciduria results from loss-of-function mutations in the D2HGDH gene, which encodes a dehydrogenase acting on d-2HG (7, 8). Other cases of d-2-hydroxyglutaric aciduria are caused by gain-of-function mutations in the mitochondrial isocitrate dehydrogenase gene IDH2 (9). Interestingly, somatic IDH1 (cytosolic isocitrate dehydrogenase) and IDH2 mutations leading to increased d-2HG production have also been identified in a number of cancers, including gliomas and acute myelogenous leukemia (10). Increased 2HG levels have in addition been detected in human breast tumors with wild-type isocitrate dehydrogenase (IDH) (11). d-2HG is therefore suspected to act as an oncometabolite (10). Combined d,l-hydroxyglutaric aciduria is caused by mutations in the mitochondrial citrate carrier SLC25A1 (12).
The cancer-associated mutant IDH1 and IDH2 enzymes actively convert α-ketoglutarate to d-2HG leading to high (millimolar) intracellular concentrations of the latter. It is less clear which is the main source of d-2HG in normal mammalian tissues. Hydroxyacid-oxoacid transhydrogenase (ADHFE1) catalyzes the conversion of γ-hydroxybutyrate to succinic semialdehyde with concomitant reduction of α-ketoglutarate to d-2HG (13). Human phosphoglycerate dehydrogenase was also recently found to convert α-ketoglutarate to d-2HG, using NADH as a cofactor (14). Finally, there is evidence that wild-type IDH1 and IDH2 also slowly form d-2HG from α-ketoglutarate (15). A known source of l-2HG in mammalian cells is a low side activity of malate dehydrogenase, converting α-ketoglutarate to l-2HG at the expense of NADH (16, 17). Such an l-2HG-producing side activity has also been demonstrated for the mitochondrial malate dehydrogenases of Arabidopsis thaliana (18). Lactate dehydrogenase A has recently been described as a major source of l-2HG in mammalian cells under hypoxia (19). Isotopic labeling studies using [13C]glucose and [2H]glutamate strongly indicate that, in the mammalian system, d-2HG and l-2HG are derived from the mitochondrial pool of α-ketoglutarate (20, 21). In agreement with this, both the mammalian d- and l-2HG dehydrogenases were shown to be localized in the mitochondria (22, 23).
In contrast to the mammalian system, very little is known about the occurrence of 2-hydroxyglutarate and its metabolism in yeast. 2HG was detected in Saccharomyces cerevisiae cultures grown anaerobically in the presence of glucose and of glutamate as the sole nitrogen source (24). 14C labeling of either the supplemented glucose or glutamate revealed that 2HG was entirely derived from glutamate under these conditions. The purpose of the present work was to determine whether S. cerevisiae produces the l and/or the d enantiomer of 2HG and to identify the enzymes involved in the formation and degradation of this organic acid in yeast. We show that S. cerevisiae only forms detectable amounts of the d form of 2HG under standard yeast cultivation conditions (aerobic batch cultures in minimal defined medium supplemented with glucose) and that the main enzyme responsible for its degradation is encoded by the DLD3 gene, currently annotated as a d-lactate dehydrogenase. We found that this cytosolic protein actually converts d-2HG to α-ketoglutarate with concomitant reduction of pyruvate to d-lactate, qualifying this enzyme as a transhydrogenase rather than a dehydrogenase. Finally, we show that the Ser3 and Ser33 proteins, known to catalyze the oxidation of 3-phosphoglycerate in the first step of the main yeast serine biosynthesis pathway, in addition catalyze the NADH-dependent conversion of α-ketoglutarate to d-2HG. The physiological significance of these reactions is discussed in the light of our findings.
Experimental Procedures
Materials
Reagents, of analytical grade whenever possible, were acquired from Sigma, unless otherwise indicated. LC-MS grade solvents were from VWR Chemicals.
Phylogenetic Analysis
Yeast Dld2 and Dld3 protein sequences were obtained from the NCBI protein database or from the Saccharomyces Genome Database. The protein sequences were then aligned using ClustalW2 (25) with a gap-opening penalty of 10. The phylogenetic relationships between those proteins were analyzed using the Neighbor-Joining (Poisson distance model) and the Maximum-Likelihood (LG substitution model) methods. These analyses were performed with the SeaView (26) and PhyML (27) programs, respectively. Bootstrap analyses were used to assess the confidence level of each node with 100 replications for the Maximum-Likelihood method and 1000 replications for the Neighbor-Joining method.
dN/dS Ratio Calculation
Protein and nucleic acid sequences from 40 different S. cerevisiae strains were obtained from the Saccharomyces Genome Database. Nucleic acid sequences were aligned based on the protein sequence alignments using the TRANalign program (available in the EMBOSS package). The dN/dS ratios were calculated based on these nucleic acid sequence alignments using CODEML in PAMLX (28) for the DLD1, DLD2, and DLD3 genes. Briefly, for each gene, Neighbor-Joining trees were constructed using ClustalX. Those trees were then used for dN and dS calculations based on a neutral model.
Generation of Mutant, Overexpression, and Rescue Yeast Strains
The S. cerevisiae strains used in this study (supplemental Table S1), all isogenic to the reference BY4741 or BY4742 strains, were either taken from the collection of non-essential gene deletion mutants (dld2Δ and dld3Δ mutants) or generated via PCR-mediated gene replacement (dld1Δ, ser3Δ, and ser33Δ mutants) (29). For gene replacement with the KanMX cassette, primer pairs binding 200–300 bp upstream and 150–350 bp downstream of the ORF of interest were designed (supplemental Table S2). Genomic DNA of the corresponding knock-out strain from the yeast deletion collection served as a template for PCR amplification. The PCR was performed using Phusion DNA polymerase (Thermo Fisher Scientific) in the buffer provided by the manufacturer (Phusion HF Buffer) as follows: (i) denaturation for 10 min at 98 °C; (ii) 35 cycles of denaturation (30 s at 98 °C), annealing (30 s at 48 °C for dld1Δ or 53 °C for ser3Δ and ser33Δ), and elongation for 2 min at 72 °C; and (iii) final elongation for 10 min at 72 °C. The PCR products were purified and transformed into wild-type strains of both mating types (BY4741 and BY4742). Transformants were selected after 2–3 days at 30 °C on YPD plates containing 200 μg/ml of the aminoglycoside antibiotic G418. The double knock-out strain ser3Δser33Δ was generated by mating haploid single mutant cells of opposing mating types, followed by sporulation on agar plates containing potassium acetate (10 g/liter) and tetrad dissection. All the gene deletions in the knock-out strains used in this study were confirmed by PCR analysis and Sanger sequencing (Eurofins Genomics).
To generate overexpression and rescue strains, the Gateway Cloning technology was used. For each gene to overexpress (SER3, SER33) or to rescue (DLD3), Entry clones were generated in a BP reaction between the attB-flanked ORFs of interest (PCR-amplified from wild-type BY4741 genomic DNA) and the pDONR221 vector (Invitrogen) according to Ref. 30. The sequences of the PCR primers used and the plasmids obtained are given in supplemental Tables S2 and S3. The inserts were then subcloned from the sequence-verified Entry clones into the pAG416-GPD-ccdB Destination vector (Addgene ID 14148; yeast centromere plasmid containing a constitutive glyceraldehyde-3-phosphate dehydrogenase promoter and the URA3 gene for nutrient selection) in LR reactions to create the desired Expression clones (supplemental Table S3). The purified expression plasmids were transformed into yeast strains with different genetic backgrounds using the EZ-YEASTTM transformation kit (MP Biomedicals) (supplemental Table S1).
Yeast Cultivation
Yeast strains were grown in filter-sterilized minimal defined medium (6.7 g/liter Yeast Nitrogen Base from MP Biomedicals, 10 g/liter d-glucose, 80 mg/liter uracil, 80 mg/liter l-methionine, 80 mg/liter l-histidine, and 240 mg/liter l-leucine), if not otherwise indicated. The pH was adjusted to 5.5 by addition of sodium hydroxide. Batch cultures were shaken in flasks at 200 rpm and 30 °C, with a working volume corresponding to 10% of the volume of the Erlenmeyer flasks. Liquid batch cultures were started by inoculation with glycerol stocks (0.05–0.1% of the final cultivation volume) that were prepared as follows: liquid yeast cultures (100 ml) were inoculated from single colonies and grown for at least 18 h in minimal defined medium, concentrated 10-fold by centrifugation, enriched with glycerol to a final concentration of 20% (v/v), and stored at −80 °C until further use.
Growth Characterization
To monitor the growth of yeast cultures, biovolume and cell concentration were determined in aliquots taken from the yeast cultivations using a Multisizer Z3 equipped with a 30-μm measurement capillary (Beckman Coulter) after dilution in ISOTON II solution (Beckman Coulter).
Metabolite Extraction and Sample Preparation for Metabolite Analyses
10-ml aliquots of yeast cultivations were quenched by adding 30 ml of 60% (v/v) methanol at −60 °C. This mixture was shaken by tube inversion, and cells were pelleted by a 10-min centrifugation at 10,000 × g and −10 °C. Pellets were stored at −80 °C until further processing. Intracellular metabolites were extracted from the frozen cell pellets (of known biovolume) by addition of a 55.5-fold volume of extraction fluid (50% methanol, 50% TE buffer at pH 7.0 containing 10 mm Tris-HCl and 1 mm EDTA) at −20 °C and an equal volume of chloroform at −20 °C. The mixture was incubated at −20 °C in a shaking device (Vortexer or Eppendorf shaker) for 2 h and then centrifuged for 10 min at 10,000 × g and −10 °C. The upper aqueous phase was taken up, filtered (0.22 μm, cellulose acetate membrane) into an HPLC glass vial, and stored at −20 °C until LC-MS analysis. For the analysis of extracellular metabolites, 0.5–2-ml aliquots of the yeast cultivations were filtered on 0.22-μm cellulose acetate membranes and stored at −20 °C until further processing.
LC-MS Analysis
Two different types of LC-MS analyses were performed. To differentially quantify d- and l-2-hydroxyglutarate in our samples, a first method was adapted from a previously published MRM-based LC-MS/MS method that allows separation of both 2HG enantiomers by reverse-phase HPLC (3). Deuterated 2HG, prepared by reduction of [3,3,4,4-2H4]α-ketoglutaric acid as described previously (2), was added as an internal standard (10 nmol) to the cell extracts, prepared as described above. Samples were dried in a CentriVap Vacuum Concentrator (LabConco) at −4 °C and resuspended in 50 μl of dichloromethane/acetic acid (4:1, v/v) containing 50 mg/ml of the derivatizing chiral compound diacetyl-l-tartaric anhydride. The samples were heated for 30 min at 75 °C, dried down again in the vacuum concentrator at ambient temperature, and resuspended in 500 μl of water. HPLC separation was performed on an Agilent 1260SL instrument using an Agilent Poroshell 120 EC-C18 column (150 × 2.1 mm, 2.7 μm particle size) with an isocratic flow (0.25 ml/min) of a mobile phase composed of 90% aqueous ammonium acetate (5 mm) containing 0.1% (v/v) formic acid and 10% acetonitrile. The temperature was kept constant at 30 °C. The target compounds were detected on an ABSciex 4500 QTRAP mass spectrometer in negative MRM mode. Detection and quantification were based on the peak areas of specific MRM transitions (2HG, 363 > 147 m/z; [2H4]2HG, 367 > 151 m/z) and one transition was used for confirmation of the 2HG isomers (363 > 129 m/z). The l- and d-2HG derivatives eluted at a retention time of 1.84 and 2.03 min, respectively.
A second targeted LC-MS method was used for the quantification of a range of physiological organic acids (including total 2HG) and all 20 amino acids (supplemental Table S4). For this analysis, samples were not derivatized, and hence the l and d enantiomers of 2HG were not separated during liquid chromatography. Chromatographic separation was performed on a Dionex UltiMate 3000 (Thermo Fisher Scientific) coupled to a Q Exactive Orbitrap mass spectrometer equipped with a HESI electrospray ion source (Thermo Fisher Scientific). Nitrogen was supplied by a Genius 1022 high purity generator (Peak Scientific Instruments, Ltd.). A ZicHILIC SeQuant column (150 × 2.1 mm, 3.5 μm particle size, 100 Å pore size) connected to a ZicHILIC guard column (20 × 2.1 mm, 5 μm particle size) (Merck KgAA) was used for separation. LC was performed at 15 °C and a constant flow of 0.4 ml/min in gradient mode. Using buffers A (water/acetonitrile/formic acid, 99:1:0.1 by volume) and B (acetonitrile/water/formic acid, 99:1:0.1 by volume), a 25-min chromatographic gradient was applied. An isocratic step at 95% B for 2.7 min was followed by a progressive decrease to 93% B over 3.3 min, 90% B over 3 min, 80% B over 4 min, 50% B over 7 min, and a final decrease to 10% B over 1 additional min. The gradient was followed by an isocratic step at 10% B for 2 min, before the initial conditions were restored within 0.1 min and kept constant for additional 1.9 min. Mass spectral data were obtained in positive and negative electrospray ionization modes via scheduled selected ion monitoring. The sheath gas flow rate was set to 10, the spray voltage to 4 kV and the capillary temperature to 320 °C. Intra- and extracellular concentrations were determined using external calibration curves ranging from 0.01 to 50 μm for each metabolite.
Other Small Molecule Assays
Extracellular glucose levels were measured using a YSI 2950D (Yellow Springs Instruments) biochemistry analyzer. l- and d-lactate concentrations in culture media and in transhydrogenase activity assays (see below) were measured spectrophotometrically (TECAN M200 Pro) at 30 °C by determining the change of absorbance due to NADH formation at 340 nm after addition of either l-LDH (from rabbit muscle, Sigma) or d-LDH (from Lactobacillus leichmannii, Sigma). The assay mixture (200 μl total volume) contained a glycine/hydrazine buffer at pH 9.5 (100 mm glycine, 40 mm hydrazine sulfate), 3 mm NAD+, and l-LDH (4 units) or d-LDH (6.6 units for the transhydrogenase assays and 10 units for extracellular d-lactate measurements).
Protein Expression and Purification of Dld2, Dld3, Ser3, and Ser33
Sequence-verified Gateway Entry clones containing the ORFs of the DLD2, DLD3, SER3, and SER33 genes were generated as described above. For Dld2, the 44 N-terminal amino acids of the coding sequence (mitochondrial targeting sequence (MTS)) were omitted for plasmid construction. All inserts were subcloned into the Destination Vector pDest-527 (Addgene ID 11518; isopropyl 1-thio-β-d-galactopyranoside-inducible T7 promoter, N-terminal His6 tag). The resulting Expression clones were transformed into Escherichia coli BL21(DE3) cells for protein overexpression (supplemental Tables S1 and S3). Precultures of transformed E. coli BL21(DE3) cells were inoculated (at a 1:50 ratio) into 500–1000 ml of LB medium containing 100 μg/ml ampicillin and cultivated at 37 °C with shaking until the optical density measured at 600 nm (Biochrom WPA CO8000 Cell density meter) reached 0.5–1.0. Cultures were then put on ice for 30 min and induced by adding isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 0.5 mm for Dld3 and 0.1 mm for Dld2, Ser3, and Ser33 production. The induced cultures were left with shaking for 16–21 h at 18 °C, and cells were harvested by a 15-min centrifugation at 4500 × g and 4 °C. The pellets were resuspended in lysis buffer (25 mm Tris-HCl, pH 8.0, 300 mm NaCl, 0.5 mm PMSF, 1 mm DTT, 1× EDTA-free Complete Ultra protease inhibitor mixture from Roche Applied Science) in a volume corresponding to 5% of the original culture volume. Cell suspensions were sonicated for 2 min at an amplitude of 25–30% with 0.5-s pulses separated by 1–2.5-s breaks to minimize sample heat-up, and then centrifuged for 35 min at 17,000 × g and 4 °C to separate cell debris from the soluble protein fraction.
Protein purifications and protein desalting were conducted at 4 °C on an ÄKTA protein purifier (GE Healthcare). Cleared cell lysates were filtered on cellulose acetate membranes (0.45- or 1.2-μm pore size, Minisart), and buffer B (25 mm Tris-HCl, pH 8.0, 300 mm NaCl, 300 mm imidazole) was added to the filtrate to reach a final imidazole concentration of 10 mm. The latter preparation was loaded onto a 1-ml HisTrap HP column (GE Healthcare), which was then washed with at least 10 column volumes of buffer A (25 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10 mm imidazole). This was followed by another 10-min washing step at a higher imidazole concentration (18.7 mm) and a 20-min linear imidazole gradient (18.7–300 mm) during which 1-ml fractions were collected. A flow rate of 1 ml/min was used throughout the purification procedure. Protein fractions of this and subsequent purification steps were analyzed by SDS-PAGE and Western blotting using a monoclonal primary anti-His antibody (mouse-derived, 1:1000 dilution in PBS with 0.1% (v/v) Tween 20; GE Healthcare) and a polyclonal red fluorescent secondary antibody (goat-derived, 1:5000–1:10,000 dilution in ODYSSEY blocking buffer; Westburg, Leusden, Netherlands). Fractions containing the protein of interest in high concentration and high purity were pooled and desalted on 2 HiTrap desalting columns (2 × 5 ml, GE Healthcare) connected in series using buffer C (20 mm Tris-HCl, pH 7.5, 25 mm NaCl) at a flow rate of 2.5 ml/min.
Recombinant His-tagged Ser3 and Ser33 proteins were purified further by anion exchange chromatography on a 1-ml HiTrap Q HP column (GE Healthcare). The desalted protein preparations were diluted 3.5-fold with buffer D (25 mm Hepes, pH 7.1, 1 mm DTT) and loaded onto the column equilibrated with this same buffer. After washing with 4 ml of buffer D and then 4 ml of the same buffer containing 20 mm NaCl, proteins were eluted with a linear NaCl gradient (20–750 mm) over 20 min. A flow rate of 1 ml/min was applied throughout the purification procedure.
Protein concentrations were estimated by measuring A280 with a Nanodrop 200C spectrophotometer (Thermo Fisher Scientific). Protein purity was estimated based on band intensity of the proteins of interest compared with the sum of the intensities of all the bands detected in the corresponding lanes in SDS-polyacrylamide gels. Analysis was done with the ImageJ software (version 1.47v). Purified protein fractions were stored at −80 °C in the presence of 10% glycerol (Dld2) and additionally 100 μg/ml BSA (Dld3, Ser3, and Ser33).
Spectral Characterization of Dld2 and Dld3
The UV-visible absorbance spectra of the purified recombinant Dld2 and Dld3 proteins were recorded in a SPECORD 210 PLUS spectrophotometer (Analytik Jena) in 2-nm steps and with a slit width of 1 nm. Absorbance spectra were determined at 30 °C for a mixture (1 ml) composed of 20 mm Tris-HCl, pH 8.0, 5 μm ZnCl2, and 0.7 mg of Dld2 or Dld3 contained in a Quartz cuvette, before and after addition of d-2HG at a final concentration of 200 μm. To minimize interference with oxygen, the cuvette with the reaction mixture and the substrate solution were kept under a constant flow of nitrogen for at least 2 min, and the cuvette was then sealed with parafilm for the remainder of the experiment, except during addition of the substrate.
Enzymatic Activity Assays
All enzymatic assays were performed in a TECAN M200 Pro plate reader at 30 °C. The total reaction volume for the assays was 200 μl. Polystyrene flat-bottom 96-well plates (Greiner Bio-One) and CellCarrier-96 Black plates with clear bottoms (PerkinElmer Life Sciences) were used for spectrophotometric and fluorometric assays, respectively. The light path length (0.56–0.58 cm depending on the assay) was determined according to the manufacturer's instructions (TECAN) and used for activity calculations. Reactions were started by addition of the enzyme, and control reactions without substrate were run for background correction. The kinetic constants for each enzyme were calculated, unless otherwise indicated, from at least three independent saturation curves using nonlinear regression fitting in the GraphPad Prism Software (version 6.05).
The d-2HG dehydrogenase activity of Dld2 and Dld3 was assayed by monitoring the reduction of the artificial electron acceptor 2,6-dichlorophenol indophenol (DCIP) spectrophotometrically at 600 nm. The reaction mixture (200 μl total volume) contained 50 mm Tris-HCl, pH 8.0, 40 μm DCIP, 100 μg/ml BSA, 5 μm ZnCl2, and 500 μm d-2HG, unless otherwise indicated. We determined the extinction coefficient of DCIP at 600 nm to be 19,310 m−1 cm−1 under our assay conditions, and we used this value for enzymatic activity calculations. To measure the d-2HG oxidase activity of Dld2 and Dld3, DCIP was omitted from the reaction mixture described above and 0.4 mm o-dianisidine, 0.5 mm NaN3, and 2 units/ml horseradish peroxidase (Sigma) were added (22, 31). The expected H2O2 formation was determined spectrophotometrically at 440 nm and an extinction coefficient of 11,300 m−1 cm−1 for o-dianisidine was used to calculate d-2HG oxidase activity.
The transhydrogenase activity of Dld2 and Dld3 was measured by incubating the enzymes for 10 min with d-2HG in a reaction mixture containing 50 mm Tris-HCl, pH 8.0, 100 μg/ml BSA, 5 μm ZnCl2, and 2 mm pyruvate, unless otherwise indicated. Reactions were stopped by heating for 3 min at 95 °C. The samples were centrifuged for 10 min at 16,000 × g and 4 °C, and the d-lactate concentration in the supernatant was determined by the enzymatic spectrophotometric assay described above using d-LDH.
The α-ketoglutarate reductase activity of Ser3 and Ser33 was assayed by monitoring the oxidation of NADH spectrophotometrically at 340 nm. The reaction mixture contained 45 mm Hepes, pH 7.4, 1 mm DTT, 0.25 mm NADH, and 1.6–3.3 μg of purified Ser3 or Ser33. For activity calculations, an extinction coefficient of 6220 m−1 cm−1 for NADH was used.
The 3-phosphoglycerate dehydrogenase activity of Ser3 and Ser33 was assayed by measuring the formation of NADH over time by fluorescence, with excitation at 340 nm and emission at 460 nm. The reaction mixture contained 50 mm Tris-HCl, pH 8.0, 100 mm hydrazine sulfate, 0.9 mm NAD+, and 1 mm DTT. The substrate (3-phosphoglycerate) concentration was varied from 0 to 100 μm to determine kinetic parameters. Activity calculations were based on a standard curve allowing conversion of fluorescence units into NADH concentration. All measurements on which these calculations were based were in the linear range (0–50 μm) of the standard curve.
Results
Post-duplication Yeast Species Encode Two Putative d-2HG Dehydrogenase Paralogs
BLAST searches (using the blastp algorithm) with the human d- and l-2HG dehydrogenases (NCBI reference sequences NP_689996.4 and NP_079160.1 were used as queries for D2HGDH and L2HGDH, respectively) against the S. cerevisiae S288c genome identified two putative homologs of D2HGDH, namely Dld2 (NP_010103.1; 53% amino acid sequence identity and E-value = 9e-177) and Dld3 (NP_010843.1; 47% amino acid sequence identity and E-value = 5e-152) but did not identify any L2HGDH homolog in this yeast species. Other budding yeasts (e.g. Zygosaccharomyces rouxii, Candida albicans, and Yarrowia lipolytica) encode a protein with about 30% amino acid sequence identity with human L2HGDH. The S. cerevisiae Dld1 protein (NP_010107.1) also aligned with the human D2HGDH protein, but with a much lower score (28% amino acid sequence identity and E-value = 8e-45). A multiple sequence alignment of D2HGDH and related sequences, including also Dld1, is shown in supplemental Fig. S1.
The mammalian D2HGDH enzyme is a mitochondrial protein (22), and as expected, the generally poorly conserved predicted N-terminal mitochondrial targeting sequences (MTS) of the mammalian proteins did not align with the N terminus of any of the S. cerevisiae or other non-mammalian sequences (supplemental Fig. S1). However, the S. cerevisiae Dld2 protein also localizes to the mitochondria (32, 33), in agreement with the presence of an N-terminal MTS (predicted by the TargetP program (34)), whereas the Dld3 protein lacks an MTS and displays cytosolic localization (see also supplemental Fig. S1) (32, 33). Although Dld1, Dld2, and Dld3 are all currently annotated as d-lactate dehydrogenases in the Saccharomyces Genome Database, only Dld1 seems to be required for utilization of d-lactate in S. cerevisiae. Indeed, as shown previously by others and as confirmed by us during this study (data not shown) (35), dld1Δ single knock-out cells cannot grow in a medium containing d-lactate as the sole carbon source, indicating that the physiological role of Dld2 and Dld3 does not consist in the catabolism of d-lactate. In agreement with these observations and with the above BLAST analyses, a phylogenomic study of members of the FAD-binding oxidoreductase/transferase type 4 family, which includes the yeast Dld1, Dld2, and Dld3 proteins, revealed that yeast Dld1 sequences and yeast Dld2 and Dld3 sequences belong to two divergent phylogroups (36). The Dld1 phylogroup showed a close phylogenetic relationship to animal and plant d-lactate dehydrogenases, whereas the Dld2/3 phylogroup displayed a strong phylogenetic connection to animal and plant d-2HG dehydrogenases.
Here, we further analyzed the Dld2/Dld3 phylogroup within the Hemiascomycetes class. A Neighbor-Joining phylogenetic tree was built based on Dld2- and Dld3-like protein sequences of 20 Hemiascomycetes species (and one Schizosaccharomyces pombe sequence). As shown in Fig. 1, two groups can clearly be distinguished in the generated tree as follows: a large group containing sequences more closely related to S. cerevisiae DLD2 and a smaller group containing sequences with higher similarity to S. cerevisiae DLD3. With the exception of S. pombe, which would be expected to appear as an outgroup, but is branched to Y. lipolytica, the phylogeny observed based on the Dld2 sequences follows the general Hemiascomycetes phylogeny with the distinction into three main groups: the Saccharomycetaceae, the CTG group, and the Dipodascaceae (37, 38). A phylogenetic tree with a highly similar topology was generated when the Maximum-Likelihood method was used (data not shown). A whole genome duplication event occurred during the evolution of the Saccharomycetaceae family (39). Our phylogenetic analysis indicates that only post-duplication species possess a DLD3 gene in addition to the DLD2 gene. Interestingly, the only post-duplication strain among the ones analyzed that does not encode the Dld3 protein is the pathogenic strain Candida glabrata.
FIGURE 1.

Sequence evolution of yeast d-2-hydroxyglutarate dehydrogenase candidate proteins. Phylogenetic tree obtained by Neighbor-Joining analysis of Dld2- and Dld3-like protein sequences of Hemiascomycetes yeasts and the fission yeast Schizosaccharomyces pombe (green background). Numbers represent the bootstrap values from 1000 replicates. The protein identifiers of the sequences used for generating the phylogenetic tree are indicated in parentheses. The protein sequences were all extracted from the NCBI protein database, except for those indicated by an asterisk (Saccharomyces paradoxus, Saccharomyces mikatae, and Saccharomyces uvarum), which were retrieved from the Saccharomyces Genome Database. The three main groups that are commonly distinguished within the Hemiascomycetes species are highlighted by colored backgrounds (red, Saccharomycetaceae; orange, CTG group; blue, Dipodascaceae). Within the Saccharomycetaceae group, two subgroups are further distinguished here as follows: the pre-duplication or protoploid species (light red background) and the post-duplication species (dark red background). The Whole Genome Duplication occurrence is indicated by a red star.
Analysis of the syntenic context of the DLD2 gene using the Yeast Gene Order Browser (40) shows that the gene order around DLD2 is highly conserved across post-duplication species as well as protoploid (or pre-duplication) species. By contrast, no synteny was found for the DLD3 gene. The latter is localized in the subtelomeric region (at 16 kb from the telomere) of the left arm of chromosome V in S. cerevisiae. Genes in subtelomeric regions are evolving faster than the rest of the genome (41) leading to frequent duplication or gene loss events, notably in the case of large gene families such as the PAU, HXT, or MAL gene families (42–44). To quantify the selective pressure on the DLD3 gene, we compared the rate of substitutions at non-synonymous sites (dN) and at synonymous sites (dS) based on pairwise alignments of nucleic acid sequences from 40 S. cerevisiae species. The calculated dN/dS ratios indicate that, despite its subtelomeric localization, the DLD3 gene remains under a strong purifying selection, with a mean dN/dS ratio (0.12) that is only slightly higher than those for the DLD1 and DLD2 genes (0.036 and 0.037, respectively).
Detection of d-2HG, and Not l-2HG, in Wild-type Yeast and Deletion Mutants of d-2HG Dehydrogenase Candidate Genes
On the experimental level, our first aim was to confirm the presence of 2-hydroxyglutarate in a wild-type lab yeast strain (S. cerevisiae BY4741) and to determine the configuration of the detected metabolite. We therefore prepared intracellular metabolite extracts and filtered culture supernatants at different growth stages (mid-exponential and early stationary phase) and derivatized the samples with a chiral compound (diacetyl-l-tartaric anhydride) to achieve separation of the l- and d-2-hydroxyglutarate enantiomers on a non-chiral reverse phase LC column. Using a sensitive LC-MS/MS-based MRM method (3), d-2HG, but not l-2HG, could be detected in the cell extracts (Fig. 2A, left panel) as well as in the spent medium (data not shown) of our wild-type strain. Based on the above bioinformatics analyses, we also measured d-2HG and l-2HG levels in deletion mutants of the DLD1, DLD2, and DLD3 genes. As for the wild-type strain, d-2HG could be detected in all of these strains, but l-2HG levels were below the quantification limit (0.5 μm for l-2HG and 0.1 μm for d-2HG) in all of the analyzed samples.
FIGURE 2.
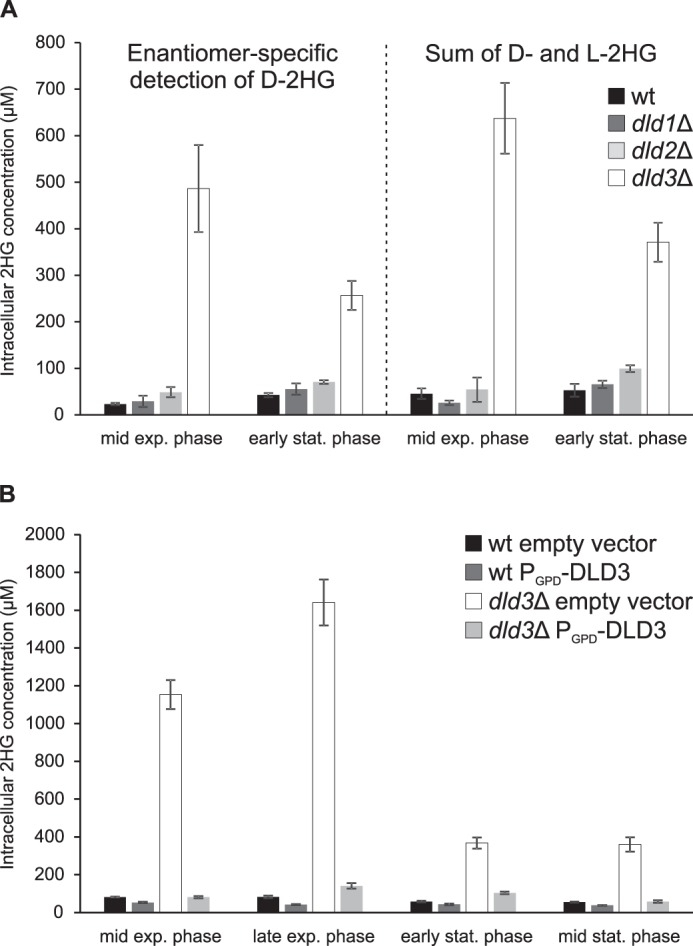
2-Hydroxyglutarate levels in wild-type, dld1Δ, dld2Δ, and dld3Δ deletion strains. The indicated yeast strains were cultivated in minimal defined medium with 1% glucose, and intracellular metabolites were extracted at different stages of growth. A, left panel, 2HG was measured by an MRM-based LC-MS/MS method allowing for differential quantification of l-2HG and d-2HG. Only d-2HG concentrations are shown as l-2HG levels were below the quantification limit in all the samples measured. Right panel, total 2HG was quantified using an HRAM LC-MS method in which d-2HG and l-2HG cannot be distinguished. B, the latter method was also used to measure 2HG in wild-type or dld3Δ mutant strains transformed with an “empty vector” (PGPD-ccdB) or a plasmid expressing DLD3 from a strong constitutive promoter (PGPD-DLD3). For this rescue experiment, uracil was omitted from our standard cultivation medium for plasmid maintenance. Means and standard deviations of three biological replicates are shown. exp., exponential; stat., stationary.
Similar low intracellular levels (ranging from 20 to 60 μm) of d-2HG were measured in the wild-type and dld1Δ strains, and up to two times higher levels were found in the dld2Δ mutant (Fig. 2A, left panel). Interestingly, up to 20-fold higher concentrations were detected in the dld3Δ mutant (about 0.5 mm at mid-exponential phase) than in the wild-type strain. Increased d-2HG levels were also found in the extracellular medium for the dld3Δ mutant as compared with the wild-type strain. As at none of the time points and in none of the yeast strains tested we could detect l-2HG, we did not use the differential LC-MS/MS assay for the subsequent experiments described in this study. Instead, we measured total 2HG levels (which we assume to be exclusively d-2HG in our experimental system for the rest of this study) using a derivatization-free HILIC-based targeted high-resolution accurate-mass (HRAM) LC-MS method that allowed for the additional quantification of all amino acids and other physiological organic acids in the same samples. As shown in Fig. 2A, highly similar results were obtained when quantifying intracellular d-2HG after derivatization by the differential LC-MS/MS assay or total 2HG (sum of d-2HG and l-2HG) without derivatization by the HRAM LC-MS method.
Constitutive expression of DLD3 in the dld3Δ mutant background by transformation of a low copy number centromere-based expression plasmid decreased d-2HG levels almost back to wild-type levels (Fig. 2B), confirming that d-2HG accumulation is caused specifically by the DLD3 gene deletion. When DLD3 was overexpressed using this same plasmid in the wild-type strain, the basal levels of d-2HG present in this strain were further decreased by around 50% (Fig. 2B). Although all of the above observations were consistent with the in silico predictions based on amino acid sequence analyses (putative d-2HG dehydrogenase activity for Dld2 and Dld3 but not for Dld1), the large differences in d-2HG levels observed between the dld2Δ and dld3Δ mutants were surprising and suggested a major role for Dld3 in d-2HG degradation in the living yeast cell.
Effect of DLD1, DLD2, and DLD3 Deletion on Growth, Glucose Consumption, and on the Intracellular and Extracellular Concentrations of d-2HG and Other Metabolites
To investigate how DLD1, DLD2, and DLD3 deficiency impacts growth and primary metabolism more generally, we monitored cell concentration, extracellular glucose, and ethanol (data not shown) levels as well as intra- and extracellular concentrations of d-2HG and other metabolites (organic acids and amino acids listed in supplemental Table S4) within a 9-h period starting 15 h after inoculation of the cultures (Fig. 3). None of the studied gene deletions affected cell growth or glucose uptake (Fig. 3, A and B) in our standard cultivation medium (minimal defined medium with 10 g/liter glucose). For all strains (wild-type and dld deletion strains), intracellular d-2HG concentrations increased during the exponential growth phase and then decreased during the stationary phase (Figs. 2B and 3C). Although the dld3Δ mutant showed highly increased intra- and extracellular d-2HG levels throughout the assay period, the concentration of this metabolite in the dld1Δ and dld2Δ mutants was not significantly different from the wild-type strain, for most of the time points measured (Fig. 3, C and D). During the exponential growth, intracellular d-2HG concentrations of up to 1.1 mm were measured in the dld3Δ strain, whereas in the wild-type, dld1Δ, and dld2Δ strains the concentrations did not exceed 0.12 mm. Although the extracellular concentration of d-2HG stabilized after 19 h of cultivation, a rapid decrease in the intracellular d-2HG concentration from 1.1 to 0.37 mm was measured upon total glucose consumption in the dld3Δ mutant, suggesting the existence of a d-2HG-induced or glucose-repressed alternative secretion or catabolic mechanism for d-2HG. Consistent with a role of Dld3 in the oxidation of d-2HG to α-ketoglutarate, decreased levels of the latter metabolite were measured in the extracellular medium of dld3Δ mutant cultivations (supplemental Fig. S2).
FIGURE 3.

Growth and metabolic characterization of the wild-type and dld mutant strains. Cell concentration (A), extracellular glucose concentration (B), intra- and extracellular 2HG concentrations (C and D), and intra- and extracellular lactate concentrations (E and F) were measured at the indicated times during batch cultivations in minimal defined medium containing 1% glucose. For cell concentration and intracellular metabolite concentrations, means and standard deviations of three biological replicates are shown. Extracellular metabolite concentrations represent single measurements.
As expected from what is known about the function of Dld1, we found increased lactate levels, both intra- and extracellularly for the dld1Δ mutant (Fig. 3, E and F). Although the dld2Δ mutant had intracellular lactate levels similar to the wild-type strain, increased extracellular lactate levels were observed for this strain, indicating that Dld2 contributes to lactate catabolism, although to a lesser extent than Dld1. Interestingly, and in strong contrast to the dld1Δ and dld2Δ mutants, lactate levels were greatly decreased in the dld3Δ strain (Fig. 3, E and F). This result was intriguing, given the d-lactate dehydrogenase activity reported for yeast extracts overexpressing DLD3 in a cyb2Δdld1Δdld2Δdld3Δ quadruple knock-out background (CYB2 encodes an l-lactate dehydrogenase) (32); based on the latter observation, one would have expected an increase rather than a decrease in lactate levels upon deletion of the DLD3 gene. The lactate levels found in this study based on the LC-MS assay were confirmed using spectrophotometric enzymatic assays (total extracellular lactate concentrations of up to 302, 473, 439, and 55 μm were detected in wild-type, dld1Δ, dld2Δ, and dld3Δ cultivations, respectively, using the enzymatic assays). For the latter assays, specific commercial d- and l-lactate dehydrogenases were used. As expected, more than 90% of the total lactate measured for the wild-type strain corresponded to d-lactate at all time points (S. cerevisiae does normally not produce significant amounts of l-lactate (45)). The same was true for the dld1Δ and dld2Δ strains. For the dld3Δ strain, a higher proportion of the measured lactate corresponded to l-lactate (up to 29%).
In addition to the changes in d-2HG and d-lactate levels (intra- and extracellular) and in α-ketoglutarate levels (extracellular) in the dld3Δ strain compared with the wild-type strain, the extracellular succinate levels were slightly increased, and the extracellular malate, fumarate, and serine levels were slightly decreased in this strain (supplemental Fig. S2). The other organic acid and amino acid concentrations measured were not affected by the DLD3 gene deletion (supplemental Figs. S2 and S3).
DLD2 and DLD3 Act as d-2HG Dehydrogenases in Vitro
Sequence analyses performed by us (this study) and others (36, 46) as well as our above metabolite measurements in mutant yeast strains strongly suggested that the Dld2 and Dld3 proteins can oxidize d-2HG to α-ketoglutarate as reported previously for homologous proteins in rat (22) and A. thaliana (46). To characterize the enzymatic properties of the yeast proteins, we recombinantly produced N-terminally His-tagged Dld2 and Dld3 in E. coli and purified the two enzymes by nickel-affinity chromatography. First expression attempts of full-length Dld2 yielded only very low amounts of soluble protein. Expression of a shorter form of Dld2 (truncation of the 44 N-terminal amino acids predicted to correspond to the MTS by the Mitoprot tool (47)) allowed us to considerably increase the production of soluble protein and it is this N-terminally truncated form of Dld2 that we purified and characterized in this study, along with the full-length Dld3 protein. SDS-PAGE analysis showed that for both Dld3 and truncated Dld2, the one-step purification procedure yielded protein preparations that were more than 90% pure (Fig. 4).
FIGURE 4.
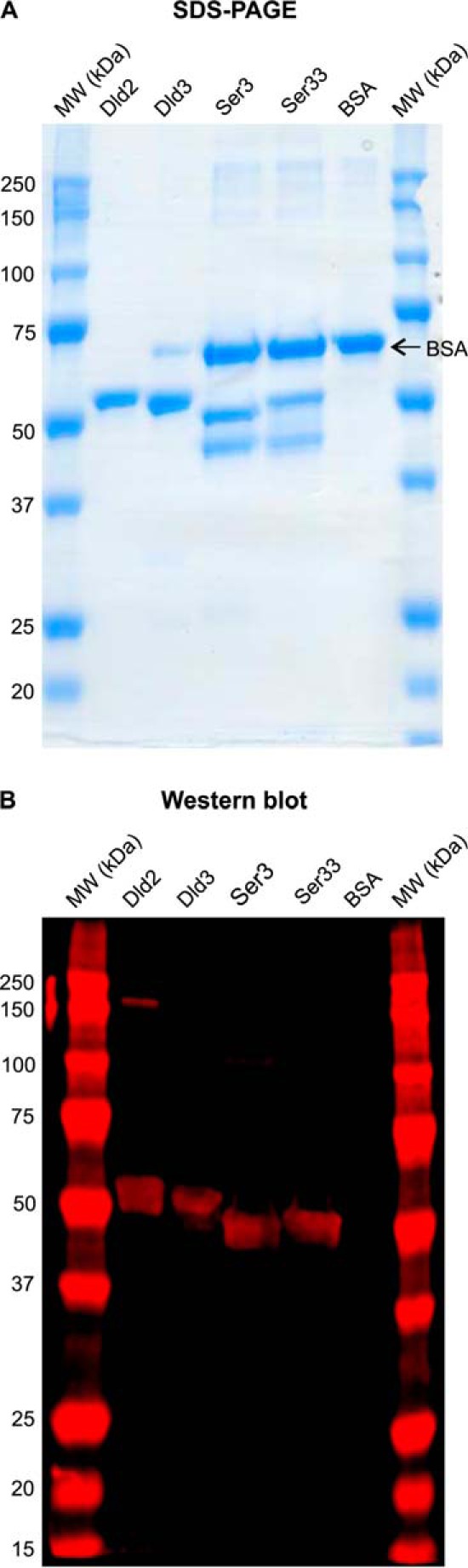
SDS-PAGE and Western blot analyses of purified recombinant Dld2, Dld3, Ser3, and Ser33. Purified preparations of recombinant Dld2 (1.2 μg of protein), Dld3 (1.2 μg of protein), Ser3 (1.0 μg of protein), and Ser33 (0.7 μg of protein) used in this study were analyzed by SDS-PAGE using Coomassie Blue staining (A) and Western blotting using an antibody directed against the N-terminal polyhistidine tag fused to each of the recombinant proteins (B). Commercial BSA (Sigma), which had been added to the Dld3, Ser3, and Ser33 preparations for improved protein preservation during storage, was also loaded alone (1.2 μg of protein) in a control lane. BSA (66.43 kDa) is therefore also present in the Dld3 lane (0.12 μg) and in the Ser3 and Ser33 lanes (1.2 μg). The expected molecular weights of the recombinant proteins are 57.64 kDa for Dld2 (truncated MTS), 58.80 kDa for Dld3, and 54.77 kDa for Ser3 and Ser33. Only the upper band of the double band revealed by Coomassie staining for the Ser3 and Ser33 preparations was detected by the anti-His antibody, indicating that the lower band corresponds to a proteolytic cleavage product lacking the N-terminal His tag.
We tested the dehydrogenase activity of recombinant Dld2 and Dld3 in the presence of the artificial electron acceptor DCIP on a range of 2-hydroxyacids at 0.5 and 5 mm (Fig. 5). For the low substrate concentration used, Dld2 showed the highest activity with d-2HG, whereas Dld3 showed the highest activity with both d-2HG and d-malate, a close structural homolog of d-2HG (four carbon backbone instead of five). Dld2 also acted on d-malate, but with a 2-fold lower activity than on d-2HG. Furthermore, Dld2 showed a low but detectable activity on l-2HG, d-lactate, and l-lactate. In addition to d-2HG and d-malate, Dld3 showed substantial activity with d-lactate and a lower activity with d-glycerate. The assays done at 5 mm substrate concentration confirmed this substrate specificity profiles of Dld2 and Dld3 and, taken together, the results indicate that Dld2 has a relatively high specificity for d-2HG, whereas Dld3 is more promiscuous, using d-2HG and d-malate as the preferred substrates and d-lactate with lower affinity. In addition, our results suggest, at first sight, that Dld3 displays a higher stereoselectivity for d-2-hydroxyacids over l-2-hydroxyacids than Dld2. However, using the differential LC-MS/MS assay, we determined that our commercial preparation of l-2HG contains around 0.3% of d-2HG (and that the commercial preparation of d-2HG contains around 1.8% of l-2HG). Combined with the kinetic properties determined below for the d-2HG dehydrogenase activity of Dld2, we can conclude that the Dld2 activity measured in the presence of l-2HG (see Fig. 5) can be fully accounted for by the low amounts of d-2HG contaminating the commercial l-2HG preparation.
FIGURE 5.

Substrate specificity of the dehydrogenase activities of Dld2 and Dld3. The dehydrogenase activity of Dld2 (A) and Dld3 (B) was assayed in the presence of the artificial electron acceptor DCIP and of the indicated 2-hydroxyacids at a final concentration of 0.5 mm (black bars) or 5 mm (white bars). Control reactions without substrate were run for background correction. Dehydrogenase activities are represented relative to the activities measured in the presence of 0.5 or 5 mm d-2HG. For Dld2, dehydrogenase activities of 0.186 ± 0.003 and 0.20 ± 0.02 μmol·min−1·mg protein−1 were measured in the presence of 0.5 and 5 mm d-2HG, respectively. For Dld3, dehydrogenase activities of 4.0 ± 0.8 and 4.2 ± 0.5 μmol·min−1·mg protein−1 were measured in the presence of 0.5 and 5 mm d-2HG, respectively. The values shown are means ± S.D. of three independent replicates. The structures of the 2-hydroxyacids tested as substrates are shown in C.
The purified Dld2 and Dld3 protein fractions showed a yellow color, suggesting the presence of a bound flavin cofactor. The identity of the prosthetic group of the A. thaliana homolog of Dld2 and Dld3 had experimentally been confirmed to correspond to FAD previously (46). The d-2HG dehydrogenase activities of Dld2 and Dld3 were not affected by the addition of NAD+, NADP+, or FMN (tested at 0.2 and 1 mm). Externally added FAD also did not affect Dld2 activity, but stimulated the Dld3 activity by almost 2-fold when older enzyme preparations that had been frozen and thawed several times were used (no effect was observed with NAD+, NADP+, or FMN). These observations, as well as the presence of a conserved FAD-binding domain in both protein sequences, strongly suggested that Dld2 and Dld3 tightly bind FAD as a cofactor, although Dld3 seemed to slowly release it upon prolonged storage or repeated freeze-thaw cycles.
The UV-visible absorbance spectra of purified Dld2 and Dld3 displayed maxima centered at 378 and 450 nm, further supporting the presence of a flavin cofactor bound to both proteins (Fig. 6). For the Dld3 protein, we detected an additional absorption maximum at 360 nm. Upon addition of d-2HG under aerobic conditions, only a very modest and transient decrease in the absorbance at 450 nm could be observed (data not shown), but a more robust and persistent decrease was detected when the reaction mixture was flushed with nitrogen prior to spectral analysis (Fig. 6, C and D). For the Dld2 protein, but not for Dld3, a decrease of absorbance was also observed for the 378-nm peak upon addition of d-2HG. Due most likely to the fact that we could not work under strictly anaerobic conditions, the absorbance spectra for both Dld2 and Dld3 reverted back to the fully oxidized spectra after more prolonged incubation times even after prior nitrogen flushing (data not shown). These observations showed that the flavin cofactor bound to Dld2 and Dld3 acts as an intermediate electron acceptor during the oxidation of d-2HG by the two enzymes, but it also indicated that electrons can be further transferred to oxygen in the absence of another final electron acceptor.
FIGURE 6.

Spectral analysis of purified recombinant Dld2 and Dld3. A 1-ml mixture containing 20 mm Tris-HCl, pH 8.0, 5 μm ZnCl2, and purified Dld2 or Dld3 (0.7 mg) was flushed for 5 min with N2 in a quartz glass cuvette. The absorbance spectra were recorded at 30 °C for both Dld2 (A) and Dld3 (B). After addition of d-2HG at a final concentration of 200 μm, the change of absorbance at 378 and 450 nm was followed over time for Dld2 (C) and Dld3 (D) until no further decrease was observed.
We could indeed detect H2O2 formation (spectrophotometrically at 440 nm) during incubation of Dld2 and Dld3 with d-2HG in the presence of o-dianisidine and horseradish peroxidase (31). This d-2HG oxidase activity of Dld2 and Dld3 (determined in the presence of 200 μm d-2HG) was, however, about 30- and 10-fold lower, respectively, than the d-2HG dehydrogenase activity determined at the same d-2HG concentration in the presence of 40 μm DCIP (data not shown).
As rat liver d-2HG dehydrogenase had previously been shown to be stimulated by bivalent metal cations (22), we tested the effect of several metal ions on the d-2HG dehydrogenase activity of Dld2 and Dld3 (Fig. 7). At a substrate concentration of 500 μm, we found that 5 μm Zn2+ increases the basal d-2HG activity of Dld2 more than three times, whereas Co2+, Mn2+, Mg2+, and Ca2+ did not stimulate the activity at this concentration (Fig. 7A). At a higher metal concentration (50 μm), the stimulatory effect of Zn2+ on Dld2 activity decreased. For Dld3, Zn2+ and Co2+ stimulated the d-2HG dehydrogenase activity to a similar extent at the low and high metal concentrations tested, whereas Mn2+, Mg2+, and Ca2+ did not significantly affect the activity (Fig. 7B). Very similar metal effects were found for both enzymes when d-2HG was added at a lower concentration of 100 μm, except that 5 μm Co2+ did not show the stimulatory effect observed for Dld3 at the higher substrate concentration (data not shown). EDTA (1 mm) inhibited the basal d-2HG activity of Dld2 and Dld3 by more than 75% (Fig. 7). Based on these results, Zn2+ was added at a final concentration of 5 μm in the reaction mixture used throughout this study for assaying the dehydrogenase activity of Dld2 or Dld3. Taken together, our observations suggest that Dld2 and Dld3 are Zn2+-dependent enzymes using tightly bound FAD as an intermediate electron acceptor to oxidize d-2HG and other d-2-hydroxyacids.
FIGURE 7.

Effect of bivalent metal ions on the d-2HG dehydrogenase activity of Dld2 and Dld3. The d-2HG dehydrogenase activities of recombinant purified Dld2 (A) and Dld3 (B) were assayed spectrophotometrically in the absence or presence of 5 or 50 μm of the chloride salts of the indicated metal ions and with 500 μm d-2HG as substrate. The effect of EDTA at a final concentration of 1 mm was also tested. Control reactions without substrate were run for background correction. The values shown correspond to means ± S.D. from three independent replicates.
Given the current annotations of Dld2 and Dld3 as d-lactate dehydrogenases based on the observations by Chelstowska et al. (32) and our metabolomic characterization of dld2Δ and dld3Δ mutant strains (this study), we analyzed the kinetic properties of the Dld2 and Dld3 dehydrogenase activities more in detail for the d-lactate and d-2HG substrates. As shown in Table 1, both Dld2 and Dld3 have a higher affinity for d-2HG than for d-lactate. This difference is, however, much more pronounced for Dld2 than for Dld3. In contrast, both enzymes show a higher turnover number with d-lactate than with d-2HG. The catalytic efficiencies show that Dld2 is a 28-fold better d-2HG dehydrogenase than a d-lactate dehydrogenase, whereas this difference is much less pronounced for Dld3 (1.6-fold higher catalytic efficiency for d-2HG than for d-lactate). Considering only the activity on d-2HG, Dld2 appears like a high affinity enzyme, whereas Dld3 has a more than 30-fold higher turnover number than Dld2 with this substrate. The low affinity of Dld2 for d-lactate (Km >5 mm) indicates that its d-lactate dehydrogenase activity may only be of minor physiological relevance. From the kinetic properties of Dld3, it seems that both d-2HG and d-lactate represent plausible physiological substrates. The catalytic efficiencies of the d-lactate dehydrogenase activities determined here for Dld2 and Dld3 are more than 1500- and 10-fold lower than the catalytic efficiency determined previously for Dld1 (Table 1) (48), further supporting that Dld1 is the major yeast d-lactate dehydrogenase.
TABLE 1.
Kinetic properties of the d-2HG and d-lactate dehydrogenase activities of Dld2 and Dld3
The dehydrogenase activities of recombinant purified Dld2 and Dld3 proteins were measured spectrophotometrically in the presence of the artificial electron acceptor DCIP by monitoring the change in absorbance at 600 nm. Substrate concentrations were varied between 0 and 1.2 mm for d-2HG and up to 22.5 mm for d-lactate. Absorbance changes measured in the presence of substrate were corrected by subtracting absorbance changes measured in the absence of substrate. Kinetic parameters were estimated using non-linear regression fitting in the GraphPad Prism software (version 6.05). The values shown are means ± S.D. obtained from three independent saturation curves.
| Km | kcat | kcat/Km | |
|---|---|---|---|
| μm | s−1 | s−1 m−1 | |
| d-2HG | |||
| Dld2 | 28 ± 8 | 0.18 ± 0.03 | 7.0 ± 1.2 × 103 |
| Dld3 | 130 ± 9 | 6.6 ± 0.5 | 5.0 ± 0.2 × 104 |
| d-Lactate | |||
| Dld2 | 5215 ± 231 | 1.31 ± 0.03 | 2.5 ± 0.1 × 102 |
| Dld3 | 533 ± 138 | 16.5 ± 1.4 | 3.2 ± 0.8 × 104 |
| Dld1a | 4450 | 1820 | 4.1 × 105 |
a The kinetic properties shown for Dld1 were adapted from Gregolin and Singer (48), using a predicted molecular mass for Dld1 of 65.29 kDa and the Km and Vmax values reported in this study using a DCIP-based spectrophotometric assay. The reaction mixture used by Gregolin and Singer (48) contained 50 mm imidazole buffer, pH 7.5, 7 μm DCIP, and 2.2 mm phenazine methosulfate.
To determine the identity of the product formed from d-2HG by Dld2 and Dld3, the purified enzymes were incubated in the presence of DCIP and 50 μm d-2HG, and the reaction mixtures were analyzed at different time points by HRAM LC-MS. We observed that for both Dld2 and Dld3, the d-2HG consumption over time correlated with the production of a compound that had the same retention time and the same accurate mass as α-ketoglutarate (Fig. 8). When the same incubations were conducted in the absence of enzyme, no α-ketoglutarate formation was observed.
FIGURE 8.

Dld2 and Dld3 convert d-2HG into α-ketoglutarate in the presence of DCIP. A reaction mixture containing 50 mm Tris-HCl, pH 8.0, 120 μm DCIP, 100 μg/ml BSA, 5 μm ZnCl2, and 50 μm d-2HG was incubated at 30 °C in the absence or presence of recombinant purified Dld2 (6.6 μg/ml) or Dld3 (0.25 μg/ml). Aliquots were heat-inactivated (3 min at 95 °C) after a reaction time of 0, 20, 40, or 60 min and analyzed with a targeted quantitative HRAM LC-MS method. Substrate and product of the enzymatic reaction were identified by comparison of the retention time (A) and the mass spectrum (B) with a mixed standard containing 50 μm d-2HG and 50 μm α-ketoglutarate. Exemplary extracted ion chromatograms and mass spectra are shown after a reaction time of 60 min for both proteins. Considering theoretical masses (82) of 145.01370 for α-ketoglutarate and 147.02935 for d-2HG, the error on the detected masses in the standard as well as in the experimental samples corresponded to 6–7 ppm. Presumably due to ion suppression effects induced by the artificial electron acceptor DCIP, absolute quantifications of d-2HG and α-ketoglutarate were not possible in these experiments. For this reason, peak areas (C) are shown instead of concentrations. The values represented correspond to single point measurements. α-KG, α-ketoglutarate; cps, counts/s; Enz, enzyme; RT, retention time; t, time.
Dld3 Is a Hydroxyacid-Oxoacid Transhydrogenase Using Pyruvate as a Hydrogen Acceptor
Given our intriguing observation of greatly decreased d-lactate concentrations in the dld3Δ mutant strain, we tested whether the cytosolic Dld3 protein could actually use pyruvate as the final hydrogen acceptor during the oxidation of d-2HG to α-ketoglutarate and thus act as a transhydrogenase. Purified Dld3 was incubated in the presence of d-2HG and pyruvate, and after heat inactivation of the enzyme, d-lactate was assayed spectrophotometrically in the reaction mixture using d-LDH from L. leichmannii. Under these conditions, we could indeed detect a formation of d-lactate that was dependent on the presence of pyruvate (as well as Dld3 and d-2HG). This pyruvate-dependent d-lactate formation increased linearly with time and with the concentration of Dld3 protein (data not shown). Similarly to the d-2HG dehydrogenase activity measured with Dld3 in the presence of DCIP, the transhydrogenase activity of Dld3 assayed in the presence of pyruvate was stimulated by low concentrations of Zn2+ and to a lesser extent by Co2+ and Mn2+ (data not shown). Subsequent transhydrogenase assays were therefore performed systematically in the presence of 5 μm Zn2+.
Using our targeted HRAM LC-MS method, we could show that the consumption of d-2HG in the presence of pyruvate was accompanied by a stoichiometric formation of two compounds that were identified as α-ketoglutarate and lactate based on exact mass and identical retention time as standards (Fig. 9A). We also found that incubation of Dld3 in the presence of α-ketoglutarate and d-lactate, but not l-lactate, leads to the formation of pyruvate by coupling this reaction to l-LDH in the presence of NADH and monitoring the decrease of A340 (data not shown). Given the reduction of DCIP observed when incubating Dld3 in the presence of d-malate, we tested in addition whether oxaloacetate could be used as a hydrogen acceptor by Dld3 instead of pyruvate to oxidize d-2HG to α-ketoglutarate. We could indeed detect formation of α-ketoglutarate and malate after incubation of d-2HG and oxaloacetate in the presence of Dld3 using the targeted HRAM LC-MS method (Fig. 9B). Possibly because of spontaneous breakdown of pyruvate and more importantly oxaloacetate, the concentrations of lactate and malate produced during the transhydrogenase reaction with pyruvate and oxaloacetate, respectively, decreased after longer incubation times, while the α-ketoglutarate concentration reached a plateau in both reactions (Fig. 9).
FIGURE 9.
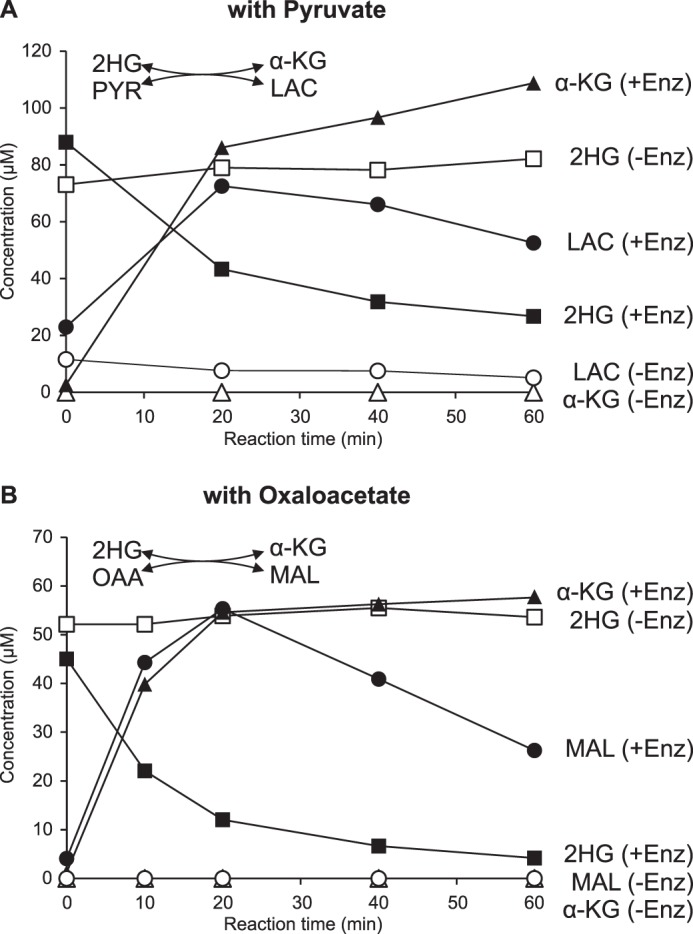
Dld3 can use pyruvate and oxaloacetate as electron acceptors and acts as a transhydrogenase. A reaction mixture containing 50 mm Tris-HCl, pH 8.0, 100 μg/ml BSA, 5 μm ZnCl2, and either 100 μm d-2HG and 100 μm pyruvate (A) or 50 μm d-2HG and 100 μm oxaloacetate (B) was incubated at 30 °C in the absence or presence of recombinant purified Dld3. Aliquots were heat-inactivated at the indicated times and analyzed with a targeted quantitative HRAM LC-MS method. α-KG, α-ketoglutarate; Enz, enzyme; LAC, lactate; MAL, malate; OAA, oxaloacetate; PYR, pyruvate.
With Dld2 we also detected a pyruvate-dependent formation of d-lactate in the presence of d-2HG. We determined the kinetic properties for the d-2HG-pyruvate transhydrogenase activity of Dld2 and Dld3 (Table 2). While the Kmvalue of Dld2 and Dld3 for d-2HG was similar (74 and 111 μm for Dld2 and Dld3, respectively; Table 2), Dld3 displayed a more than 30-fold higher turnover rate for the transhydrogenase reaction than Dld2. Also, although for Dld3 we could determine a Km of around 450 μm for pyruvate, the transhydrogenase activity of Dld2 did not show saturation when keeping the d-2HG concentration fixed at 2 mm and increasing the pyruvate concentration up to 1.5 mm. These results obtained in vitro with the recombinant purified Dld2 and Dld3 enzymes together with the metabolite levels measured in the dld2Δ and dld3Δ mutants suggested that, physiologically, Dld3, but not Dld2, functions as a cytosolic transhydrogenase that oxidizes d-2HG to α-ketoglutarate in the first half-reaction and reduces pyruvate to d-lactate in the second half-reaction. The low affinity of Dld2 for pyruvate and its mitochondrial localization favor the hypothesis that this enzyme functions as a d-2HG dehydrogenase that transfers electrons to the respiratory chain via the electron transfer flavoprotein (ETF), as it was suggested for the mammalian D2HGDH (22). Another possibility, which would depend on a targeting of Dld2 to the mitochondrial intermembrane space, is that the enzyme transfers electrons directly to cytochrome c.
TABLE 2.
Kinetic properties of the d-2HG-pyruvate transhydrogenase activity of Dld2 and Dld3
Recombinant purified Dld2 or Dld3 proteins were incubated for 10 min at 30 °C in a reaction mixture containing d-2HG and pyruvate. One substrate concentration was varied between 0 and 2 mm, and the other one was kept constant at 2 mm. After heat-inactivation of the enzyme, d-lactate was assayed spectrophotometrically by using a commercial d-LDH. The values obtained were corrected for control reactions run without substrate. Kinetic parameters were estimated using non-linear regression fitting in the GraphPad Prism software (version 6.05). The values shown are means ± S.D. obtained from three independent saturation curves. For Dld2, kinetic parameters for pyruvate as the varied substrate were not determined (ND) as saturation was not reached with the range of pyruvate concentrations tested.
| Km | kcat | kcat/Km | |
|---|---|---|---|
| μm | s−1 | s−1 m−1 | |
| d-2HG | |||
| Dld2 | 74 ± 14 | 0.11 ± 0.00 | 1.5 ± 0.3 × 103 |
| Dld3 | 111 ± 14 | 4.0 ± 0.4 | 3.6 ± 0.5 × 104 |
| Pyruvate | |||
| Dld2 | ND | ND | ND |
| Dld3 | 450 ± 180 | 4.9 ± 1.1 | 1.2 ± 0.2 × 104 |
Effect of Modulating Intracellular Yeast Phosphoglycerate Dehydrogenase Activity on d-2HG Levels
The results obtained with the dld3Δ mutant showed that S. cerevisiae is producing significant amounts of d-2HG under standard yeast cultivation conditions. It has previously been shown that E. coli as well as human phosphoglycerate dehydrogenase can reduce α-ketoglutarate (a close structural analog of 3-phosphohydroxypyruvate) to d-2HG in addition to catalyzing their main physiological reaction, which consists of the conversion of 3-phosphoglycerate to 3-phosphohydroxypyruvate in the first step of serine biosynthesis (14, 49). Furthermore, in S. cerevisiae, the gene expression profiles of DLD3 and SER33, one of the two yeast phosphoglycerate dehydrogenase isoforms, are highly correlated across several datasets (SPELL version 2.0.3 (50)). These observations, in addition to the cytosolic localization of Ser3, Ser33, and Dld3, qualified yeast phosphoglycerate dehydrogenases as strong candidates for contributing to the observed d-2HG formation. To test this hypothesis, we first overexpressed the SER3 and SER33 genes in both wild-type and dld3Δ background using a plasmid allowing for constitutive expression (glyceraldehyde-3-phosphate dehydrogenase promoter) and for selection through the use of a uracil drop-out medium (plasmidic URA3 gene compensating for the uracil auxotrophy of the parental strains). As shown in Fig. 10A, SER3 overexpression led to a more than 2-fold increase in intracellular d-2HG levels, independently of the genetic background. A smaller, but still significant increase, was observed upon SER33 overexpression. As the effects of SER3 or SER33 overexpression on intracellular serine (end product of the main pathway initiated by the expression products of these two genes) levels (Fig. 10B) virtually mirrored those on d-2HG levels, the differential effect of the two genes may be a consequence of differing expression levels from the plasmid constructs rather than reflecting different enzyme properties. The observation that the relative effects of SER3 or SER33 overexpression on d-2HG levels were similar in the wild-type and in the dld3Δ backgrounds suggests that the capacity of the Dld3 enzyme is limited in wild-type cells under the cultivation conditions used.
FIGURE 10.
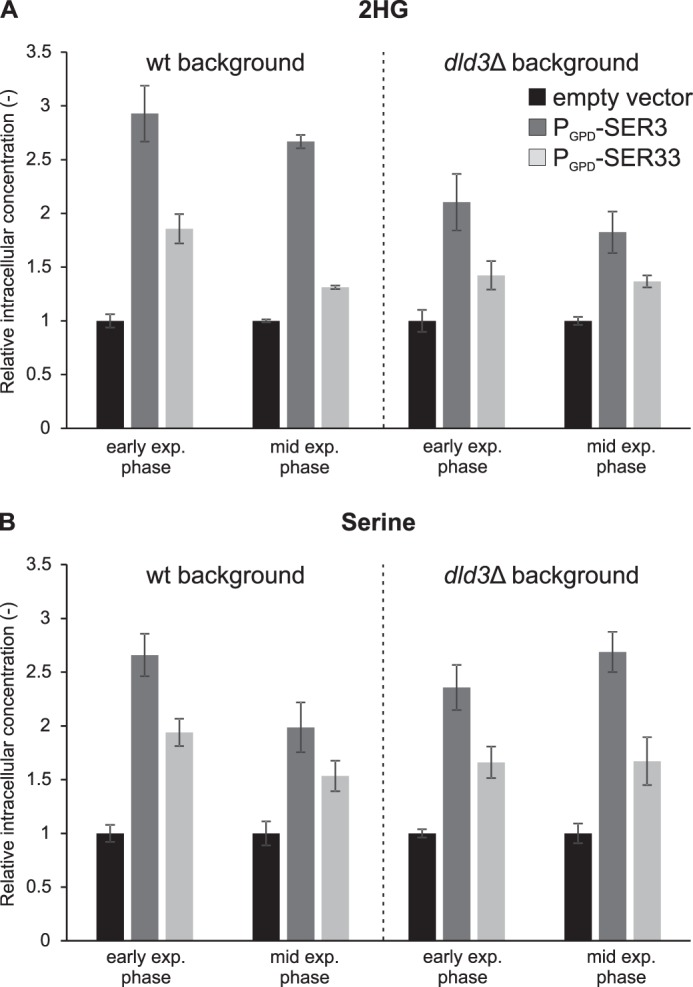
2HG and serine levels in yeast strains overexpressing SER3 or SER33. SER3 and SER33 were overexpressed in wild-type and dld3Δ mutant strains from a low copy number plasmid under the control of the GPD promoter (PGPD-SER3 and PGPD-SER33, respectively). The overexpression strains were cultivated along with control strains transformed with an “empty vector” (PGPD-ccdB) in minimal medium with 1% glucose and without uracil. Metabolites were extracted from yeast cells at different stages of growth and 2HG (A) and serine (B) concentrations were measured by HRAM LC-MS. Intracellular concentrations normalized to the concentrations measured in the respective control strains are shown. Values are means ± S.D. of three biological replicates. Intracellular 2HG levels of the controls were, in the wild-type background, 64 ± 5 and 64 ± 3 μm at early and mid-exponential phase, respectively, and in the dld3Δ mutant background 392 ± 28 and 584 ± 18 μm at early and mid-exponential phase, respectively. Intracellular serine levels of the controls were, in the wild-type background, 1438 ± 202 and 1382 ± 73 μm at early and mid-exponential phase, respectively, and in the dld3Δ mutant background, 1001 ± 28 and 1191 ± 61 μm at early and mid-exponential phase, respectively. exp., exponential.
We also did the reverse experiment and tested the effect of SER3 and/or SER33 gene deletion on d-2HG levels. Individual deletion of SER3 or SER33 led to an ∼40% decrease in intracellular d-2HG levels as compared with the wild-type strain when metabolites were extracted at mid-exponential phase (Fig. 11A). A similar decrease in intracellular serine levels was observed in those mutants (Fig. 11B), validating the gene deletions through impairment of the main pathway associated with these genes. A less pronounced reduction in d-2HG levels (10–15%) was found in the ser3Δ and ser33Δ single mutants when metabolites were extracted at later growth stages (Fig. 11A). As expected, the ser3Δser33Δ double knock-out strain did not grow in a medium lacking serine. Serine supplementation rescued growth but led to much lower d-2HG levels in both the wild-type and mutant strains. It should be noted that, due to different auxotrophies in the ser3Δser33Δ strain (isogenic to BY4742) as in the other strains used in this study (isogenic to BY4741), lysine had to be supplemented in addition to serine to the medium used for the double knock-out experiment shown in Fig. 11A (right panel). Despite the decreased d-2HG levels measured under these conditions in the wild-type strain, we could show a more than 80% reduction in d-2HG levels in the ser3Δser33Δ mutant as compared with the wild-type strain at mid-exponential phase (Fig. 11A). As for the single mutants, the relative decrease in d-2HG levels became less pronounced at later growth stages, indicating that this trend is not due, in the single mutants, to compensatory effects by the non-deleted paralogous enzyme. The more pronounced decrease in d-2HG concentration observed in the double knock-out strain as compared with the single ser3Δ and ser33Δ mutants supports that both genes contribute to d-2HG formation. Our results suggest that under the conditions tested, Ser3 and Ser33 are major sources for d-2HG formation in S. cerevisiae during exponential growth on glucose but that alternative sources, whose contribution becomes more important under stationary phase conditions, exist. The greatly decreased d-2HG levels in the wild-type strain grown under serine supplementation could be due to the previously reported inhibitory effect of serine on yeast phosphoglycerate dehydrogenase (51), representing therefore an additional argument in favor of this enzyme being a major source of d-2HG in yeast.
FIGURE 11.
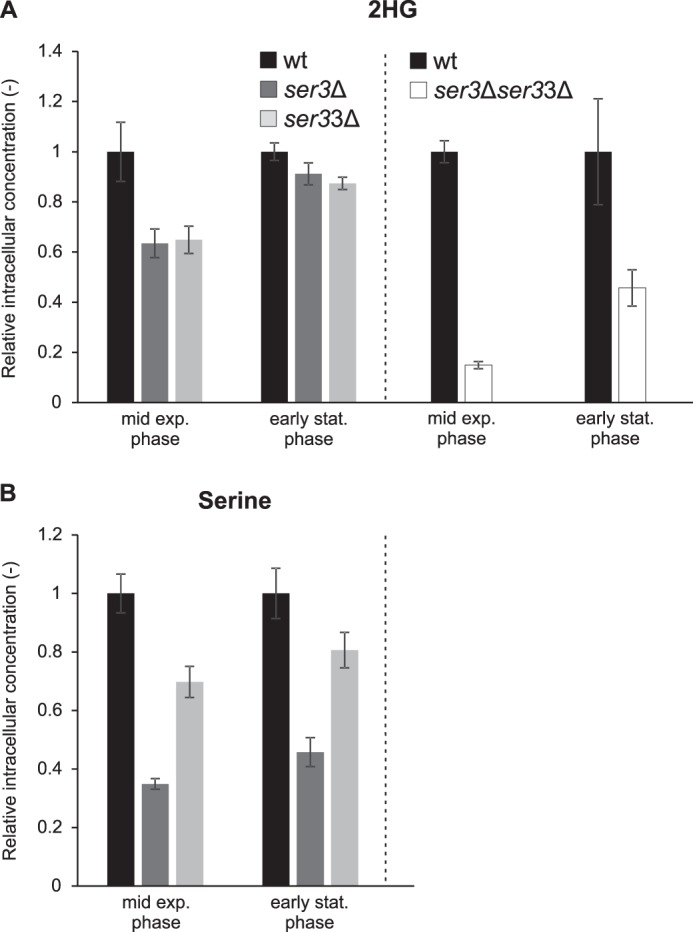
2HG and serine levels in yeast deletion strains of SER3 and/or SER33. Single knock-out mutants (ser3Δ and ser33Δ) and the wild-type control strain were cultivated in minimal defined medium with 1% glucose, while for the double knock-out strain (ser3Δser33Δ) and the wild-type control strain this medium was supplemented with serine and lysine at a final concentration of 240 mg/liter each. All the strains were isogenic to BY4741, except for the double knock-out strain, which was isogenic to BY4742. Metabolites were extracted at the indicated growth stages, and 2HG (A) and serine (B) concentrations were measured by targeted HRAM LC-MS. Intracellular concentrations normalized to the concentrations measured in the control wild-type strain are shown. Values are means ± S.D. of three biological replicates. The intracellular wild-type 2HG concentrations corresponded to 114 ± 10 μm (mid exp. phase) and 119 ± 3 μm (early stat. phase) without serine and lysine supplementation (A, left panel) and to 15 ± 2 μm (mid exp. phase) and 24 ± 4 μm (early stat. phase) with serine and lysine supplementation (A, right panel). The intracellular wild-type serine concentrations were 1191 ± 55 μm (mid exp. phase) and 1217 ± 73 μm (early stat. phase) without serine and lysine supplementation (B). exp., exponential; stat., stationary.
Ser3 and Ser33 Catalyze the Conversion of α-Ketoglutarate to d-2HG in Vitro
The yeast phosphoglycerate dehydrogenase paralogs Ser3 and Ser33 were overexpressed as N-terminally His-tagged proteins in E. coli and purified extensively by Ni2+ affinity chromatography followed by anion exchange chromatography. SDS-PAGE analysis of the purified fractions revealed two major bands around the expected molecular weight for the His-tagged proteins (Fig. 4A). Western blot analysis of the same fractions using an anti-His antibody detected only the upper band in both the Ser3 and Ser33 preparations (Fig. 4B). However, the double bands co-eluted during the chromatographic procedure, and peptide analysis of the purified fractions by LC-MS/MS after tryptic digestion did not reveal the presence of any E. coli protein contaminants having a similar molecular weight as Ser3 or Ser33 (data not shown). Taken together, these observations suggested that after the affinity purification step, proteolysis in the proximity of the N terminus occurred for both proteins. Taking the double band into account, the purity of both the Ser3 and Ser33 preparations exceeded 90% based on the SDS-PAGE analysis (Fig. 4A). For specific activity calculations of Ser3 and Ser33, we assumed that both protein forms retained catalytic activity, but we cannot formally exclude that the partial proteolytic cleavage led to a decrease of enzyme activity.
As shown in Fig. 12, both proteins reduced α-ketoglutarate to 2-hydroxyglutarate in a time-dependent manner in the presence of NADH, as determined by HRAM LC-MS. Analysis of the reaction product by the derivatization-based LC-MS/MS method confirmed that both Ser3 and Ser33 convert α-ketoglutarate into the d enantiomer of 2-hydroxyglutarate (data not shown). Using a spectrophotometric assay to monitor the conversion of NADH to NAD+ at 340 nm, we found that Ser3 and Ser33 display very similar kinetic properties for the reduction of α-ketoglutarate to d-2HG, both possessing a relatively robust d-2HG forming activity (Table 3). For reasons that remain unclear, it was much more difficult to detect the 3-phosphoglycerate dehydrogenase activity of the Ser3 and Ser33 proteins in vitro. As the forward reaction is thermodynamically highly unfavorable, we first tried to detect the reverse reaction (reduction of 3-phosphohydroxypyruvate or PHP to 3-phosphoglycerate or PGA) using the same assay as the one used for measuring the conversion of α-ketoglutarate to d-2HG. As a strong salt-sensitive substrate inhibition was reported for mammalian orthologs of Ser3 and Ser33 (52), we tested the activity of Ser3 and Ser33 also at low PHP concentrations (down to 5 μm) and in the presence of high salt concentrations (100–400 mm KCl). A340 was monitored in a plate reader, but also in a conventional cuvette-based spectrophotometer during those attempts to detect the PHP reductase activity. Under none of the conditions tested could an NADH-dependent conversion of PHP to PGA be detected with either the Ser3 or the Ser33 protein, or a mixture of both. In the physiological direction, we could detect an NAD+-dependent oxidation of PGA to PHP (as well as of d-2HG to α-ketoglutarate) by Ser3 and Ser33 using a fluorometric assay performed in the presence of hydrazine and at basic pH. However, a transient decrease in fluorescence after enzyme addition in the absence of substrate made it difficult to measure initial velocities and to accurately determine the kinetic properties of the PGA dehydrogenase activity. Based on this assay, we estimated the Km value of the yeast phosphoglycerate dehydrogenases Ser3 and Ser33 to be ∼1 and 4 μm, respectively, for PGA. In contrast to those high substrate affinities, the turnover numbers estimated for the PGA dehydrogenase activities of Ser3 and Ser33 were more than 100-fold lower than the ones determined for the α-ketoglutarate reductase activity with the spectrophotometric assay (Table 3). In conclusion, although we had no difficulties measuring and characterizing the “side activity” (d-2HG forming activity) that we hypothesized to exist for Ser3 and Ser33, detection of the presumed main activity (PGA oxidation) was much less straightforward with our recombinant purified yeast proteins.
FIGURE 12.
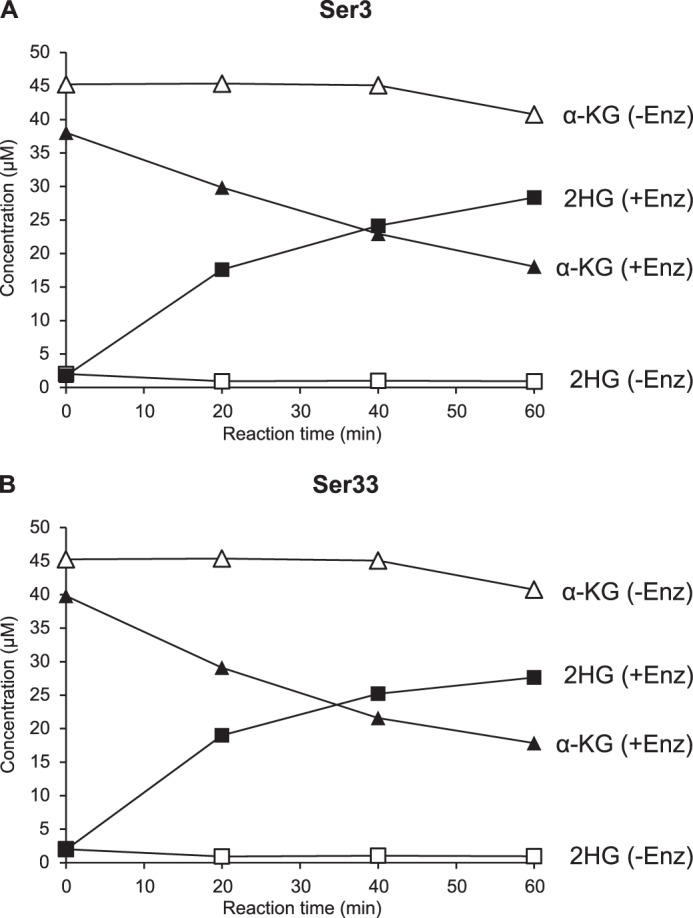
Ser3 and Ser33 reduce α-ketoglutarate to 2HG. A reaction mixture containing 45 mm Hepes, pH 7.4, 1 mm DTT, 0.25 mm NADH, and 50 μm α-ketoglutarate was incubated at 30 °C in the absence or presence of recombinant purified Ser3 (A) or Ser33 (B). Aliquots were heat-inactivated at the indicated times and analyzed with a targeted quantitative HRAM LC-MS method. α-KG, α-ketoglutarate; Enz, enzyme.
TABLE 3.
Kinetic properties of the α-ketoglutarate reductase activity of Ser3 and Ser33
Recombinant purified Ser3 or Ser33 proteins were incubated in a reaction mixture containing NADH (250 μm) and α-KG concentrations varying from 0 to 500 μm. The reductase activity was assayed spectrophotometrically by monitoring the change of absorbance at 340 nm. All values obtained were corrected by subtracting absorbance changes measured in control reactions run without substrate. Kinetic parameters were estimated using non-linear regression fitting in the GraphPad Prism software (version 6.05). The values shown are means ± S.D. obtained from three and four independent saturation curves for Ser3 and Ser33, respectively.
| Km | kcat | kcat/Km | |
|---|---|---|---|
| μm | s−1 | s−1 m−1 | |
| Ser3 | 49 ± 8 | 0.20 ± 0.03 | 4.3 ± 1.4 × 103 |
| Ser33 | 54 ± 37 | 0.25 ± 0.03 | 6.6 ± 4.2 × 103 |
Discussion
The S. cerevisiae Genome Harbors Two Putative d-2HG Dehydrogenase Paralogs but No l-2HG Dehydrogenase Candidate
Sequence analyses indicate that S. cerevisiae cells contain enzymes (encoded by DLD2 and DLD3) with d-2HG dehydrogenase activity but lack l-2HG dehydrogenase activity. This may not be generally true, as a protein with about 30% amino acid sequence identity to human l-2HG dehydrogenase can be found in other yeast species. Further analyses would be necessary to verify whether this protein indeed possesses l-2HG dehydrogenase activity and whether the corresponding yeast species produce l-2HG.
Others have previously reported that the S. cerevisiae Dld1, Dld2, and Dld3 proteins display d-lactate dehydrogenase activity, explaining the current annotation of all three proteins as such. However, sequence analyses, both by Cristescu and Egbosimba (36) and now in this study, strongly suggest another function for Dld2 and Dld3. In addition, an S. cerevisiae strain with a single DLD1 gene deletion did not grow on a medium containing d-lactate as the sole carbon source (35), further supporting the existence of physiological functions other than d-lactate oxidation for Dld2 and Dld3.
Our phylogenetic analyses based on Dld2- and Dld3-like protein sequences within the Hemiascomycetes class suggest that DLD2 is the ancestral gene, from which DLD3 derived by gene duplication, during or after the Whole Genome Duplication (WGD) that occurred in this class of yeast species. Indeed, although a Dld2-like sequence was found in all analyzed yeast species, Dld3-like sequences were only present in post-duplication species. DLD2 and DLD3 are not among the 551 paralogous gene pairs identified by Byrne and Wolfe (40) as having arisen from a WGD (also called ohnologs). It may be argued that the subtelomeric localization of DLD3 could have erased the traces of synteny with pre-WGD species, which is a commonly used criterion to identify ohnologs (39, 40, 53). Subtelomeric regions evolve generally faster than other genomic regions (41) leading to more frequent gene duplication and gene loss events. For instance, one of the adjacent genes of DLD3 is HXT13, a gene encoding a member of the hexose transporter family and known to have a high evolutionary rate (43). Interestingly, comparison of the DLD3 gene sequence from 40 different S. cerevisiae species revealed that this gene remains under a strong purifying selection (mean dN/dS ratio of 0.12) thus indicating that preserving its function is beneficial for the organism. This contrasts with the considerably higher dN/dS ratio found for the adjacent HXT13 gene (we found an average value of 0.89 based on the pairwise comparison of gene sequences from 10 different S. cerevisiae species), indicating a quasi-neutral selection and in line with the HXT family expansion (43). Finally, it is also interesting to note that the only post-duplication species, of those that we analyzed, not encoding the Dld3 protein is the pathogenic species C. glabrata. Similarly, for the ohnologous gene pair IFH1 and CRF1 (both genes are involved in the regulation of expression of ribosomal proteins), only IFH1 is maintained in the C. glabrata genome (54).
All of the Dld3-like sequences found in the post-duplication species lack the MTS that is present in the Dld2-like sequences in yeasts but also in other eukaryotic species such as mammals. Experimental evidence in S. cerevisiae further supports the differential subcellular localization of the Dld2 (mitochondrial) and Dld3 (cytosolic) proteins (32, 33). Also, the DLD3 gene expression is under retrograde regulation by the RTG transcription factors (32), and the DLD3 and SER33 genes show a highly correlated expression as revealed by the SPELL search engine (50). DLD2 is not a member of the retrograde regulon, and there is no evidence for a co-regulation of the DLD2 and SER33 genes. Taken together, all these observations suggest that DLD3 arose by gene duplication from DLD2, during or after the WGD, and that a slightly higher evolutionary rate for the DLD3 gene subsequently led to a subfunctionalization characterized by a new subcellular localization, a different regulation of gene expression, and, as shown in this study, a new enzymatic mechanism for the conversion of d-2-hydroxyglutarate to α-ketoglutarate.
Both Dld2 and Dld3 Oxidize d-2HG to α-Ketoglutarate in Vitro, but Only DLD3 Deficiency Leads to Intracellular Accumulation of d-2HG
In agreement with our in silico analyses, we detected d-2HG in wild-type S. cerevisiae cells but not l-2HG. The formation of 2HG had previously been reported in S. cerevisiae, but the configuration of this 2-hydroxyacid had not been determined (24, 55). Intriguingly, although sequence analyses and subcellular localization evidence suggested Dld2 as the most plausible ortholog of the mammalian d-2HG dehydrogenase, our experimental results show that the cytosolic Dld3 protein is the major enzyme responsible for in vivo degradation of d-2HG in S. cerevisiae, and presumably other post-duplication yeast species, at least under the cultivation conditions used. That Dld2 and Dld3 play distinct roles in S. cerevisiae metabolism is consistent with a previous study showing that DLD3, but not DLD2, is a retrograde-responsive gene (32). Retrograde regulation designates a process in which changes in the functional state of the mitochondria trigger changes in the expression of certain nuclear genes, leading to metabolic adaptations in the cell. The corresponding signaling pathway has been extensively investigated in yeast (56) where it involves the Rtg1 and Rtg3 transcription factors as well as the cytoplasmic signaling protein Rtg2. Two Rtg target genes are strongly induced in ρ0 cells (i.e. cells lacking mitochondrial DNA), namely CIT2 and DLD3 (32, 56). CIT2 encodes the peroxisomal isoform of citrate synthase and, together with other Rtg target genes (CIT1, ACO1, IDH1, and IDH2), is induced to maintain production of a sufficient level of α-ketoglutarate for glutamate synthesis and ammonia assimilation under fermentative growth, in respiration-deficient cells, or other conditions in which the tricarboxylic acid cycle is inoperative as a whole (56, 57).
Because the physiological function of DLD3 was unknown, its role in the retrograde response has remained obscure. It had been proposed to be involved in the re-oxidation of NADH potentially accumulating in respiratory-deficient cells (56). The identification of the d-2HG transhydrogenase activity of Dld3 in this study, producing α-ketoglutarate and d-lactate from d-2HG and pyruvate, supports this possibility: Dld3 is indirectly involved in NADH recycling to NAD+ by removing the potentially toxic d-2HG produced by dehydrogenases that reduce α-ketoglutarate to d-2HG using NADH (Fig. 13). The contribution of this “d-2HG pathway” to NADH re-oxidation should, however, be minor in S. cerevisiae as the flux to d-2HG formation is certainly low compared with the ones leading to glycerol-3-P and, most importantly, ethanol formation under fermentative conditions. In our batch cultivations for example, in minimal media supplemented with 1% glucose, extracellular ethanol concentrations reached 122 and 133 mm in the wild-type strain and the dld3Δ mutant, respectively, whereas the extracellular d-lactate concentrations remained below 400 μm in the wild-type strain and the extracellular d-2HG concentrations below 140 μm in the dld3Δ mutant. In this context it is worth mentioning that, although d-lactate is so far considered as a metabolite specifically derived from methylglyoxal detoxification in yeast (58), the important drop in d-lactate levels measured in the dld3Δ mutant strain in this study suggests that d-lactate actually derives mainly from the reduction of pyruvate that accompanies the newly identified transhydrogenase reaction catalyzed by Dld3 on d-2HG. As discussed below, rather than NADH recycling, the critical role of Dld3 may be mainly the reconversion of the useless and potentially toxic metabolite d-2HG to the important metabolic intermediate α-ketoglutarate.
FIGURE 13.

d-2HG formation and degradation in yeast. A metabolic network, including the d-2HG formation and degradation reactions identified in this study, is represented. We showed here that yeast produces the d enantiomer of 2-hydroxyglutarate, and we identified Dld3 as the major d-2HG-degrading enzyme in S. cerevisiae. Unlike in other organisms, d-2HG is degraded via a transhydrogenase reaction in yeast in which Dld3 oxidizes d-2HG to α-ketoglutarate in the first half-reaction and reduces pyruvate to d-lactate in the second half-reaction. d-2HG is produced by the S. cerevisiae phosphoglycerate dehydrogenases Ser3 and Ser33, which catalyze the conversion of α-ketoglutarate to d-2HG in addition to their known activity in the serine biosynthesis pathway. Although we found that Dld2 acts as a d-2HG dehydrogenase in vitro, deletion mutants of this gene displayed only a moderate increase in intracellular d-2HG levels if any, which, together with the subcellular localization information available for Dld2 (mitochondrial) and Dld3, Ser3, and Ser33 (cytosolic), indicates that d-2HG is mainly produced and degraded in the cytosol of yeast cells. The newly identified reactions seem to couple d-2HG degradation to the shuttling of reducing equivalents from cytosolic NADH to the mitochondrial respiratory chain via the d-lactate dehydrogenase Dld1. DLD3 expression is induced together with CIT2, ACO1, IDH1, and IDH2 by the retrograde transcription factors Rtg1 and Rtg3 under conditions of mitochondrial dysfunction to preserve intracellular α-ketoglutarate pools. Idh1 and Idh2, but also cytosolic Idp2, may be additional sources of d-2HG formation.
Identification of a New Type of Enzymatic Activity for Degradation of d-2HG
Our in vitro enzyme characterizations show that Dld2 and Dld3 act as d-2HG dehydrogenases in the presence of the artificial electron acceptor DCIP. Dld2 and Dld3 both contain an FAD-binding p-cresol methylhydroxylase-type domain (amino acids 98–277 in Dld2 and amino acids 64–243 in Dld3 according to the Prosite database) (59), and our observations with the recombinant purified enzymes support the hypothesis that Dld2 and Dld3 tightly bind FAD as a prosthetic group. As for other members of the p-cresol methylhydroxylase family of FAD-binding proteins (FAD-dependent d-lactate dehydrogenases (48, 60) and mammalian d-2HG dehydrogenase (22)), we found that Dld2 and Dld3 are metalloenzymes dependent on Zn2+.
Although the mitochondrial localization of Dld2 suggests that electrons collected by this enzyme may be transferred to the respiratory chain by the ETF complex (Aim45 and Cir1 in yeast) and ETF dehydrogenase (Cir2 in yeast), the question about the identity of the electron acceptor for the cytosolic Dld3 flavoprotein remained open. The depletion of d-lactate levels in the dld3Δ mutant cells pointed to the possibility that Dld3 may actually use pyruvate as an electron acceptor. We could confirm this hypothesis by using the recombinant enzyme, and we thereby identified a new type of enzymatic activity (transhydrogenase activity) for the conversion of d-2HG to α-ketoglutarate; Dld3 catalyzes the transfer of reducing equivalents from d-2HG to pyruvate, leading to the formation of α-ketoglutarate and d-lactate. Relatively few transhydrogenases are currently known as follows: the mammalian ADHFE1 or HOT enzyme (1.1.99.24) that converts 4-hydroxybutyrate to succinate semialdehyde with the concomitant conversion of α-ketoglutarate to d-2HG (13), a bacterial l-lactate-oxaloacetate transhydrogenase (1.1.99.7) that produces pyruvate and l-malate (61), and a bacterial glucose-fructose transhydrogenase (1.1.99.28) that produces gluconolactone and sorbitol (62). In addition, the enzymatic activity of the E. coli erythronate-4-phosphate dehydrogenase PdxB has been shown to depend on α-hydroxyacids such as α-ketoglutarate, oxaloacetate, or pyruvate as hydride acceptors and therefore also qualifies as a transhydrogenase (63). While lactate-oxaloacetate transhydrogenase (61), PdxB (63), and presumably also ADHFE1 (13) bind NAD as a prosthetic group and glucose-fructose transhydrogenase binds NADP (62), Dld3 appears to be the first example of a transhydrogenase using FAD as the intermediate hydrogen acceptor. We found that Dld3 can also use oxaloacetate as an electron acceptor instead of pyruvate and then form d-malate instead of d-lactate. The lower intracellular concentrations of oxaloacetate as compared with pyruvate (64) and the much more pronounced effect of DLD3 gene deletion on intra- and extracellular d-lactate levels than on malate levels are, however, strong arguments in favor of pyruvate being the physiological electron acceptor for Dld3. This is further supported by comparison of the Km value of the Dld3 transhydrogenase for pyruvate (about 0.5 mm in the presence of a saturating concentration of d-2HG) with the pyruvate concentrations measured intracellularly (0.6–1.1 mm; supplemental Fig. S3).
Possible Sources of d-2HG in Yeast
Our results obtained with overexpression strains, gene deletion strains, as well as in vitro with purified recombinant enzymes demonstrate that, as suggested previously for other organisms (14, 49), 3-phosphoglycerate dehydrogenase is a source for d-2HG formation in S. cerevisiae. Comparing the kinetic properties of the α-ketoglutarate reductase activity of E. coli SerA (49), human PHGDH (14), and yeast Ser3 and Ser33 (this study), it appears that the bacterial protein is a much more efficient enzyme for α-ketoglutarate to d-2HG conversion (Km = 0.088 mm and kcat = 33.3 s−1 (49)) than the human enzyme (Km = 10.1 mm and kcat = 0.078 s−1 (14)), with the yeast enzymes showing a similarly high affinity as the bacterial enzyme for α-ketoglutarate (Km = 0.05 mm) but with a turnover number (kcat = 0.2 s−1) situated in between the values found for the bacterial and the human enzymes. Co-regulated gene expression for the SER33 and DLD3 genes in S. cerevisiae (50) as well as gene clustering of phosphoglycerate dehydrogenase genes and d-2HG dehydrogenase genes in bacterial genomes (65) are other arguments supporting a conserved functional association between those two enzymes and the existence of a d-2HG forming side activity for phosphoglycerate dehydrogenases across different species. It remains difficult to understand why we only detected a very low 3-phosphoglycerate dehydrogenase activity and no 3-phosphohydroxypyruvate reductase activity with the recombinant purified Ser3 and Ser33 yeast proteins. It is possible that we are missing an activator in the in vitro system that is present in the cell (Ser3 and Ser33 overexpression led to increased intracellular serine levels, and ser3Δ and ser33Δ mutants produced less serine, supporting that those enzymes do convert PGA to PHP in the yeast cell) and that is specifically required for the PGA dehydrogenase (and not the α-ketoglutarate reductase) activity of Ser3 and Ser33. We tested the effect of glutamate, which is used in the transamination reaction that follows the PGA to PHP conversion in the serine synthesis pathway, on the PGA dehydrogenase activity of Ser3, but we did not find a stimulatory effect.
As residual d-2HG was detected in a ser3Δser33Δ double knock-out strain, additional sources for d-2HG formation must exist. In addition to phosphoglycerate dehydrogenase, two other enzymes are known to catalyze the reduction of α-ketoglutarate to d-2HG in mammals: isocitrate dehydrogenase and the 4-hydroxybutyrate-α-ketoglutarate transhydrogenase ADHFE1 (13). While point mutations in the isocitrate dehydrogenases IDH1 and IDH2 are well known to lead to a loss of the normal catalytic activity and a gain of d-2HG forming activity both in d-2-hydroxyglutaric acidurias and in a number of cancers (9, 66), there is also evidence that wild-type IDH1 and IDH2 contribute to d-2HG formation in mammalian cells (11, 15). The yeast orthologs of the human NADP-dependent IDH1 and IDH2 enzymes are Idp1 (mitochondrial), Idp2 (cytosolic), and Idp3 (peroxisomal). As for humans, yeast also encodes mitochondrial NAD-dependent isocitrate dehydrogenases (which are designated IDH1 and IDH2 in yeast, whereas the human gene name is IDH3). As mentioned above, yeast IDH1 and IDH2 are retrograde-responsive genes. From what is known from the mammalian enzymes, the NADP-dependent yeast Idp1, Idp2, and Idp3 enzymes would be the best candidates for presenting a d-2HG-forming side activity (queries using the SPELL tool show a co-regulation of expression for DLD3 and IDP1), but as mentioned above, IDH1 and IDH2 are, as DLD3, retrograde-regulated genes. It would therefore be tempting to speculate that while the retrograde genes CIT2, ACO1, and IDH1/2 are up-regulated to maintain sufficient α-ketoglutarate levels under certain conditions, the concomitant up-regulation of DLD3 is necessary for reconverting the d-2HG side product that is potentially formed by yeast Idh1 and/or Idh2 to α-ketoglutarate (Fig. 13). Although the existence of these multiple isoforms of isocitrate dehydrogenases in yeast would make it more complicated to investigate their implication in d-2HG formation using deletion mutants, testing whether they can convert α-ketoglutarate to d-2HG in vitro should be more straightforward. There is no clear homolog of human ADHFE1 in yeast (the most closely related protein is alcohol dehydrogenase Adh4, which shares 24% identity with ADHFE1 at the amino acid level), and it therefore seems less likely that a 4-hydroxybutyrate-α-ketoglutarate transhydrogenase is involved in d-2HG formation in yeast cells. Albers et al. (55) had previously reported an α-ketoglutarate reductase activity in crude S. cerevisiae extracts and measured actually higher activities in extracts of a ser3Δser33Δ double knock-out strain than in a wild-type strain. This led them to hypothesize that enzymes other than Ser3 and Ser33 are responsible for the detected α-ketoglutarate reductase activity. Although interference by contaminating activities in the crude extracts might have influenced the results, these observations also support that Ser3 and Ser33 are not the only enzymes responsible for d-2HG formation in yeast.
A 2HG synthase activity, producing 2HG from propionyl-CoA and glyoxylate, has been described in E. coli and Aspergillus glaucus grown on propionate (67, 68). The S. cerevisiae genome does not encode a propionyl-CoA carboxylase and therefore cannot catabolize propionyl-CoA via the methylmalonyl-CoA pathway. However, there is evidence that propionyl-CoA is catabolized via the methylcitrate cycle in S. cerevisiae (69, 70). No gene has been associated with the reported bacterial 2HG synthase activity yet, hampering the search for a 2HG synthase candidate gene in other genomes.
In one of the very few studies that included investigations on 2HG formation in S. cerevisiae (24), an industrial baker's strain was cultivated under anaerobic conditions in the presence of glucose as the sole carbon source and glutamate as the sole nitrogen source. During labeling experiments with [14C]glucose or [14C]glutamate, 2HG (as well as α-ketoglutarate) was found to incorporate radioactivity only from glutamate, not from glucose. In our experiments, we clearly detect 2HG formation in a minimal controlled medium containing also glucose but with ammonium as the main nitrogen source and no glutamate. A critical difference between our experiments and the ones by Albers et al. (24) may be the activation status of the retrograde pathway (activated in our experiments, inhibited by glutamate in Albers et al. (24); see also Ref. 56). Under our aerobic fermentation conditions, the retrograde enzymes Cit2, Aco1, Idh1, and Idh2 presumably produce α-ketoglutarate from acetyl-CoA and oxaloacetate. If this takes place in the mitochondria (Aco1, Idh1, and Idh2 are annotated as mitochondrial enzymes, but experimental evidence for a dual mitochondrial/cytosolic localization of Aco1 exists (71)), a transport system exists for α-ketoglutarate translocation to the cytosol (72), where the latter can then be converted to d-2HG by Ser3 and Ser33 (Fig. 13). In addition, Idh1 and Idh2 may directly produce d-2HG as a side product. As, under our cultivation conditions, a major fraction of the produced d-2HG seems to be degraded in the cytosol by Dld3, a transport system for mitochondrially produced d-2HG would be required. The physiological role of the mitochondrial Dld2 protein, shown in this study to be a high affinity d-2HG dehydrogenase in vitro, remains unclear. The sharp drop in intracellular d-2HG levels upon total glucose consumption in the dld3Δ mutant indicates that Dld2 might be a glucose-repressed enzyme and therefore contribute to d-2HG degradation only when cells are growing on other carbon sources.
Physiological Relevance of d-2HG Formation and Degradation in Yeast
S. cerevisiae can grow on d-lactate as the sole carbon source as it possesses a mitochondrial d-lactate dehydrogenase (Dld1) that oxidizes d-lactate to pyruvate using cytochrome c as the electron acceptor (Fig. 13) (35). The concomitant action of Ser3 or Ser33 (or other NAD-dependent dehydrogenases converting α-ketoglutarate to d-2HG), Dld3, and Dld1 should therefore lead to the shuttling of reducing equivalents from cytosolic NADH to the respiratory chain at the level of cytochrome c (Fig. 13). Under conditions where the respiratory chain is blocked, d-lactate would be the end product of this pathway. Under conditions where the respiratory chain is active, one would expect d-2HG to serve as a potential carbon source for S. cerevisiae cells. The drop in the intracellular d-2HG concentration observed upon complete glucose consumption (Fig. 3) could, as discussed before, indicate that Dld2 replaces Dld3 under specific conditions, but this decrease could also be caused by induction of Dld1, which is a known glucose-repressed enzyme (73). In contrast to this intracellular decrease of d-2HG concentration, the extracellular d-2HG concentrations remained stable after reaching a plateau about 19 h after inoculation of dld3Δ mutant cultures. This indicates that extracellular d-2HG is not taken up by yeast cells. Therefore, the membrane-permeable octyl ester derivative of d-2HG could instead be used to test whether S. cerevisiae can grow on d-2HG.
Although the contribution of the proposed “d-2HG shuttle” to NADH recycling is probably minor, the important role of Dld3 in the cell may consist of keeping d-2HG at very low levels. Over the past years, a number of so-called “metabolite repair enzymes” have been identified whose apparently exclusive role is to convert useless and potentially toxic side products formed endogenously by metabolic enzymes (65, 74). These side products are formed because metabolic enzymes are not perfectly specific, and they can act on compounds that resemble their main physiological substrate or they can catalyze more than one type of reaction on the main physiological substrate. In humans, deficiency of the metabolite repair enzyme D2HGDH (dehydrogenase specifically acting on d-2HG) leads to d-2HG aciduria, a neurometabolic disorder encompassing clinical features such as epilepsy, hypotonia, and psychomotor retardation (7). Another cause for d-2HG aciduria are gain-of-function mutations in IDH1 or IDH2 that lead to a high α-ketoglutarate reductase activity of those enzymes. Such mutations have also been detected in certain forms of cancer, leading to the notion that d-2HG may act as an oncometabolite. Although some mechanisms have been proposed by which d-2HG could stimulate tumor progression (activation of the prolyl 4-hydroxylase EGLN that acts on HIF (75) and inhibition of DNA and histone demethylation (76, 77)), the better outcome of glioma patients with IDH mutations compared with glioma patients without IDH mutations indicates that the connection between d-2HG and cancer is more complex than what is currently understood. Very recently, d-2HG has been shown to inhibit ATP synthase and signaling through the serine/threonine-protein kinase mTOR in U87 glioblastoma cells, suggesting a growth-suppressive function of this metabolite (78). Except for these latter observations, most of the cellular d-2HG effects identified so far involve activity modulation of α-ketoglutarate-dependent dioxygenases such as prolyl 4-hydroxylase, Jmj histone demethylases, and ten-eleven translocation (TET) 5-methylcytosine hydroxylases (75–77, 79).
Given these multiple effects observed with d-2HG in mammalian cell systems, one could expect a yeast dld3Δ knock-out strain to display cellular impairments. In our experimental system, we did not observe obvious phenotypic changes due to d-2HG accumulation. The dld3Δ mutant cells showed no growth phenotype in our shaking flask cultivations with minimal controlled medium containing 1% glucose, despite the important increase in intracellular d-2HG levels and the drop in intracellular d-lactate levels measured in those conditions. Cell morphology (single cell size) as well as glucose uptake and ethanol production were similar in the dld3Δ mutant and the wild-type strain. It would be interesting to extend our growth, morphology, and metabolome studies to different cultivation conditions, especially using different (respiratory) carbon and nitrogen sources, to reveal phenotypic changes associated with d-2HG accumulation and to elucidate which functions this metabolite impacts intracellularly. Given the important up-regulation of DLD3 gene expression in ρ0 cells (32), it would be interesting to investigate the phenotypic effects of DLD3 deficiency in this background. From what is currently known concerning the intracellular targets of d-2HG, another promising direction for investigations is the analysis of chromatin structure and gene expression profiles in the dld3Δ mutant. JmjC domain containing histone demethylases have been identified in yeast (80, 81) and may be inhibited by high intracellular d-2HG concentrations. Further experiments are designed to clarify which cellular functions are affected by d-2HG in yeast and whether Dld3 acts as a metabolite repair enzyme to protect the yeast cell against adverse effects of this dicarboxylic hydroxyacid.
Author Contributions
J. B. K., N. P., and C. L. L. designed the yeast cultivation experiments. D. P. K. created yeast mutant strains. J. B. K., D. P. K., and N. P. performed the cultivations and did the metabolite extractions. N. P. and C. G. performed LC-MS analyses of the samples. J. B. K. and C. L. L. designed the biochemical experiments, and J. F. C. and J. B. K. performed the biochemical experiments. J. B. K., N. P., and C. L. L. analyzed and interpreted the data. P. P. J. and C. L. L. did the phylogenetic analysis. J. B. K., N. P., and C. L. L. wrote and revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Acknowledgments
pAG416GPD-ccdB was a gift from Susan Lindquist (Addgene plasmid ID 14148), and pDest-527 was a gift from Dominic Esposito (Addgene plasmid ID 11518). We sincerely thank Dr. Aimée Dudley for providing us with the dld2Δ and dld3Δ mutant strains; Zaya Aïnouch for help with generating Gateway Entry clones and bacterial expression vectors as well as with LC-MS method setup; Charandeep Singh for LC-MS/MS analysis of 3-phosphohydroxypyruvate; Céline Leclercq and Jenny Renaut (Luxembourg Institute of Science and Technology) for LC-MS/MS analysis of purified Ser3 and Ser33 proteins; and Laurence Joly and Emmanuelle Cocco (Luxembourg Institute of Science and Technology) for their extensive help with the differential LC-MS/MS analyses of d-2HG and l-2HG. Finally, we are very grateful to Prof. Emile Van Schaftingen for inspiring discussions and critical reading of the manuscript.
This work was supported by the Fonds National de la Recherche Luxembourg Grant CORE C13/BM/5773107, AFR Ph.D. Fellowship 4044610 (to J. B. K.), and a Fondation du Pélican de Mie et Pierre Hippert-Faber scholarship (to J. B. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1–S4 and Figs. S1–S3.
- 2HG
- 2-hydroxyglutarate
- DCIP
- 2,6-dichlorophenol indophenol
- d-2HG
- d-2-hydroxyglutarate
- D2HGDH
- d-2-hydroxyglutarate dehydrogenase
- ETF
- electron transfer flavoprotein
- GPD
- glyceraldehyde-3-phosphate dehydrogenase
- HRAM
- high-resolution accurate-mass
- IDH
- isocitrate dehydrogenase
- l-2HG
- l-2-hydroxyglutarate
- MRM
- multiple reaction monitoring
- MTS
- mitochondrial targeting sequence
- PGA
- 3-phosphoglycerate
- PHP
- 3-phosphohydroxypyruvate
- WGD
- Whole Genome Duplication.
References
- 1. Gregersen N., Ingerslev J., and Rasmussen K. (1977) Low molecular weight organic acids in the urine of the newborn. Acta Paediatr. Scand. 66, 85–89 [DOI] [PubMed] [Google Scholar]
- 2. Gibson K. M., ten Brink H. J., Schor D. S., Kok R. M., Bootsma A. H., Hoffmann G. F., and Jakobs C. (1993) Stable-isotope dilution analysis of d- and l-2-hydroxyglutaric acid: application to the detection and prenatal diagnosis of d- and l-2-hydroxyglutaric acidemias. Pediatr. Res. 34, 277–280 [DOI] [PubMed] [Google Scholar]
- 3. Struys E. A., Jansen E. E., Verhoeven N. M., and Jakobs C. (2004) Measurement of urinary d- and l-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-l-tartaric anhydride. Clin. Chem. 50, 1391–1395 [DOI] [PubMed] [Google Scholar]
- 4. Chalmers R. A., Lawson A. M., Watts R. W., Tavill A. S., Kamerling J. P., Hey E., and Ogilvie D. (1980) d-2-Hydroxyglutaric aciduria: case report and biochemical studies. J. Inherit. Metab. Dis. 3, 11–15 [DOI] [PubMed] [Google Scholar]
- 5. Duran M., Kamerling J. P., Bakker H. D., van Gennip A. H., and Wadman S. K. (1980) l- 2-Hydroxyglutaric aciduria: an inborn error of metabolism? J. Inherit. Metab. Dis. 3, 109–112 [DOI] [PubMed] [Google Scholar]
- 6. Muntau A. C., Röschinger W., Merkenschlager A., van der Knaap M. S., Jakobs C., Duran M., Hoffmann G. F., and Roscher A. A. (2000) Combined d-2- and l-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria? Neuropediatrics 31, 137–140 [DOI] [PubMed] [Google Scholar]
- 7. Kranendijk M., Struys E. A., Salomons G. S., Van der Knaap M. S., and Jakobs C. (2012) Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 35, 571–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Schaftingen E., Rzem R., and Veiga-da-Cunha M. (2009) l-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 32, 135–142 [DOI] [PubMed] [Google Scholar]
- 9. Kranendijk M., Struys E. A., van Schaftingen E., Gibson K. M., Kanhai W. A., van der Knaap M. S., Amiel J., Buist N. R., Das A. M., de Klerk J. B., Feigenbaum A. S., Grange D. K., Hofstede F. C., Holme E., Kirk E. P., et al. (2010) IDH2 mutations in patients with d-2-hydroxyglutaric aciduria. Science 330, 336. [DOI] [PubMed] [Google Scholar]
- 10. Losman J. A., and Kaelin W. G. Jr. (2013) What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terunuma A., Putluri N., Mishra P., Mathé E. A., Dorsey T. H., Yi M., Wallace T. A., Issaq H. J., Zhou M., Killian J. K., Stevenson H. S., Karoly E. D., Chan K., Samanta S., Prieto D., et al. (2014) MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 124, 398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nota B., Struys E. A., Pop A., Jansen E. E., Fernandez Ojeda M. R., Kanhai W. A., Kranendijk M., van Dooren S. J., Bevova M. R., Sistermans E. A., Nieuwint A. W., Barth M., Ben-Omran T., Hoffmann G. F., de Lonlay P., et al. (2013) Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined d-2- and l-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 92, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kardon T., Noël G., Vertommen D., and Schaftingen E. V. (2006) Identification of the gene encoding hydroxyacid-oxoacid transhydrogenase, an enzyme that metabolizes 4-hydroxybutyrate. FEBS Lett. 580, 2347–2350 [DOI] [PubMed] [Google Scholar]
- 14. Fan J., Teng X., Liu L., Mattaini K. R., Looper R. E., Vander Heiden M. G., and Rabinowitz J. D. (2015) Human phosphoglycerate dehydrogenase produces the oncometabolite d-2-hydroxyglutarate. ACS Chem. Biol. 10, 510–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsunaga H., Futakuchi-Tsuchida A., Takahashi M., Ishikawa T., Tsuji M., and Ando O. (2012) IDH1 and IDH2 have critical roles in 2-hydroxyglutarate production in d-2-hydroxyglutarate dehydrogenase depleted cells. Biochem. Biophys. Res. Commun. 423, 553–556 [DOI] [PubMed] [Google Scholar]
- 16. Rzem R., Achouri Y., Marbaix E., Schakman O., Wiame E., Marie S., Gailly P., Vincent M. F., Veiga-da-Cunha M., and Van Schaftingen E. (2015) A mouse model of l-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS ONE 10, e0119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rzem R., Vincent M. F., Van Schaftingen E., and Veiga-da-Cunha M. (2007) l-2-Hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 30, 681–689 [DOI] [PubMed] [Google Scholar]
- 18. Hüdig M., Maier A., Scherrers I., Seidel L., Jansen E. E., Mettler-Altmann T., Engqvist M. K., and Maurino V. G. (2015) Plants possess a cyclic mitochondrial metabolic pathway similar to the mammalian metabolic repair mechanism involving malate dehydrogenase and l-2-hydroxyglutarate dehydrogenase. Plant Cell Physiol. 56, 1820–1830 [DOI] [PubMed] [Google Scholar]
- 19. Intlekofer A. M., Dematteo R. G., Venneti S., Finley L. W., Lu C., Judkins A. R., Rustenburg A. S., Grinaway P. B., Chodera J. D., Cross J. R., and Thompson C. B. (2015) Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 22, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Struys E. A., Gibson K. M., and Jakobs C. (2007) Novel insights into l-2-hydroxyglutaric aciduria: mass isotopomer studies reveal 2-oxoglutaric acid as the metabolic precursor of l-2-hydroxyglutaric acid. J. Inherit. Metab. Dis. 30, 690–693 [DOI] [PubMed] [Google Scholar]
- 21. Struys E. A., Verhoeven N. M., Brunengraber H., and Jakobs C. (2004) Investigations by mass isotopomer analysis of the formation of d-2-hydroxyglutarate by cultured lymphoblasts from two patients with d-2-hydroxyglutaric aciduria. FEBS Lett. 557, 115–120 [DOI] [PubMed] [Google Scholar]
- 22. Achouri Y., Noël G., Vertommen D., Rider M. H., Veiga-Da-Cunha M., and Van Schaftingen E. (2004) Identification of a dehydrogenase acting on d-2-hydroxyglutarate. Biochem. J. 381, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rzem R., Van Schaftingen E., and Veiga-da-Cunha M. (2006) The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie 88, 113–116 [DOI] [PubMed] [Google Scholar]
- 24. Albers E., Gustafsson L., Niklasson C., and Lidén G. (1998) Distribution of 14C-labelled carbon from glucose and glutamate during anaerobic growth of Saccharomyces cerevisiae. Microbiology 144, 1683–1690 [DOI] [PubMed] [Google Scholar]
- 25. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 26. Gouy M., Guindon S., and Gascuel O. (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 [DOI] [PubMed] [Google Scholar]
- 27. Guindon S., and Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 28. Xu B., and Yang Z. (2013) PAMLX: a graphical user interface for PAML. Mol. Biol. Evol. 30, 2723–2724 [DOI] [PubMed] [Google Scholar]
- 29. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 30. Alberti S., Gitler A. D., and Lindquist S. (2007) A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24, 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duley J., and Holmes R. S. (1975) A spectrophotometric procedure for determining the activity of various rat tissue oxidases. Anal. Biochem. 69, 164–169 [DOI] [PubMed] [Google Scholar]
- 32. Chelstowska A., Liu Z., Jia Y., Amberg D., and Butow R. A. (1999) Signalling between mitochondria and the nucleus regulates the expression of a new d-lactate dehydrogenase activity in yeast. Yeast 15, 1377–1391 [DOI] [PubMed] [Google Scholar]
- 33. Koh J. L., Chong Y. T., Friesen H., Moses A., Boone C., Andrews B. J., and Moffat J. (2015) CYCLoPs: a comprehensive database constructed from automated analysis of protein abundance and subcellular localization patterns in Saccharomyces cerevisiae. G3 5, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emanuelsson O., Brunak S., von Heijne G., and Nielsen H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971 [DOI] [PubMed] [Google Scholar]
- 35. Lodi T., and Ferrero I. (1993) Isolation of the DLD gene of Saccharomyces cerevisiae encoding the mitochondrial enzyme d-lactate ferricytochrome c oxidoreductase. Mol. Gen. Genet. 238, 315–324 [DOI] [PubMed] [Google Scholar]
- 36. Cristescu M. E., and Egbosimba E. E. (2009) Evolutionary history of d-lactate dehydrogenases: a phylogenomic perspective on functional diversity in the FAD binding oxidoreductase/transferase type 4 family. J. Mol. Evol. 69, 276–287 [DOI] [PubMed] [Google Scholar]
- 37. Dujon B. (2010) Yeast evolutionary genomics. Nat. Rev. Genet. 11, 512–524 [DOI] [PubMed] [Google Scholar]
- 38. Kurtzman C. P., and Robnett C. J. (2003) Phylogenetic relationships among yeasts of the 'Saccharomyces complex' determined from multigene sequence analyses. FEMS Yeast Res. 3, 417–432 [DOI] [PubMed] [Google Scholar]
- 39. Kellis M., Birren B. W., and Lander E. S. (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428, 617–624 [DOI] [PubMed] [Google Scholar]
- 40. Byrne K. P., and Wolfe K. H. (2005) The yeast gene order browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 15, 1456–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kellis M., Patterson N., Endrizzi M., Birren B., and Lander E. S. (2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423, 241–254 [DOI] [PubMed] [Google Scholar]
- 42. Brown C. A., Murray A. W., and Verstrepen K. J. (2010) Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr. Biol. 20, 895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin Z., and Li W. H. (2011) Expansion of hexose transporter genes was associated with the evolution of aerobic fermentation in yeasts. Mol. Biol. Evol. 28, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luo Z., and van Vuuren H. J. (2009) Functional analyses of PAU genes in Saccharomyces cerevisiae. Microbiology 155, 4036–4049 [DOI] [PubMed] [Google Scholar]
- 45. Lee J. Y., Kang C. D., Lee S. H., Park Y. K., and Cho K. M. (2015) Engineering cellular redox balance in Saccharomyces cerevisiae for improved production of l-lactic acid. Biotechnol. Bioeng. 112, 751–758 [DOI] [PubMed] [Google Scholar]
- 46. Engqvist M., Drincovich M. F., Flügge U. I., and Maurino V. G. (2009) Two d-2-hydroxy-acid dehydrogenases in Arabidopsis thaliana with catalytic capacities to participate in the last reactions of the methylglyoxal and β-oxidation pathways. J. Biol. Chem. 284, 25026–25037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Claros M. G., and Vincens P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786 [DOI] [PubMed] [Google Scholar]
- 48. Gregolin C., and Singer T. P. (1963) The lactic dehydrogenase of yeast. III. d(−)Lactic cytochrome c reductase, a zinc-flavoprotein from aerobic yeast. Biochim. Biophys. Acta 67, 201–218 [DOI] [PubMed] [Google Scholar]
- 49. Zhao G., and Winkler M. E. (1996) A novel α-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J. Bacteriol. 178, 232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hibbs M. A., Hess D. C., Myers C. L., Huttenhower C., Li K., and Troyanskaya O. G. (2007) Exploring the functional landscape of gene expression: directed search of large microarray compendia. Bioinformatics 23, 2692–2699 [DOI] [PubMed] [Google Scholar]
- 51. Ulane R., and Ogur M. (1972) Genetic and physiological control of serine and glycine biosynthesis in Saccharomyces. J. Bacteriol. 109, 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Achouri Y., Rider M. H., Schaftingen E. V., and Robbi M. (1997) Cloning, sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem. J. 323, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dietrich F. S., Voegeli S., Brachat S., Lerch A., Gates K., Steiner S., Mohr C., Pöhlmann R., Luedi P., Choi S., Wing R. A., Flavier A., Gaffney T. D., and Philippsen P. (2004) The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304, 304–307 [DOI] [PubMed] [Google Scholar]
- 54. Wapinski I., Pfiffner J., French C., Socha A., Thompson D. A., and Regev A. (2010) Gene duplication and the evolution of ribosomal protein gene regulation in yeast. Proc. Natl. Acad. Sci. U.S.A. 107, 5505–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albers E., Laizé V., Blomberg A., Hohmann S., and Gustafsson L. (2003) Ser3p (Yer081wp) and Ser33p (Yil074cp) are phosphoglycerate dehydrogenases in Saccharomyces cerevisiae. J. Biol. Chem. 278, 10264–10272 [DOI] [PubMed] [Google Scholar]
- 56. Liu Z., and Butow R. A. (2006) Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159–185 [DOI] [PubMed] [Google Scholar]
- 57. Tate J. J., and Cooper T. G. (2003) Tor1/2 regulation of retrograde gene expression in Saccharomyces cerevisiae derives indirectly as a consequence of alterations in ammonia metabolism. J. Biol. Chem. 278, 36924–36933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stewart B. J., Navid A., Kulp K. S., Knaack J. L., and Bench G. (2013) d-Lactate production as a function of glucose metabolism in Saccharomyces cerevisiae. Yeast 30, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dym O., and Eisenberg D. (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci. 10, 1712–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Reed D. W., and Hartzell P. L. (1999) The Archaeoglobus fulgidus d-lactate dehydrogenase is a Zn2+ flavoprotein. J. Bacteriol. 181, 7580–7587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Allen S. H., and Patil J. R. (1972) Studies on the structure and mechanism of action of the malate-lactate transhydrogenase. J. Biol. Chem. 247, 909–916 [PubMed] [Google Scholar]
- 62. Zachariou M., and Scopes R. K. (1986) Glucose-fructose oxidoreductase, a new enzyme isolated from Zymomonas mobilis that is responsible for sorbitol production. J. Bacteriol. 167, 863–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rudolph J., Kim J., and Copley S. D. (2010) Multiple turnovers of the nicotino-enzyme PdxB require α-keto acids as cosubstrates. Biochemistry 49, 9249–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petersen S., Mack C., de Graaf A. A., Riedel C., Eikmanns B. J., and Sahm H. (2001) Metabolic consequences of altered phosphoenolpyruvate carboxykinase activity in Corynebacterium glutamicum reveal anaplerotic regulation mechanisms in vivo. Metab. Eng. 3, 344–361 [DOI] [PubMed] [Google Scholar]
- 65. Linster C. L., Van Schaftingen E., and Hanson A. D. (2013) Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 9, 72–80 [DOI] [PubMed] [Google Scholar]
- 66. Vissers L. E., Fano V., Martinelli D., Campos-Xavier B., Barbuti D., Cho T. J., Dursun A., Kim O. H., Lee S. H., Timpani G., Nishimura G., Unger S., Sass J. O., Veltman J. A., Brunner H. G., et al. (2011) Whole-exome sequencing detects somatic mutations of IDH1 in metaphyseal chondromatosis with d-2-hydroxyglutaric aciduria (MC-HGA). Am. J. Med. Genet. A 155, 2609–2616 [DOI] [PubMed] [Google Scholar]
- 67. Bleiweis A. S., Reeves H. C., and Ajl S. J. (1967) Formation of α-hydroxyglutaric acid by Aspergillus glaucus. J. Bacteriol. 94, 1560–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reeves H. C., and Ajl S. J. (1962) α-Hydroxyglutaric acid synthetase. J. Bacteriol. 84, 186–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luttik M. A., Kötter P., Salomons F. A., van der Klei I. J., van Dijken J. P., and Pronk J. T. (2000) The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J. Bacteriol. 182, 7007–7013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pronk J. T., van der Linden-Beuman A., Verduyn C., Scheffers W. A., and van Dijken J. P. (1994) Propionate metabolism in Saccharomyces cerevisiae: implications for the metabolon hypothesis. Microbiology 140, 717–722 [DOI] [PubMed] [Google Scholar]
- 71. Regev-Rudzki N., Karniely S., Ben-Haim N. N., and Pines O. (2005) Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol. Biol. Cell 16, 4163–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Palmieri L., Agrimi G., Runswick M. J., Fearnley I. M., Palmieri F., and Walker J. E. (2001) Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J. Biol. Chem. 276, 1916–1922 [DOI] [PubMed] [Google Scholar]
- 73. Lodi T., Alberti A., Guiard B., and Ferrero I. (1999) Regulation of the Saccharomyces cerevisiae DLD1 gene encoding the mitochondrial protein d-lactate ferricytochrome c oxidoreductase by HAP1 and HAP2/3/4/5. Mol. Gen. Genet. 262, 623–632 [DOI] [PubMed] [Google Scholar]
- 74. Van Schaftingen E., Rzem R., Marbaix A., Collard F., Veiga-da-Cunha M., and Linster C. L. (2013) Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 36, 427–434 [DOI] [PubMed] [Google Scholar]
- 75. Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G., et al. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., et al. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fu X., Chin R. M., Vergnes L., Hwang H., Deng G., Xing Y., Pai M. Y., Li S., Ta L., Fazlollahi F., Chen C., Prins R. M., Teitell M. A., Nathanson D. A., Lai A., et al. (2015) 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 22, 508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Wang P., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kwon D. W., and Ahn S. H. (2011) Role of yeast JmjC-domain containing histone demethylases in actively transcribed regions. Biochem. Biophys. Res. Commun. 410, 614–619 [DOI] [PubMed] [Google Scholar]
- 81. Tu S., Bulloch E. M., Yang L., Ren C., Huang W. C., Hsu P. H., Chen C. H., Liao C. L., Yu H. M., Lo W. S., Freitas M. A., and Tsai M. D. (2007) Identification of histone demethylases in Saccharomyces cerevisiae. J. Biol. Chem. 282, 14262–14271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Patiny L., and Borel A. (2013) ChemCalc: a building block for tomorrow's chemical infrastructure. J. Chem. Inf. Model. 53, 1223–1228 [DOI] [PubMed] [Google Scholar]