

Abstract
Fibrotic diseases display mesenchymal cell (MC) activation with pathologic deposition of matrix proteins such as collagen. Here we investigate the role of mTOR complex 1 (mTORC1) and mTORC2 in regulating MC collagen expression, a hallmark of fibrotic disease. Relative to normal MCs (non-Fib MCs), MCs derived from fibrotic human lung allografts (Fib-MCs) demonstrated increased phosphoinositide-3kinase (PI3K) dependent activation of both mTORC1 and mTORC2, as measured by increased phosphorylation of S6K1 and 4E-BP1 (mTORC1 substrates) and AKT (an mTORC2 substrate). Dual ATP-competitive TORC1/2 inhibitor AZD8055, in contrast to allosteric mTORC1-specific inhibitor rapamycin, strongly inhibited 4E-BP1 phosphorylation and collagen I expression in Fib-MCs. In non-Fib MCs, increased mTORC1 signaling was shown to augment collagen I expression. mTORC1/4E-BP1 pathway was identified as an important driver of collagen I expression in Fib-MCs in experiments utilizing raptor gene silencing and overexpression of dominant-inhibitory 4E-BP1. Furthermore, siRNA-mediated knockdown of rictor, an mTORC2 partner protein, reduced mTORC1 substrate phosphorylation and collagen expression in Fib-, but not non-Fib MCs, revealing a dependence of mTORC1 signaling on mTORC2 function in activated MCs. Together these studies suggest a novel paradigm where fibrotic activation in MCs increases PI3K dependent mTORC1 and mTORC2 signaling and leads to increased collagen I expression via the mTORC1-dependent 4E-BP1/eIF4E pathway. These data provide rationale for targeting specific components of mTORC pathways in fibrotic states and underscore the need to further delineate mTORC2 signaling in activated cell states.
Keywords: collagen, extracellular matrix protein, fibrosis, mammalian target of rapamycin (mTOR), pulmonary fibrosis, Bronchiolitis obliterans syndrome, Chronic allograft rejection, mesenchymal cell
Introduction
Tissue fibrosis, marked by mesenchymal cell (MC)2 infiltration and collagen deposition, continues to be a major cause of organ failure and death both before and after transplantation. Within five years of lung transplantation, 50% of the recipients demonstrate a decline in their lung function termed bronchiolitis obliterans syndrome which arises from progressive airway fibrosis (1, 2). Histologic samples reveal pathologic collagen matrix deposition with interspersed MCs (3). Relentless fibro-proliferation is the hallmark of chronic allograft failure across all solid organ transplants, and targeting mechanisms of MC activation and matrix deposition is key to improving long term outcomes.
Mammalian target of rapamycin (mTOR), an evolutionarily conserved serine/threonine protein kinase, links multiple upstream signaling cascades to downstream translational activation (4, 5). mTOR forms the catalytic core of two distinct complexes known as mTOR complex 1 (mTORC1) and mTORC2, which are defined by their partner proteins raptor and rictor, respectively (6). mTORC1 promotes cap-dependent mRNA translation via phosphorylation of its effectors S6K1 (ribosomal protein S6 Kinase 1) and 4E-BP1 (eukaryotic initiation factor 4E-binding protein1) (7–11). mTORC2 functions as the major kinase for AKT (12). Constitutive activation of the mTORC1 pathway and increased cap dependent translation has been shown to be key in the development and progression of various cancers (13). A potential role for mTORC1 signaling in modulating MC function and promoting fibrosis has also been suggested, predominantly from studies utilizing rapamycin, a first-generation mTORC1 specific allosteric inhibitor that suppresses mTORC1 function by binding a domain N-terminal to the kinase domain (14–19). It is important to note that not all mTORC1 substrates show similar sensitivity to rapamycin; for example, rapamycin reduces the phosphorylation of S6K1 potently while it has modest inhibitory effect on the phosphorylation of 4E-BP1. As roles for mTORC1 and mTORC2 in modulating fibrotic responses in MCs remain poorly defined, we have studied MCs derived from fibrotic and normal human lung allografts and demonstrate that the mTORC1 and mTORC2 pathways function as key signaling intermediates in MC activation and fibrosis.
Experimental Procedures
Isolation of Mesenchymal Cell from Human Lung Allografts
MCs were isolated from bronchoalveolar lavage fluid obtained by bronchoscopy from human lung transplant recipients with and without bronchiolitis obliterans syndrome as previously described under a protocol approved by the University of Michigan Institutional Review Board (20). Briefly, cell pellet recovered from bronchoalveolar lavage fluid by centrifugation (1,000 rpm for 5 min) was seeded and maintained in tissue culture dishes. MCs were identified by their growth as fibroblastoid colony forming units and isolated by trypsinization and cultured by further passage in plastic adherent conditions. MCs grown from each individual BAL sample were treated as a separate cell line. Cells at passages three through six were used for all experiments performed. The mesenchymal phenotype of the cells was confirmed by immunofluorescent staining for the mesenchymal marker vimentin utilizing mouse mAbs against vimentin (Sigma-Aldrich). Cells were visualized and photographed using a Zeiss fluorescence microscope. Expression of N-cadherin and E-cadherin was also evaluated. Total RNA was prepared from isolated MCs and human lung bronchial epithelial cells using an RNeasy minikit (Qiagen, Inc., Valencia, CA), per manufacturer's instructions. Real-time qPCR analysis was performed on an ABI Prism 7000 SDS (Applied Biosystems, Foster City, CA) using TaqMan PCR Master Mix (Applied Biosystems). The TaqMan real-time PCR primers included Hs00983056_ml for N-cadherin, and Hs01023894_m1 for E-cadherin (Applied Biosystems).
Tissue Culture, Protein Isolation, and Western Blotting
MCs were plated and grown to 80% confluence in 60-mm dishes prior to serum-deprivation for 24 h. Following serum deprivation, cells were treated in serum-free medium containing the indicated doses of either rapamycin (Cayman Chemical, Ann Arbor, MI) or AZD8055 (Selleck Chemicals, Houston, TX) for 24 h. Total protein was collected from treated or untreated cells by cell lysis on ice. Lysates were separated on 10% or 4–12% gradient Bis-Tris gels prior to Western blot analysis phosphorylated S6 kinase (Thr-389), phosphorylated 4E-BP1 (Thr-37/46), total 4E-BP1, phosphorylated AKT (Ser-473), total AKT, rictor, raptor, and TSC2 (all from Cell Signaling Technologies, Boston, MA). Plasmid transfection efficiency was determined by blotting for HA or FLAG M2, both purchased from Sigma-Aldrich. Scraped cell lysates which contains both intracellular and extracellular proteins were also utilized for quantitating collagen I expression using an Anti-Human Collagen Type I, purified IgG (Polyclonal) (rabbit IgG) antibody (Cedarlane Laboratories, Ontario, Canada). This Collagen I antibody (CL50111AP) detects bands between molecular mass of 150 and 250 kDA, which correspond to detected epitopes located in the procollagen and collagen portions and the predominant band we detect at 200 kDA corresponds to collagen beta 1,1 and beta 1,2.
siRNA-mediated Silencing and Plasmid Transfection
siGENOME SMARTpool, siRNA targeting human rictor (253260), raptor (57521), or TSC2 were obtained from Dharmacon/GE Healthcare (Lafayette, CO). Non-Targeting scrambled siRNA-A was purchased from Santa Cruz Biotechnology (Dallas, TX). Briefly, cells were plated to 50% confluency in 6-well plates followed by transient transfection utilizing oligofectamine (Invitrogen, Carlsbad, CA) for 24 h. Following transfection cells were maintained in serum free media for 72 h prior to protein harvesting. pACTAG2-HA-human wild-type 4E-BP1, pACTAG2-HA-AA-4E-BP1 (Thr37Ala/Thr46Ala), and pACTAG2-HA-TOS-4E-BP1, pcDNA3-HA-AKT-WT-mouse and pRK7-FLAG-Rheb was kindly provided by Dr. D. C. Fingar (University of Michigan). For plasmid transfection, cells were plated to 90% confluence in 6-well plates followed by transient transfection utilizing Lipofectamine LTX and 250 ng of plasmid DNA (Invitrogen, Carlsbad, CA) for 24 h. Following transfection, cells were maintained in opti-MEM medium for 24 h prior to protein harvesting.
Sircol Assay
40,000 cells per well were plated in triplicate into 48-well plates and allowed to adhere overnight. Cells were then serum deprived for 24 h followed by treatment with either 250 nm rapamycin or 250 nm AZD8055 for 16 h. Cells and cell supernatants were scraped and harvested into micro-centrifuge tubes and assay was commenced per manufacturer's protocol. Each sample was run on the assay plate in replicates of 6 and normalized to a standard curve generated used acid-soluble bovine collagen.
pcDNA3-HA-AKT-S473D Plasmid Generation
Site-directedmutagenesis was performed on pcDNA3-HA-WT mouse to introduce the S473D mutation using Q5 Site-Directed Mutagenesis Kit (New England BioLabs). Mutagenesis PCR reactions were carried out per manufacturer's protocol utilizing a 71° annealing temperature and the following primers: Forward primer: 5′-cccccagttcGACtactcagccagtggc-3′; reverse primer: 5′-aagtgcggcctccgctca-3′. All plasmids were fully sequenced prior to use in transient transfection experiments.
Lenti-viral Silencing
V2L pGIPZ Rictor clone 120392 and scrambled pGIPZ control plasmids were obtained from Open Biosystems. Cells were plated to 60% confluency in 6-well plates followed by infection with 1× lentivirus in serum-free DMEM containing 8 μg/ml polybrene. Lentivirus was removed 24 h later and replaced with DMEM containing 10% FBS for an additional 48 h. Transient transfection of HA-AKT and HA-AKT-S473D plasmids was performed on lentiviral silenced MCs using Lipofectamine LTX and 500 ng of plasmid DNA.
Statistical Analysis
Data are presented as mean values ± S.E. Statistical significance was analyzed using GraphPad Prism 6 software (La Jolla, CA). Significance was assessed with a Student t test for comparisons of two groups, or with analysis of variance and a post hoc Neuman-Keuls test for three or more groups. p less than 0.05 were considered significant.
Results
Mesenchymal Cells Derived from Fibrotic Lung Allografts Demonstrate mTORC1/mTORC2 Pathway Activation
Mesenchymal phenotype of cells isolated from lung allografts was confirmed by immunofluorescence staining for vimentin (Fig. 1A) and by realtime PCR demonstrating presence of N-cadherin and absence of E-cadherin (Fig. 1B). Baseline protein expression in MCs derived from fibrotic lung allografts (Fib-MCs) were compared with mesenchymal cells derived from normal lung allografts (non-Fib MCs). As previously reported (21), Fib-MCs demonstrated elevated expression of collagen I protein compared with non-Fib MCs (Fig. 1, C and D). Interestingly, phosphorylation of S6K1 (Thr-389) and 4E-BP1 (Thr-37/46), two important substrates of mTORC1, was elevated in Fib-MCs compared with non-Fib MCs (Fig. 1, C and D). To determine the extent of mTORC2 signaling in fibrotic MCs compared with normal counterparts, we monitored the phosphorylation of AKT on its hydrophobic motif site, Ser-473. We found that Fib-MCs also displayed increased AKT phosphorylation compared with non-Fib MCs (Fig. 1, C and D), indicating increased mTORC2 function. Treatment of Fib-MCs with the PI3K inhibitor wortmannin decreased phosphorylation of S6K1 (Thr-389), 4E-BP1 (Thr-37/46), and AKT (Ser-473) (Fig. 1E), demonstrating the dependence of mTORC1 and mTORC2 signaling on PI3K in Fib-MCs.
FIGURE 1.

A and B, characterization of mesenchymal cells from bronchalveolar lavage of lung transplant recipients. Mononuclear cells obtained from bronchoalveolar lavage of human lung allografts were plated and maintained in culture. On subsequent trypsinization and culturing, a homogeneous population of MCs was obtained by passage 2 as demonstrated by immunofluorescence staining for vimentin (an intermediate filament protein). N-cadherin and E-cadherin mRNA expression in MCs as compared with bronchial epithelial cells by real-time PCR is shown (n = 3) **, p < 0.01; ***, p < 0.001. C and D, mTOR activation in fibrotic mesenchymal cell. Baseline protein expression of fibrotic (collagen I), mTORC1 (phosphorylated S6Kinase at Thr-389 and 4E-BP1 at Thr-37/46), and mTORC2 (phosphorylated AKT at Ser-473) activation markers was measured by Western blot analysis in MCs isolated from normal (non-Fib MCs) or fibrotic lung allografts (Fib-MCs). Quantitation by densitometric analysis is shown in D. Each data point represents MCs from an individual lung transplant recipient for each group (n = 8). ***, p < 0.001. E, mTOR activation in Fib-MCs is dependent of PI3kinase (PI3K) activity. Fib-MCs were treated with the PI3K inhibitor wortmannin (500 nm) for one hour and protein expression measured by Western blot analysis. Representative blots are shown.
mTOR Kinase Inhibitors Reduce Collagen I Expression in Fibrotic Mesenchymal Cells
To investigate a potential link between the mTORC1 and mTORC2 pathways and control of collagen I expression in fibrotic lung-resident MCs, a pharmacological approach was first utilized in which Fib-MCs were treated with rapamycin, an allosteric mTORC1 inhibitor. As has been shown for other cell types (22–24), rapamycin reduced S6K1 phosphorylation (Thr-389) strongly but failed to reduce 4E-BP1 phosphorylation (Thr-37/46) (Fig. 2A). Furthermore, rapamycin increased AKT phosphorylation in a dose-dependent manner in Fib-MCs (Fig. 2A), likely due to previously described mTORC1-mediated negative feedback toward PI3K (25–27). Rapamycin treatment at various doses did not significantly affect collagen I protein expression in Fib-MCs cells. Non-Fib MCs treated with rapamycin demonstrated an increase in AKT (Ser-473) and 4E-BP1 (Thr-37/46) phosphorylation over time and an increased collagen I expression was noted at 48 h (Fig. 2B). These data suggested that rapamycin not only selectively inhibits mTORC1-mediated S6K1 phosphorylation in lung MCs but also activates mTORC2 and can potentially induce 4E-BP1 phosphorylation and collagen I expression in non-activated cells.
FIGURE 2.

Specific mTOR activity signals collagen I expression in fibrotic mesenchymal cells with suppression of collagen I by AZD8055 but not rapamycin. A, Fib-MCs (n = 7) were treated with rapamycin (varying doses) for 24 h, with collagen I and phosphorylation of S6K1 (Thr-389), 4E-BP1 (Thr-37/46), and AKT (Ser-473) measured by Western blot analysis. B, Non-Fib MCs (n = 6) were treated with rapamycin (250 nm) for varying times (24–72 h) and 4E-BP1 (Thr-37/46), AKT (Ser-473), and collagen I expression measured by Western blot analysis. C, Fib-MCs (n = 7) were treated with AZD8055 for 24 h and phosphorylation of S6K1 (Thr-389), 4E-BP1 (Thr-37/46), and AKT (Ser-473) measured by Western blot analysis. D, effect of AZD8055 treatment on collagen I protein expression in Fib-MCs over varying time intervals of treatment is shown (n = 7). Densitometric analysis here is presented as percent of untreated control. Approximately 80% suppression in collagen I protein expression was induced by treatment with AZD8055, and this effect was persistent at 48 and 72 h. E, time course of collagen I suppression by AZD8055 in Fib-MCs (n = 8). F, acid-soluble collagen produced by Fib-MCs in in vitro tissue culture condition was quantitated by Sircol Collagen Assay in the presence of AZD8055 (n = 8). **, p < 0.01; ***, p < 0.001.
This led us to investigate the effect of an ATP-competitive mTOR catalytic inhibitor (AZD8055), a robust mTORC1 and mTORC2 inhibitor (28), on collagen expression. We found that AZD8055 reduced the phosphorylation of S6K1 (Thr-389), 4E-BP1 (Thr-37/46), and AKT (Ser-473) (Fig. 2C) as well as collagen expression (by 80%) in Fib-MCs (Fig. 2D). The strong reduction in collagen expression caused by AZD8055 persisted for up to 72 h (Fig. 2D). Investigation of earlier time points demonstrated that 1 h of AZD8055 (250 nm) treatment resulted in a 66% reduction in collagen expression, suggesting that the effect is likely not due to reduced transcription of collagen (Fig. 2E). Collagen produced by Fib-MCs was also studied by Sircol collagen assay, a quantitative dye-binding method designed for the analysis of acid-soluble collagens. This assay confirmed that AZD8055 treatment results in reduced expression of collagen (Fig. 2F). The effect of mTOR inhibition on other matrix proteins was also evaluated. A significant decrease in fibronectin (p = 0.017) was noted with AZD8055 with no change in vimentin (p = 0.737), demonstrating a role for mTOR in modulating specific fibrotic functions (data not shown).
mTORC1 Signaling Promotes Pro-fibrotic Activation of Mesenchymal Cells
To further investigate the differential roles of the two mTOR complexes in fibrotic functions of mesenchymal cells, we employed siRNA to knockdown raptor and rictor, which inactivate mTORC1 and mTORC2 function, respectively. As expected, raptor knockdown decreased phosphorylation of S6K1 (Thr-389) and 4E-BP1 (Thr-37/46) in Fib-MCs; raptor knockdown also decreased expression of collagen (Fig. 3, A and B). We next asked if activation of mTORC1 would impart a fibrotic phenotype to normal MCs. We first utilized a FLAG-tagged Rheb construct to overexpress wild type Rheb protein. Rheb is a small GTPase (guanosine triphosphatase) that activates mTORC1 on lysosomal membranes when GTP-bound (29, 30). When overexpressed, wild type Rheb, increases mTORC1 signaling in diverse cell types (30). In non-Fib MCs, overexpression of FLAG-Rheb elevated mTORC1 signaling (as measured by increased S6K1 (Thr-389) and 4E-BP1 (Thr-37/46) phosphorylation) and increased collagen I protein expression (Fig. 3C). We also elevated mTORC1 signaling in non-Fib MCs by knocking down TSC2 with siRNA. The TSC1-TSC2 complex functions as a GAP (GTPase activating protein) for Rheb (30). Indeed, patients with the benign tumor syndrome tuberous sclerosis (TSC) possess inactivating mutations in either TSC1 or TSC2, and their cells and tissues possess elevated mTORC1 signaling (31). As TSC1 and TSC2 function as obligate partners, knocking down either one inactivates TSC1/2 function. Consistent with our other data, we found that TSC2 knockdown increases mTORC1 signaling (increased S6K1 (Thr-389) and 4E-BP1 (Thr-37/46) phosphorylation) and leads to higher collagen I expression in non-Fib MCs (Fig. 3D).
FIGURE 3.

mTORC1 regulates collagen I expression in lung mesenchymal cells. A and B, mesenchymal cells derived from fibrotic lung allografts (Fib-MCs) were transfected with Raptor or scrambled siRNA (100 nm) for 48 h followed by protein collection and Western blot analysis. Efficacy of raptor silencing was studied by measuring the effect of the siRNA on Raptor protein expression. Effect of Raptor silencing on phosphorylated S6K1 (Thr-389), 4E-BP1 (Thr-37/46), AKT (Ser-473), and collagen I was measured by Western blot analysis and quantitated (n = 5–10). *, p < 0.05; ***, p < 0.001. C, mesenchymal cells derived from normal lung allografts (non-Fib MCs) were transfected with pRK7-Flag-Rheb followed by protein collection and Western blot analysis at 48 h (n = 5). Representative blots are shown. D, Non-Fib MCs were transfected with TSC2 siRNA and protein expression quantitated at 48 h (n = 5). Representative blots are shown.
Dependence of mTORC1 Signaling on mTORC2 Function in Fibrotic Mesenchymal Cells
As the mTOR inhibitor AZD8055 reduced collagen I expression profoundly in MCs derived from fibrotic lungs, we evaluated the role of mTORC2 in MC activation further. To specifically inactive mTORC2 without affecting mTORC1, we employed siRNA to knockdown expression of rictor in Fib-MCs. As expected, rictor knockdown reduced AKT phosphorylation at Ser-473 and PKC phosphorylation at Ser-657, confirming inactivation of mTORC2 function (Fig. 4A). Rictor knockdown also reduced expression of collagen I by 72% (Fig. 4A). Somewhat unexpectedly, rictor knockdown reduced phosphorylation of S6K1 (Thr-389) and 4E-BP1 (Thr-37/46) (Fig. 4A). These data indicate that in human lung MCs, mTORC1 signaling requires upstream mTORC2 function, likely by boosting AKT signaling to mTORC1. Consistently, a known AKT target TSC-2 demonstrated decreased phosphorylation with rictor silencing (Fig. 4A).
FIGURE 4.

mTORC2 signaling mediates collagen I expression and lies upstream of mTORC1 in activated mesenchymal cells. A, Fib-MCs were transfected with Rictor siRNA (100 nm) or scrambled siRNA for 48 h followed by protein collection and Western blot analysis. Effect of Rictor silencing on baseline collagen I, P-S6K1 (Thr-389), P-4E-BP1 (Thr-37/46), P-AKT (Ser-473), P-TSC2 (Thr-462), and P-PKC (Ser-657) expression is shown (n = 6–8). B, Non-Fib MCs transfected with Rictor (100 nm) or scrambled siRNA were treated with or without 10 μm lysophosphatidic acid (LPA) for 6 h. Effect of rictor silencing in presence and absence of LPA on P-S6K1 (Thr-389), P-4E-BP1 (Thr-37/46), P-AKT (Ser-473), and collagen I expression is shown (n = 6–8). C, Fib-MCs were treated with AKT inhibitor MK-2206 (5um) or PKC inhibitor Go6796 (1um) for 24 h, and protein expression of mTORC substrates and collagen I was quantitated by Western blot analysis (n = 5). D, non-Fib MCs were transfected with pcDNA3 backbone, pcDNA3 HA-AKT or pcDNA3 HA-AKT S473D plasmids followed by protein collection at 24 h and Western blot analysis. (n = 6). E, Fib-MCs were infected with lentivirus containing pGIPZ Rictor shRNA for 72 h followed by transient transfection with HA-AKT plasmids and subsequent protein collection and Western blot analysis. Representative blots from all experiments are shown.
It is important to note that mTORC2-mediated AKT phosphorylation on its hydrophobic motif site Ser-473 boosts AKT activity while PDK1-mediated phosphorylation on its activation loop site (Thr-308) is absolutely required for AKT activity. Moreover, while many reports indicate that mTORC1 signaling does not require mTORC2-mediated AKT Ser-473 phosphorylation (32, 33), a more limited set of reports show dependence of mTORC1 signaling on mTORC2 function (34). For example, it has been shown that under conditions of elevated PI3K activity, mTORC2-mediated AKT Ser-473 phosphorylation boosts AKT activity significantly (34).
To investigate if mTORC2 positively regulates mTORC1 in activated lung MCs, we studied the effect of rictor silencing on mTORC1 substrate phosphorylation in non-Fib MCs at baseline and in the presence of lysophosphatidic acid (LPA), an activator of the PI3K pathway. In non-Fib cells, rictor silencing had no effect on the phosphorylation of 4E-BP1 at Thr-37/46 or phosphorylation of S6K1at Thr-389 (Fig. 4B). LPA treatment increased mTORC1 signaling to S6K1 and 4E-BP1, and resulted in increased collagen I expression. In these activated cells, rictor silencing reduced P-S6K1-Thr389, P-4E-BP1-Thr37/46, and Collagen I expression (Fig. 4B), further confirming that mTORC1 signaling depends on mTORC2 function in both activated and fibrotic MCs.
To elucidate the mechanism by which mTORC2 function promotes mTORC1 signaling, Fib-MCs were treated with AKT inhibitor (MK-2206) and PKC inhibitor (Go6796). Fib-MCs treated with MK-2206 (AKT inhibitor) demonstrated decreased phosphorylation of S6K1 (Thr-389) and 4E-BP1 (Thr-37/46) where as Go6796 (PKC inhibitor) had no effect on mTORC1 substrate phosphorylation (Fig. 4C). Collagen I expression was also significantly attenuated in the presence of AKT inhibitor MK-2206.
To further determine if AKT Ser-473 phosphorylation is sufficient to induce the activation of mTORC1 by mTORC2, non-Fib MCs were transfected with HA-AKT WT and HA-AKT S473D plasmids. In cells where AKT was up-regulated, a robust increase in phospho 4E-BP1 (Thr-37/46) and a moderate increase in phospho S6K1 (Thr-389) was noted (Fig. 4D). Phosphorylated TSC2 (Thr-1462) and collagen I protein expression was also noted to be higher in non-Fib cells transfected with AKT plasmids (Fig. 4D). In the final experiment, we silenced rictor in Fib-MCs by lentiviral shRNA and concomitantly transfected them with AKT plasmids. Overexpression of AKT robustly rescued phosphorylation of 4E-BP1 (Thr-37/46) and also led to a moderate increase in pS6K1 (Thr-389) in rictor silenced cells (Fig. 4E). In Fib-MCs transfected with HA-AKT S473D plasmid, rictor silencing failed to attenuate Collagen 1 expression. Together, these data suggest that AKT phosphorylation is the likely intermediary pathway linking mTORC2 to mTORC1 activation in lung MCs.
Regulation of Collagen I Expression by the mTORC1-mediated 4E-BP1/eIF4E Signaling Axis in Fibrotic Mesenchymal Cells
We next investigated a role for the mTORC1-S6K1 and mTORC1–4E-BP1/eIF4E signaling axes in control of collagen I expression in MCs. As rapamycin completely inhibits mTORC1-mediated phosphorylation of S6K1, yet has minimal effect on collagen I expression, we hypothesized that the mTORC1-controlled 4E-BP1/eIF4E pathway may prove to be the more important modulator. Indeed, the S6K1 inhibitor PF-4708671 yielded no effect on collagen I expression (Fig. 5A). On the contrary, chemical inhibition of eIF4E downstream signaling with 4EGI-1, a synthetic molecule that displaces eIF4G association with eIF4E, thus preventing formation of the translation initiation complex, resulted in a significant decrease in collagen I expression in Fib-MCs (Fig. 5B). To further investigate if matrix protein expression in Fib-MCs is dependent on eIF4E-dependent translation, we inhibited mTORC1/elF4E signaling by overexpressing the dominant-inhibitory HA-4E-BP1 alleles Phe114Ala TOS (mTOR signaling-motif mutant) and HA-4E-BP1-Thr37Ala/Thr46Ala (phosphorylation site-defective). These alleles encode mTOR-insensitive mutants of 4E-BP1 that dominantly bind to and constitutively inhibit eIF4E and therefore cap-dependent translation. In Fib-MCs transfected with the mutant alleles, collagen I expression was reduced significantly (Fig. 5, C and D). Interestingly, overexpression of wild type 4E-BP1 also decreased collagen I expression (Fig. 5, C and D). Taken together, these data suggest that the TORC1-dependent 4E-BP1/eIF4E pathway, which is insensitive to rapamycin but sensitive to mTOR kinase inhibitors, drives collagen expression in fibrotic MCs.
FIGURE 5.
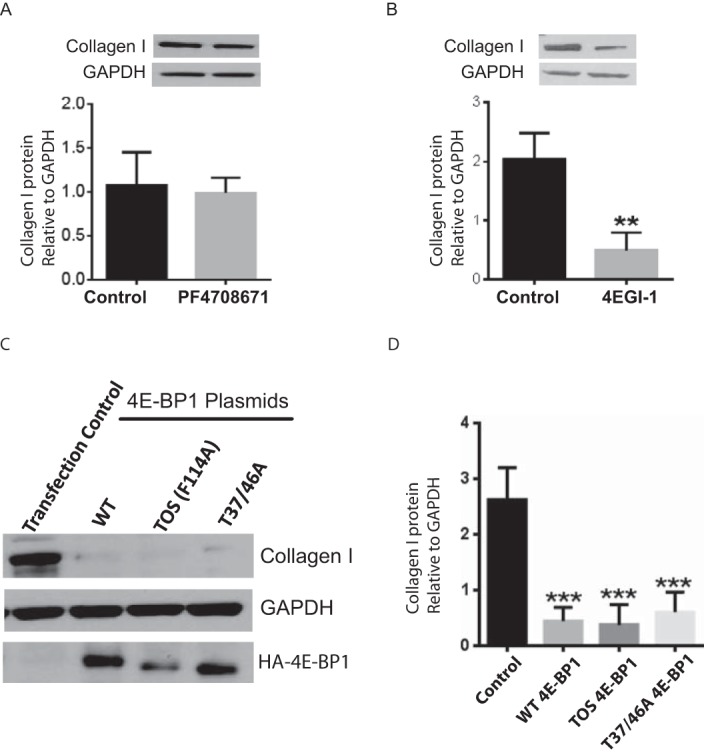
Collagen I expression in fibrotic mesenchymal cells is dependent on the 4EB-P1/eIF4E signaling axis. A and B, collagen I protein expression was measured by Western blot analysis in Fib-MCs treated with selective p70 ribosomal S6 kinase (S6K1) inhibitor PF-4708671 (100 nm) or eIF4E:eIF4G interaction inhibitor 4EGI-1 (10 μm) for 24 h (n = 4). C and D, Fib-MCs were transfected with pACTAG2-HA human wild-type 4E-BP1 (WT), pACTAG2-HA-AA-4E-BP1-Thr37Ala/Thr46Ala (Thr-37/46A), and pACTAG2-HA-TOS-4E-BP1 plasmids (TOS F114A). 4E-BP1 and collagen I expression was measured by Western blot analysis. Densitometric quantitation of collagen I from 6 cell lines is shown in panel D. **, p < 0.01; ***, p < 0.001.
Discussion
Numerous upstream biological pathways have been implicated in mesenchymal cell activation and matrix deposition during fibrosis, underscoring a need to identify and target final common pathways utilized by MCs in regulating its fibrotic functions (35). Here, we provide evidence for crucial roles for mTORC1 and mTORC2, key integrators of diverse intracellular and extracellular stimuli, in regulating collagen I expression in fibrotic human MCs. We have identified mTORC1-mediated phosphorylation of 4E-BP1 as a key mechanism involved in increased collagen I expression by fibrotic MCs. Our investigations reveal a unique dependence of mTORC1 on mTORC2 activity under conditions of MC activation and fibrosis but not under normal conditions, identifying mTORC2 as a positive regulator of mTORC1 and MC fibrotic functions. These data suggest a potential therapeutic role for mTORC kinase inhibitors in fibrotic diseases and emphasize the need to delineate mTORC2 signaling in MCs, as targeting this pathway could represent a strategy to selectively modulate activated MCs.
Our results suggest that the mTORC1/4E-BP1/eIF4E pathway, which is insensitive to rapamycin but sensitive to ATP-competitive mTOR catalytic inhibitors, is critical for driving increased collagen I expression in MCs during fibrosis. Translational control, characterized by differential use of pre-exiting mRNAs, regulates cellular processes in response to various stimuli (36). The primary target of regulation of translation in eukaryotes is the initiation step, and mTORC1 is a key controller of translation-initiation complex component eIF4E (37, 38). The mTORC1 downstream target, 4E-BP1, binds to and negatively regulates eIF4E. Phosphorylation of 4E-BP1 by mTORC1 dissociates it from eIF4E, promoting translation. mTORC pathway activation and dysregulated translation has been shown in autonomous cancer cells (39–41). While a similar unchecked mesenchymal cell activity characterizes fibrosis and contributes to progressive aberrant tissue remodeling, mTOR activation in human fibrotic mesenchyme has not been previously described. Both pharmacologic and small-interfering RNA approaches confirmed that the increased mTORC1 and mTORC2 activity in fibrotic mesenchymal cells was important in mediating increased expression of collagen I, a key fibrotic extracellular matrix protein. Furthermore, mTORC1 activation was sufficient to cause fibrotic differentiation of normal MCs. Downstream of mTORC1, 4E-BP1 was recognized as an integrator of the effects of mTORC activation on collagen I expression. elF4E hyper-activation has also been shown previously to be important in translation of adhesion molecules and extracellular matrix proteins such as fibronectin and CD44 in cancer cells (40). However, the role of 4E-BP1 in regulating collagen I expression in MCs has not been previously identified. The dependence of collagen I expression in fibrotic mesenchymal cells on eIF4E-dependent translation was provided by blunted collagen I expression noted in Fib-MCs overexpressing alleles encoding mTOR-insensitive mutants of 4E-BP1 that dominantly bind to and constitutively inhibit eIF4E and therefore cap-dependent translation. Our findings suggesting mTOR-mediated translational activation in MCs during allograft fibrosis are also strengthened by recent reports of greater differences in matrix expression by translational rather than transcriptional control in MCs from patients with idiopathic pulmonary fibrosis (42).
Comparison of MCs derived from fibrotic and normal lung allografts yielded novel insights into the role of the mTORC2 signaling pathway in diseased and non-diseased states. In normal lung-derived MCs, rictor silencing inhibited AKT Ser-473 phosphorylation but had no effect on basal levels of S6K1 or 4E-BP1 phosphorylation. This finding was consistent with previous studies in several types of cultured mammalian cells and in Drosophila, where it has been shown that, under physiological conditions, TORC2-mediated phosphorylation of AKT on its hydrophobic motif (Ser-473) is not essential for AKT kinase activity and signaling (32–34). In MCs derived from fibrotic lung allografts, however, rictor silencing inhibited mTORC1 substrate phosphorylation potently, suggesting that in the pathological context of fibrosis, mTORC2-mediated AKT Ser-473 phosphorylation promotes AKT-dependent mTORC1 function. It has been recently demonstrated that under conditions of higher-than-normal PI3K activity, rictor/mTORC2-mediated hydrophobic motif phosphorylation boosts AKT activity significantly and mTORC2 activity is needed to permit high-level PI3K/AKT signaling (34). The PI3K-dependent AKT-mTORC1 cascade can be activated by many extracellular stimuli, including ligands for growth factor receptors, G- protein coupled receptors (GPCRs), and cytokine receptors. As fibrotic MCs are exposed to such stimuli, it can be hypothesized that an activated PI3K pathway in fibrotic mesenchymal cells renders mTORC1 activation dependent on mTORC2 in these cells. This proposed paradigm was strengthened by the observation that active PI3K signaling was required for mTORC1 and mTORC2 function in Fib-MCs, with wortmannin leading to decreased mTORC1 and mTORC2 signaling. Activation of the PI3K pathway in normal MCs by LPA, an exogenous ligand, rendered mTORC1 effector phosphorylation susceptible to rictor silencing. A role for AKT phosphorylation as an intermediary step was further suggested by the ability of AKT plasmids to upregulate mTORC1 signaling in non-Fib MCs and to rescue mTORC1 inhibition by rictor silencing in Fib-MCs. mTORC2 regulation of mTORC1 in the context of diseases states also has recent precedence in the field of pulmonary hypertension where mTORC2 was shown to be required for mTORC1-mediated S6K1 activity in pulmonary arterial vascular smooth muscle cells (43). These findings underscore the need for further investigation into mTORC2 signaling as it can potentially provide a means of targeting cells in diseased/activated rather than basal states.
Of significant clinical relevance to the field of lung transplantation is the finding that rapamycin, a drug commonly utilized in clinical practice, does not target the pathway relevant to collagen I expression in lung MCs. While rapamycin is mostly utilized in the transplant field for its immunosuppressive action, its potential anti-fibrotic effects has garnered significant interest for prevention and treatment of chronic allograft rejection in heart, lung, and kidney transplantation. We confirm resistance of the mTORC1–4E-BP1 axis, the signaling axis found to modulate collagen I expression in human lung-allograft MCs, to rapamycin. The differential effect on phosphorylation of mTORC1 downstream targets 4E-BP1 and S6K1 by rapamycin has been shown to be cell specific and is thought to be the reason behind variability in the effect of rapamycin on cap-dependent translation (22). Rapamycin can also induce a rebound increase in phosphorylation of AKT on Ser-473 by interrupting mTORC1 dependent feedback inhibition of IRS phosphorylation and expression (25–27). In fact, rapamycin treatment in both fibrotic and normal lung-derived MCs led to a marked up-regulation of phospho AKT Ser-473. Furthermore, an induction in collagen I expression was noted over a longer time course of rapamycin treatment in non-Fib MCs coinciding with an increase in phospho AKT Ser-473 and phospho 4E-BP1 (Thr-37/46). Also of interest in this context is that clinically, rapamycin treatment is known to be associated with fibrotic pneumonitis (44), and a recent clinical trial of rapamycin derivative everolimus was associated with worsening outcomes in patients with idiopathic pulmonary fibrosis (45). ATP-competitive mTOR kinase inhibitors offer an advantage by effectively targeting the rapamycin resistant mTORC1 pathway and preventing any potential negative feedback by directly inhibiting mTORC2. These compounds have attracted significant attention in the cancer field, and several of these drugs are in clinical trials (46, 47). The profound decrease in collagen I protein expression (near 80%) by the mTOR kinase inhibitor AZD8055 in Fib-MCs suggests a potential role for mTORC1/2 targeting in MCs with fibrotic diseases.
In conclusion, activation of the mTORC1 and mTORC2 pathways is a key feature of fibrotic MCs characterized by pathologic matrix production. mTORC1 and mTORC2 activation was PI3K-dependent, suggesting that multiple upstream mediators can likely utilize this signaling mechanism and mTOR signaling might be the final common pathway. We also demonstrate for the first time that direct activation of mTORC1 is sufficient for increasing collagen I expression in normal MCs and that the increased collagen I expression in fibrotic mesenchymal cells is dependent on the 4E-BP1-eIF4E pathway. Furthermore, these studies establish an obligatory requirement of the mTORC2 complex for mTORC1 activity and collagen I expression in fibrotic MCs.
Author Contributions
Conception and design: N. M. W., L. S., V. N. L.; Acquisition of data, data analysis, and interpretation: N. M. W., K. M. C., E. A. B., J. L., W. L., A. C., D. C. F., V. N. L.; Drafting of the manuscript: V. N. L., N. M. W., D. C. F., S. M. M.
This work was supported by National Institutes of Health Grants R01 HL118017 and R01 HL094622 (to V. N. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MC
- mesenchymal cell
- mTOR
- mammalian target of rapamycin
- Fib-MC
- fibrotic lung allografts
- mTORC
- mechanistic target of rapamycin complex.
References
- 1. Estenne M., Maurer J. R., Boehler A., Egan J. J., Frost A., Hertz M., Mallory G. B., Snell G. I., and Yousem S. (2002) Bronchiolitis obliterans syndrome 2001: an update of the diagnostic criteria. J. Heart Lung Transplant. 21, 297–310 [DOI] [PubMed] [Google Scholar]
- 2. Yusen R. D., Christie J. D., Edwards L. B., Kucheryavaya A. Y., Benden C., Dipchand A. I., Dobbels F., Kirk R., Lund L. H., Rahmel A. O., and Stehlik J. (2013) The Registry of the International Society for Heart and Lung Transplantation: thirtieth adult lung and heart-lung transplant report–2013; focus theme: age. J. Heart Lung Transplant 32, 965–978 [DOI] [PubMed] [Google Scholar]
- 3. Lama V. N., Harada H., Badri L. N., Flint A., Hogaboam C. M., McKenzie A., Martinez F. J., Toews G. B., Moore B. B., and Pinsky D. J. (2006) Obligatory role for interleukin-13 in obstructive lesion development in airway allografts. Am. J. Pathol. 169, 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fingar D. C., and Blenis J. (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171 [DOI] [PubMed] [Google Scholar]
- 5. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster K. G., and Fingar D. C. (2010) Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wullschleger S., Loewith R., and Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 8. Fonseca B. D., Smith E. M., Lee V. H., MacKintosh C., and Proud C. G. (2007) PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 282, 24514–24524 [DOI] [PubMed] [Google Scholar]
- 9. Hara K., Maruki Y., Long X., Yoshino K., Oshiro N., Hidayat S., Tokunaga C., Avruch J., and Yonezawa K. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 [DOI] [PubMed] [Google Scholar]
- 10. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 11. Kim D. H., Sarbassov D. D., Ali S. M., Latek R. R., Guntur K. V., Erdjument-Bromage H., Tempst P., and Sabatini D. M. (2003) GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 11, 895–904 [DOI] [PubMed] [Google Scholar]
- 12. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 13. Sabatini D. M. (2006) mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6, 729–734 [DOI] [PubMed] [Google Scholar]
- 14. Goc A., Choudhary M., Byzova T. V., and Somanath P. R. (2011) TGFβ- and bleomycin-induced extracellular matrix synthesis is mediated through Akt and mammalian target of rapamycin (mTOR). J. Cell. Physiol. 226, 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White E. S., Sagana R. L., Booth A. J., Yan M., Cornett A. M., Bloomheart C. A., Tsui J. L., Wilke C. A., Moore B. B., Ritzenthaler J. D., Roman J., and Muro A. F. (2010) Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp. Cell Res. 316, 2644–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen G., Chen H., Wang C., Peng Y., Sun L., Liu H., and Liu F. (2012) Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PloS one 7, e33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao X. M., Wong G., Wang B., Kiriazis H., Moore X. L., Su Y. D., Dart A., and Du X. J. (2006) Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J. Hypertens. 24, 1663–1670 [DOI] [PubMed] [Google Scholar]
- 18. Lian H., Ma Y., Feng J., Dong W., Yang Q., Lu D., and Zhang L. (2012) Heparin-binding EGF-like growth factor induces heart interstitial fibrosis via an Akt/mTor/p70s6k pathway. PloS one 7, e44946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu S. Y., Liu L., Li P., and Li J. (2013) Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac. Cardiovasc. Surg. 61, 223–228 [DOI] [PubMed] [Google Scholar]
- 20. Lama V. N., Smith L., Badri L., Flint A., Andrei A. C., Murray S., Wang Z., Liao H., Toews G. B., Krebsbach P. H., Peters-Golden M., Pinsky D. J., Martinez F. J., and Thannickal V. J. (2007) Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J. Clin. Investig. 117, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walker N., Badri L., Wettlaufer S., Flint A., Sajjan U., Krebsbach P. H., Keshamouni V. G., Peters-Golden M., and Lama V. N. (2011) Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am. J. Pathol. 178, 2461–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choo A. Y., Yoon S. O., Kim S. G., Roux P. P., and Blenis J. (2008) Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U.S.A. 105, 17414–17419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., and Gray N. S. (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thoreen C. C., and Sabatini D. M. (2009) Rapamycin inhibits mTORC1, but not completely. Autophagy 5, 725–726 [DOI] [PubMed] [Google Scholar]
- 25. O'Reilly K. E., Rojo F., She Q. B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., and Rosen N. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi Y., Yan H., Frost P., Gera J., and Lichtenstein A. (2005) Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 4, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 27. Sun S. Y., Rosenberg L. M., Wang X., Zhou Z., Yue P., Fu H., and Khuri F. R. (2005) Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 65, 7052–7058 [DOI] [PubMed] [Google Scholar]
- 28. Yu K., Toral-Barza L., Shi C., Zhang W. G., Lucas J., Shor B., Kim J., Verheijen J., Curran K., Malwitz D. J., Cole D. C., Ellingboe J., Ayral-Kaloustian S., Mansour T. S., Gibbons J. J., Abraham R. T., Nowak P., and Zask A. (2009) Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 69, 6232–6240 [DOI] [PubMed] [Google Scholar]
- 29. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., and Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tee A. R., Manning B. D., Roux P. P., Cantley L. C., and Blenis J. (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 31. Inoki K., and Guan K. L. (2009) Tuberous sclerosis complex, implication from a rare genetic disease to common cancer treatment. Human Mol. Genet. 18, R94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., and Sabatini D. M. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 33. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., and Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 [DOI] [PubMed] [Google Scholar]
- 34. Hietakangas V., and Cohen S. M. (2007) Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 21, 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wynn T. A., and Ramalingam T. R. (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garcia-Sanz J. A., Mikulits W., Livingstone A., Lefkovits I., and Müllner E. W. (1998) Translational control: a general mechanism for gene regulation during T cell activation. FASEB J. 12, 299–306 [DOI] [PubMed] [Google Scholar]
- 37. Merrick W. C. (1992) Mechanism and regulation of eukaryotic protein synthesis. Microbiol. Rev. 56, 291–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rhoads R. E. (1993) Regulation of eukaryotic protein synthesis by initiation factors. J. Biol. Chem. 268, 3017–3020 [PubMed] [Google Scholar]
- 39. Bitterman P. B., and Polunovsky V. A. (2012) Attacking a nexus of the oncogenic circuitry by reversing aberrant eIF4F-mediated translation. Mol. Cancer Ther. 11, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., Weissman J. S., Shokat K. M., Rommel C., and Ruggero D. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsieh A. C., and Ruggero D. (2010) Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 16, 4914–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larsson O., Diebold D., Fan D., Peterson M., Nho R. S., Bitterman P. B., and Henke C. A. (2008) Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PloS one 3, e3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goncharov D. A., Kudryashova T. V., Ziai H., Ihida-Stansbury K., DeLisser H., Krymskaya V. P., Tuder R. M., Kawut S. M., and Goncharova E. A. (2014) Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129, 864–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pham P. T., Pham P. C., Danovitch G. M., Ross D. J., Gritsch H. A., Kendrick E. A., Singer J., Shah T., and Wilkinson A. H. (2004) Sirolimus-associated pulmonary toxicity. Transplantation 77, 1215–1220 [DOI] [PubMed] [Google Scholar]
- 45. Malouf M. A., Hopkins P., Snell G., and Glanville A. R. (2011) An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 16, 776–783 [DOI] [PubMed] [Google Scholar]
- 46. Schenone S., Brullo C., Musumeci F., Radi M., and Botta M. (2011) ATP-competitive inhibitors of mTOR: an update. Curr. Med. Chem. 18, 2995–3014 [DOI] [PubMed] [Google Scholar]
- 47. Sun S. Y. (2013) mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 340, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]