

Abstract
Mitochondrial dysfunction is one of the major contributors to neurodegenerative disorders including Parkinson disease. The mitochondrial permeability transition pore is a protein complex located on the mitochondrial membrane. Under cellular stress, the pore opens, increasing the release of pro-apoptotic proteins, and ultimately resulting in cell death. MicroRNA-7 (miR-7) is a small non-coding RNA that has been found to exhibit a protective role in the cellular models of Parkinson disease. In the present study, miR-7 was predicted to regulate the function of mitochondria, according to gene ontology analysis of proteins that are down-regulated by miR-7. Indeed, miR-7 overexpression inhibited mitochondrial fragmentation, mitochondrial depolarization, cytochrome c release, reactive oxygen species generation, and release of mitochondrial calcium in response to 1-methyl-4-phenylpyridinium (MPP+) in human neuroblastoma SH-SY5Y cells. In addition, several of these findings were confirmed in mouse primary neurons. Among the mitochondrial proteins identified by gene ontology analysis, the expression of voltage-dependent anion channel 1 (VDAC1), a constituent of the mitochondrial permeability transition pore, was down-regulated by miR-7 through targeting 3′-untranslated region of VDAC1 mRNA. Similar to miR-7 overexpression, knockdown of VDAC1 also led to a decrease in intracellular reactive oxygen species generation and subsequent cellular protection against MPP+. Notably, overexpression of VDAC1 without the 3′-UTR significantly abolished the protective effects of miR-7 against MPP+-induced cytotoxicity and mitochondrial dysfunction, suggesting that the protective effect of miR-7 is partly exerted through promoting mitochondrial function by targeting VDAC1 expression. These findings point to a novel mechanism by which miR-7 accomplishes neuroprotection by improving mitochondrial health.
Keywords: microRNA (miRNA), mitochondrial membrane potential, mitochondrial permeability transition (MPT), Parkinson disease, voltage-dependent anion channel (VDAC), 1-methyl-4-phenylpyridinium
Introduction
The mitochondrial permeability transition pore (PTP),2 which spans both the outer and the inner mitochondrial membranes, is a conductance channel responsible for maintaining the mitochondrial membrane potential. The mitochondrial PTP consists of three proteins: voltage-dependent anion channel 1 (VDAC1), adenine nucleotide transporter (ANT), and cyclophilin D (1). VDAC1 and ANT are integral membrane proteins of the outer and inner mitochondrial membranes, respectively. Together, these two proteins form the conductance channel of the mitochondrial PTP. Cyclophilin D is a mitochondrial matrix protein that associates with ANT and modulates pore function. Under normal physiological conditions, the mitochondrial PTP is maintained in a closed state, allowing mitochondria to remain polarized. Stressful stimuli, such as oxidative stress and accumulation of unfolded proteins, lead to the opening of the mitochondrial PTP (1). As a result, the mitochondrial membrane potential is dissipated, causing depolarization of mitochondria, decrease in ATP production, swelling of mitochondria, efflux of mitochondrial calcium, generation of reactive oxygen species (ROS), and release of pro-apoptotic proteins (cytochrome c and apoptosis-inducing factor (AIF)) (2). These events trigger the apoptotic cascade, resulting in cell death.
Numerous studies have implicated mitochondrial dysfunction as a causative factor in neurodegenerative diseases including Parkinson disease (PD) (3–5). The level of the NADPH ubiquinone reductase component of complex I in the mitochondrial electron transport chain is significantly reduced in the substantia nigra of PD patients (6, 7). Additionally, the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) that recapitulates Parkinsonian symptoms in humans, non-human primates, and mice acts by inhibiting complex I of the electron transport chain after being converted into its active metabolite 1-methyl-4-phenylpyridinium (MPP+) (8). Further, MPP+ administration to cultured cells results in depolarization of mitochondria due to the opening of the mitochondrial PTP (9). Exaggerated opening of the mitochondrial PTP has also been reported in response to α-synuclein overexpression (10) and 6-hydroxydopamine (11). Opening of the mitochondrial PTP therefore appears to be one of the most prominent cell death-triggering mechanisms in PD models. Strategies aimed at preventing the opening of the mitochondrial PTP might have therapeutic potential in PD.
MicroRNAs (miRs) belong to a class of small, non-coding, regulatory RNAs that bind to the 3′-UTR of target mRNA, thereby reducing target protein level. miRs are important regulators of neuronal function, both in normal physiological state as well as in pathological conditions. Differential expression and function of miRs have been linked to the pathogenesis of several neurodegenerative diseases (12–14). For example, miR-7 has been suggested as a player in the pathogenesis of PD. miR-7 could functionally target 3′-UTR of α-synuclein mRNA, resulting in decrease in its mRNA and protein levels, which could potentially help to prevent aggregation of α-synuclein, a prominent feature of PD pathology (15, 16). In addition, miR-7 has been shown to protect neuronal cells against the cytotoxic effect of MPP+ by down-regulating another target, RelA, through promoting glycolysis (17, 18).
Herein, we show that miR-7 regulates the function of mitochondrial PTP by targeting the 3′-UTR of VDAC1 mRNA, which resulted in a decrease of VDAC1 mRNA and protein levels. Consequently, miR-7 prevents MPP+-induced opening of mitochondrial PTP, thereby conferring neuroprotection.
Experimental Procedures
Materials
MPP+ was purchased from Sigma. 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1), 2,7-dichlorodihydrofluorescein diacetate (DCF-DA), and propidium iodide (PI) were purchased from Molecular Probes. Rhod2-AM was purchased from Invitrogen.
Animals
All animals were housed and handled in accordance with the Institutional Animal Care guidelines of Rutgers-Robert Wood Johnson Medical School. C57BL/6J mice purchased from The Jackson Laboratory were used in this study.
Cloning and Site-directed Mutagenesis
VDAC1 3′-UTR was amplified from genomic DNA using the following primers: forward primer, 5′-CAATATCTAGAATGAATACTGTACAATTGTTT-3′, and reverse primer, 5′-GGCCGTCTAGAGTTTATTTATATTTTATTAAT-3′. The PCR product was cloned into pGL4.51 luciferase reporter vector (Promega). The predicted miR-7 binding site in VDAC1 3′-UTR was mutated with QuikChange site-directed mutagenesis kit (Agilent Technology) using the following primers: 5′-GGGTACATTTTAGAGTCTAGCATTTTGTTGGAATTAGA-3′ and 5′-TCTAATTCCAACAAAATGCTAGACTCTAAAATGTACCC-3′. VDAC1 coding sequence was amplified from human brain cDNA library using the following primers: forward primer, 5′-ATAAAGGATCCATGGCTGTGCCACCCACGTAT-3′, and reverse primer, 5′-CGGGCCTCGAGTTATGCTTGAAATTCCAGTCC-3′. The PCR product was cloned into pcDNA 3.1 vector using BamHI and XhoI restriction sites, and then named pcDNA3.1-VDAC1. All sequences were confirmed by DNA sequencing.
Cell Culture and Transfections
SH-SY5Y neuroblastoma cells obtained from ATCC were maintained in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen). Cells were transfected with pre-miR-scrambled (miR-SC) (Ambion), pre-miR-7 (Ambion), siRNA-NT (non-targeting siRNA; Ambion), siRNA-human VDAC1 (Ambion), anti-miR-SC (Ambion), and anti-miR-7 (Ambion) by using Lipofectamine RNAiMAX according to the manufacturer's instructions. Lipofectamine 2000 (Invitrogen) was used for transfection of pcDNA3.1-VDAC1, pGL4.51-VDAC1-3′-UTR, and pGL4.51-mutant VDAC1-3′-UTR plasmids according to the manufacturer's instructions. Primary mouse cortical neurons were isolated from embryonic day 18 embryos and cultured as described previously (15). Neurons were allowed to differentiate in Neurobasal medium (Life Technologies) containing B27 supplement (Life Technologies), penicillin/streptomycin (Life Technologies), and GlutaMAX (Life Technologies) for 10 days before experiments. Primary neurons were transfected with siRNA-mouse VDAC1 (Ambion) or siRNA-NT using Lipofectamine RNAiMAX.
Lentivirus Production
Lenti-miR-SC and lenti-miR-7 were packaged in HEK293T cells using the packaging vectors pDMG.2 and psPAX as described previously (18). Lentiviral particles were concentrated using Lenti-X Concentrator (Clontech) according to the manufacturer's protocol. Lentiviral titer was estimated by counting the number of RFP-positive cells after the transduction of serially diluted virus. Approximately 1 × 108 transduction units/ml were used for experiments.
Cell Viability Assay
Cell viability was measured using the CellTiter 96 AQueous cell proliferation assay (Promega) according to the manufacturer's protocol.
Isobaric Tag for Relative and Absolute Quantification (iTRAQ) Labeling and Mass Spectrometry
iTRAQ labeling and mass spectrometry were performed as described previously (18).
RNA Isolation and Quantitative Real-time PCR
Total RNA was extracted from SH-SY5Y cells using the miRNeasy kit (Qiagen) following the manufacturer's instructions. cDNA was obtained by reverse transcription using the Superscript RT III kit (Invitrogen). Quantitative real-time PCR was performed using SYBR Select Master Mix (Life Technologies) using the Applied Biosystems 7500 real-time PCR system. The following PCR primers were used: VDAC1, 5′-CGGAAGGCAGAAGATGGCTGT-3′ and 5′-CCCGTCACTTTGGTGGTCTC-3′; 18S, 5′-CGGCTACCACATCCAAGGAA-3′ and 5′-GCTGGAATTACCGCGGCT-3′. Relative mRNA expression level was normalized to 18S rRNA and calculated by the 2−ΔΔCt method.
Reporter Gene Assay
Cells were lysed with the Glo lysis buffer (Promega), and luciferase activity was measured using the Steady-Glo luciferase assay system (Promega) with a Wallac 1420 multilabel counter (PerkinElmer). β-Galactosidase activity was determined using chlorophenol Red-β-d-galactopyranoside (Roche Applied Science) and reaction buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 1 mm MgSO4, 10 mm KCl, 50 mm 2-mercaptoethanol, pH 7.0). Luciferase activity was normalized to β-galactosidase activity.
Western Blotting
Cells were lysed with PBS containing 2% SDS with protease inhibitor cocktail and phosphatase inhibitors (Roche Applied Science). Cell lysates were sonicated for 30 s, and protein concentration in the lysates was quantified using the BCA protein assay kit (Thermo Scientific). Proteins were resolved using 4–20% SDS-PAGE gels, followed by transfer to PVDF membrane (Millipore). Membrane was blocked with 5% skim milk. The following primary antibodies were used for Western blotting: anti-VDAC1 (Santa Cruz Biotechnologies, N-18), anti-β-actin (catalogue number A5316, Sigma-Aldrich), followed by incubation with horseradish peroxidase-conjugated anti-goat (catalogue number HAF008, R&D Systems) or anti-mouse antibody (catalogue number HAF007, R&D Systems). Western blots were quantified by densitometry using ImageJ (National Institutes of Health).
Subcellular Fractionation
Mitochondrial and cytosolic fractions were isolated using the mitochondrial fractionation kit (Active Motif) according to the manufacturer's protocol. Protein extracts from the mitochondria and cytosol were resolved using SDS-PAGE. The following antibodies were used for detection of protein levels by Western blotting analysis: anti-cytochrome c (Santa Cruz Biotechnologies, H-104), anti-AIF (Santa Cruz Biotechnologies, D-20), anti-TOM20 (Santa Cruz Biotechnologies, FL-145), and anti-β-tubulin (Sigma, catalogue number T-4026).
Immunocytochemistry
Immunocytochemistry was performed as described previously (4). In brief, cells grown on glass coverslips were fixed in 4% paraformaldehyde and permeabilized with 0.4% Triton X-100 (Sigma) in PBS, followed by blocking with 1% bovine serum albumin (Sigma) and 0.25% Triton X-100 in PBS. Cells were incubated with anti-TOM20 antibody (Santa Cruz Biotechnology, FL-145) for 1 h at room temperature, followed by FITC-conjugated anti-rabbit antibody (Sigma). Nuclei were counterstained with DAPI. Coverslips were mounted using Prolong Diamond mounting media (Life Technologies) and imaged using a Zeiss LSM 710 confocal microscope equipped with a built-in camera using a 63× water immersion objective at room temperature. To quantify mitochondrial fragmentation, cells were categorized in two groups according to their mitochondrial morphology: tubular (interconnected) and fragmented (clumping) as reported previously (19). Five distinct microscopic fields containing at least 20 cells were analyzed for each sample.
Measurement of Intracellular ROS Level
Intracellular ROS levels were measured with DCF-DA as described previously (20). Briefly, the cells were incubated with 10 μm DCF-DA for 60 min at 37 °C in the dark. The cells were then washed with PBS. The fluorescence intensities were measured using excitation and emission filters of 485 and 535 nm, respectively, in a Wallac1420 Victor 2 microplate reader (PerkinElmer).
JC-1 Labeling
JC-1 labeling was performed as described previously (21). In brief, cells were loaded with JC-1 dye (5 μg/ml) for 30 min at 37 °C in the dark. Cells were washed in PBS and immediately imaged using an Axiovert 2000 fluorescent microscope (Carl Zeiss) equipped with an AxioCam MRm camera using 20× air objective (numerical aperture 0.55). Images were imported into ImageJ and converted to 8 bits, and fluorescent intensity was measured.
Rhod2-AM Labeling
Cells were seeded onto glass bottom 35-mm dish (MatTek). After transfection and treatment, Rhod2-AM labeling was performed according to the manufacturer's protocol. In brief, cells were loaded with Rhod2-AM at a concentration of 5 μm for 30 min at 37 °C in the dark. Live cell imaging was performed using a Zeiss LSM 710 confocal microscope equipped with an environmental chamber to maintain temperature at 37 °C and CO2 concentration at 5%. Imagings were performed using 63× water immersion objective. Images were quantified using Zen lite software.
PI Staining
SH-SY5Y cells seeded on glass coverslips were co-transfected as indicated in the figure. Notably, a 10 times higher amount of pcDNA3.1-VDAC1 was co-transfected with EGFP-C1 into SH-SY5Y cells to ensure that nearly all GFP-positive cells express the transfected VDAC1 construct. Forty-eight hours after transfection, cells were treated with 2 mm MPP+ for 24 h. Cells were then labeled with PI (10 μg/ml), washed, and imaged using an Axiovert 2000 fluorescent microscope equipped with an AxioCam MRm camera using a 20× air objective (numerical aperture 0.55). For quantifying the number of dead cells in each sample, cells that were successfully transfected (GFP-positive) and labeled with PI were counted. Five distinct microscopic fields were quantified for each sample.
Statistical Analysis
Data were representative of three sets of independent experiments performed in triplicates for each group. Data are presented as means ± S.E. and were analyzed by two-way analysis of variance followed by Bonferroni's post hoc test using GraphPad Prism software unless otherwise stated. The level of significance was set at p < 0.05.
Results
miR-7 Regulates the Expression of Mitochondrial Proteins
We performed proteomic analysis to determine the miR-7 target profile. Human neuroblastoma cells, SH-SY5Y, were transfected with miR-7 or a scrambled control, miR-SC. Changes in protein expression were quantified using an iTRAQ-based proteomic platform. As miRs mostly down-regulate the expression of their target proteins, we focused on the proteins that were significantly (p < 0.05) reduced in the miR-7-transfected cells. One hundred and eighty-five proteins were found to be significantly down-regulated with a -fold change of <0.8 in the miR-7-transfected cells (supplemental Table 1). To identify over-represented groups of proteins, we performed gene ontology (GO) analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) (22, 23). Three out of the top ten enriched GO terms pertained to the mitochondria, namely, mitochondrion, mitochondrial part, and mitochondrial large ribosome (Fig. 1A). Nineteen down-regulated proteins, belonging to the GO term mitochondrial part, are listed (Table 1). Therefore, we postulate that miR-7 regulates the expression of mitochondrial proteins and could play a crucial role in modulating mitochondrial function.
FIGURE 1.
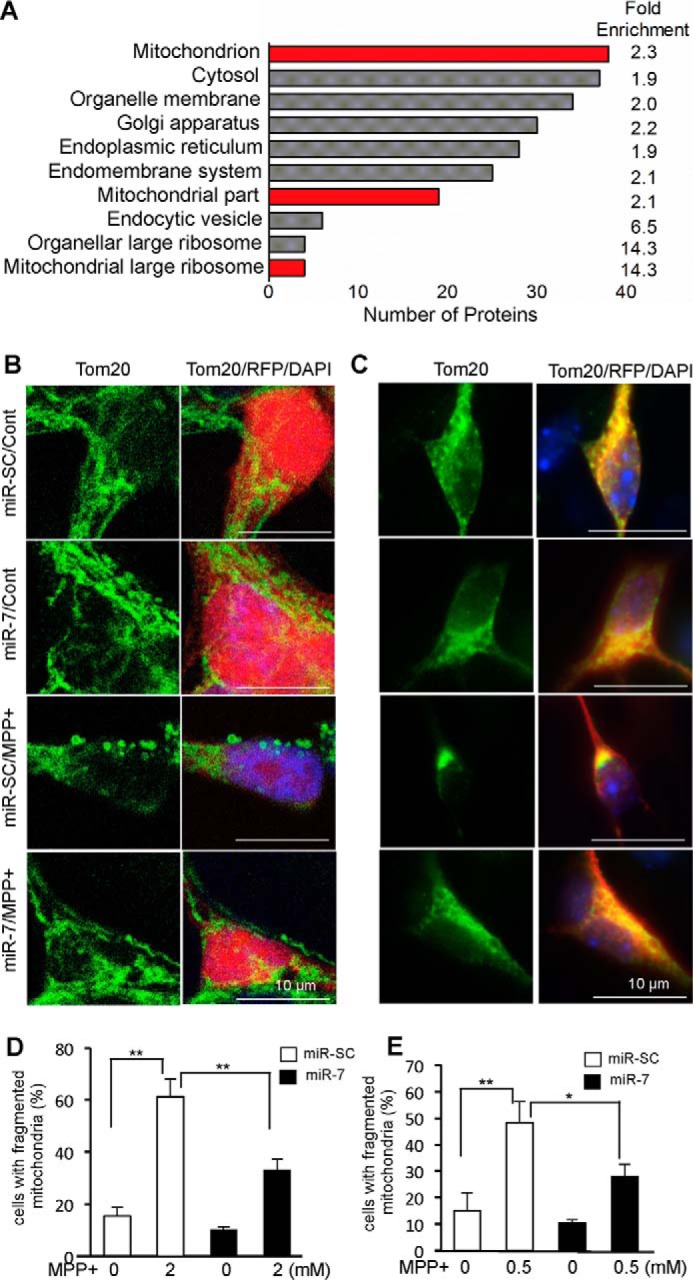
miR-7 regulates mitochondrial function. A, gene ontology analysis of proteins significantly down-regulated by miR-7. Mitochondrial proteins were significantly enriched. GO terms related with mitochondria are indicated in red. B and C, miR-7 inhibits the fragmentation/clumping of mitochondria in response to MPP+. SH-SY5Y cells (B) or primary mouse cortical neurons (C) were transduced with lenti-miR-SC or lenti-miR-7 (red). Three days after transduction, cells were treated with 2 mm (SH-SY5Y) or 0.5 mm MPP+ (primary neurons) for 12 h. Cells were stained with anti-TOM20 (green). D and E, quantification of mitochondrial fragmentation (clumping) was performed in SH-SY5Y (D) and primary neurons (E). These results (B–E) are representatives of three sets of independent experiments. *, p < 0.05, **, p < 0.01.
TABLE 1.
List of proteins significantly downregulated by miR-7 belonging to the GO term “mitochondrial part”
| Gene I.D. | Gene name | -Fold change (miR-7/miR-SC) | p value |
|---|---|---|---|
| AGK | Homo sapiens multiple substrate lipid kinase | 0.6 | 0.005 |
| COX11 | Cytochrome c oxidase assembly protein COX11, mitochondrial | 0.6 | 0.03 |
| TIMM9 | Mitochondrial import inner membrane translocase subunit Tim9 | 0.6 | 0.03 |
| ACO2 | Aconitate hydratase, mitochondrial | 0.7 | 0.005 |
| ACSL4 | Long-chain fatty-acid-CoA ligase 4 | 0.7 | 0.005 |
| SRGAP2 | SLIT-ROBO Rho GTPase-activating protein 2 | 0.7 | 0.01 |
| ABCB10 | ATP-binding cassette sub-family B member 10, mitochondrial | 0.8 | 0.03 |
| APEX2 | DNA-apurinic or -apyrimidinic site lyase 2 | 0.8 | 0.04 |
| BCKDHA | H. sapiens branched chain keto acid dehydrogenase E1, α-polypeptide | 0.8 | 0.03 |
| COX6B1 | Cytochrome c oxidase subunit 6B1 | 0.8 | 0.005 |
| CYB5R3 | NADH-cytochrome b5 reductase 3 | 0.8 | 0.03 |
| MRPL10 | Mitochondrial ribosomal protein L10 | 0.8 | 0.03 |
| MRPL12 | 39S ribosomal protein L12, mitochondrial | 0.8 | 0.005 |
| MRPL27 | 39S ribosomal protein L27, mitochondrial | 0.8 | 0.005 |
| MRPL35 | 39S ribosomal protein L35, mitochondrial | 0.8 | 0.03 |
| MRPL52 | 39S ribosomal protein L52, mitochondrial | 0.8 | 0.005 |
| NLRX1 | NLR family member X1 | 0.8 | 0.02 |
| SARM1 | Sterile α and TIR motif-containing protein 1 | 0.8 | 0.005 |
| VDAC1 | Voltage-dependent anion-selective channel protein 1 | 0.8 | 0.04 |
miR-7 Modulates Mitochondrial Morphology
Our proteomics analysis prompted us to investigate the mitochondrial biology in response to miR-7. First, we observed the mitochondrial morphology. To this end, we employed MPP+, which is well known to induce mitochondrial fragmentation by blocking complex I activity of the mitochondrial electron transport chain (24). Although a small percentage (15 ± 3.4%) of cells demonstrated fragmented mitochondria as evidenced by the appearance of small and round mitochondria in control SH-SY5Y cells (serum-free condition for 12 h), MPP+ (2 mm) exposure for 12 h led to a significant increase in mitochondrial fragmentation in 61.2 ± 6.8% cells (Fig. 1, B and D). However, overexpression of miR-7 by transducing lenti-miR-7 significantly reduced the percentage of cells having fragmented mitochondria to 33.2 ± 4.0% in response to MPP+ (Fig. 1, B and D). We also confirmed this effect of miR-7 using mouse primary cortical neurons. MPP+ (0.5 mm) treatment resulted in mitochondrial fragmentation and clumping in 48.3 ± 7.9% of mouse primary cortical neurons infected with lenti-miR-SC (Fig. 1, C and E). However, lenti-miR-7 infection exhibited significantly less mitochondrial fragmentation (27.9 ± 4.6%) when compared with lenti-miR-SC (Fig. 1, C and E).
miR-7 Regulates Mitochondrial Membrane Potential
Next, we investigated whether miR-7 affects mitochondrial membrane potential by using JC-1. JC-1 is a lipophilic, cationic dye that can selectively enter into mitochondria of healthy cells and forms J-aggregates with red fluorescence (emission 590 nm). Following exposure to cytotoxic stimuli such as MPP+, mitochondria are depolarized. As a result, J-aggregates fail to form and JC-1 remains in the cytosol as a diffuse green staining (emission 529 nm). The ratio of red/green fluorescent intensity therefore indicates the polarization state of mitochondria, with healthy mitochondria having a higher red/green intensity ratio. Exposure to MPP+ for 12 h leads to depolarization of mitochondria in SH-SY5Y cells, observed as an increase in green fluorescence and a decrease in red/green intensity ratio (Fig. 2A, second row, quantified in Fig. 2C). However, overexpression of miR-7 prevented mitochondrial depolarization after MPP+ treatment (Fig. 2A, bottom panel) as evidenced by a significant increase in red/green fluorescent intensity ratio when compared with cells transfected with miR-SC (Fig. 2, A and C). To confirm that miR-7 helps to maintain mitochondrial membrane potential in post-mitotic neurons, we transduced primary mouse cortical neurons with lenti-miR-SC or lenti-miR-7. As the lentiviral constructs express an RFP reporter, we could only quantify the increase in JC-1 green fluorescence intensity as a marker for mitochondrial depolarization upon exposure to MPP+. Similar to our observation in SH-SY5Y cells, treatment with MPP+ led to mitochondrial depolarization in primary cortical neurons, as evidenced by an increase in green signal (Fig. 2B, second row, quantified in Fig. 2D), which was prevented by miR-7 (Fig. 2, B and D).
FIGURE 2.

miR-7 regulates the function of mitochondrial PTP. A–D, miR-7 prevents depolarization of mitochondria in response to MPP+. A, SH-SY5Y cells were transfected with miR-SC or miR-7. Forty-eight hours after transfection, cells were treated with 2 mm MPP+ for 12 h. Cells were labeled with JC-1 and imaged. Upon exposure to MPP+, mitochondria of cells transfected with miR-SC were depolarized (green). B, primary mouse neurons were transduced with lenti-miR-SC or lenti-miR-7 (red). Three days after transduction, cells were treated with 0.5 mm MPP+ for 12 h. Upon exposure to MPP+, mitochondria of cells transduced with lenti-miR-SC were depolarized (green). Red fluorescence indicates the lentiviral transduction. C, quantification of JC-1 fluorescence intensities from A. The ratio of red (healthy mitochondria) to green (depolarized mitochondria) fluorescent intensities was calculated from five different microscopic fields for each sample. D, quantification of JC-1 fluorescence intensities from B. Green (depolarized mitochondria) fluorescent intensity was measured from at least 10 transduced (RFP-positive) neurons for each sample. These results (A–D) are representatives of three sets of independent experiments. E, miR-7 prevents release of pro-apoptotic proteins from mitochondria to cytosol. SH-SY5Y cells were transduced with lenti-miR-SC or lenti-miR-7. Three days after transduction, cells were treated with 0.5 mm MPP+ for 6 or 12 h. Cells were lysed, and mitochondrial and cytosolic fractions were isolated. Western blotting analysis was performed. The arrow denotes the position of cytochrome c (Cyt C). This result is a representative of two sets of independent experiments. F and G, miR-7 lowers ROS level in response to MPP+ in SH-SY5Y cells (F) and primary mouse cortical neurons (G). Forty-eight hours after transfection, cells were treated with the indicated concentrations of MPP+ for 12 h in SH-SY5Y cells. Primary mouse neurons were transduced with lenti-miR-SC or lenti-miR-7. Three days after transduction, cells were treated with 0.5 mm MPP+ for 12 h. Data are shown as the means ± S.E. *, p < 0.05, ***, p < 0.005. These results (F and G) are representatives of three sets of independent experiments performed in triplicates for each group.
miR-7 Regulates Function of Mitochondrial PTP
Depolarization of the mitochondria in response to cytotoxic stimuli occurs due to opening of the mitochondrial PTP (25). As miR-7 significantly inhibited mitochondrial depolarization following MPP+ treatment, we investigated whether miR-7 inhibits the opening of the mitochondrial PTP. For this, we isolated the mitochondrial and cytosolic fractions, followed by Western blotting analysis to detect release of mitochondrial proteins through the mitochondrial PTP. Exposure to MPP+ led to an increase in the pro-apoptotic proteins, cytochrome c and AIF, in the cytosolic fraction. However, overexpression of miR-7 attenuated the release of these proteins as evidenced by lower cytosolic levels of AIF and cytochrome c when compared with miR-SC-transfected cells (Fig. 2E), suggesting that miR-7 inhibits the opening of mitochondrial PTP. TOM20 was used as a marker for mitochondrial fraction, and β-tubulin was used as a marker for the cytosolic fraction. In addition, opening of mitochondrial PTP also increases the level of ROS (25). We quantified intracellular ROS levels using DCF-DA, an ROS-sensor probe. SH-SY5Y cells transfected with miR-7 or mouse primary cortical neurons transduced with lenti-miR-7 appeared to have a lower basal level of intracellular ROS (Fig. 2, F and G). Treatment with MPP+ led to a dose-dependent increase in ROS generation. However, in cells overexpressing miR-7, this increase was abrogated and these cells showed significantly lower intracellular ROS levels with all doses of MPP+ tested. Taken together, these results indicate that miR-7 regulates mitochondrial PTP function and prevents its opening.
miR-7 Targets the Mitochondrial PTP Component Protein, VDAC1
As our results demonstrated that miR-7 regulates function of the mitochondrial PTP, we reviewed our proteomics data to identify whether any of the 19 “mitochondrial part” proteins that were significantly down-regulated by miR-7 were associated with the PTP. Indeed, we found that VDAC1 was down-regulated with a -fold change of 0.8 in the proteomic data. VDAC1 is an integral protein of the mitochondrial outer membrane and forms the channel for the mitochondrial PTP. Overexpression of miR-7 reduced the level of VDAC1 protein by 55% (Fig. 3A) in SH-SY5Y cells, confirming the observation from our proteomic study. Further, overexpression of miR-7 in mouse primary cortical neurons also led to a significant decrease in VDAC1 expression by 42% (Fig. 3B). We performed quantitative real-time PCR analysis to determine whether overexpression of miR-7 resulted in degradation of VDAC1 mRNA as well. Certainly, miR-7 led to a 60% decrease in VDAC1 mRNA levels (Fig. 3C). Further, we wanted to determine whether endogenous miR-7 is responsible for regulation of VDAC1 expression by transfecting SH-SY5Y cells with miR-7 inhibitor (anti-miR-7) or control inhibitor (anti-miR-SC). Inhibition of miR-7 dramatically increased VDAC1 protein levels (Fig. 3D), suggesting that endogenous miR-7 represses VDAC1 expression. To identify the potential miR-7 binding site in the VDAC1 3′-UTR, we employed a prediction algorithm from TargetScan. We found a potential miR-7 target site in the 3′-UTR of VDAC1 mRNA, which is conserved in the human, chimpanzee, rhesus, rat, and mouse (Fig. 3E). To investigate whether miR-7 directly targets the 3′-UTR of VDAC1, this 3′-UTR was inserted downstream of the firefly luciferase reporter gene. Co-expression of miR-7 along with VDAC1 3′-UTR luciferase reporter vector led to a significant decrease in luciferase activity when compared with co-expression of this vector with miR-SC. Also, miR-7 significantly decreased luciferase activity from the VDAC1 3′-UTR construct, but had no effect on pGL4.51 construct devoid of VDAC1 3′-UTR (Fig. 3F). To further verify that the predicted miR-7 binding site on VDAC1 3′-UTR is essential for its function, we mutated this site and performed the luciferase reporter assay. As expected, miR-7 was unable to suppress luciferase reporter expression from the mutated VDAC1 3′-UTR, confirming the authenticity of the predicted binding site. Expression of VDAC1 was reported to increase in several PD models, including the MPP+ model (26, 27). To determine whether miR-7 could prevent the increase in VDAC1 expression in the pathological context, SH-SY5Y cells transfected with miR-SC or miR-7 were treated with 2 mm MPP+ for 12 h. Indeed, MPP+-induced increase in VDAC1 protein level was abrogated in the miR-7-transfected cells (Fig. 3G). Therefore, we concluded that miR-7 directly targets VDAC1 and reduces its expression even after exposure to MPP+.
FIGURE 3.

miR-7 down-regulates the mitochondrial PTP component VDAC1. A and B, miR-7 down-regulates VDAC1 protein expression. SH-SY5Y cells (A) were transfected with miR-SC or miR-7. Mouse primary cortical neurons (B) were infected with lenti-miR-SC or lenti-miR-7 for 72 h. Cells overexpressing miR-7 had significantly lower VDAC1 levels. Representative figure and quantification are shown. VDAC1 band intensity was normalized to that of β-actin, whose expression is not affected by miR-7. C, miR-7 down-regulates VDAC1 mRNA levels. SH-SY5Y cells were transfected with miR-SC or miR-7. D, inhibition of endogenous miR-7 increases VDAC1 protein expression. SH-SY5Y cells were transfected with anti-miR-SC or anti-miR-7. Representative figure and quantification are shown. E, schematic diagram showing conserved miR-7 seed match in the 3′-UTR of VDAC1 mRNA. F, miR-7 targets VDAC1 3′-UTR. SH-SY5Y cells were transfected with miR-SC or miR-7 along with pGL4.51 vector or VDAC1 3′-UTR or mutant VDAC1 3′-UTR. miR-7-transfected cells suppressed luciferase expression from VDAC1 3′-UTR, but failed to do so when the miR-7 binding site was mutated in the VDAC1 3′-UTR. G, miR-7 abrogates MPP+-induced increase in VDAC1 protein expression. SH-SY5Y cells were transfected with miR-SC or miR-7. After 48 h, cells were treated with 2 mm MPP+ for 12 h followed by Western blotting analysis for VDAC1. β-Actin was used as loading control. Data are shown as the means ± S.E. Student's t test was used for data analysis in A–C. *, p < 0.05, ***, p < 0.005. Results are representatives of two independent experiments (A, B, D, and G) and three separate experiments performed in triplicates for each group (C and F).
Knockdown of VDAC1 Decreases ROS Production and Cell Death against MPP+
As miR-7 targets the expression of VDAC1, we set out to determine whether silencing VDAC1 expression confers protection against MPP+. Similar to overexpression of miR-7, knockdown of VDAC1 prevented ROS generation (Fig. 4A) and protected cells from the cytotoxic effect of MPP+ (Fig. 4B) in SH-SY5Y cells. Successful knockdown of VDAC1 by transfection of siRNA-VDAC1 was confirmed using Western blotting (Fig. 4C). Similarly, silencing VDAC1 expression in mouse primary cortical neurons led to a significant increase in cell survival upon MPP+ (Fig. 4D). In addition, sensitized cell death resulting from inhibition of endogenous miR-7 in response to MPP+ was significantly mitigated in SH-SY5Y when VDAC1 expression was silenced (Fig. 4E).
FIGURE 4.

Knockdown of VDAC1 protects cells against MPP+. A, VDAC1 knockdown reduces intracellular ROS production in response to MPP+. SH-SY5Y cells were transfected with siRNA-NT or siRNA-VDAC1. Forty-eight hours after transfection, cells were treated with the indicated concentrations of MPP+ for 12 h. B, VDAC1 knockdown protects cells from the cytotoxic effect of MPP+. SH-SY5Y cells were transfected with siRNA-NT or siRNA-VDAC1. Forty-eight hours after transfection, cells were treated with the indicated concentrations of MPP+ for 24 h, and cell viability was measured. C, a representative Western blot showing effective down-regulation of VDAC1 after transfection of siRNA-VDAC1. β-Actin was used as loading control. This result is a representative of three separate experiments. NT, non-targeting. D, VDAC1 knockdown increases cell survival in mouse primary cortical neurons following 1 mm MPP+ exposure for 24 h. E, VDAC1 knockdown prevents anti-miR-7-induced sensitization against the cytotoxic effect of MPP+. SH-SY5Y cells were transfected as indicated above. Forty-eight hours after transfection, cells were treated with the indicated concentrations of MPP+ for 24 h, and cell viability was measured. Data are shown as the means ± S.E., *, p < 0.05, **, p < 0.01, ***, p < 0.005. These results (A, B, D, and E) are representatives of three separate experiments performed in triplicates for each group.
Overexpression of VDAC1 Abrogates Protective Effect of miR-7 on Cell Death and Mitochondrial Function
To study whether miR-7-mediated decrease in VDAC1 expression underlies the cytoprotective effect of miR-7 against MPP+, SH-SY5Y cells were transfected with plasmid containing VDAC1 cDNA without its 3′-UTR (pcDNA3.1-VDAC1), along with pre-miR-7. This approach restores VDAC1 levels despite down-regulation of endogenous VDAC1 by miR-7. We performed PI staining to determine cell death in SH-SY5Y cells transfected as indicated. We observed that the proportion of PI-positive (dead) cells dramatically increases to 67% upon MPP+ treatment, whereas overexpression of miR-7 decreases PI-positive cells to 18% (Fig. 5, A and B). Notably, overexpression of VDAC1 partly abolished the protective effect of miR-7 against MPP+ (Fig. 5, A and B), as evidenced by an increase in PI-positive cells from 18% in miR-7 and pcDNA3.1 co-transfected cells to 55% in miR-7 and VDAC1 co-transfected cells after exposure to MPP+. This result demonstrates that the cytoprotective effect of miR-7 against MPP+ in part requires the down-regulation of VDAC1 expression. Additionally, we measured mitochondrial calcium efflux using Rhod2-AM. Rhod2-AM is a calcium sensor dye that preferentially compartmentalizes to the mitochondria. Treatment with MPP+ triggers opening of mitochondrial PTP and efflux of calcium through the pore. This event can be detected as a decrease in red fluorescence after Rhod2-AM labeling. Transfection of miR-7 prevented efflux of calcium from mitochondria after treatment of MPP+ (Fig. 5, C and D). However, overexpression of VDAC1 abrogated this effect of miR-7. Further, overexpression of VDAC1 mitigated the miR-7-induced decrease in intracellular ROS generation in response to MPP+ (Fig. 5E). Taken together, we conclude that miR-7 regulates the function of mitochondrial PTP and protects against MPP+-induced cytotoxicity by targeting VDAC1.
FIGURE 5.

Overexpression of VDAC1 abrogates the mitochondrial protective function of miR-7. A and B, overexpression of VDAC1 abolishes the cytoprotective effect of miR-7 against MPP+-induced cell death. SH-SY5Y cells were co-transfected as indicated above. After 48 h, cells were treated with 2 mm MPP+ for 24 h followed by labeling of dead cells with PI. Representative images of MPP+-treated samples are shown in A. Arrows indicate dual (GFP and PI) positive cells that were counted as dead cells. Quantification of images is shown in B. C and D, overexpression of VDAC1 inhibits miR-7-induced decrease in mitochondrial calcium efflux. Plasmid pEGFP-C1 was also co-transfected in all samples, followed by treatment with 2 mm MPP+ for 12 h. Quantification of Rhod2-AM fluorescent intensities was performed from at least 10 cells among GFP-positive cells in each sample. These results (in A–D) are representatives of three separate experiments. E, overexpression of VDAC1 abolishes miR-7-induced decrease in intracellular ROS production. SH-SY5Y cells were transfected as indicated above. Forty-eight hours after transfection, cells were treated with the indicated concentrations of MPP+ for 12 h, and ROS levels were measured. This result (in E) is a representative of three separate experiments performed in triplicates for each group. Data are shown as the means ± S.E. ***, p < 0.005.
Discussion
In the present study, we have demonstrated that miR-7 prevents depolarization of mitochondria in response to MPP+ by directly down-regulating VDAC1. Consequently, MPP+-induced calcium efflux and cytochrome c release from mitochondria to cytosol were attenuated by miR-7. This effect of miR-7 contributes to cytoprotection.
VDAC1 is an integral protein of the mitochondrial outer membrane. Along with ANT in the inner membrane of the mitochondria and cyclophilin D in the mitochondrial matrix, VDAC1 forms the mitochondrial PTP (1). The amino-terminal domain of VDAC1 is thought to function as the gating voltage sensor for the pore (28). Opening of the mitochondrial PTP occurs in response to cytotoxic stimuli, leading to dissipation of mitochondrial membrane potential as well as release of calcium and pro-apoptotic proteins that ultimately trigger cell death (1). miR-7 is able to attenuate this course of events by down-regulating VDAC1. We also showed that overexpression of VDAC1 partly mitigates the cytoprotective effect of miR-7, indicating that down-regulation of VDAC1 is a crucial component of the cytoprotective effect of miR-7.
Mitochondrial dysfunction, including fragmentation of the mitochondrial network, opening of PTP, and elevated VDAC1 expression, appears to be a common condition in several cellular and animal models of neurodegenerative disorders including PD (26, 29–34). VDAC1 protein and mRNA levels were reported to be elevated following administration of mitochondrial complex I inhibitors, such as rotenone and MPP+ (26, 27). VDAC1 expression was also increased in striatal extracts of 6-hydroxydopamine-treated rats (35). Another study reported that VDAC1 protein level is elevated in the striatum and cortex of parkin null mice (36). It was reported that VDAC1 is a substrate for Parkin whose loss of function mutations have been linked to familial forms of PD (37). As VDAC1 is a Parkin substrate, it is normally ubiquitinated and targeted for degradation in the presence of Parkin. Loss of function mutation of Parkin may therefore result in increased VDAC1 expression, thereby making neurons more vulnerable to mitochondrial toxins. Therefore, it appears that increased level of VDAC1 is a potential early marker of cell death in PD models and silencing of VDAC1 by miR-7 can attenuate the initiation of apoptotic events.
Aggregation of misfolded proteins has been reported to cause opening of the mitochondrial PTP (1). Interestingly, human α-synuclein was found to associate with VDAC1 in the brain stem, striatum, and cortex of transgenic mice expressing a pathogenic mutant form (A53T) of human α-synuclein (10). The mitochondria associated with human α-synuclein A53T appeared to be swollen, an indication of opening of the mitochondrial PTP (10). This finding suggests that increased level of α-synuclein in vivo could play a role in opening the mitochondrial PTP. Notably, we and others have previously reported the neuroprotective role of miR-7 by showing its ability to target α-synuclein expression (15, 16). Therefore, down-regulation of both proteins, VDAC1 and α-synuclein, by miR-7 could synergistically result in inhibition of opening of mitochondrial PTPs in transgenic α-synucleinopathy models.
In previous studies, we have also demonstrated that miR-7 attenuates neurotoxic effects of MPP+ by down-regulating RelA, which belongs to the nuclear factor κB (NF-κB) family of transcription factors (17, 18). In the absence of any stimulus, RelA is sequestered in the cytosol by a protein, inhibitor of κB (IκB). Interestingly, a recent study demonstrated that IκB also localizes to the mitochondria, where it associates with VDAC1 and stabilizes the closed state of the PTP (38). It would be interesting to investigate whether miR-7, by down-regulating RelA, makes more IκB available to the mitochondria. This might represent an additional mechanism by which miR-7 exerts its mitochondrial protective function.
In addition to PD, VDAC1 has also been implicated in the pathogenesis of other neurodegenerative diseases as well (30). Significantly increased VDAC1 level was reported in Alzheimer disease patients relative to control subjects (39). Mouse neuronal cells expressing a variant of human TATA box-binding protein harboring an expanded polyglutamine tract form intranuclear aggregates. These cells also show up-regulation of the VDAC1 and consequently increased cytochrome c release from the mitochondria (29). Silencing of VDAC1 expression by miR-7 could therefore be a generalized cellular mechanism to maintain healthy mitochondria and improve neuronal physiology. In support of this, VDAC+/− mice have higher levels of the synaptic genes, such as synaptophysin, synapsin, synaptobrevin, neurogranin, and PSD95, when compared with wild type. Additionally, these mice showed lower free radical production and lipid peroxidation levels, whereas cytochrome oxidase activity and ATP levels were elevated (30). These findings indicate that VDAC1+/− mice have improved mitochondrial functional profile and reducing VDAC1 expression may be beneficial for neurons.
In conclusion, our study demonstrates that miR-7 maintains mitochondrial health in the face of toxic insults such as MPP+. This beneficial effect of miR-7 is mediated by down-regulation of VDAC1, thereby decreasing the opening of mitochondrial PTPs. As a result, dissipation of mitochondrial membrane potential is prevented and initiation of apoptotic cascade is prevented. Therefore, miR-7 could be used as a therapeutic strategy to maintain mitochondrial health in PD.
Author Contributions
A. D. C. performed and analyzed experiments shown in all figures, and wrote the paper. D. C. C. performed and analyzed experiments in Figures 2, 3, and 4. S. K. performed and analyzed experiments in Figure 3. A. T. performed and analyzed experiment in Figure 3. E. J. conceived and designed the study, analyzed the data, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We appreciate Sue Junn for critical reading of the manuscript.
This work was supported by National Institutes of Health Grant NS070898 (to E. J.). The following author has disclosed actual or potential conflicts: E. J. (patent). The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table 1.
- PTP
- permeability transition pore
- AIF
- apoptosis-inducing factor
- ANT
- adenine nucleotide transporter
- DCF-DA
- 2,7-dichlorodihydrofluorescein diacetate
- GO
- Gene Ontology
- iTRAQ
- isobaric tag for relative and absolute quantification
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide
- miR-7
- microRNA-7
- miR-SC
- scrambled microRNA control
- MPP+
- 1-methyl-4-phenylpyridinium
- PD
- Parkinson disease
- PI
- propidium iodide
- ROS
- reactive oxygen species
- VDAC1
- voltage-dependent anion channel 1
- siRNA-NT
- non-targeting siRNA
- IκB
- inhibitor of κB.
References
- 1. Abou-Sleiman P. M., Muqit M. M., and Wood N. W. (2006) Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 7, 207–219 [DOI] [PubMed] [Google Scholar]
- 2. Duchen M. R. (2004) Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol. Aspects Med. 25, 365–451 [DOI] [PubMed] [Google Scholar]
- 3. Exner N., Lutz A. K., Haass C., and Winklhofer K. F. (2012) Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dias V., Junn E., and Mouradian M. M. (2013) The role of oxidative stress in Parkinson's disease. J. Parkinsons Dis. 3, 461–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Subramaniam S. R., and Chesselet M. F. (2013) Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog. Neurobiol. 106–107, 17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schapira A. H., Cooper J. M., Dexter D., Clark J. B., Jenner P., and Marsden C. D. (1990) Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 54, 823–827 [DOI] [PubMed] [Google Scholar]
- 7. Keeney P. M., Xie J., Capaldi R. A., and Bennett J. P. Jr. (2006) Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26, 5256–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Langston J. W., Ballard P., Tetrud J. W., and Irwin I. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 [DOI] [PubMed] [Google Scholar]
- 9. Cassarino D. S., Parks J. K., Parker W. D. Jr., and Bennett J. P. Jr. (1999) The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim. Biophys. Acta 1453, 49–62 [DOI] [PubMed] [Google Scholar]
- 10. Martin L. J., Semenkow S., Hanaford A., and Wong M. (2014) Mitochondrial permeability transition pore regulates Parkinson's disease development in mutant α-synuclein transgenic mice. Neurobiol. Aging 35, 1132–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gomez-Lazaro M., Galindo M. F., Concannon C. G., Segura M. F., Fernandez-Gomez F. J., Llecha N., Comella J. X., Prehn J. H., and Jordan J. (2008) 6-Hydroxydopamine activates the mitochondrial apoptosis pathway through p38 MAPK-mediated, p53-independent activation of Bax and PUMA. J. Neurochem. 104, 1599–1612 [DOI] [PubMed] [Google Scholar]
- 12. Junn E., and Mouradian M. M. (2012) MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 133, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harraz M. M., Dawson T. M., and Dawson V. L. (2011) MicroRNAs in Parkinson's disease. J. Chem. Neuroanat. 42, 127–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yelamanchili S. V., and Fox H. S. (2010) Defining larger roles for “tiny” RNA molecules: role of miRNAs in neurodegeneration research. J. Neuroimmune Pharmacol. 5, 63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Junn E., Lee K. W., Jeong B. S., Chan T. W., Im J. Y., and Mouradian M. M. (2009) Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doxakis E. (2010) Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chaudhuri A. D., Kabaria S., Choi D. C., Mouradian M. M., and Junn E. (2015) MicroRNA-7 promotes glycolysis to protect against 1-methyl-4-phenylpyridinium-induced cell death. J. Biol. Chem. 290, 12425–12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi D. C., Chae Y. J., Kabaria S., Chaudhuri A. D., Jain M. R., Li H., Mouradian M. M., and Junn E. (2014) MicroRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J. Neurosci. 34, 12725–12737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lutz A. K., Exner N., Fett M. E., Schlehe J. S., Kloos K., Lämmermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A. S., Bouman L., Vogt-Weisenhorn D., Wurst W., Tatzelt J., Haass C., and Winklhofer K. F. (2009) Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Junn E., Taniguchi H., Jeong B. S., Zhao X., Ichijo H., and Mouradian M. M. (2005) Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. U.S.A. 102, 9691–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu J. H., Gusdon A. M., Cimen H., Van Houten B., Koc E., and Chu C. T. (2012) Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 3, e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 23. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu M., Li W.-W., and Lu C.-Z. (2014) Histone deacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS Neurosci. Ther. 20, 308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lemasters J. J., Nieminen A. L., Qian T., Trost L. C., Elmore S. P., Nishimura Y., Crowe R. A., Cascio W. E., Bradham C. A., Brenner D. A., and Herman B. (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1366, 177–196 [DOI] [PubMed] [Google Scholar]
- 26. Burté F., De Girolamo L. A., Hargreaves A. J., and Billett E. E. (2011) Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J. Proteome Res. 10, 1974–1986 [DOI] [PubMed] [Google Scholar]
- 27. Xiong Y., Ding H., Xu M., and Gao J. (2009) Protective effects of Asiatic acid on rotenone- or H2O2-induced injury in SH-SY5Y cells. Neurochem. Res. 34, 746–754 [DOI] [PubMed] [Google Scholar]
- 28. Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., and Arbel N. (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285 [DOI] [PubMed] [Google Scholar]
- 29. Ghosh T., Pandey N., Maitra A., Brahmachari S. K., and Pillai B. (2007) A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS ONE 2, e1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manczak M., Sheiko T., Craigen W. J., and Reddy P. H. (2013) Reduced VDAC1 protects against Alzheimer's disease, mitochondria, and synaptic deficiencies. J. Alzheimers Dis. 37, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hajnóczky G., Csordás G., and Yi M. (2002) Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32, 363–377 [DOI] [PubMed] [Google Scholar]
- 32. Lemasters J. J., and Holmuhamedov E. (2006) Voltage-dependent anion channel (VDAC) as mitochondrial governator: thinking outside the box. Biochim. Biophys. Acta 1762, 181–190 [DOI] [PubMed] [Google Scholar]
- 33. Vyssokikh M. Y., and Brdiczka D. (2003) The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol. 50, 389–404 [PubMed] [Google Scholar]
- 34. Das S., Wong R., Rajapakse N., Murphy E., and Steenbergen C. (2008) Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 103, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lessner G., Schmitt O., Haas S. J., Mikkat S., Kreutzer M., Wree A., and Glocker M. O. (2010) Differential proteome of the striatum from hemiparkinsonian rats displays vivid structural remodeling processes. J. Proteome Res. 9, 4671–4687 [DOI] [PubMed] [Google Scholar]
- 36. Periquet M., Corti O., Jacquier S., and Brice A. (2005) Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J. Neurochem. 95, 1259–1276 [DOI] [PubMed] [Google Scholar]
- 37. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., and Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 38. Pazarentzos E., Mahul-Mellier A. L., Datler C., Chaisaklert W., Hwang M. S., Kroon J., Qize D., Osborne F., Al-Rubaish A., Al-Ali A., Mazarakis N. D., Aboagye E. O., and Grimm S. (2014) Iκβα inhibits apoptosis at the outer mitochondrial membrane independently of NF-κB retention. EMBO J. 33, 2814–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun Y., Rong X., Lu W., Peng Y., Li J., Xu S., Wang L., and Wang X. (2015) Translational study of Alzheimer's disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J. Alzheimers Dis. 45, 269–282 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.