

Abstract
The transcription factor, X-box-binding protein-1 (XBP1), controls the development and maintenance of the endoplasmic reticulum (ER) in multiple secretory cell lineages. We show here that Hepatocyte Nuclear Factor 4α (HNF4α) directly induces XBP1 expression. Mutations in HNF4α cause Mature-Onset Diabetes of the Young I (MODYI), a subset of diabetes characterized by diminished GSIS. In mouse models, cell lines, and ex vivo islets, using dominant negative and human- disease-allele point mutants or knock-out and knockdown models, we show that disruption of HNF4α caused decreased expression of XBP1 and reduced cellular ER networks. GSIS depends on ER Ca2+ signaling; we show that diminished XBP1 and/or HNF4α in β-cells led to impaired ER Ca2+ homeostasis. Restoring XBP1 expression was sufficient to completely rescue GSIS in HNF4α- deficient β-cells. Our findings uncover a transcriptional relationship between HNF4α and Xbp1 with potentially broader implications about MODYI and the importance of transcription factor signaling in the regulation of secretion.
Keywords: beta cell (B-cell), endoplasmic reticulum (ER), hepatocyte nuclear factor 4 (HNF-4), insulin secretion, X-box binding protein 1 (XBP1)
Introduction
Cells use transcription factors to regulate expression of gene cohorts that coordinate response to stress, determine specific developmental fates, and scale intracellular architecture during development and disease(1, 2). When the biosynthetic load of a cell is increased and misfolded proteins accumulate in the endoplasmic reticulum (ER),2 the volume and composition of the ER is altered to facilitate the synthesis and processing of nascent polypeptides via the unfolded protein response (UPR) pathway. One of the principal components of the UPR is the transcription factor X-box-binding protein 1 (XBP1), which is canonically activated via IRE1 splicing of the XBP1 transcript during ER stress (3, 4). However, XBP1 also establishes and maintains the subcellular machinery for synthesizing large quantities of protein during the normal development of professional secretory cells (2, 5). While the majority of genes activated by XBP1 are involved in ER biogenesis, up to 40% of its targets are not directly linked to the ER-stress response (6), further supporting its functions outside the UPR. In addition, the increased XBP1 that coordinates the scaling up of the secretory apparatus during development and homeostasis of dedicated secretory cells does not seem to require activation of the UPR (7). How XBP1 is induced and maintained during differentiation of secretory cells even in the absence of substantial ER stress is unclear, but a potential alternative mechanism is that Xbp1 may also be transcriptionally regulated. Hepatocyte Nuclear Factor 4-alpha (HNF4α) is a highly conserved transcription factor responsible for orchestrating the early development and maintenance of multiple adult organs. As a master developmental regulator, HNF4α likely acts upstream of the factors that establish the extensive cellular machinery required in professional secretory cell lineages within those organs. Despite overlapping expression and function, no direct relationship between HNF4α and Xbp1 has yet been described.
HNF4α is vital for β-cell function, and indeed, human mutations in HNF4α cause Mature-Onset Diabetes of the Young 1 (MODYI), a subset of diabetes characterized by diminished glucose-stimulated insulin secretion (GSIS) in pancreatic β-cells, and susceptibility to type II diabetes (8, 9). While we know that β-cells require HNF4α to function, we understand little about the mechanistic/physiological role of HNF4α in these cells. Previous work showed that disrupting HNF4α expression in vivo in mouse islets resulted in diminished GSIS similar to that observed in MODY patients with HNF4α mutations (10, 11). Loss of HNF4α also was observed to disrupt Ca2+ signaling, though the mechanisms underlying those defects remain unclear. Decreased ER function is a plausible mechanism for the loss of function in MODYI β-cells, because insulin secretion in β-cells is diminished if ER homeostasis is disturbed (12, 13). Defects in ER-related proteins contribute to multiple diabetic phenotypes in humans (14), and HNF4α is known to be important for maintaining ER stress response (15). In addition, knocking down XBP1 specifically in β-cells also leads to significantly reduced GSIS (16). Finally, disruption of calcium homeostasis in the ER also leads to impaired GSIS (17). Here we have characterized how XBP1 expression is governed at the transcriptional level and establish HNF4α as a direct transcriptional regulator of its expression. This implicates HNF4α in the maintenance and establishment of secretory cell ER networks. Accordingly, we report that both HNF4α and XBP1 are required to maintain ER calcium homeostasis and GSIS in β-cells. In addition, we show that restoration of XBP1 expression alone in islets deficient for HNF4α is sufficient to rescue impaired GSIS. Thus, the results may provide new insight toward discerning why dysfunction in HNF4α causes the pathophysiological findings in MODYI patients.
Experimental Procedures
Cell Lines and Transient Transfection
Min6 cells were routinely maintained in Dulbecco's modified Eagle's medium (DMEM) containing 25 mm glucose, supplemented with 10% fetal calf serum, 2 mm l-glutamine, 25 mm Hepes, and 285 μm 2-mercaptoethanol, and penicillin and streptomycin. INS-1 832/13 cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS), penicillin, and streptomycin, sodiumpyruvate and β-mercaptoethanol. Human embryonic kidney (HEK)-293 cells (ATCC) were cultured in DMEM containing 10% FBS and penicillin and streptomycin. INS-1 cells containing doxycycline inducible dnHNF4α were treated with 500 ng/ml doxycycline to induce expression as previously described(36). All cells were passaged at 90% confluency using trypsin-EDTA. For overexpression of HNF4α in INS-1 cells coding regions of human HNF4α2 (obtained from Addgene) were subcloned into a pcDNA3.1expression vector, and 5 μg of each plasmid or the pmaxGFP (lonza) control plasmid were transiently transfected using TransIT-2020 (Mirus, Madison, WI). For mutation analysis, site-directed mutagenesis was performed using the HNF4α overexpression vector described above as an initial template. Mutations were introduced for R154X using primers (F-GAGGTCCTGTCCTGACAGATCACCTC, R- GAGGTGATCTGTCAGGACAGGACCTC) and for R127W using primers (F-GAATGAGCGGGACTGGATCAGCACTC,R- GAGTGCTGATCCAGTCCCGCTCATTC).
Constructs were verified to be correct by DNA sequencing. For siRNA we transfected MIN6 and INS-1 cells with 10 nm HNF4α siRNA (silencer select, Invitrogen) using Lipofectamine 2000 according to the manufacturer's protocol.
In Silico Identification of HNF4α Binding Sites in XBP1 Promoter
Areas of high conservation among multiple mammalian species (human, rhesus, mouse, rat, dog) 10 kb upstream and downstream of the XBP1 transcription start site were identified using ECR browser. These areas were then scanned with the Transfac transcription factor binding database for known transcription factor binding site sequences (18). Two consensus HNF4α sequences were identified; one 1.2-kb upstream (hg19 chr22:29,198,941) of the XBP1 transcription start site and one 2.4-kb upstream (hg19 chr22:29,197,731).
qRT-PCR and Western Blot
RNA was isolated using RNeasy (Qiagen) per the manufacturer's protocol. RNA was treated with DNase I (Invitrogen) and then reverse transcribed using the SuperScript III (Invitrogen) standard protocol (most cDNA syntheses started with 1 μg of total RNA). Measurements of cDNA levels were performed by qRT-PCR using a Stratagene MX3000P detection system. Absolute QPCR SYBR green mix (Thermo Scientific) fluorescence was used to quantify relative amplicon amounts of each gene.
Cells for Western blot analysis were lysed in RIPA buffer. Proteins were quantified by DC protein assay (Bio-Rad) and then separated on NuPAGE Bis-Tris gels (Invitrogen), transferred onto Amersham Biosciences Hybond ECL nitrocellulose (GE Healthcare) membranes, and detected by Immobilon chemiluminescence (Millipore). Primary antibodies used were rabbit anti-XBP1 (Santa Cruz Biotechnology), mouse anti-myc-tag (Cell Signaling), and rabbit anti-α- and β-tubulin (Cell Signaling). Secondary antibodies were horseradish peroxidase-conjugated donkey anti-rabbit and anti-mouse Ig (Santa Cruz Biotechnology).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described previously(47). Approximately 100 mg of tissue from the pancreata of 5, 6–8-week-old WT mice were homogenized and used for this ChIP experiment. Ten microliters of anti-HNF4α (Santa Cruz Biotechnology) or whole rabbit serum (preimmune control) together with protein A/G plus-agarose (Santa Cruz Biotechnology) was added to the homogenized tissue for immunoprecipitation. Quantitative real-time PCR (qRT-PCR) was performed (primer sequences are available from corresponding author) to assess the quantity of genomic sequences immunoprecipitated by either preimmune control or HNF4α antiserum, as well as a 1:10 dilution of the cell extract prior to immunoprecipitation (input). The two predicted HNF4α binding sites described above were probed in addition to an intronic control region with no predicted HNF4α binding sites nearby. Data are graphed as a percentage of precipitated DNA:total input (genomic DNA).
Beta-cell Morphological Characterization using Immunofluorescence
Pancreata were prepared and stained as described previously(49). Briefly, they were fixed with freshly prepared formalin and suspended in fixative for 24 h at room temperature, followed by multiple rinses in 70% ethyl alcohol (EtOH), arrangement in 2% agar in a tissue cassette, and routine paraffin processing. Sections (5 μm) were deparaffinized and rehydrated, and then antigen retrieval was performed by boiling in 50 mm Tris-HCl, pH 9.0. Slides were blocked in 1% bovine serum albumin (BSA) and 0.3% Triton X-100 in phosphate-buffered saline (PBS) and then incubated in goat anti-Calregulin (Santa Cruz Biotechnology) followed by AlexaFluor594 antigoat. Fluorescence microscopy and imaging were performed using a Zeiss Axiovert 200 microscope with Axiocam MRM camera and Apotome II instrument for grid-based optical sectioning.
For morphological analysis, three 5-μm sections taken 100-μm apart were stained with hematoxylin and eosin to allow identification of islets. Whole slides were scanned with the Nanozoom microscope and the cross-sectional area of islet/total pancreas tissue was measured across each slide using Nanozoom Digital Pathology and software (Hamamatsu). Samples were randomized, and the scorer was blinded to ensure unbiased quantification. Values are expressed as %β-cell area.
Mouse Studies
All experiments involving animals were performed according to protocols approved by the Washington University School of Medicine Animal Studies Committee. Floxed Hnf4α, CAGGCreERTM transgenic mice were generated by crossing Hnf4αfloxed/floxed mice (a gift from Frank Gonzalez, NIH)(23), with CAGGCreERTM;Hnf4αfloxed/+(48) mice to allow systemic, tamoxifen-inducible knock out of HNF4α. 6–8-week-old CAGGCreERTM;Hnf4αfloxed/floxed mice and CAGGCreERTM;Hnf4αfloxed/+ littermate controls were injected intraperitoneally with tamoxifen (5 mg/20g body weight, 5 consecutive days) to induce cre-mediated Hnf4α deletion. Mice were sacrificed 4 weeks after first tamoxifen injection. No mouse samples were excluded from analysis in this study. Immunofluorescent Quantification: For quantification of ER in islets, the pancreata of HNF4α KO and littermate control heterozygous mice were fixed, mounted, and stained as described above. 16-bit images captured in Zeiss Axiovision software were analyzed with ImageJ software as follows; Insulin positive regions (Santa Cruz Biotechnology rabbit anti-insulin) were measured as regions of interest, and mean fluorescence intensity of the global ER marker Calregulin in each region was determined and then subtracted from the median fluorescent intensity of acinar cell regions in the same image to normalize fluorescence intensity on each slide. The mean fluorescent intensity was measured in every islet in a 5-μm section (three mice/condition). Analysis of Calregulin fluorescence was restricted to the β-cell cytoplasm by excluding Hoechst-positive (nuclear) regions. After capture, each image was assigned a random number so that subsequent fluorescent quantification was blind relative to condition. Nuclear areas were identified by Hoescht staining, and pixels with an intensity of >30 gray value as determined by the “plot profile” tool in ImageJ were excluded from measurement. Hoescht-negative, cytoplasmic pixels were measured and normalized by subtracting the mean fluorescence of the surrounding acinar tissue.
Endoplasmic Reticulum Calcium FRET Measurement
INS-1 832/13 cells stably expressing the D1ER calcium sensor (12, 34), were cultured as described above. Cells were transiently transfected with 10 nm HNF4α siRNA or scrambled control siRNA to knockdown HNF4α expression. Cells were also treated with vehicle or 16 μm 4-methyl umbelliferone 8-carbaldehyde (4μ8C) to inhibit XBP1 splicing as previously described (50). Five days post-transfection/treatment, 1 × 105 cells were seeded in transparent bottom 96-well plates to achieve 70% confluency 6 h pre-measurement. Cells were washed twice in PBS and incubated in Krebs-Ringer buffer supplemented with 3 mm glucose immediately before measurement. To establish maximum and minimum ER Ca2+ levels, at this time, cells were respectively treated with 10 μm membrane-permeabalizing ionomycin and subsequently 10 mm CaCl2 or 5 mm EGTA. As an additional control for low ER Ca2+, cells were treated with 1 μm thapsigargin to specifically diminish ER Ca2+. FRET ratio of the D1ER cameleon was measured using the Tecan Infinite M1000Pro microplate reader. Fluorophores were excited at 434 nm, and emission was quantified at 530 nm (YFP) and 477 nm (CFP). The ratios were measured across 4 fields/well, and the values were averaged from 4 wells per experimental condition. After FRET microplate measurement, RNA was isolated as described above and quantified by qPCR to measure Hnf4α and Xbp1 knockdown efficiency.
Ratiometric Calcium Imaging and Data Analysis
Studies were performed 24 h after plating INS-1 823/13 cells at 50% confluency and carried out at 37 °C with 5% CO2 in a perifusion chamber with a flowrate of 2 ml/min. Cells were loaded with Fura-2AM by incubation at 37 °C in Krebs-Ringer buffer supplemented with 3 mm glucose, 1 μm Fura-2AM, and 0.1% Pluronic F-127 for 30 min, washed in HBSS, and incubated for another 30 min to allow for ester hydrolysis. After loading, cells were imaged on an inverted microscope (Till Photonics; Munich, Germany) equipped with a cooled CCD camera (Cooke, Auburn Hill, MI) using a ×20/0.45 Plan Fluor objective (Nikon). The fluorescence excitation (340 and 380 nm) was provided by a Polychrome V Monochromator (Till Photonics). After the matching background was subtracted, the image intensities from each pair of images, measured at 520 nm, were divided by one another to yield ratio values for individual cells. [Ca2+]i in individual cells was estimated based on the formula: [Ca2+]i = KD×B×(R − Rmin)/(Rmax − R), where KD is the indicator's dissociation constant for Ca2+ (0.22 μm); R is ratio of fluorescence intensity at two different wavelengths (340/380 nm); Rmax and Rmin are the ratios of Ca2+-free and Ca2+-bound Fura-2, respectively; and B is the ratio of the fluorescence intensity of the second excitation wavelength at zero and saturating Ca2+ concentrations. The calibration constants were determined as previously described(51), and the ratio values were plotted against time.
Islet Isolation and Culture
Pancreatic islets from CAGGCreERTM;Hnf4αfloxed/floxed mice and CAGGCreERTM;Hnf4αfloxed/+ littermate controls were isolated as previously described (52), by pancreatic duct injection of 1000 U/ml of collagenase solution (Sigma) followed by digestion at 37 °C for 15 min with mild shaking. Islets were washed several times with Hanks' balanced salt solution, separated from acinar cells by straining through a 100-μm filter, viewed under a dissecting microscope, and handpicked for culture (yield = 200–300 islets/mouse). Isolated Islets were maintained in RPMI1640 supplemented with 10% FBS and penicillin and streptomycin at 37° with 5% CO2. All islets were allowed to recover from isolation for 24 h before analysis. Islets were isolated from mice in random order relative to condition.
Adenoviral Transduction
Unspliced XBP1 adenovirus (Applied Biological Materials), LacZ Adenovirus (Applied Biological Materials), and spliced XBP1 (a gift from Laurie Glimcher) (53), were amplified in HEK293 cells, cultured as described above. Infected cells were lysed by three cycles of freezing and thawing and then centrifuged. Viral titer was determined by infecting HEK-293 cells with serially diluted viral stock and overlaying with agar and subsequently counting the resulting plaques. INS-1 cells containing dnHNF4α were treated with doxycycline or vehicle to induce expression of dnHNF4α as described above. Five days post-treatment, cells were infected with either XBP1u or LacZ adenovirus at a multiplicity of infection (MOI) of 100. Viral stock was replaced with complete medium after 2 h of infection. Isolated murine islets were infected as described previously(54). Briefly, 70 islets/condition were washed in cold PBS, pretreated with HBSS containing 2 mm EGTA at 37° with 5% CO2 for 15 min, then infected with adenovirus in serum-free RPMI 1640. Following a 15-min incubation, complete medium was added to islet culture. Islets were infected for 24 h before GSIS assay and harvesting RNA. Adenovirus was used in isolated islets at the following MOIs: LacZ MOI = 50, XBP1u MOI = 50, XBP1s MOI = 10.
Glucose-stimulated Insulin Secretion Measurement
For INS-1 GSIS assay, 2 days post- adenoviral infection cells were washed with PBS, then incubated for 1 h in Krebs-Ringer Buffer containing 3 mm glucose. After 1 h, cells were washed with PBS, and basal insulin secretion was measured by incubating cells for 1 h in Krebs- Ringer Buffer containing 3 mm glucose. Medium was sampled, then replaced with medium containing 16.7 mm glucose, or 200 μm Tolbutamide for 1 h. Medium was collected and analyzed for insulin content by ELISA using the Singulex Erenna platform by the Washington University Diabetes Research Center Immunoassay Core. Static GSIS was similarly measured in isolated islets as previously described(55). 24 h post-infection, fifty islets were placed in Krebs-Ringer Buffer containing 3 mm glucose to measure basal insulin secretion, then stimulated with 16 mm glucose. Insulin secretion was measured in each condition as described above.
Graphing and Statistics
All graph values represent the mean of the sample, and error bars represent S.E. where indicated. Significance was determined using Student's t test or ANOVA with Dunnet's posthoc comparison as indicated. Wherever possible, samples were randomized and measurements were blinded to prevent the introduction of experimental bias. Sample sizes were determined based on statistical significance and practicality.
Results
Regulation of Xbp1 by HNF4α
To elucidate the potential transcriptional regulation of XBP1, we identified evolutionarily conserved binding sites in the human XBP1 promoter by aligning regions of synteny, then screening them with the Transfac transcription factor binding site database(18). Two regions with high conservation containing putative HNF4α binding sites, 1.4 and 2.6 kilobases upstream of the Xbp1 transcription start site, were identified using first the Transfac transcription factor binding library and then affirmed using a previously published algorithm developed to search for sites of high HNF4α binding affinity (19). These putative binding sites were constitutively occupied by HNF4α in mouse pancreas, measured via chromatin immunoprecipitation (Fig. 1A). Overexpression and knockdown experiments in vitro showed HNF4α was both sufficient and necessary for normal Xbp1 expression in pancreatic β-cell derived-cell lines. Disrupting HNF4α either by siRNA knockdown (Fig. 1, B and C) or by overexpressing a doxycycline-inducible dominant-negative version of HNF4α that lacks a DNA-binding domain and has been shown to bind to endogenous HNF4α (Fig. 1, D and E) (36), resulted in a 65–75% decrease in Xbp1 expression in INS-1 and MIN-6 cells. Conversely, overexpression of HNF4α via transient transfection caused a 5-fold increase in Xbp1 expression (Fig. 1F). To further substantiate this transcriptional relationship, we analyzed the effects of Hnf4α deletion on Xbp1 expression in other tissues by mining published microarray studies and by direct qRT-PCR analysis of adult (Fig. 1G) and embryonic liver (20) as well as adult small intestine (data not shown but available on Gene Expression Omnibus under accession numbers GSE34581, GSE3124, GSE3126) (21–23). Again, the results showed a consistent trend toward correlation of Xbp1 expression decrease with loss of Hnf4α in multiple secretory tissues.
FIGURE 1.

HNF4α is a direct transcriptional regulator of XBP1. A, immunoprecipitation of chromatin from 5 C57/B6 mouse pancreata with anti-HNF4α followed by qPCR (ChIP assay) showed significant occupancy at 2 predicted binding sites in the Xbp1 promoter (but not at a downstream intronic control site lacking a predicted HNF4α-binding motif) when compared with normal preimmune serum controls. B, MIN6 and C, INS-1 cells were transfected with siRNA targeting Hnf4α or scrambled control siRNA. Hnf4α and Xbp1 mRNA were quantified by RT-qPCR at 48 h post-transfection and normalized to 18S. (means ± S.E. of n = 6 experiments depicted, statistical significance by one-tailed Student's t test). D, INS-1 cells stably expressing a doxycycline-inducible Hnf4α construct that acts as a dominant negative were incubated in the presence or absence of doxycycline for 7 days (means ± S.E. of n = 12 experiments depicted, statistical significance by one-tailed Student's t test). E, representative Western blot following treatment of doxycycline inducible myc-tagged dnHNF4α in INS-1 cells containing the inducible dnHNF4α or WT INS-1 cells. F, transient transfection of INS-1 cells with an HNF4α expression vector or a GFP control plasmid and quantification of XBP1 mRNA 48 h post-transfection. (means ± S.E. of n = 3 experiments depicted, statistical significance by one-tailed Student's t test). G, quantification of mRNA in ΔHNF4α KO embryonic (E18.5) liver tissue relative to wildtype littermates by qRT-PCR (20). (Significance was determined using Student's t test; error bars represent S.E. of 3 mice/condition.) H, Xbp1 mRNA expression levels in INS-1 cells with HNF 4α constructs harboring point mutations isolated from MODYI patients that would cause the indicated amino acid variation or a GFP expression plasmid as a control. (Error bars represent standard deviation of three biological replicates. Significance determined by ANOVA with Dunnet's comparison.) For all figures the following symbols mean: “***”, p < 0.001; “**”, p < 0.01; “*”, p < 0.05.
Various single point mutations in the HNF4α locus have been identified in human patients afflicted with impaired β-cell function. To better understand the impact of these mutations, we designed two HNF4α expression vectors, each containing one of the most prevalent mutations (24, 25). Overexpression of each of these individual mutants in INS-1 cells resulted in a ∼4-fold decrease in the expression of XBP1 mRNA (Fig. 1H). Interestingly, although we could detect the transfection-mediated increase in mRNA for both mutant and wild-type HNF4α qPCR, we could detect only wild-type HNF4α protein by Western blot (Fig. 4D and data not shown). We speculate the mutant genes may encode protein products that are abnormally folded and/or truncated and degraded. In any case, all the data support the conclusion that common HNF4α mutations found in human patients decrease abundance of Xbp1.
FIGURE 4.

HNF4α and XBP1 are required for ER Ca2+ homeostasis. A, qRT-PCR performed on RNA harvested from ΔHnf4α islets as for (Fig. 2) for transcripts for the KATP channel genes KIR6.2 (Kcnj11) and SUR1 (Abcc8) and the ER Ca2+ pump SERCA2b (Atp2a2) (means ± S.E. from 6 mice/condition, significance determined by one-tailed Student's t test). B, cartoon of the ER Ca2+ FRET Sensor D1ER. In low Ca2+ conditions, calmodulin is conformed in a manner that does not allow photon transfer between the 405 nm excitable CFP and the unexcitable YFP fluorophore. In high Ca2+ conditions, the CFP domain emits at a frequency to excite YFP. Measuring the YFP(FRET):CFP ratio determines relative Ca2+ levels in the ER as opposed to other cellular compartments. C, FRET:CFP ratio in INS-1 D1ER cells was determine to quantify relative ER Ca2+ levels under the following conditions: 5 days after transfection of Hnf4α or scrambled siRNA; 5 days after incubation with vehicle or 4μ8c, an inhibitor of splicing-mediated XBP1 activation; or combinations thereof. To determine maximal detectable Ca2+ levels, FRET:CFP was determined 1 h following treatment with the Ca2+ ionophore ionomycin + CaCl2. The FRET/CFP of that condition was set to 1.0, and all other conditions were normalized to it. To determine the minimal detectable ER Ca2+ levels using this assay, cells were treated with 10 μm ionomycin + EGTA for 1 h or 1 μm thapsigargin. (means ± S.E. of n = 6 experiments depicted, statistical significance by one-tailed Student's t test). D, representative Western blot quantifying the abundance of HNF4α, XBP1, or β-tubulin in INS-1 D1ER cells treated as described above. Arrow indicates XBP1s band. E, cytoplasmic [Ca2+] of individual INS-1 cells 5 days after transfection of Hnf4α or scrambled siRNA determined by Fura-2AM emission levels in response to 16.7 mm glucose. Plots are representative of (n>50 cells;4 biological replicates. F, cytoplasmic [Ca2+] of individual INS-1 cells 5 days after transfection of Hnf4α or scrambled siRNA determined by Fura-2AM emission levels in response to 20 mm caffeine, an agonist of the ryanodine receptor, added to induce release of ER Ca2+ stores. Plots are representative of (n > 50 cells; four biological replicates).
HNF4α Is Required for ER Maintenance in Vivo
The mechanisms whereby disruptions of HNF4α in MODYI cause β-cell dysfunction remain an area of open debate. As XBP1 is critical in scaling up and maintaining the ER of professional secretory cells, we hypothesized mutations in HNF4α may impair β-cell function via dysregulation of Xbp1 and consequent ER dysfunction. Previous studies of effects of loss of Hnf4α in islets focused on deletion of the gene during development. As we were concerned here with the effects of adult onset of aberrant Hnf4α on islet function, we induced deletion of Hnf4αflox/flow mice using a global, tamoxifen-inducible Cre in 6–8-week-old mice. As predicted, deleting Hnf4α in existing islets resulted in significant loss (∼60%) of both Xbp1 expression in islets and, in turn, of XBP1 transcriptional targets like Edem1 (Fig. 2A) when compared with littermate Hnf4αflox/+controls (referred to hereafter as “WT”) (26). Supporting previous findings, insulin and Hnf1α mRNA levels were unaffected by loss of Hnf4α(10, 11). While XBP1 establishes secretory cell machinery and maintains cell architecture(2, 27), likely in large part independent of activation of the UPR (28), in chronic/long-term ER stress conditions, XBP1 is a fundamental component of the UPR (29). We sought to characterize the ER-stress state of ΔHNF4α β-cells to determine whether other branches of the UPR were activated to compensate for loss of Xbp1. One of the most reliable methods of measuring UPR activation in cells is by quantifying the expression of genes whose transcription is enhanced by activated master regulators of the UPR (30). Decreased Xbp1 in Hnf4αΔ/Δ mice did not cause increase in such UPR transcripts like Chop, Bip, or Atf4, nor a decrease in mRNA levels of the ER-marking Calregulin (CRP55) (Fig. 2B). However, loss of XBP1 following deletion of Hnf4α did correlate with a nearly 7-fold reduction in ER area in β- cells (Fig. 2, C and D). Despite decreased ER in each cell, overall islet mass, as measured by microscopic area through cross-sectioned pancreata, was not changed in Hnf4αΔ/Δ pancreata (Fig. 2E), indicating that Hnf4α is not required to maintain islet number or size. Thus, loss of Hnf4α caused diminished ER network, a phenotype similar to that caused by deleting Xbp1 from existing adult secretory cells (2, 5).
FIGURE 2.

HNF4α is required for XBP1 expression in vivo. Hnf4αfloxed/floxed mice under the control of a ubiquitously expressed CAGGCreERT promoter were treated with tamoxifen to induce Hnf4α deletion. A, pancreatic islets were isolated 28 days after beginning tamoxifen treatment (see “Experimental Procedures”) and their RNA harvested. Expression of Xbp1, Hnf4α, and the downstream XBP1 target, Edem1, was assayed by qRT-PCR. B, expression of other unfolded protein response pathway transcripts, Chop, Bip, and Atf4 was assayed as for A. C, immunofluorescent images of the ER marker Calregulin reveal altered ER structure in ΔHnf4α mouse islets. Scale bars = 20 μm (white box shown at higher magnification immediately below). D, mean fluorescent intensity (arbitrary units from 16-bit images) of non-nuclear, β-cell-specific Calregulin staining in mouse islets was determined after normalizing each tissue section to neighboring acinar Calregulin mean fluorescence. Data represent means ± S.E. from three mice/condition, significance determined by one-tailed Student's t test. E, total β-cell area in the pancreas was quantified by anti-insulin immunofluorescence from sections by completely sectioning through whole tissue blocks of the entire embedded pancreata from ΔHnf4α and controls. (Means ± S.E. from 6 mice/condition, significance determined by one-tailed Student's t test.)
HNF4α and XBP1 Are Necessary to Maintain ER Calcium Homeostasis
In previous reports, constitutive deletion of Hnf4α from islets early in development, as opposed to in the adult, caused impaired GSIS(10, 11). The mechanism of decreased GSIS was hypothesized to be dysregulated cytoplasmic Ca2+ signaling in response to glucose, but the molecular mechanism driving this impairment has remained unclear. Ca2+ signaling depends on the ATP-dependent closure of KATP channels, triggering membrane depolarization and opening voltage-gated Ca2+ channels(31). Thus, one mechanism that could mediate how loss of Hnf4α could cause GSIS could be via disruption of those channels. However the sulfonylurea Tolbutamide, which closes KATP channels leading to membrane depolarization, stimulated less insulin secretion in INS-1 cells expressing dominant negative-HNF4α (dnHNF4α, described above) than in normal INS-1 cells (Fig. 3). The inhibition of Tolbutamide-stimulated insulin secretion caused by disrupted HNF4α was rescued by transduction of Xbp1, though transducing Xbp1 alone did not increase tolbutamide- stimulated insulin secretion (Fig. 3). Additionally, qPCR analysis showed abundance of transcripts for the KATP channel subunits Sur1 (Abbc8) and Kir6.2 (Kcnj11) in Hnf4αΔ/Δ islets were unchanged relative to littermate controls (Fig. 4A). These data, along with previous work showing impaired insulin secretion even upon depolarization with KCl (10), suggest the defect in the glucose response pathway due to disrupted HNF4α is distal to KATP channel dependent-membrane depolarization and that it depends, in part, on loss of XBP1. Though KATP channel expression was unchanged in Hnf4α islets, expression of the putative XBP1 transcriptional target, Serca2b(Atp2a2), was significantly reduced (Fig. 4A) (32). SERCA2b is an ER Ca2+ pump, responsible for establishing and maintaining the large calcium gradient between the ER and cytoplasm (33). Intracellular stores of Ca2+ are critical for GSIS, because Ca2+ release from these stores triggers secretion of insulin granules. Accordingly, decreasing ER Ca2+ stores and/or flux has been shown to disrupt GSIS (17). Thus, in Hnf4αΔ/Δ mice, the decreased expression of a key molecular driver of the ER Ca2+ gradient suggested that disruption of ER Ca2+ stores may play a critical role in the GSIS abnormalities seen in the absence of normal HNF4α. We used an ER-specific FRET sensor to measure ER [Ca2+] in ΔHnf4α β-cells (12, 34). The D1ER cameleon construct encodes two fluorophores conjugated to a calmodulin molecule that is targeted specifically to the ER lumen with a KDEL sequence. When it binds Ca2+, the cameleon undergoes a conformational change that approximates the fluors to produce FRET activity quantitatively proportional to ER [Ca2+] (Fig. 4B). Changes in FRET:No FRET ratios correspond with changes in ER [Ca2+] as observed when treating cells permeabilized by ionomycin with either CaCl2 to increase ER [Ca2+] or EGTA/thapsigargin to diminish it (Fig. 4C). In accordance with the mechanism of lost GSIS in MODYI being disruption of XBP1-mediated ER Ca2+ stores, both knockdown of HNF4α and pharmacological inhibition of XBP1 activation (using 4μ8C, which blocks IRE1-mediated conversion of the XBP1 transcript to the active spliced form) in INS-1 cells resulted in decreased ER [Ca2+] (Fig. 4C) and loss of HNF4α or XBP1 protein levels as expected (Fig. 4D). Simultaneously knocking down HNF4α and pharmacologically inhibiting XBP1 did not augment the decreased ER [Ca2+]. Thus, it is likely that HNF4α and XBP1 work via the same pathway to maintain high ER [Ca2+] in adult β-cells. To further explore the requirement of HNF4α for proper Ca2+ signaling in β-cells, we observed changes in cytoplasmic Ca2+ levels in response to various stimuli in INS-1 cells transiently transfected with scrambled siRNA or siRNA targeting HNF4a using Fura 2AM-based Ca2+ imaging. Expectedly, ΔHnf4α β-cells exhibited a diminished response to stimulation with 16.7 mm glucose, as observed in other MODYI β-cell models (Fig. 4E). To identify the cause of this deficit in Ca2+ signaling, and further explore our previous results indicating HNF4α is required for ER Ca2+ homeostasis, we exposed these cells to 20 mm Caffeine. Caffeine is an agonist of the ryanodine receptor that stimulates release of Ca2+ from stores in the ER and thus an increase in cytoplasmic [Ca2+] (35). Caffeine induced diminished cytoplasmic [Ca2+] increase in ΔHnf4α β-cells (Fig. 4F), indicating that ER Ca2+ homeostasis is disrupted. In short, loss of ER Ca2+ in ΔHnf4α β-cells may underlie their impaired GSIS.
FIGURE 3.
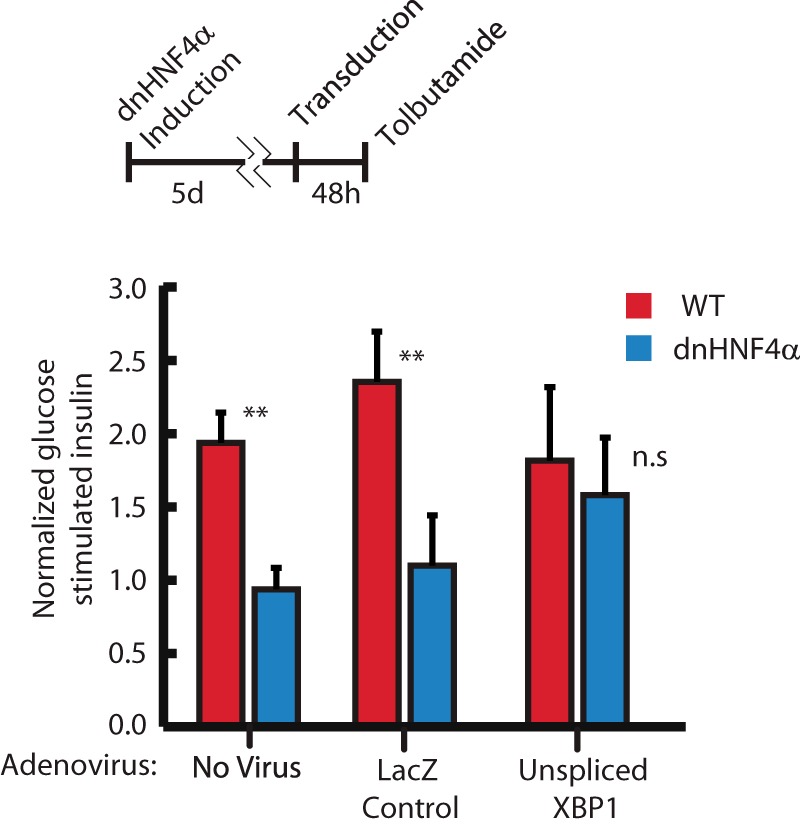
Tolbutamide-induced insulin secretion. Doxycycline-induced expression of the dominant negative form of HNF4α from Fig. 1 (DE) in INS-1 cells completely abrogates increased insulin secretion in response to tolbutamide exposure, suggesting the MODY1 secretion defect is distal to β-cell membrane depolarization in the GSIS signaling cascade. Infection with an adenovirus carrying Xbp1u expression vector is sufficient to rescue this GSIS defect. (Means ± S.E. of n = 3 biological replicates depicted, statistical significance by one-tailed Student's t test.)
XBP1 Is Sufficient to Rescue Insulin Secretion in ΔHnf4α β-cells
We next sought to confirm the physiological relevance of the HNF4α → XBP1 relationship by determining if we could rescue aberrant GSIS in the absence of HNF4α simply by restoring XBP1. If XBP1 were decreased in the absence of HNF4α due to direct loss of transcriptional up-regulation by HNF4α, as we hypothesized, then restoration specifically of the unspliced XBP1 (XBP1u) form should rescue GSIS, because XBP1u is the unmodified mRNA directly generated from transcription of the XBP1 gene (i.e. prior to activation by IRE1-mediated splicing). Restoring the deficit of XBP1u transcript caused by loss of HNF4α would allow cells the freedom to use the normal IRE1 splicing mechanism to produce only as much active XBP1s transcript as they require for ER homeostasis. We returned to our in vitro model employing transgenic INS-1 cells containing a doxycycline inducible dnHNF4α described above (Figs. 1 and 3) (36). As shown above, the functional loss of HNF4α activity in insulin-secreting cells when doxycycline is added to induce dnHNF4α, caused both decreased XBP1 (Fig. 1, D and E) and loss of GSIS (Fig. 5A). This decreased GSIS was completely rescued by adenoviral transduction of Xbp1u. As discussed above, forced expression of Xbp1u was also sufficient to rescue impaired insulin secretion in response to sulfonylurea treatment in these dnHNF4α β-cells (Fig. 3). We repeated this study ex vivo, using Hnf4αΔ/Δ islets isolated 3 weeks following tamoxifen-induced Hnf4αΔ/Δ deletion. As expected, cultured islets had impaired GSIS due to HNF4α deficiency. Xbp1u restoration increased the direct XBP1 transcriptional targets Edem1 and Serca2b (Fig. 5B) confirming that transduction of Xbp1u restored functional XBP1-mediated transcriptional activity to scale up expression of its normal transcriptional targets. Remarkably, as predicted, restoring unspliced Xbp1 expression in these ex vivo cultured, HNF4α-deficient β-cells was also sufficient to completely rescue their GSIS, indicating that the impaired GSIS in the absence of HNF4α depends on transcriptional maintenance of XBP1 expression by HNF4α (Fig. 5C). Because of the direct transcriptional regulation of XBP1 by HNF4α and the lack of GSIS enhancement in WT β-cells transduced with Xbp1u, the ability of Xbp1u to rescue the phenotype caused by loss of HNF4α is likely because it corrects the diminished basal Xbp1 expression in ΔHNF4α β-cells.
FIGURE 5.

XBP1 is sufficient for GSIS in ΔHnf4α β-cells. A, glucose-stimulated insulin secretion (GSIS) was determined by harvesting supernatant from wildtype INS-1 and doxycycline-inducible dnHNF4α INS-1 cells following incubation for one hour under high (16 mm) glucose conditions. Induction of dominant negative HNF4α abrogates GSIS (insulin secretion of ∼1 means no induction relative to baseline insulin secretion with 3 mm glucose). All cells were transduced by adenovirus carrying either unspliced Xbp1 or LacZ control vectors. Note that Xbp1 transduction rescues GSIS in dnHNF4α cells (means±S.E. of n = 6 experiments depicted, statistical significance by one-tailed Student's t test), B, ΔHNF4α or heterozygote control islets were cultured for 24 h after isolation and then transduced with either LacZ or Xbp1 vector-containing adenovirus. 24 h later, RNA was harvested and qRT-PCR performed for transcripts from Hnf4α, Xbp1 and two downstream transcriptional targets of XBP1, Serca2b, and Edem11 (data represent means ± S.E. from three individual islet isolations and transduction experiments). C, normalized GSIS was determined as for panel A with transduction of either LacZ or Xbp1 vector-containing adenovirus into isolated islets. Note isolated islets from Hnf4αΔ/Δ mice exhibit a complete lack of GSIS. (Data represent means ± S.E. from three individual islet isolations and transduction experiments.) D, model of MODYI pathology. XBP1 regulates transcription of multiple genes like SERCA2B that induce and maintain normal ER and ER Ca2+ function. Loss of HNF4α in patients with MODYI causes reduced XBP1 expression.
We also transduced spliced XBP1 (Xbp1s) in isolated ΔHNF4α mouse islets, bypassing the normal regulation of transcriptionally regulated Xbp1u by IRE1α splicing. Transduction of Xbp1s rescued the XBP1 targets, Edem1 and Serca2b (Fig. 6A) but resulted in GSIS roughly 50% lower than that in control WT islets (Fig. 6B). That result is consistent with previous reports that β-cell homeostasis is compromised by forced expression of spliced Xbp1 because cells must be able to dynamically regulate XBP1 levels via the endogenous IRE1 splicing mechanism, with direct forced expression of high levels of already activated Xbp1 potentially being toxic (37). Accordingly, Hnf4αΔ/Δ islets infected with Xbp1s exhibited GSIS rescue to the levels observed in WT islets infected with Xbp1s, suggesting that, while forced expression of XBP1s is detrimental to β-cell health, it is still able to compensate for GSIS defects in β-cells caused by the absence of HNF4α.
FIGURE 6.
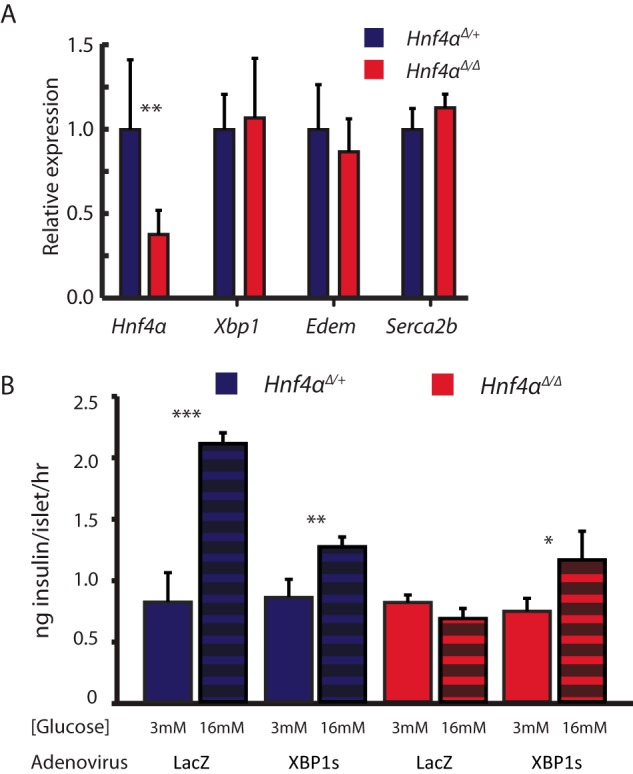
Rescue of GSIS in ΔHNF4α islets with spliced Xbp1. A, ΔHnf4α and heterozygote control islets were isolated and treated as in (Fig. 5B) but infected with adenovirus harboring plasmid a transcript for spliced XBP1 (XBP1s). XBP1s expression restores mRNA abundance of downstream targets, Edem and Serca2b, to normal levels in ΔHNF4α islets (means ± S.E. of n = 3 experiments depicted). B, restoring expression of XBP1s sufficient to partially rescue GSIS in isolated ΔHNF4α islets, but GSIS (i.e. insulin release following switching of islets to 16 mm glucose) is diminished in both WT and ΔHnf4α islets upon transducing expression of XBP1s when compared with WT GSIS. LacZ vector-containing adenovirus was used as a control for transduction (means ± S.E. of n = 3 experiments depicted, statistical significance was determined by one-tailed Student's t test).
Discussion
We report that Xbp1 is a direct transcriptional target of HNF4α in multiple secretory tissues. Given the importance of HNF4α mutations in diabetes, we have focused on the relationship between HNF4α and XBP1 specifically in insulin-secreting β-cells. Deletion of HNF4α in β-cells causes them to lose XBP1, which in turn causes dismantling of ER. HNF4α point mutants designed to match mutations that cause human diabetes also resulted in loss of XBP1 in vitro. Loss of either HNF4α or XBP1 leads to diminished ER Ca2+, which in turn impairs GSIS, a defect that can be completely rescued by reestablishing normal Xbp1 transcript levels (Fig. 5D). Together, our results identify a new transcriptional relationship between evolutionarily-conserved genes, Xbp1 and Hnf4αΔ/Δ, involved in fundamental development and disease in multiple tissues.
We also demonstrate specific cellular contexts during which Xbp1 expression is functionally regulated at the transcriptional level. XBP1 is induced in response to accumulation of unfolded protein in the ER by splicing of its message via the endonuclease IRE1α, and the canonical view of how XBP1 abundance is modulated concern that mechanism. However, the transcriptional regulation of Xbp1 may be as important to its role in secretory cell development as its activation by IRE1. Xbp1 expression is significantly induced and maintained at much higher levels in secretory cells, and large pools of unspliced Xbp1 mRNA are required to restore homeostasis in chronic ER stress conditions (38). Additionally, IRE1 is basally activated in dedicated secretory cells at levels comparable to those observed during acute ER stress in non-secretory cells (>40%) (39). When the presumably UPR- activating biosynthetic load-stimulus is removed in Β-lymphocytes differentiating to plasma cells by disrupting expression of IgM, XBP1 transcript levels still increase, indicating that XBP1 activation is differentiation-dependent rather than UPR-dependent in certain secretory cells (40). Together, these studies suggest that transcription of Xbp1 is the rate-limiting step in its activation during secretory cell development and maintenance.
It is somewhat surprising that HNF4α, which is largely studied in developmental contexts as a master regulator of differentiation in endodermal organs, is required for continued maintenance of XBP1 in differentiated, adult cells. However, unbiased, comprehensive screens for genes whose expression depends on HNF4α have previously identified XBP1 as a potential target (14–17), and chromatin immunoprecipitation followed by sequencing (ChIP-Seq) has also shown peaks indicating potential binding of HNF4α to the putative XBP1 promoter (41). Thus, though the results of the previous screens have not been validated and the direct relationship between HNF4α and XBP1 has apparently never been specifically studied prior to the current work, our results are not entirely unprecedented. Indeed, HNF4α has also been shown to regulate expression of ankyrin repeat and sterile α motif domain containing 4b (Anks4b), a protein that binds ER chaperones and augments the ER stress response, supporting our hypothesis that HNF4α is required for the establishment and maintenance of the ER in β-cells (42).
In ongoing work in our lab, we also observe that Xbp1 expression depends on HNF4α in the stomach, and, exactly as in β-cells, loss of HNF4α in mature gastric chief cells causes dismantling of the ER.3 The results are consistent with work by us and others showing that continued XBP1 is required for ER in gastric chief cells(2) and in β cells (16). On the other hand, though we observe Xbp1 expression clearly depends on HNF4α in the liver (see Fig. 1G plus previous studies available in GEO that we have analyzed (21–23)), we have not been able to detect substantial effects on ER in that organ when HNF4α is deleted from mature hepatocytes (data not shown). Actually, this is in agreement with previous studies showing that direct depletion of XBP1 itself from mature hepatocytes is not sufficient to cause ER dismantling in existing cells (53). Together, the results suggest that there are tissue-specific differences in the role of XBP1 in regulating the ER: perhaps XBP1 is generally required for establishment of an elaborate ER network in dedicated secretory cells but is only required in certain cell types for its continued, homeostatic maintenance.
While previous studies suggest alterations in the KATP channel may play a role in MODYI pathology (10, 11) they also show that MODYI β-cells have impaired GSIS even upon membrane depolarization with KCL, suggesting the cause of the diminished GSIS is distal to the KATP channel/membrane depolarization event. ER Ca2+ homeostasis is important for myriad cellular processes and plays a pivotal role in intracellular Ca2+ signaling. Our data indicate that XBP1 and HNF4α are required for maintaining this homeostasis. Though inflammation models have shown that activation of XBP1 leads to expansion of ER calcium stores in bronchial secretory cells (43), to our knowledge we present the first evidence that XBP1 is required to maintain ER Ca2+ stores. The targets whose expression are dictated by XBP1/HNF4α and help maintain ER homeostasis are not clear, but one such candidate target may be SERCA2b. Depleting ER calcium stores by inhibiting SERCA2b with thapsigargin reduces GSIS, similar to our MODYI model (33). Transduction of XBP1u or XBP1s in β-cells increased expression of SERCA2b, the ER Ca2+ transporter, in isolated islets (Figs. 4B and 5A), confirming previous studies performed in vivo in the liver(32). Thus, our results indicate that knocking down HNF4α causes disruption of the expression of the Ca2+ pump responsible for establishing the high [Ca2+] in the ER, a functional decrease in ER [Ca2+], and finally, a diminished release of ER Ca2+ when cells are stimulated with caffeine (Fig. 4, A, C, F). These data outline a potential mechanism wherein altered ER Ca2+ homeostasis from loss of HNF4α function disrupts Ca2+ signaling and insulin release in β-cells of patients with MODYI.
There are no faithful animal models of MODYI. Previously mouse models have completely disrupted HNF4α expression in β-cells constitutively, characterizing its role in development (10, 11), rather than in the adult mouse, highlighting its role in β-cell maintenance as we show above. Some mutations in HNF4α in humans result in alleles thought to cause diabetes via a dominant negative mechanism (24, 25), whereas others would be expected to act via haploinsufficiency (44, 45). Thus, it may be premature to reach any firm conclusions about MODYI mechanisms based on our results using known MODYI-inducing human polymorphisms and adult-onset knock-out of Hnf4α in mice. However, if we are to speculate on the implications of our findings, we might suggest that they indicate consideration of a new angle on MODYI therapy. Currently, like type II diabetes, MODYI is responsive to treatment with sulfonylureas, though treatment often eventually involves insulin therapy to manage hyperglycemia, presumably because β-cells eventually become dysfunctional or die (46). Our results suggest that various therapies targeting ER homeostasis, a rapidly developing therapeutic avenue with several drugs at various stages of development, may augment or improve existing approaches to managing MODYI.
Author Contributions
B. D. M. designed and performed experiments, analyzed data, and wrote the manuscript; R. U. J. designed experiments and analyzed data; H. L. designed and performed experiments; M. J. designed and performed experiments; H. W. provided unique reagents; M. B. provided unique reagents and edited the manuscript; C. B. W. provided reagents and edited the manuscript; F. U. provided reagents, designed and guided experiments; J. C. M. designed experiments, analyzed data, and wrote the manuscript.
Acknowledgments
We thank the Digestive Disease Core Centers (DDRCC) Advanced Imaging and Tissue Analysis (AITA) Core for histological processing of tissues (Grant P30 DK052574). We also thank the Immunoassay Core of the Washington University Diabetes Research Center for insulin quantification Supported by DRC, Grant No. P30 DK020579. We also thank Center for Investigation of Membrane Excitability Diseases (CIMED) Live Cell Imaging Center for use of Ratiometric Ca2+ Imaging facilities.
This work was supported by National Institutes of Health NIDDK awards DK094989 and DK052574 (to J. C. M.), DK087873 (to M. A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
B. D. Moore and J. C. Mills, unpublished observations.
- ER
- endoplasmic reticulum
- XBP
- X-box-binding protein
- HNF
- hepatocyte nuclear factor
- MODY
- Mature-Onset Diabetes of the Young
- GSIS
- glucose-stimulated insulin secretion
- UPR
- unfolded protein response.
References
- 1. Mills J. C., and Taghert P. H. (2012) Scaling factors: transcription factors regulating subcellular domains. BioEssays 34, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huh W. J., Esen E., Geahlen J. H., Bredemeyer A. J., Lee A. H., Shi G., Konieczny S. F., Glimcher L. H., and Mills J. C. (2010) XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology 139, 2038–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen X., Ellis R. E., Lee K., Liu C. Y., Yang K., Solomon A., Yoshida H., Morimoto R., Kurnit D. M., Mori K., and Kaufman R. J. (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903 [DOI] [PubMed] [Google Scholar]
- 4. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 5. Lee A. H., Chu G. C., Iwakoshi N. N., and Glimcher L. H. (2005) XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Acosta-Alvear D., Zhou Y., Blais A., Tsikitis M., Lents N. H., Arias C., Lennon C. J., Kluger Y., and Dynlacht B. D. (2007) XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 [DOI] [PubMed] [Google Scholar]
- 7. Iwakoshi N. N., Lee A. H., Vallabhajosyula P., Otipoby K. L., Rajewsky K., and Glimcher L. H. (2003) Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321–329 [DOI] [PubMed] [Google Scholar]
- 8. Yamagata K., Furuta H., Oda N., Kaisaki P. J., Menzel S., Cox N. J., Fajans S. S., Signorini S. Stoffel M., and Bell G. I. (1996) Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature 384, 458–460 [DOI] [PubMed] [Google Scholar]
- 9. Bagwell A. M., Bento J. L., Mychaleckyj J. C., Freedman B. I., Langefeld C. D., and Bowden D. W. (2005) Genetic analysis of HNF4A polymorphisms in Caucasian-American type 2 diabetes. Diabetes 54, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 10. Gupta R. K., Vatamaniuk M. Z., Lee C. S., Flaschen R. C., Fulmer J. T., Matschinsky F. M., Duncan S. A., and Kaestner K. H. (2005) The MODY1 gene HNF-4α regulates selected genes involved in insulin secretion. J. Clin. Invest. 115, 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miura A., Yamagata K., Kakei M., Hatakeyama H., Takahashi N., Fukui K., Nammo T., Yoneda K., Inoue Y., Sladek F. M., Magnuson M. A., Kasai H., Miyagawa J., Gonzalez F. J., and Shimomura I. (2006) Hepatocyte nuclear factor-4α is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J. Biol. Chem. 281, 5246–5257 [DOI] [PubMed] [Google Scholar]
- 12. Hara T., Mahadevan J., Kanekura K., Hara M., Lu S., and Urano F. (2014) Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 155, 758–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cardozo A. K., Ortis F., Storling J., Feng Y. M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., and Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 14. Inoue H., Tanizawa Y., Wasson J., Behn P., Kalidas K., Bernal-Mizrachi E., Mueckler M., Marshall H., Donis-Keller H., Crock P., Rogers D., Mikuni M., Kumashiro H., Higashi K., Sobue G., Oka Y., and Permutt M. A. (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 20, 143–148 [DOI] [PubMed] [Google Scholar]
- 15. Luebke-Wheeler J., Zhang K., Battle M., Si-Tayeb K., Garrison W., Chhinder S., Li J., Kaufman R. J., and Duncan S. A. (2008) Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology 48, 1242–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heidtman K., Hotamisligil G. S., and Glimcher L. H. (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 108, 8885–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacobo S. M., Guerra M. L., and Hockerman G. H. (2009) Cav1.2 and Cav1.3 are differentially coupled to glucagon-like peptide-1 potentiation of glucose-stimulated insulin secretion in the pancreatic beta-cell line INS-1. J. Pharmacol. Exp. Ther. 331, 724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ovcharenko I., Loots G. G., Giardine B. M., Hou M., Ma J., Hardison R. C., Stubbs L., and Miller W. (2005) Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 15, 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolotin E., Liao H., Ta T. C., Yang C., Hwang-Verslues W., Evans J. R., Jiang T., and Sladek F. M. (2010) Integrated approach for the identification of human hepatocyte nuclear factor 4α target genes using protein binding microarrays. Hepatology 51, 642–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Battle M. A., Konopka G., Parviz F., Gaggl A. L., Yang C., Sladek F. M., and Duncan S. A. (2006) Hepatocyte nuclear factor 4α orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc. Natl. Acad. Sci. U.S.A. 103, 8419–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonzo J. A., Ferry C. H., Matsubara T., Kim J. H., and Gonzalez F. J. (2012) Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J. Biol. Chem. 287, 7345–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cattin A. L., Le Beyec J., Barreau F., Saint-Just S., Houllier A., Gonzalez F. J., Robine S., Pinçon-Raymond M., Cardot P., Lacasa M., and Ribeiro A. (2009) Ribeiro, Hepatocyte nuclear factor 4α, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol. Cell. Biol. 29, 6294–6308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayhurst G. P., Lee Y. H., Lambert G., Ward J. M., and Gonzalez F. J. (2001) Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furuta H., Iwasaki N., Oda N., Hinokio Y., Horikawa Y., Yamagata K., Yano N., Sugahiro J., Ogata M., Ohgawara H., Omori Y., Iwamoto Y., and Bell G. I. (1997) Organization and partial sequence of the hepatocyte nuclear factor-4α/MODY1 gene and identification of a missense mutation, R127W, in a Japanese family with MODY. Diabetes 46, 1652–1657 [DOI] [PubMed] [Google Scholar]
- 25. Lindner T., Gragnoli C., Furuta H., Cockburn B. N., Petzold C., Rietzsch H., Weiss U., Schulze J., and Bell G. I. (1997) Hepatic function in a family with a nonsense mutation (R154X) in the hepatocyte nuclear factor-4α/MODY1 gene. J. Clin. Invest. 100, 1400–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee A. H., Iwakoshi N. N., and Glimcher L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Todd D. J., McHeyzer-Williams L. J., Kowal C., Lee A. H., Volpe B. T., Diamond B., McHeyzer-Williams M. G., and Glimcher L. H. (2009) XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 206, 2151–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwakoshi N. N., Lee A. H., and Glimcher L. H. (2003) The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 194, 29–38 [DOI] [PubMed] [Google Scholar]
- 29. Leber J. H., Bernales S., and Walter P. (2004) IRE1-independent gain control of the unfolded protein response. PLoS biology 2, E235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oslowski C. M., and Urano F. (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashcroft F. M., Harrison D. E., and Ashcroft S. J. (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312, 446–448 [DOI] [PubMed] [Google Scholar]
- 32. Park S. W., Zhou Y., Lee J., Lee J., and Ozcan U. (2010) Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 19320–19325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vangheluwe P., Raeymaekers L., Dode L., and Wuytack F. (2005) Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium 38, 291–302 [DOI] [PubMed] [Google Scholar]
- 34. Palmer A. E., Jin C., Reed J. C., and Tsien R. Y. (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U.S.A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verkhratsky A., and Shmigol A. (1996) Calcium-induced calcium release in neurones. Cell Calcium 19, 1–14 [DOI] [PubMed] [Google Scholar]
- 36. Wang H., Maechler P., Antinozzi P. A., Hagenfeldt K. A., and Wollheim C. B. (2000) Hepatocyte nuclear factor 4α regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem. 275, 35953–35959 [DOI] [PubMed] [Google Scholar]
- 37. Allagnat F., Christulia F., Ortis F., Pirot P., Lortz S., Lenzen S., Eizirik D. L., and Cardozo A. K. (2010) Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 53, 1120–1130 [DOI] [PubMed] [Google Scholar]
- 38. Ogawa N., and Mori K. (2004) Autoregulation of the HAC1 gene is required for sustained activation of the yeast unfolded protein response. Genes Cells 9, 95–104 [DOI] [PubMed] [Google Scholar]
- 39. Yang L., Xue Z., He Y., Sun S., Chen H., and Qi L. (2010) A Phos-tag-based approach reveals the extent of physiological endoplasmic reticulum stress. PloS one 5, e11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu C. C., Dougan S. K., McGehee A. M., Love J. C., and Ploegh H. L. (2009) XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 28, 1624–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boyd M., Bressendorff S., Møller J., Olsen J., and Troelsen J. T. (2009) Mapping of HNF4α target genes in intestinal epithelial cells. BMC Gastroenterology 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato Y., Hatta M., Karim M. F., Sawa T., Wei F. Y., Sato S., Magnuson M. A., Gonzalez F. J., Tomizawa K., Akaike T., Yoshizawa T., and Yamagata K. (2012) Anks4b, a novel target of HNF4α protein, interacts with GRP78 protein and regulates endoplasmic reticulum stress-induced apoptosis in pancreatic beta-cells. J. Biol. Chem. 287, 23236–23245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martino M. E., Olsen J. C., Fulcher N. B., Wolfgang M. C., O'Neal W. K., and Ribeiro C. M. (2009) Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 284, 14904–14913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alam M. S., Kim I. J., Ling Z., Mahmood A. H., O'Neill J. J., Severini H., Sun C. R., Wappler F., Crawford G., Daubenmier C. M., Fulton R., Fujino D., Gan K. K., Honscheid K., Kagan H., Kass R., Lee J., Sung M., White C., Wolf A., Zoeller M. M. et al. (1995) First measurement of the rate for the inclusive radiative penguin decay b–>s gamma. Phys. Rev. Letts. 74, 2885–2889 [DOI] [PubMed] [Google Scholar]
- 45. Thomas H., Jaschkowitz K., Bulman M., Frayling T. M., Mitchell S. M., Roosen S., Lingott-Frieg A., Tack C. J., Ellard S., Ryffel G. U., and Hattersley A. T. (2001) A distant upstream promoter of the HNF-4α gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum. Mol. Genet. 10, 2089–2097 [DOI] [PubMed] [Google Scholar]
- 46. Pearson E. R., Pruhova S., Tack C. J., Johansen A., Castleden H. A., Lumb P. J., Wierzbicki A. S., Clark P. M., Lebl J., Pedersen O., Ellard S., Hansen T., and Hattersley A. T. (2005) Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4α mutations in a large European collection. Diabetologia 48, 878–885 [DOI] [PubMed] [Google Scholar]
- 47. Im H., Grass J. A., Johnson K. D., Boyer M. E., Wu J., and Bresnick E. H. (2004) Measurement of protein-DNA interactions in vivo by chromatin immunoprecipitation. Methods Mol. Biol. 284, 129–146 [DOI] [PubMed] [Google Scholar]
- 48. Hayashi S., and McMahon A. P. (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 [DOI] [PubMed] [Google Scholar]
- 49. Tian X., Jin R. U., Bredemeyer A. J., Oates E. J., Błazewska K. M., McKenna C. E., and Mills J. C. (2010) RAB26 and RAB3D are direct transcriptional targets of MIST1 that regulate exocrine granule maturation. Mol. Cell. Biol. 30, 1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cross B. C., Bond P. J., Sadowski P. G., Jha B. K., Zak J., Goodman J. M., Silverman R. H., Neubert T. A., Baxendale I. R., Ron D., and Harding H. P. (2012) The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. U.S.A. 109, E869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grynkiewicz G., Poenie M., and Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 52. Li D. S., Yuan Y. H., Tu H. J., Liang Q. L., and Dai L. J. (2009) A protocol for islet isolation from mouse pancreas. Nature protocols 4, 1649–1652 [DOI] [PubMed] [Google Scholar]
- 53. Lee A. H., Scapa E. F., Cohen D. E., and Glimcher L. H. (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Muniappan L., and Ozcan S. (2009) Adenoviral gene transfer into isolated pancreatic islets. Methods Mol. Biol. 590, 131–142 [DOI] [PubMed] [Google Scholar]
- 55. Nolan A. L., and O'Dowd J. F. (2009) The measurement of insulin secretion from isolated rodent islets of Langerhans. Methods Mol. Biol. 560, 43–51 [DOI] [PubMed] [Google Scholar]