

Abstract
Pyroglutamate-modified amyloid-β (pE-Aβ) is a highly neurotoxic amyloid-β (Aβ) isoform and is enriched in the brains of individuals with Alzheimer disease compared with healthy aged controls. Pyroglutamate formation increases the rate of Aβ oligomerization and alters the interactions of Aβ with Cu2+ and lipids; however, a link between these properties and the toxicity of pE-Aβ peptides has not been established. We report here that Aβ3pE-42 has an enhanced capacity to cause lipid peroxidation in primary cortical mouse neurons compared with the full-length isoform (Aβ(1–42)). In contrast, Aβ(1–42) caused a significant elevation in cytosolic reactive oxygen species, whereas Aβ3pE-42 did not. We also report that Aβ3pE-42 preferentially associates with neuronal membranes and triggers Ca2+ influx that can be partially blocked by the N-methyl-d-aspartate receptor antagonist MK-801. Aβ3pE-42 further caused a loss of plasma membrane integrity and remained bound to neurons at significantly higher levels than Aβ(1–42) over extended incubations. Pyroglutamate formation was additionally found to increase the relative efficiency of Aβ-dityrosine oligomer formation mediated by copper-redox cycling.
Keywords: Alzheimer disease, amyloid-beta (Aβ), lipid peroxidation, oligomer, reactive oxygen species (ROS), dityrosine, pyroglutamate
Introduction
Aβ3 peptides are found in every human brain; however, the concentration and composition of Aβ peptide isoforms are distinctly different in healthy individuals and people with AD (1–3). Amino-truncated Aβ peptides are abundant in the AD brain (4, 5) and increase in prevalence with disease progression (6). The process of Aβ amino-truncation can occur via the actions of aminopeptidases on full-length Aβ peptides (7, 8), via altered cleavage of amyloid precursor protein in the generation of Aβ (9–11), and potentially by Aβ-copper-redox cycling reactions (12). As a consequence, amino-truncation can expose glutamate residues (positions 3 and 11 of Aβ) to cyclization by the action of glutaminyl cyclase (QC), forming the highly amyloidogenic pyroglutamate-Aβ (pE-Aβ) peptides Aβ3pE-40, Aβ3pE-42, Aβ11pE-40, and Aβ11pE-42 (7, 13).
Pyroglutamate formation significantly increases the hydrophobicity of Aβ, causing the peptide to aggregate more rapidly and form oligomers at lower concentration thresholds (5, 14, 15). pE-Aβ peptides also demonstrate increased β-sheet (aggregate structure) stability (16, 17), differences in fibril ultrastructure (18, 19), and altered interactions with copper ions (20, 21) and synthetic lipid membranes (22, 23). Notably, trace quantities of Aβ3pE-42 have been observed to dramatically enhance the aggregation and neurotoxicity of Aβ(1–42) (24), prompting descriptions of pE-Aβ as “prion-like.” Still, it remains unclear as to the cytotoxic potency of pE-Aβ peptides compared with their full-length Aβ counterparts. Some studies have demonstrated pE-Aβ peptides to have enhanced toxicity (24–26), although others have reported no difference in toxicity between the isoforms (27–30). Methodological differences may account somewhat for variability in the relative toxicities reported (Table 1), yet molecular mechanisms to explain changes in cytotoxicity have not been defined.
TABLE 1.
Overview of publications comparing the cytotoxicity of pE-Aβ and full-length Aβ peptides in vitro
The following abbreviations are used in table: CSF, cerebrospinal fluid; DMSO, dimethyl sulphoxide; HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol; LDH, lactate dehydrogenase; PBS, phosphate buffered saline.

One mechanism through which Aβ peptides cause cytotoxicity is by production of reactive oxygen species (ROS) via facile copper-redox cycling (31–33), which can in turn effect oxidative damage to neuronal proteins and lipids (34). Imbalances in ROS production and detoxification are strongly implicated in AD neurodegeneration, reflected by cerebral elevations in oxidized DNA, lipids, and proteins (35–37). Pyroglutamate formation alters Aβ-Cu2+ coordination modes (20, 21), although it is not known whether this affects the capacity of pE-Aβ peptides to undertake redox cycling and produce cytotoxic ROS. We therefore aimed to determine whether full-length Aβ and pE-Aβ possess differences in their capacity to alter ROS flux and cause oxidative damage to neurons in vitro. Additionally, Aβ isoforms were compared for their capacity to form oligomers and covalent tyrosine-tyrosine bonds (dityrosine) as a result of Aβ-copper-redox cycling. The capacity for Aβ to form dityrosine has previously been correlated with neurotoxicity (38), although recent reports have found that Aβ fibrils within amyloid plaques contain intense dityrosine immunoreactivity (39), indicating that dityrosine formation may be associated with AD amyloidogenesis. Further comparisons were made between the peptides for their capacity to perturb neuronal membranes and induce changes in neuronal ion homeostasis (Ca2+ flux).
Experimental Procedures
Materials
Aβ peptides were purchased from the ERI Amyloid Laboratory, LLC, and purified by reversed-phase HPLC to >95% purity. All chemicals used were analytical grade and purchased from ChemSupply (Australia) unless otherwise stated. Thioflavin-T, coumarin-3-carboxylic acid (3-CCA), butylated hydroxytoluene, bovine serum albumin, and paraformaldehyde were purchased from Sigma. Diamsar (1,8-diaminosarcophagine) was synthesized as reported previously (40).
Preparation of Aβ Solutions and Cu2+-oxidized Aβ Oligomers
Aβ stock solutions were prepared by dissolving lyophilized peptides to 5 mg/ml in NaOH (60 mm) and incubating at ambient temperature for 5 min to dissociate aggregated material. Solutions were then diluted to 1 mg/ml in MilliQ H2O and 10× PBS (PBS is defined as 50 mm sodium phosphate, 2.7 mm KCl, 137 mm NaCl) at a v/v/v ratio of 2:7:1 (NaOH, H2O, 10× PBS). The preparation was sonicated for 10 min in an iced water bath and then centrifuged at 16,500 × g for 10 min at 4 °C. Supernatants (upper 80% of solution) were removed to pre-chilled tubes on ice; pH was adjusted by addition of NaH2PO4 (0.5 m) to 1.0% v/v and then kept on ice for immediate use. Aβ stock concentrations were determined by UV spectrometry by measuring absorbance at 214 nm (A214) and applying the following extinction coefficients (m−1 cm−1) determined from UV scans and amino acid analysis: Aβ(1–40) = 91,462; Aβ3pE-40 = 89,705; Aβ(1–42) = 94,526; and Aβ3pE-42 = 90,925.
Aβ oligomers were generated by reacting Aβ (10 μm) with Cu2+ (10 μm, as CuCl2·glycine6) and ascorbate (100 μm) at 37 °C on a vertically rotating wheel at 20 rpm, in 2-ml round-bottom tubes (catalog no. 0030123.344, Eppendorf). Reactions were halted with the addition of EDTA (to 250 μm) and placed on ice. Control incubations of Aβ only were incubated and sampled under identical conditions for comparison. Aβ fibrillization assays (thioflavin-T assays) were performed as described previously (41).
Detection of Cell-free Hydroxyl Radical Production
Hydroxyl radical production in Aβ-copper-redox cycling reactions was assessed with the fluorescent probe coumarin-3-carboxylic acid (3-CCA) using methodology adapted from Manevich et al. (42). Reactions containing Aβ peptides (20 μm) and 3-CCA (100 μm) in the presence or absence of Cu2+ (20 μm, as CuCl2·glycine6) and ascorbate (300 μm) in PBS, pH 7.4, were incubated in black-walled clear bottom microplates (PerkinElmer Life Sciences) at 37 °C in a heated Flexstation III spectrophotometer (Molecular Devices). Fluorescence signals were read from the bottom of the microplate every 30 s using excitation λ 395 nm and measuring emission at λ 450 nm. Rate constants for the 3-CCA reactions were determined from one-phase association regressions of logY transformed data (data manipulations performed in GraphPad Prism version 6.0).
Aβ-Dityrosine Determination and Hydrophobic Index Calculation
The dityrosine content of Cu2+-reacted Aβ samples was determined by fluorescence spectrophotometry (λ 320 nm excitation and λ 420 nm emission), as described previously (43). Reaction half-times and rate constants (K = min−1) were calculated from one-phase association regressions of fluorescence data (GraphPad Prism version 6.0). Calculation of Aβ hydrophobic scores were determined from the sum of the amino acid residue hydropathic indexes (44). Pyroglutamyl residues were assigned a hydropathy value of −1.0.
Aβ Detection by Western Blot
Tissue extracts and synthetic peptides were separated by SDS-PAGE using 4–12% XT BisTris gels (Criterion, Bio-Rad) according to the manufacturer's instructions. Samples were transferred to pre-assembled PVDF membrane stacks using a Trans-Blot® semi-dry transfer apparatus (Bio-Rad). Blots were blocked in TBS-T (10 mm Tris-HCl, 50 mm NaCl, 0.1% v/v Tween 20, pH 8.0) containing 5% w/v skim milk. Primary antibodies were incubated on blots for at least 1 h at room temperature or overnight at 4 °C. HRP-conjugated rabbit anti-mouse or goat anti-rabbit immunoglobulins (Dako) were diluted 1:10,000 in TBST and incubated for 1 h at room temperature. All antibodies were diluted in TBS-T containing 5% skim milk and 0.05% w/v sodium azide. Blots were washed four times for 10 min in TBS-T after each primary and secondary antibody binding step. Chemiluminescence signals were captured after application of ECL (Immobilon, Millipore) with an LAS3000 detector and analyzed using MultiGauge software (Fujifilm). Aβ peptides were detected using the monoclonal mouse antibodies 4G8 (Covance) or 6E10 (Signet Laboratories) diluted to 1 μg/ml. The dityrosine modification was detected using the 1C3 antibody raised against synthetic dityrosine (catalog no. NWA-DIY020, Northwest Life Science Specialties), at a 1:1000 dilution.
Size-exclusion Chromatography (SEC) and Atomic Force Microscopy (AFM)
Aβ oligomers in HBSS buffer were prepared as above but at twice the concentration (20 μm Aβ, 20 μm Cu2+, 200 μm ascorbate) to provide adequate signals for measurement.
SEC analysis was performed using a BioLogic DuoFlow QuadTec 40 system (Bio-Rad) fitted with a Superdex 75 10/300 column (catalog no. 17-5174-01, GE Healthcare). Both equilibration and operation of the column were in Tris-buffered saline (20 mm Tris, 200 mm NaCl, pH 8.0, 0.2 μm filtered and de-gassed) at a flow rate of 0.5 ml/min and ambient temperature. The absorbance at 214, 260, and 280 nm was monitored, collecting 5 data points/s. Samples were injected onto the column (0.5 ml per run, ∼45 μg of Aβ) immediately after collection at indicated time points.
AFM analyses were performed on Aβ reactions prepared in Neurobasal medium (catalog no. 21103-049, Gibco) at 10 μm. Samples (10 μl) were collected and spotted on freshly cleaved mica, dried at room temperature for 5 min in a laminar flow hood, and rinsed with 2 ml of de-ionized water (Milli-Q, Millipore). The sample was blown dry with nitrogen (Coregas Nitrogen 4.0) before being transferred to the AFM sample stage (Asylum Research Cypher AFM). Images were acquired in alternating current (tapping) mode in air using Tap300-G silicon AFM probes (Budget Sensors) with scan rates of 1.5–2.5 Hz; drive amplitude was kept to a minimum with amplitude set-points of 60–80%. The mask threshold was set to 250 pm for image analysis.
Primary Neuronal Cell Culture and Aβ Clearance Assays
All experiments involving animals were conducted in accordance with the Australian Code of Practice for the Use of Laboratory Animals and were approved by the Institutional Animal Experimentation Ethics Committee.
Mouse cortical neuronal cultures isolated from C57Bl/6 E14 embryos were prepared as described previously (45). Cells were plated in poly-d-lysine-coated 48- or 96-well plates at a density of 150,000 cells/cm2. All cell culture materials were purchased from Gibco/Thermo Fisher unless otherwise stated. Cells were grown in Neurobasal medium (NB) containing B27 supplements, gentamicin, and 0.5 mm GlutaMAXTM. Fresh NB medium containing B27 minus antioxidants (B27-AO) and cytosine-β-d-arabinofuranoside (2 μm; catalog no. C1768, Sigma) were applied after 6 days in vitro (DIV). Neurons were further incubated until DIV 8 or 9 prior to applying treatments.
Levels of cell-bound Aβ were measured following exposure to Aβ peptides applied in NB medium containing B27-AO and cytosine-β-d-arabinofuranoside. Cells (DIV 8 or 9) were treated for 48 h with Aβ mixtures (10 μm total). The media were removed, and cells were washed twice with Dulbecco's PBS (catalog no. 14190-144, Gibco), and then the cells were extracted with M-PER reagent (catalog no. 78501, Thermo-Scientific). For fractionation studies, cells were scraped into TBS, pH 7.5, containing protease inhibitors (cOmplete, catalog no. 11873580001, Roche Applied Science) and probe-sonicated by two rounds of 10 bursts (40% power, 0.5 s each) with a Sonifier S-250D (Branson). Lysates were centrifuged at 100,000 × g, and supernatants (TBS phase) were collected to fresh tubes. The pellets were extracted with an equal volume of Na2CO3 (100 mm, pH 12) for 1 h of incubation on ice and then briefly vortexed and centrifuged again to collect supernatants (carbonate phase). Pellets were resuspended in an equal volume of urea buffer (7 m urea, 2 m thiourea, 4% CHAPS, 1% DTT, 50 mm Tris, pH 8.0). Western blots were performed on 5 μg of total protein based on BCA assays (catalog no. 23225, Thermo-Scientific) of TBS phases, loading equal volumes for carbonate and urea phases.
Cell Viability Measurement
M17 neuroblastomas were cultured in DMEM containing fetal bovine serum (FBS; 10% v/v) and penicillin/streptomycin (catalog no. 15140, Gibco). Cells were plated in 6-well culture plates (catalog no. 140675, Nunc) at 40,000 cells/cm2 and incubated overnight (37 °C, 5% CO2). Fresh medium supplemented with retinoic acid (10 μm) was applied, and the cells were allowed to differentiate for 48 h. Freshly prepared Aβ solutions (10 μm) or synthetic lipid peroxide (1 μm) were applied to cells for 4 h and then the cells were collected using trypsin and resuspended in 1 ml of PBS containing 2% FBS. Propidium iodide was added to a final concentration of 1 μg/ml. Flow cytometric analysis was performed on a Beckman Coulter CyAn ADP analyzer with Summit 4.0.3 acquisition software. Debris and cell aggregates were excluded by gating. Live and dead cells were then identified based on exclusion and inclusion, respectively, of propidium iodide. Analysis of data were performed using FCS Express 4 analysis software.
Detection of Neuronal ROS Flux and Lipid Peroxidation
Neuronal ROS flux was measured using the redox-sensitive probe 2,7-dichlorofluorescin diacetate (catalog no. D399, Molecular Probes), essentially as described previously (46). Briefly, 2,7-dichlorofluorescin diacetate (100 μm) was applied to cells (DIV 9) in NB medium supplemented with B27-AO (37 °C, 5% CO2) for 60 min; the media were then removed, and the cells were washed with Hanks' balanced salt solution (HBSS; catalog no. 14175-079, Gibco). Freshly prepared Aβ peptides were diluted to 10 μm in HBSS and applied to the neurons for a further 60 min of incubation (37 °C, 5% CO2, light protected) until reading DCF fluorescence (λ 485 nm excitation, λ 530 nm emission, and λ 515 nm emission cutoff filter) with a Flexstation III spectrophotometer (Molecular Devices) equilibrated to 37 °C.
Lipid peroxidation was measured using the lipid hydroperoxide probe diphenyl-1-pyrenylphosphine (DPPP; catalog no. 62237, Cayman Chemicals), with methodology adapted from Ref. 47. DPPP (stock dissolved in N2-purged DMSO to 25.88 mm) was diluted to 50 μm in NB media and applied to cells (DIV 8–9) in 96-well black-walled microplates (Greiner) and then incubated for 60 min at 37 °C, 5% CO2 in the dark. The DPPP-containing media were removed; the cells were rinsed twice with HBSS and then equilibrated in HBSS at 37 °C and 5% CO2 for 30 min before treatment. Freshly prepared Aβ peptides were applied to cells for 4 h until reading fluorescence (λ 340 nm excitation and λ 380 nm emission) with an EnSpire multimode spectrophotometer (PerkinElmer Life Sciences). Positive control wells were treated with a synthetic lipid hydroperoxide standard (catalog no. 705014; Cayman Chemicals) for comparison. Care was taken at all steps to minimize artifactual oxidation of DPPP by reducing exposure to light.
Neuronal Calcium Flux Measurement
Cortical neurons plated at 230,000 cells/cm2 in 96-well black-walled microplates (Greiner) were loaded with the fluorescent Ca2+ sensor Fluo4-AM (catalog no. F14201, Molecular Probes) at DIV9 for 30 min at 37 °C, 5% CO2, and then equilibrated to room temperature for 30 min. Fluorescence (excitation λ 490 nm and emission λ 520 nm) was measured at base line for 10 reads at 27-s intervals followed by a further 10 reads after injection of glutamate or Aβ (final concentrations of 1 and 10 μm, respectively) using a Fluostar plate reader (BMG Labtech). Ca2+ flux values (ΔF/F0) were expressed as the difference between mean baseline and immediately following glutamate or Aβ application. Experiments determining the effect of metal depletion utilized neuronal cultures pre-treated for 1 h with Diamsar (10 μm). To assess the contribution of NMDAR signaling in Aβ-induced Ca2+ flux, cultures were pre-treated for 15 min with the NMDAR antagonist MK-801 (1 mm). In separate experiments the supplied assay buffer (HBSS) was replaced with Ca2+/Mg2+-free HBSS to determine the source of Ca2+ entering the cytosol.
Results
Pyroglutamate Formation Alters the Production of ROS and Dityrosine by Aβ
pE-Aβ peptides demonstrated greatly increased fibrillization rates compared with the respective full-length isoforms (Fig. 1A), consistent with previous reports (14, 18). Aβ(1–40), Aβ(1–42), Aβ3pE-40, and Aβ3pE-42 were also compared for their capacity to produce ROS upon reaction with Cu2+ and ascorbate. The rate of •OH production was significantly higher for Aβ(1–42) compared with Aβ3pE-42 (Fig. 1B). In contrast, Aβ3pE-40 produced more •OH than Aβ(1–40) and indeed all other Aβ isoforms. The kinetics of •OH production showed a similar sigmoidal pattern for all peptides with signals reaching plateau after ∼20 min.
FIGURE 1.

Comparison of the aggregation, redox cycling, and dityrosine formation capacities of Aβ peptides. A, Aβ (5 μm) fibrillization kinetics measured by thioflavin-T assay (data cropped at start of plateau due to signal decay as Aβ sediments in microplate wells). B, kinetics of •OH formation in reactions of Aβ (20 μm) with CuCl2 (20 μm) and ascorbate (300 μm) as measured by 3-CCA fluorescence assay. Kinetics of dityrosine formation in reactions of Aβ (10 μm) with CuCl2 (10 μm) and ascorbate (100 μm) Aβ(1–40) and Aβ3pE-40 peptides (C), Aβ(1–42) and Aβ3pE-42 peptides (D), measured by fluorescence spectrometry of dityrosine bond (320 nm excitation and 420 nm emission). E, relationship between Aβ hydrophobicity and dityrosine formation rate. F, relative efficiency of Aβ-dityrosine formation was further calculated as a ratio of the amount of dityrosine formed per unit of •OH produced, plotted against Aβ hydrophobicity. All measurements were repeated a minimum of three times over separate days using freshly prepared Aβ solutions. Data shown are mean ± S.E.
Differences in redox cycling and •OH production between Aβ peptides were further studied to compare formation of dityrosine bonds. Dityrosine formation rates partially reflected •OH production rates whereby Aβ3pE-40 formed dityrosine more rapidly than Aβ(1–40) (half-times of 5.44 and 33.64 min, respectively), whereas Aβ(1–42) formed dityrosine more rapidly than Aβ3pE-42 (half-times of 5.38 and 7.31 min, respectively) (Fig. 1, C and D). Total Aβ-dityrosine content, however, did not mirror the levels of •OH produced by each peptide; Aβ(1–42) formed more than double the amount of dityrosine than all other peptides. A direct relationship was not observed between the dityrosine formation rate and relative Aβ hydrophobicity (Fig. 1E); however, the efficiency of dityrosine formation (calculated as a ratio of dityrosine formed per unit of •OH generated) was increased for both pE-Aβ peptides relative to their full-length counterparts (Fig. 1F).
Comparison of the Oligomerization of Pyroglutamate-Aβ and Full-length Aβ in the Presence and Absence of Cu2+
Previous studies have shown differences between Aβ(1–40) and Aβ(1–42) in oligomerization and dityrosine formation when reacted with Cu2+ and biological reductants (48). We compared the profiles of full-length Aβ and pE-Aβ oligomers generated in the presence or absence of Cu2+ and ascorbate. Aβ(1–42) and Aβ3pE-42 rapidly formed SDS-stable oligomers within 5 min of reaction with Cu2+, whereas increases in Aβ(1–40) and Aβ3pE-40 oligomers were observed at 15 min (Fig. 2A). Significant formation of SDS-stable oligomers was not observed for Aβ incubated in the absence of Cu2+ and ascorbate over the same time period.
FIGURE 2.

A, Western blots stained with 4G8 anti-Aβ antibody demonstrating rapid formation of SDS-stable low molecular weight Aβ oligomers catalyzed by reaction with Cu2+ and ascorbate (Asc.). B, comparison of Aβ(1–40) and Aβ3pE-40 dityrosine oligomer profiles. Western blots were stained with 1C3 anti-dityrosine antibody.
Dityrosine cross-links were also detected in Aβ(1–40) and Aβ3pE-40 reactions by immunostaining with a dityrosine-specific antibody (1C3), revealing predominantly dimer and trimer-sized (∼8/12 kDa) oligomers and confirming the enhanced capacity for Aβ3pE-40 dityrosine formation compared with Aβ(1–40) (Fig. 2B). The 1C3 antibody, however, did not show significant immunoreactivity to Western blots of Cu2+-reacted Aβ(1–42) and Aβ3pE-42 peptides (data not shown).
We additionally examined the oligomer states of Aβ(1–42) and Aβ3pE-42 in non-denaturing conditions by SEC and AFM. After a 4-h incubation in PBS, the Aβ(1–42) oligomer profile did not change considerably from the freshly prepared solution, resolving predominantly as a single low-mass peak (<14 kDa) by size exclusion, with minor signals corresponding to oligomers of ∼40–75 kDa (Fig. 3A). Addition of Cu2+ and ascorbate to Aβ(1–42) caused a sizable increase in the abundance of oligomers but did not appear to alter their mass. By comparison, Aβ3pE-42 ternary states changed considerably during the 4-h incubation, observed as a dramatic loss of low-mass peak (<14 kDa) in either the presence or absence of Cu2+ and ascorbate (Fig. 3B). After a 4-h incubation, residual signals remaining in the Aβ3pE-42 reactions without Cu2+ resolved predominantly as an oligomeric peak at ∼30–45 kDa. In the Aβ3pE-42 reaction containing Cu2+ there was some preservation of the monomeric signal after the 4-h incubation, with an additional polydisperse peak across the 30–75-kDa range.
FIGURE 3.
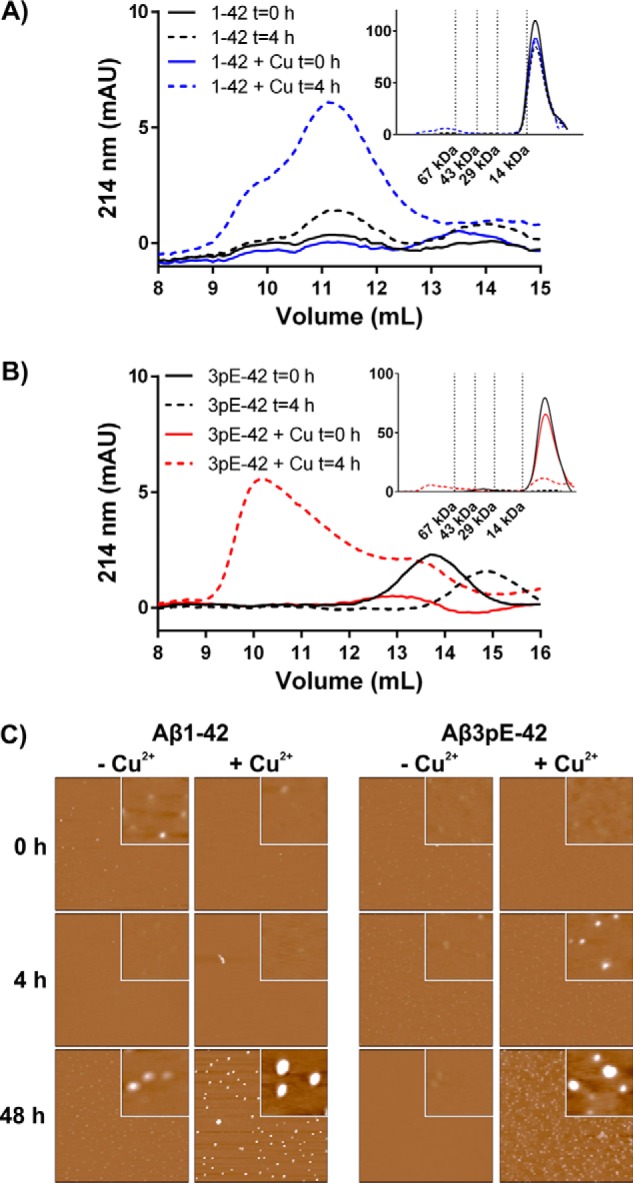
Comparison of oligomer structures formed in the presence or absence of Cu2+ and ascorbate. Aβ(1–42) (20 μm) (A) and Aβ3pE-42 (20 μm) (B) solutions incubated at 37 °C for 4 h in HBSS, pH 7.4, ± Cu2+ (20 μm) and ascorbate (200 μm) were assessed for oligomer formation by SEC using a Superdex 75 column. Main figure shows the region of >14 kDa; insets show the full chromatogram with molecular mass standards (214 nm UV detection). C, AFM images of Aβ peptides (10 μm) incubated for indicated times in Neurobasal medium. Each image displays a 2 × 2 μm field and 10 nm total z-range; insets display a 200 × 200 nm field and 2 nm total z-range. SECs represent the same profile from a minimum of n = 3 independent replicates. AFM analyses were repeated on independent reactions over separate days; representative data are shown.
To simulate Aβ oligomerization in cell culture conditions, reactions were conducted in neurobasal media over an extended incubation period, and the peptide structures were assessed by AFM (Fig. 3C). Little difference in peptide structure was observed during a 48-h incubation of Aβ(1–42) in unsupplemented neurobasal media (maximum z <1 nm). In the Aβ(1–42) reactions supplemented with Cu2+/ascorbate, there was similarly no noticeable change after 4-h incubation, although a large increase in the size and abundance of oligomers was observed after 48 h (maximum z = 4.9 nm). Likewise, Aβ3pE-42 structures did not demonstrate a noticeable size difference over a 48-h incubation in neurobasal media in the absence of supplemental Cu2+/ascorbate (maximum z <1 nm), although the reactions supplemented with Cu2+/ascorbate showed a stepwise size increase over the incubation period (maximum z = 0.4, 1.0, and 1.8 nm at 0, 4, and 48 h, respectively).
Aβ3pE-42 Predominantly Associates with Neuronal Membranes and Is Resistant to Clearance
pE-Aβ has been found to resist proteolytic clearance in astrocyte cultures (49, 50). We therefore compared the residual levels of Aβ(1–42) and Aβ3pE-42 following extended exposures to cortical neuron cultures, additionally assessing the capacity for minor quantities of Aβ3pE-42 to affect the clearance of Aβ(1–42). Neuronal Aβ levels were strikingly different after 48 h of treatment with the two peptides; relative levels of Aβ3pE-42 were 4.5-fold higher than cells treated with Aβ(1–42) (Fig. 4A). A trend toward higher mean levels of residual Aβ was observed when Aβ(1–42) solutions were supplemented with either 50 or 25% (mol/mol) Aβ3pE-42; however, this effect was not observed when Aβ3pE-42 represented only 5% of the total Aβ concentration (Fig. 4A).
FIGURE 4.

Freshly prepared Aβ peptides were applied to cortical mouse neurons and levels of residual Aβ remaining cell-bound and intact after 48 h of incubation were measured by Western blot (4G8 antibody) to model Aβ proteolytic clearance/resistance. A, Aβ(1–42), Aβ3pE-42, or mixtures thereof were quantified by densitometric analysis of bands at 4–6 kDa; percentages indicated in column titles represent the proportion of Aβ3pE-42 mixed with Aβ(1–42) to give a final concentration of 10 μm. Aβ levels are reported as relative densitometry signals compared with cells treated with Aβ(1–42) alone, as Aβ signals were not observed in vehicle control wells in these conditions. B, cell extracts 48 h after application of freshly prepared Aβ peptides to cortical neurons were fractionated to separate soluble Aβ (TBS extracted), peripheral membrane-bound Aβ (carbonate phase), and integral membrane-bound Aβ (urea phase). Aβ monomers are apparent at the approximate molecular mass of Aβ (4–5 kDa), and Aβ oligomeric signals are identified by arrowheads (contrast enhanced panel). 4G8-reactive signals above 35 kDa were present in all groups (including vehicle control) suggesting that these signals are not Aβ but may correspond to the amyloid precursor protein and its cleavage products in the cortical neurons. All densitometry signals were corrected for protein loading by re-probing blots for actin. Samples shown are representative of three separate fractionations per group; panels have been cropped from the same blot image to show only relevant lanes. Data are mean ± S.E., statistical significance is relative to Aβ(1–42)-treated group.** represents p ≤ 0.01.
From experiments utilizing sequential extraction of cells following Aβ exposure, both the Aβ(1–42) and Aβ3pE-42 peptides were predominantly found in membrane fractions, with relatively little Aβ in the soluble (TBS) phase observed qualitatively by Western blot (Fig. 4B). Aβ in cell extracts resolved primarily as monomers on SDS-PAGE; however, bands corresponding to low-mass oligomers (∼8–20 kDa) were observed in longer blot exposures.
Toxicity of Pyroglutamate-Aβ Is Associated with Membrane Damage but Not Cytosolic ROS Production
The capacities of each peptide to produce elevations in cytoplasmic and membrane ROS were studied using primary cortical mouse neurons. The fluorescent ROS probe DCF was used to measure cytoplasmic ROS flux over a 1-h treatment with freshly prepared Aβ peptides. Aβ(1–42) induced an approximate 10-fold increase in DCF fluorescence compared with control cells, whereas Aβ(1–40), Aβ3pE-40, or Aβ3pE-42 treatments were not found to be statistically different from controls (Fig. 5, A and B). Minor quantities of Aβ3pE-42 were found to significantly disrupt the neuronal ROS-inducing capacity of Aβ(1–42); the ROS flux induced by solutions of Aβ(1–42) was diminished by 47% when Aβ3pE-42 was present at 5% (mol/mol) of total Aβ (Fig. 5B).
FIGURE 5.

Comparison of the neurotoxicity profiles of freshly prepared Aβ peptides. Levels of ROS flux in cortical neurons loaded with the fluorescent probe dichlorodihydrofluorescein diacetate measured 60 min after treatment with Aβ(1–40) and A3pE-40 peptides (A) or Aβ(1–42) and Aβ3pE-42 peptides (B) (total Aβ = 10 μm; percentages indicate amount of Aβ3pE-40 and Aβ3pE-42 peptides seeded into solutions of Aβ(1–40) and Aβ(1–42), respectively). C, levels of cortical neuron lipid peroxidation (LPO) following 4 h of treatment with Aβ (10 μm total) or synthetic lipid peroxide (1 μm) measured with the DPPP fluorescent probe, represented as difference from baseline readings. D, cell viability (membrane integrity) of differentiated M17 neuroblastomas treated with Aβ peptides (10 μm) or lipid peroxide (1 μm) for 4 h, measured by flow cytometry analysis of propidium iodide uptake. Each data set is representative of a minimum of three experiments performed over separate days (shown as mean ± S.E.). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ns, not significant, p > 0.05.
To assess Aβ induction of ROS at neuronal membranes, we directly measured lipid peroxidation in living cortical mouse neurons using the fluorescent probe DPPP over a 4-h treatment. In direct contrast to the DCF assays, Aβ(1–42) (10 μm) did not increase neuronal DPPP fluorescence, whereas cells treated with Aβ3pE-42 (10 μm) demonstrated a statistically significant 35% increase in lipid peroxidation, comparable with levels induced by 1 μm synthetic lipid hydroperoxide (Fig. 5C). Further contrasting the DCF assays, minor quantities of Aβ3pE-42 had no effect on the lipoperoxide-inducing capacity of Aβ(1–42).
To determine whether lipid peroxidation was associated with a loss of plasma membrane integrity, differentiated M17 neuroblastomas were measured for propidium iodide uptake by flow cytometry following a 4-h Aβ treatment. Consistent with the lipid peroxidation experiments, Aβ3pE-42 induced significantly higher levels of membrane damage than Aβ(1–42) (Fig. 5D). Aβ(1–42) solutions seeded with 5% (mol/mol) Aβ3pE-42 induced practically identical levels of plasma membrane damage as Aβ(1–42) alone.
Aβ3pE-42 Induces Rapid Ca2+ Influx in Primary Cortical Neurons
Recent reports of pE-Aβ interactions with synthetic lipid membranes have suggested that pE-Aβ neurotoxicity is exerted via formation of ion-permeable membrane pores (22, 23). We compared the capacity of Aβ(1–42) and Aβ3pE-42 to alter cellular ion homeostasis by measuring Ca2+ flux in cortical neurons immediately following application of freshly prepared Aβ. Aβ3pE-42 (10 μm) effected a rapid and significant elevation in neuronal Ca2+ levels as measured with the Fluo-4 fluorescent Ca2+ sensor, comparable with levels induced by 1 μm glutamate (Fig. 6, A and B). By comparison, Aβ(1–42) did not cause significant elevation in Ca2+ levels above those of the vehicle control. When neurons were pre-treated with the NMDAR antagonist MK-801, there was an approximate 50% decrease in Ca2+ flux induced by both glutamate and Aβ3pE-42, indicating that Aβ3pE-42-induced Ca2+ flux is at least partially attributed to NMDAR activation (Fig. 6B). To determine whether Aβ3pE-42-induced Ca2+ flux was dependent on Aβ-metal interactions, the neuronal culture media were depleted of bioavailable first row transition metals with the chelator Diamsar. Rapid induction of Ca2+ flux by Aβ3pE-42 was not affected by Diamsar pre-treatment, suggesting that the membrane perturbation was independent of Aβ-copper-redox cycling (Fig. 6C). Aβ3pE-42 did not induce significant elevations in cytosolic Ca2+ when provided in Ca2+/Mg2+-free media, indicating that Aβ3pE-42 causes Ca2+ to enter from the extracellular space and not via release from intracellular stores (Fig. 6D).
FIGURE 6.

Measurement of Ca2+ flux in cortical mouse neurons using the Fluo-4 Ca2+ sensor. A, representative kinetics during the acute exposure of neurons to freshly prepared Aβ (10 μm). B, levels of Ca2+ flux induced by exposure to Aβ(1–42) and Aβ3pE-42 (10 μm each peptide) relative to glutamate-treated cells (Glu; 1 μm). Neurons were additionally pre-treated for 15 min with or without the NMDAR antagonist MK-801 (1 mm) to assess the contribution of NMDAR activation in Aβ3pE-42-induced Ca2+ flux. C, effect of transition metal depletion on Aβ-induced Ca2+ flux in neurons pre-treated for 1 h with the chelator Diamsar. D, separate Ca2+ flux assays were performed in HBSS buffer devoid of Ca2+ and Mg2+ to determine the source of Ca2+ moving into the cytosol. Each treatment was tested in a minimum of four replicate wells per assay and the experiments repeated at a minimum of three times over separate days. Data are represented by box and whiskers (Tukey method); statistical significance is shown above bars as relative to the vehicle treatment (PBS) or between treatments as indicated. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ns, not significant, p > 0.05.
Discussion
Aβ in the human brain is represented by a heterogeneous and dynamic mixture of isoforms, with significant compositional variation between individuals (1–3, 5, 51). The predominance of pE-Aβ in the central core of plaques suggests an early involvement in amyloid deposition in the AD brain (52), whereas correlation between pE-Aβ and a decline in Mini Mental State Examination scores implicate pE-Aβ cytotoxicity in AD neurodegeneration (53, 54). In parallel, markers of oxidative stress are among the earliest detectable pathological changes in transgenic AD mouse models (55) and the human AD brain (36, 56, 57), with numerous lines of evidence implicating Aβ as a central contributor to oxidative stress in AD (58, 59).
Our data indicate that full-length Aβ and pE-Aβ exert different oxidative insults upon neurons, representing different mechanisms of neurotoxicity. Aβ(1–42) was found to significantly increase cytosolic ROS levels, whereas Aβ3pE-42 induced lipid peroxidation in the absence of cytosolic ROS flux. Additionally, we found that that the neuronal membrane damage caused by Aβ3pE-42 results in a functional loss of plasma membrane integrity. These findings are consistent with recent publications demonstrating the capacity for Aβ3pE-42 to form membrane-disrupting pores in synthetic lipid bilayers (22, 23). Similarly, previous studies have shown that pE-Aβ oligomers disrupt lysosome membrane integrity in cultured neurons (50) and cause lactate dehydrogenase leakage from cultured astrocytes (49).
The earliest detectable effect of Aβ3pE-42 on neuronal homeostasis that we observed was the capacity to cause rapid Ca2+ influx, an effect that was partially ameliorated by pre-treatment of neurons with the NMDAR antagonist MK-801. The Ca2+ flux induced by Aβ3pE-42 appears to be separate from its capacity to undergo copper-redox cycling as the effect persisted when media were depleted of row 1 transition metals. Changes in cellular Ca2+ homeostasis induced by Aβ have previously been implicated in Aβ toxicity and are thought to occur via multiple mechanisms such as pore formation and NMDAR activation (60, 61). Importantly, however, the aggregation state of the Aβ preparation significantly affects the capacity to induce Ca2+ flux; monomeric (freshly prepared) and fibrillar Aβ(1–42) is not found to induce Ca2+ flux in SH-SY5Y neuroblastomas, whereas oligomeric preparations agonize NMDAR and trigger rapid Ca2+ flux (61, 62). Similarly, we found freshly prepared Aβ(1–42) to cause only modest elevation in cortical neuron Ca2+ levels above controls, which is contrasted by the much larger Ca2+ flux induced by Aβ3pE-42. Collectively, these data indicate that the unique neurotoxicity of pE-Aβ peptides is exerted through multiple interactions at the cell surface, including activation of NMDAR pathways, subsequently followed by peroxidation of membrane lipids and a loss of membrane integrity.
The potential for amino-truncated Aβ and pE-Aβ to alter the oligomerization and toxicity of full-length Aβ has been an area of recent debate and speculation. Aβ3pE-42 has a demonstrated capacity to dramatically enhance the aggregation of full-length Aβ (14), as does Aβ3–42 (41). Aβ3pE-42 has further been reported to enhance the toxicity of Aβ(1–42) in a prion-like seeding mechanism in cortical neuron cultures (24). Due to the strong membrane association of Aβ3pE-42, we initially predicted Aβ3pE-42 seeds to shift the ROS generation of Aβ(1–42) to a lipid compartment, thereby increasing Aβ(1–42)-induced lipoperoxidation; however, Aβ3pE-42 was not found to increase Aβ(1–42) ROS production in either the cytosolic or membrane fractions. In contrast, the capacity for Aβ3pE-42 seeds to significantly reduce Aβ(1–42) cytosolic ROS production indicates that Aβ3pE-42 does not enhance Aβ(1–42) toxicity via ROS production, yet it suggests significant interaction between the peptides in neuronal cultures. This effect may be a result of accelerated Aβ(1–42) aggregation in the presence of Aβ3pE-42, as previous studies have found that Aβ-ROS production decreases with aggregation (32, 63). In the study by Nussbaum et al. (24), trace quantities of Aβ3pE-42 were found to enhance the toxicity of Aβ(1–42) when co-aggregated prior to cell treatment; however, when Aβ3pE-42 was seeded into Aβ(1–42) solutions immediately before applying to cells (as in our experiments), the toxicity of the seeded mixture was identical to the individual Aβ(1–42) treatment. It is therefore likely that Aβ aggregates possess different cytotoxic properties depending on both the composition of the Aβ mixture and the timing of the Aβ isoform interactions. The possibility also exists that Aβ3pE-42 seeded mixtures cause oxidative damage that escape detection by the DCF and DPPP fluorescence ROS probes or possess different redox cycling capacities in other cerebral cell types (e.g. astrocytes and microglia). Other prevalent amino-truncated and pE-Aβ peptides found in AD brain tissues, such as Aβ(3–42), Aβ(4–42), and Aβ11pE-42 (2, 52), require further investigation as they also demonstrate enhanced amyloid-seeding capacity and could potentially alter Aβ-ROS dynamics.
AFM and SEC analysis revealed that Aβ(1–42) and Aβ3pE-42 oligomers differed not only in the rate of formation but also in size and structure. Previous studies have similarly reported differences between Aβ(1–42) and Aβ3pE-42 in the profile of oligomers and fibril ultrastructures formed in aqueous buffers (18, 30). Lee et al. (22) report that Aβ3pE-42 forms larger oligomers in synthetic lipid membranes than Aβ(1–42) and with faster kinetics of assembly, although it remains to be determined how this relates to the relative level of toxicity. Aβ3pE-42 neurotoxicity has also been demonstrated with a broad range of size fractions (monomers to >100 kDa), whereas the toxicity of Aβ(1–42) fractions was isolated to oligomers with an observed mass larger than 14 kDa (25). This is consistent with our SEC findings demonstrating the relative stability of low-mass Aβ(1–42) species (<14 kDa) in aqueous solution and the observation that freshly prepared Aβ(1–42) is less neurotoxic than Aβ3pE-42. The metastable nature of Aβ peptides, however, presents many technical challenges in delineating “toxic” fractions from “benign” fractions as purification processes undoubtedly alter oligomerization kinetics. Likewise, Aβ peptides undergo significant structural changes in extended cell culture incubations; hence, it is pertinent to correlate toxicity markers with time-matched biophysical characterizations.
The Aβ3pE-40 and Aβ3pE-42 peptides have been found to resist proteolysis in astrocyte cultures (49) and accumulate in the lysosomes of astrocytes in cell culture and the AD temporal cortex (50). Consistent with these reports, we observed Aβ3pE-42 to resist clearance in neuronal cultures, remaining at significantly higher levels than Aβ(1–42) over extended incubations. Aβ3pE-42 was, however, not found to prevent the clearance of Aβ(1–42) from neural cultures when present at minor quantities (5% mol/mol), suggesting that Aβ3pE-42 does not transfer protease resistance to Aβ(1–42) when applied to cells as fresh preparations. This does not exclude the possibility that pE-Aβ may affect the clearance of full-length Aβ when the peptides are aggregated, which will require further investigation. The capacity for Aβ3pE-42 to resist proteolytic degradation in both neurons and astrocytes is highly relevant given that pE-Aβ peptides are found in the cores of amyloid plaques in the AD brain (52), suggesting that pE-Aβ peptides, once formed, are long-lived neurotoxins.
Dityrosine is another post-translational protein modification that confers resistance to cellular catabolism. Total dityrosine levels are elevated in the AD hippocampus, neocortex, and ventricular cerebrospinal fluid compared with cognitively healthy individuals (64), yet the contribution of Aβ to cerebral dityrosine formation is not well understood. Dityrosine cross-linked Aβ fibrils are resistant to formic acid digestion, and sections of AD brains display intense dityrosine immunoreactivity within plaques (39). The pE-Aβ isoforms demonstrated increased efficiency for dityrosine oligomer formation compared with full-length Aβ. This observation can likely be attributed to increased hydrophobicity and the propensity to oligomerize; dityrosine formation is dependent on both the production of Aβ-tyrosyl radicals and the close proximity of Aβ molecules to allow tyrosine-tyrosine coupling (33). It is reasonable to speculate that the capacity for amyloid plaques to resist solubilization and clearance is due to the contribution of both the pE and dityrosine modifications to Aβ, which may account for the abundance of pE-Aβ in plaque cores (52).
Our data demonstrate clear differences in the neurotoxic mechanisms of pE-Aβ and full-length Aβ. The toxicity of pE-Aβ has recently been highlighted in mouse models overexpressing the soluble isoform of QC, demonstrating exacerbated neurodegeneration and behavioral deficits when crossed with mouse lines overexpressing Aβ or amyloid precursor protein transgenes (65, 66). The specific targeting of pE-Aβ via inhibition of QC-catalyzed pyroglutamate synthesis has demonstrated promising results; transgenic AD mice treated with a QC inhibitor show reduced plaque load, reduced gliosis, and an improvement in context memory and spatial learning (67). QC knock-out does not completely inhibit pE-Aβ formation in transgenic AD mouse models, however (68), suggesting that multiple pathways of pE-Aβ formation may exist. Antibodies currently under development as passive vaccine therapies for AD show significant differences in their capacity to bind amino-truncated Aβ species (69) and thus may fail to remove pE-Aβ and its precursors (Aβ3–40/42 and Aβ11–40/42). Individuals with AD may therefore require different therapeutic interventions to target distinct Aβ isoforms and their specific mechanisms of toxicity. Aβ ROS production, oligomerization, and dityrosine formation are potential therapeutic targets for AD; metal-protein attenuating compounds that inhibit these reactions are found to effect a marked decrease in amyloid deposition and improvement in cognitive deficits in transgenic AD mice (70, 71). Our observations indicate that the separate pathways of oxidative damage and neurotoxicity exerted by Aβ3pE-42 potentially have a unique contribution to AD pathology. These findings elicit further consideration of pE-Aβ peptides as targets in the pursuit of biomarkers and disease-modifying therapeutics for AD.
Author Contributions
A. P. G. and J. A. D. wrote the manuscript. A. P. G. designed and conducted the experiments. B. X. W. assisted in the conduct of the lipid peroxidation assays, SEC, and AFM analyses. T. J. assisted in the conduct of the calcium flux experiments. J. G. performed the AFM imaging and analysis. C. L. M., K. J. B., A. I. B., and R. A. C. provided critical revision of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Paul Donnelly (School of Chemistry, University of Melbourne, Australia) for kindly providing Diamsar and Dr. Vanta Jameson (Melbourne Brain Centre Flow Cytometry Facility, Australia) for assistance with flow cytometry analysis. AFM was performed in collaboration with the Materials Characterization and Fabrication Platform at the University of Melbourne and the Victorian node of the Australian National Fabrication Facility. We thank Dr. Lauren Hyde and Prof. Ray Dagastine for their input into the design and conduct of the AFM analyses.
This work was supported by National Health and Medical Research Council (NHMRC) of Australia Program Grant 628946 (to R. A. C., K. J. B., A. I. B., and C. L. M.), Senior Research Fellowship APP1002373 (to K. J. B.), Australia Fellowship GNT1037234 (to A. I. B.), and Operational Infrastructure Support Grant from the Victorian Government (to Florey Institute of Neuroscience and Mental Health). The authors declare that they have no conflicts of interest with the contents of this article.
- Aβ
- amyloid-β
- NMDAR
- N-methyl-d-aspartate receptor
- AD
- Alzheimer disease
- ROS
- reactive oxygen species
- QC
- glutaminyl cyclase
- pE-Aβ
- pyroglutamate-Aβ
- HBSS
- Hanks' balanced salt solution
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DIV
- days in vitro
- DPPC
- diphenyl-1-pyrenylphosphine
- DCF
- dichlorofluorescein
- 3-CCA
- coumarin-3-carboxylic acid
- SEC
- size-exclusion chromatography
- AFM
- atomic force microscopy.
References
- 1. Piccini A., Russo C., Gliozzi A., Relini A., Vitali A., Borghi R., Giliberto L., Armirotti A., D'Arrigo C., Bachi A., Cattaneo A., Canale C., Torrassa S., Saido T. C., Markesbery W., et al. (2005) β amyloid is different in normal aging and in Alzheimer disease. J. Biol. Chem. 280, 34186–34192 [DOI] [PubMed] [Google Scholar]
- 2. Portelius E., Bogdanovic N., Gustavsson M. K., Volkmann I., Brinkmalm G., Zetterberg H., Winblad B., and Blennow K. (2010) Mass spectrometric characterization of brain amyloid β isoform signatures in familial and sporadic Alzheimer's disease. Acta Neuropathol. 120, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Portelius E., Lashley T., Westerlund A., Persson R., Fox N. C., Blennow K., Revesz T., and Zetterberg H. (2015) Brain amyloid-β fragment signatures in pathological ageing and Alzheimer's disease by hybrid immunoprecipitation mass spectrometry. Neurodegener. Dis. 15, 50–57 [DOI] [PubMed] [Google Scholar]
- 4. Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., and Beyreuther K. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harigaya Y., Saido T. C., Eckman C. B., Prada C. M., Shoji M., and Younkin S. G. (2000) Amyloid β protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer's disease brain. Biochem. Biophys. Res. Commun. 276, 422–427 [DOI] [PubMed] [Google Scholar]
- 6. Güntert A., Döbeli H., and Bohrmann B. (2006) High sensitivity analysis of amyloid-β peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 143, 461–475 [DOI] [PubMed] [Google Scholar]
- 7. Schilling S., Hoffmann T., Manhart S., Hoffmann M., and Demuth H. U. (2004) Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett. 563, 191–196 [DOI] [PubMed] [Google Scholar]
- 8. Bien J., Jefferson T., Causević M., Jumpertz T., Munter L., Multhaup G., Weggen S., Becker-Pauly C., and Pietrzik C. U. (2012) The metalloprotease meprin β generates amino-terminal-truncated amyloid β peptide species. J. Biol. Chem. 287, 33304–33313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cynis H., Scheel E., Saido T. C., Schilling S., and Demuth H. U. (2008) Amyloidogenic processing of amyloid precursor protein: evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-β. Biochemistry 47, 7405–7413 [DOI] [PubMed] [Google Scholar]
- 10. Hook G., Yu J., Toneff T., Kindy M., and Hook V. (2014) Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer's disease therapeutic. J. Alzheimers Dis. 41, 129–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oberstein T. J., Spitzer P., Klafki H. W., Linning P., Neff F., Knölker H. J., Lewczuk P., Wiltfang J., Kornhuber J., and Maler J. M. (2015) Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 73, 24–35 [DOI] [PubMed] [Google Scholar]
- 12. Kowalik-Jankowska T., Ruta M., Wiśniewska K., Łankiewicz L., and Dyba M. (2004) Products of Cu(II)-catalyzed oxidation in the presence of hydrogen peroxide of the 1–10, 1–16 fragments of human and mouse β-amyloid peptide. J. Inorg. Biochem. 98, 940–950 [DOI] [PubMed] [Google Scholar]
- 13. Cynis H., Schilling S., Bodnár M., Hoffmann T., Heiser U., Saido T. C., and Demuth H. U. (2006) Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim. Biophys. Acta 1764, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 14. Schilling S., Lauber T., Schaupp M., Manhart S., Scheel E., Böhm G., and Demuth H. U. (2006) On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45, 12393–12399 [DOI] [PubMed] [Google Scholar]
- 15. D'Arrigo C., Tabaton M., and Perico A. (2009) N-terminal truncated pyroglutamyl β amyloid peptide Aβpy3–42 shows a faster aggregation kinetics than the full-length Aβ1–42. Biopolymers 91, 861–873 [DOI] [PubMed] [Google Scholar]
- 16. He W., and Barrow C. J. (1999) The Aβ 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater β-sheet forming and aggregation propensities in vitro than full-length Aβ. Biochemistry 38, 10871–10877 [DOI] [PubMed] [Google Scholar]
- 17. Sun N., Hartmann R., Lecher J., Stoldt M., Funke S. A., Gremer L., Ludwig H. H., Demuth H. U., Kleinschmidt M., and Willbold D. (2012) Structural analysis of the pyroglutamate-modified isoform of the Alzheimer's disease-related amyloid-β using NMR spectroscopy. J. Pept. Sci. 18, 691–695 [DOI] [PubMed] [Google Scholar]
- 18. Schlenzig D., Manhart S., Cinar Y., Kleinschmidt M., Hause G., Willbold D., Funke S. A., Schilling S., and Demuth H. U. (2009) Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 48, 7072–7078 [DOI] [PubMed] [Google Scholar]
- 19. Sanders H. M., Lust R., and Teller J. K. (2009) Amyloid-β peptide Aβp3–42 affects early aggregation of full-length Aβ1–42. Peptides 30, 849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alies B., Bijani C., Sayen S., Guillon E., Faller P., and Hureau C. (2012) Copper coordination to native N-terminally modified versus full-length amyloid-β: second-sphere effects determine the species present at physiological pH. Inorg. Chem. 51, 12988–13000 [DOI] [PubMed] [Google Scholar]
- 21. Drew S. C., Masters C. L., and Barnham K. J. (2010) Alzheimer's Aβ peptides with disease-associated N-terminal modifications: influence of isomerisation, truncation and mutation on Cu2+ coordination. PLoS ONE 5, e15875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J., Gillman A. L., Jang H., Ramachandran S., Kagan B. L., Nussinov R., and Teran Arce F. (2014) Role of the fast kinetics of pyroglutamate-modified amyloid-β oligomers in membrane binding and membrane permeability. Biochemistry 53, 4704–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gillman A. L., Jang H., Lee J., Ramachandran S., Kagan B. L., Nussinov R., and Teran Arce F. (2014) Activity and architecture of pyroglutamate-modified amyloid-β (AβpE3–42) pores. J. Phys. Chem. B 118, 7335–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., et al. (2012) Prion-like behaviour and τ-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galante D., Corsaro A., Florio T., Vella S., Pagano A., Sbrana F., Vassalli M., Perico A., and D'Arrigo C. (2012) Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1–42 and Aβpy3–42 β-amyloids. Int. J. Biochem. Cell Biol. 44, 2085–2093 [DOI] [PubMed] [Google Scholar]
- 26. Schlenzig D., Rönicke R., Cynis H., Ludwig H. H., Scheel E., Reymann K., Saido T., Hause G., Schilling S., and Demuth H. U. (2012) N-terminal pyroglutamate formation of Aβ38 and Aβ40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J. Neurochem. 121, 774–784 [DOI] [PubMed] [Google Scholar]
- 27. Tekirian T. L., Yang A. Y., Glabe C., and Geddes J. W. (1999) Toxicity of pyroglutaminated amyloid β-peptides 3(pE)-40 and -42 is similar to that of Aβ1–40 and -42. J. Neurochem. 73, 1584–1589 [DOI] [PubMed] [Google Scholar]
- 28. Shirotani K., Tsubuki S., Lee H. J., Maruyama K., and Saido T. C. (2002) Generation of amyloid β peptide with pyroglutamate at position 3 in primary cortical neurons. Neurosci. Lett. 327, 25–28 [DOI] [PubMed] [Google Scholar]
- 29. Youssef I., Florent-Béchard S., Malaplate-Armand C., Koziel V., Bihain B., Olivier J. L., Leininger-Muller B., Kriem B., Oster T., and Pillot T. (2008) N-truncated amyloid-β oligomers induce learning impairment and neuronal apoptosis. Neurobiol. Aging 29, 1319–1333 [DOI] [PubMed] [Google Scholar]
- 30. Bouter Y., Dietrich K., Wittnam J. L., Rezaei-Ghaleh N., Pillot T., Papot-Couturier S., Lefebvre T., Sprenger F., Wirths O., Zweckstetter M., and Bayer T. A. (2013) N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 126, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Opazo C., Huang X., Cherny R. A., Moir R. D., Roher A. E., White A. R., Cappai R., Masters C. L., Tanzi R. E., Inestrosa N. C., and Bush A. I. (2002) Metalloenzyme-like activity of Alzheimer's disease β-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J. Biol. Chem. 277, 40302–40308 [DOI] [PubMed] [Google Scholar]
- 32. Guilloreau L., Combalbert S., Sournia-Saquet A., Mazarguil H., and Faller P. (2007) Redox chemistry of copper-amyloid-β: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. Chembiochem. 8, 1317–1325 [DOI] [PubMed] [Google Scholar]
- 33. Barnham K. J., Haeffner F., Ciccotosto G. D., Curtain C. C., Tew D., Mavros C., Beyreuther K., Carrington D., Masters C. L., Cherny R. A., Cappai R., and Bush A. I. (2004) Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer's disease β-amyloid. FASEB J. 18, 1427–1429 [DOI] [PubMed] [Google Scholar]
- 34. Mark R. J., Lovell M. A., Markesbery W. R., Uchida K., and Mattson M. P. (1997) A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J. Neurochem. 68, 255–264 [DOI] [PubMed] [Google Scholar]
- 35. Greilberger J., Koidl C., Greilberger M., Lamprecht M., Schroecksnadel K., Leblhuber F., Fuchs D., and Oettl K. (2008) Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer's disease. Free Radic. Res. 42, 633–638 [DOI] [PubMed] [Google Scholar]
- 36. Sayre L. M., Zelasko D. A., Harris P. L., Perry G., Salomon R. G., and Smith M. A. (1997) 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J. Neurochem. 68, 2092–2097 [DOI] [PubMed] [Google Scholar]
- 37. Gabbita S. P., Lovell M. A., and Markesbery W. R. (1998) Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J. Neurochem. 71, 2034–2040 [DOI] [PubMed] [Google Scholar]
- 38. Smith D. P., Smith D. G., Curtain C. C., Boas J. F., Pilbrow J. R., Ciccotosto G. D., Lau T. L., Tew D. J., Perez K., Wade J. D., Bush A. I., Drew S. C., Separovic F., Masters C. L., Cappai R., and Barnham K. J. (2006) Copper-mediated amyloid-β toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281, 15145–15154 [DOI] [PubMed] [Google Scholar]
- 39. Al-Hilaly Y. K., Williams T. L., Stewart-Parker M., Ford L., Skaria E., Cole M., Bucher W. G., Morris K. L., Sada A. A., Thorpe J. R., and Serpell L. C. (2013) A central role for dityrosine crosslinking of amyloid-β in Alzheimer's disease. Acta Neuropathol. Commun. 1, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bottomley G., Clark I., Creaser I., Englehardt L., Geue R., Hagen K., Harrowfield J., Lawrance G., Lay P., Sargeson A., See A., Skelton B., White A., and Wilner F. (1994) The synthesis and structure of encapsulating ligands: properties of bicyclic hexamines. Aust. J. Chem. 47, 143 [Google Scholar]
- 41. McColl G., Roberts B. R., Gunn A. P., Perez K. A., Tew D. J., Masters C. L., Barnham K. J., Cherny R. A., and Bush A. I. (2009) The Caenorhabditis elegans Aβ 1–42 model of Alzheimer disease predominantly expresses Aβ 3–42. J. Biol. Chem. 284, 22697–22702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manevich Y., Held K. D., and Biaglow J. E. (1997) Coumarin-3-carboxylic acid as a detector for hydroxyl radicals generated chemically and by γ radiation. Radiat. Res. 148, 580–591 [PubMed] [Google Scholar]
- 43. Gunn A. P., Roberts B. R., and Bush A. I. (2012) Rapid generation of dityrosine cross-linked Aβ oligomers via Cu-redox cycling. Methods Mol. Biol. 849, 3–10 [DOI] [PubMed] [Google Scholar]
- 44. Kyte J., and Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 45. Barnham K. J., Ciccotosto G. D., Tickler A. K., Ali F. E., Smith D. G., Williamson N. A., Lam Y. H., Carrington D., Tew D., Kocak G., Volitakis I., Separovic F., Barrow C. J., Wade J. D., Masters C. L., et al. (2003) Neurotoxic, redox-competent Alzheimer's β-amyloid is released from lipid membrane by methionine oxidation. J. Biol. Chem. 278, 42959–42965 [DOI] [PubMed] [Google Scholar]
- 46. Wang H., and Joseph J. A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 27, 612–616 [DOI] [PubMed] [Google Scholar]
- 47. Takahashi M., Shibata M., and Niki E. (2001) Estimation of lipid peroxidation of live cells using a fluorescent probe, diphenyl-1-pyrenylphosphine. Free Radic. Biol. Med. 31, 164–174 [DOI] [PubMed] [Google Scholar]
- 48. Atwood C. S., Perry G., Zeng H., Kato Y., Jones W. D., Ling K. Q., Huang X., Moir R. D., Wang D., Sayre L. M., Smith M. A., Chen S. G., and Bush A. I. (2004) Copper mediates dityrosine cross-linking of Alzheimer's amyloid-β. Biochemistry 43, 560–568 [DOI] [PubMed] [Google Scholar]
- 49. Russo C., Violani E., Salis S., Venezia V., Dolcini V., Damonte G., Benatti U., D'Arrigo C., Patrone E., Carlo P., and Schettini G. (2002) Pyroglutamate-modified amyloid β-peptides–AβN3(pE)–strongly affect cultured neuron and astrocyte survival. J. Neurochem. 82, 1480–1489 [DOI] [PubMed] [Google Scholar]
- 50. De Kimpe L., van Haastert E. S., Kaminari A., Zwart R., Rutjes H., Hoozemans J. J., and Scheper W. (2013) Intracellular accumulation of aggregated pyroglutamate amyloid β: convergence of aging and Aβ pathology at the lysosome. Age 35, 673–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saido T. C., Iwatsubo T., Mann D. M., Shimada H., Ihara Y., and Kawashima S. (1995) Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 14, 457–466 [DOI] [PubMed] [Google Scholar]
- 52. Sullivan C. P., Berg E. A., Elliott-Bryant R., Fishman J. B., McKee A. C., Morin P. J., Shia M. A., and Fine R. E. (2011) Pyroglutamate-Aβ 3 and 11 colocalize in amyloid plaques in Alzheimer's disease cerebral cortex with pyroglutamate-Aβ 11 forming the central core. Neurosci. Lett. 505, 109–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morawski M., Schilling S., Kreuzberger M., Waniek A., Jäger C., Koch B., Cynis H., Kehlen A., Arendt T., Hartlage-Rübsamen M., Demuth H. U., and Rossner S. (2014) Glutaminyl cyclase in human cortex: correlation with (pGlu)-amyloid-β load and cognitive decline in Alzheimer's disease. J. Alzheimers Dis. 39, 385–400 [DOI] [PubMed] [Google Scholar]
- 54. Mandler M., Walker L., Santic R., Hanson P., Upadhaya A. R., Colloby S. J., Morris C. M., Thal D. R., Thomas A. J., Schneeberger A., and Attems J. (2014) Pyroglutamylated amyloid-β is associated with hyperphosphorylated τ and severity of Alzheimer's disease. Acta Neuropathol. 128, 67–79 [DOI] [PubMed] [Google Scholar]
- 55. Ghosh D., LeVault K. R., Barnett A. J., and Brewer G. J. (2012) A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J. Neurosci. 32, 5821–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E. K., Jones P. K., Ghanbari H., Wataya T., Shimohama S., Chiba S., Atwood C. S., Petersen R. B., and Smith M. A. (2001) Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767 [DOI] [PubMed] [Google Scholar]
- 57. Ansari M. A., and Scheff S. W. (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Duce J. A., and Bush A. I. (2010) Biological metals and Alzheimer's disease: implications for therapeutics and diagnostics. Prog. Neurobiol. 92, 1–18 [DOI] [PubMed] [Google Scholar]
- 59. Swomley A. M., Förster S., Keeney J. T., Triplett J., Zhang Z., Sultana R., and Butterfield D. A. (2014) Aβ, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim. Biophys. Acta 1842, 1248–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferreira I. L., Bajouco L. M., Mota S. I., Auberson Y. P., Oliveira C. R., and Rego A. C. (2012) Amyloid β peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 51, 95–106 [DOI] [PubMed] [Google Scholar]
- 61. You H., Tsutsui S., Hameed S., Kannanayakal T. J., Chen L., Xia P., Engbers J. D., Lipton S. A., Stys P. K., and Zamponi G. W. (2012) Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Demuro A., Mina E., Kayed R., Milton S. C., Parker I., and Glabe C. G. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300 [DOI] [PubMed] [Google Scholar]
- 63. Tabner B. J., El-Agnaf O. M., Turnbull S., German M. J., Paleologou K. E., Hayashi Y., Cooper L. J., Fullwood N. J., and Allsop D. (2005) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 280, 35789–35792 [DOI] [PubMed] [Google Scholar]
- 64. Hensley K., Maidt M. L., Yu Z., Sang H., Markesbery W. R., and Floyd R. A. (1998) Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci. 18, 8126–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wirths O., Breyhan H., Cynis H., Schilling S., Demuth H. U., and Bayer T. A. (2009) Intraneuronal pyroglutamate-Aβ 3–42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 118, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wittnam J. L., Portelius E., Zetterberg H., Gustavsson M. K., Schilling S., Koch B., Demuth H. U., Blennow K., Wirths O., and Bayer T. A. (2012) Pyroglutamate amyloid β (Aβ) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J. Biol. Chem. 287, 8154–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schilling S., Zeitschel U., Hoffmann T., Heiser U., Francke M., Kehlen A., Holzer M., Hutter-Paier B., Prokesch M., Windisch M., Jagla W., Schlenzig D., Lindner C., Rudolph T., Reuter G., et al. (2008) Glutaminyl cyclase inhibition attenuates pyroglutamate Aβ and Alzheimer's disease-like pathology. Nat. Med. 14, 1106–1111 [DOI] [PubMed] [Google Scholar]
- 68. Alexandru A., Jagla W., Graubner S., Becker A., Bäuscher C., Kohlmann S., Sedlmeier R., Raber K. A., Cynis H., Rönicke R., Reymann K. G., Petrasch-Parwez E., Hartlage-Rübsamen M., Waniek A., Rossner S., et al. (2011) Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J. Neurosci. 31, 12790–12801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Watt A. D., Crespi G. A., Down R. A., Ascher D. B., Gunn A., Perez K. A., McLean C. A., Villemagne V. L., Parker M. W., Barnham K. J., and Miles L. A. (2014) Do current therapeutic anti-Aβ antibodies for Alzheimer's disease engage the target? Acta Neuropathol. 127, 803–810 [DOI] [PubMed] [Google Scholar]
- 70. Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., Barnham K. J., Volitakis I., Fraser F. W., Kim Y., Huang X., Goldstein L. E., Moir R. D., Lim J. T., Beyreuther K., et al. (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676 [DOI] [PubMed] [Google Scholar]
- 71. Adlard P. A., Cherny R. A., Finkelstein D. I., Gautier E., Robb E., Cortes M., Volitakis I., Liu X., Smith J. P., Perez K., Laughton K., Li Q. X., Charman S. A., Nicolazzo J. A., Wilkins S., et al. (2008) Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59, 43–55 [DOI] [PubMed] [Google Scholar]