

Abstract
Fatty acid kinase (Fak) is a ubiquitous Gram-positive bacterial enzyme consisting of an ATP-binding protein (FakA) that phosphorylates the fatty acid bound to FakB. In Staphylococcus aureus, Fak is a global regulator of virulence factor transcription and is essential for the activation of exogenous fatty acids for incorporation into phospholipids. The 1.2-Å x-ray structure of S. aureus FakB2, activity assays, solution studies, site-directed mutagenesis, and in vivo complementation were used to define the functions of the five conserved residues that define the FakB protein family (Pfam02645). The fatty acid tail is buried within the protein, and the exposed carboxyl group is bound by a Ser-93-fatty acid carboxyl-Thr-61-His-266 hydrogen bond network. The guanidinium of the invariant Arg-170 is positioned to potentially interact with a bound acylphosphate. The reduced thermal denaturation temperatures of the T61A, S93A, and H266A FakB2 mutants illustrate the importance of the hydrogen bond network in protein stability. The FakB2 T61A, S93A, and H266A mutants are 1000-fold less active in the Fak assay, and the R170A mutant is completely inactive. All FakB2 mutants form FakA(FakB2)2 complexes except FakB2(R202A), which is deficient in FakA binding. Allelic replacement shows that strains expressing FakB2 mutants are defective in fatty acid incorporation into phospholipids and virulence gene transcription. These conserved residues are likely to perform the same critical functions in all bacterial fatty acid-binding proteins.
Keywords: crystal structure, fatty acid, fatty acid-binding protein, site-directed mutagenesis, Staphylococcus aureus (S. aureus), fatty acid, fatty acid kinase
Introduction
Fatty acid kinase (Fak)2 is a two-protein system that consists of a dimeric ATP-binding protein (FakA) and a monomeric fatty acid-binding protein (FakB) (1). Fak expression is widespread in Gram-positive bacteria with most strains possessing a single FakA along with multiple FakBs. Staphylococcus aureus expresses two FakBs that both function with FakA. FakB1 binds a saturated fatty acid, although FakB2 prefers an unsaturated fatty acid (1). The FakBs possess a bound fatty acid, but they function as exchange proteins that swap a bound fatty acid for a fatty acid in a detergent micelle or membrane bilayer (Fig. 1). This exchange reaction underlies the function of Fak in exogenous fatty acid incorporation into membrane phospholipids in S. aureus (2). Extracellular fatty acids are flipped to the inner aspect of the membrane where they exchange onto a FakB. The acyl chain bound to FakB is phosphorylated by FakA, and the phosphorylated FakB then exchanges the acyl-PO4 for a fatty acid in the membrane to initiate another round of phosphorylation. The deposited acyl-PO4 is used by PlsY (glycerol-3-phosphate acyltransferase) to initiate membrane phospholipid synthesis or is converted to acyl-acyl carrier protein by PlsX (Fig. 1). Acyl-acyl carrier protein is used by either PlsC in the second acyltransferase step of phospholipid synthesis or by FabF, the elongation condensing enzyme of bacterial type II fatty acid synthesis (1). In addition to the role of FakA and FakB in the activation of exogenous fatty acids, Fak inactivation abrogates the expression of a constellation of virulence factor genes (Fig. 1) (1, 3). The genes altered by Fak inactivation are the same genes that are regulated by the SaeRS two-component virulence regulatory system suggesting SaeRS is the focal point for Fak regulation of virulence factor transcription (1). Also, the inactivation of fakA increases biofilm formation in S. aureus, although the biochemical role of Fak in biofilm formation is unknown (4).
FIGURE 1.

Catalytic cycle of Fak and its role in phospholipid synthesis and virulence factor transcription. Fak consists of a dimeric ATP-binding protein (FakA) and a fatty acid-binding protein (FakB). The first step in the reaction is the binding of FakB to FakA. The fatty acid bound to FakB is then phosphorylated by FakA, and the acyl-PO4-bound FakB is released from the complex. Phosphorylated FakB participates in the activation of virulence factor transcription by an unknown biochemical cascade. In the presence of an exchangeable fatty acid pool in the cell membrane, the acyl-PO4 bound to FakB exchanges with a fatty acid to regenerate the substrate for FakA. The released acyl-PO4 is either used by PlsY to initiate phospholipid synthesis or converted to acyl-acyl carrier protein by PlsX where it can be used by the PlsC acyltransferase or enter the fatty acid elongation cycle.
The goal of this study is to define the biochemical roles of the conserved residues in the FakB bacterial fatty acid-binding protein family (Pfam02645). This protein family is also known as the DegV family named for the founder gene in Bacillus subtilis, degV. Multiple crystal structures of FakB homologs are known with sequence identities between 17 and 37% with respect to FakB2 (PDB codes 1MGP, 1VPV, 1PZX, 2G7Z, 2DT8, 3EGL, 3FDJ, 3FYS, 3JR7, 3LUP, 3NYI, and 3PL5) (5). The FakB proteins belong to the EDD superfamily, which includes the mannose transporter EIIA domain and dihydroxyacetone kinase (6–8). All crystallized FakB proteins have a fatty acid or fatty acid-like ligand bound (7, 8). The discovery of their role in Fak provided a function for these ubiquitous FakB proteins in bacterial physiology (1), but the role(s) of the highly conserved FakB residues that define the protein family in Fak biochemistry are unknown. In this study, a combination of x-ray crystallography, site-directed mutagenesis, and in vitro and in vivo functional analyses are used to determine the structural and functional roles of the five conserved residues that define the bacterial fatty acid-binding protein family. These proteins do not just bind fatty acids, they also contribute key residues to the Fak active site.
Experimental Procedures
Materials
[1-14C]Oleic acid (56.3 mCi/mmol) was from PerkinElmer Life Sciences. Antibiotics, high density nickel resin, and isopropyl β-d-thiogalactopyranoside were from GoldBio. All other reagents are denoted in the text or were from Sigma.
Plasmid and Strain Construction
The ΔfakA strain was constructed starting with strain SA178R1 (9) by the insertion of a group II intron into the fakA gene using the primer design software and plasmid system provided in the Targetron Gene Knock-out kit (Sigma) (10). The presence of the insertion was verified by PCR using primers outside the intron insertion site. A second goal was to knock out the fakB1 allele to create a strain that only expressed fakB2. However, amplification of the fakB1 locus from strain SA178R1 genomic DNA using primers attB2-FakB1 forward 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGACACGCCCGATATCATAAATG and attB1-FakB1 reverse and 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTACAACAACATCTACACGTGG yielded a product of about 1500 bp instead of the expected 2082 bp. DNA sequencing showed the presence of a 483-bp deletion that removed residues 33–193 from the translated FakB1 protein. Thus, strain SA178R1 was already a fakB1 deletion mutant. Strain RN4220 (11), the parent of strain SA178R1, also harbored the same fakB1 deletion.
Strain Construction
The knock-out and knock-in strains were derivatives of strain SA178R1 and were constructed using allelic replacement as described (12, 13). First, the fakB2 locus was knocked out and then this strain was used to knock in the mutant alleles. Briefly, a 999-bp region upstream of fakB2 and 1096-bp sequence downstream of fakB2 were cloned into plasmid pJB38, a shuttle vector with a temperature-sensitive replicon in S. aureus. Growth at 43 °C in the presence of antibiotic was used to select for plasmid integration, followed by several rounds of growth at 30 °C in the absence of antibiotic selection. Anhydrotetracycline-mediated induction of pKOR1-encoded secY antisense transcripts was used to select for chromosomal excision and loss of the plasmid (14). Single colonies were obtained and scored on antibiotic plates to identify those that have lost the plasmid. The removal of the fakB2 gene was confirmed by PCR amplification of the genomic regions with primers located outside fakB2. The knock-in strains were constructed the same way, except the ΔfakB2 strain was the parent strain for plasmids that harbored mutant fakB2 alleles cloned between the upstream and downstream regions. Knock-in strains were screened first by PCR to identify clones with a restored fakB2 gene, and the PCR products from positive colonies were sequenced to confirm the replacement of the fakB2 gene with a fakB2 allele that differed from the wild-type locus by only a single missense mutation in the coding sequence.
Protein Purification
S. aureus fakA in pET28, fakB2 in pET15, or fakB2 T61A, S93A, R170A, and H266A mutant derivatives in pET15 containing amino-terminal histidine tags were transformed into Escherichia coli strain BL21(DE3). Luria-Bertani broth (10 g of tryptone, 5 g of yeast extract, 5 g of NaCl) (1 liter) was inoculated with a single colony, grown with shaking at 37 °C to an A600 = 0.8–1.0, and induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside. Growth continued at 37 °C for 3 h with shaking except FakB2(S93A), which was grown for 24 h with shaking at 16 °C. Cells were then harvested by centrifugation at 10,816 × g for 10 min. Pellets were resuspended in 50 ml of binding buffer (0.5 m NaCl, 20 mm Tris, pH 8.0) and then lysed by microfluidic methods. Cell debris was removed by centrifugation at 38,724 × g for 30 min, and the supernatant was bound to a 3.5-ml nickel-nitrilotriacetic acid column pre-equilibrated with binding buffer. The column was washed sequentially with 25 ml of binding buffer containing 20 and then 40 mm imidazole. The protein was eluted from the column with 15 ml of binding buffer containing 400 mm imidazole and then purified further by gel filtration chromatography on HiLoad 16/60 Superdex 75 eluted with 0.2 m NaCl and 20 mm Tris, pH 7.5.
Crystallography, Data Collection, and Refinement
Wild-type FakB2 used for crystallographic studies was purified as described above except that after the affinity column the fractions containing FakB2 were pooled and desalted using Zeba Spin Desalting Columns (Thermo Scientific) according to the manufacturer's protocol. Desalted FakB2 (200 μm) was incubated with 240 μm oleate overnight at 4 °C. The protein was then purified by gel filtration chromatography and concentrated to 13.5 mg/ml. The protein crystallized at 18 °C by sitting drop vapor diffusion. The 400-nl drop contained 200 nl of FakB2 exchanged with oleate and 200 nl of reservoir solution (0.1 m citrate, pH 5.5, 20% PEG 3000). Crystals were cryopreserved by adding 25% (v/v) ethylene glycol to the reservoir solution and immediately flash-frozen in liquid nitrogen. X-ray diffraction data were collected to 1.2 Å at the SER-CAT 22-BM beamline at the Advanced Photon Source and processed using HKL2000 (15). The crystal belonged to the C2 space group and contained one monomer in the asymmetric unit. The phase information was obtained by molecular replacement using phaser (16) and the model of the fatty acid-binding protein from Thermus thermophiles (PDB code 2DT8) as a search model. The model was built manually using COOT (17) and refined with phenix.refine (16), and 2000 reflections were withheld to calculate Rfree. The final model lacks residues 1, 174–183, and the amino-terminal histidine tag. Structural figures were prepared using PyMOL (18). Crystallographic data and refinement statistics are summarized in Table 1, and the coordinates have been deposited in the Protein Data Bank (PDB code 4X9X).
TABLE 1.
Data collection and refinement statistics for FakB2
| FakB2-oleate | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 167.3, 41.9, 38.6 |
| β (°) | 94.5 |
| Resolution (Å) | 1.2 (1.24-1.2)a |
| Rsym | 0.06 (0.36) |
| I/σI | 27.4 (2.4) |
| Completeness (%) | 94.5 (77.0) |
| Redundancy | 4.0 (2.1) |
| Refinement | |
| Resolution (Å) | 1.2 |
| Total reflections | 315,167 |
| Unique reflections | 78,877 (6,196) |
| Rwork/Rfreeb | 0.118/0.144 |
| No. of atoms | |
| Protein | 2167 |
| Ligand/ion | 32 |
| Water | 285 |
| B-factors (Å2) | |
| Wilson | 9.4 |
| Protein | 13.0 |
| Ligand/ion | 13.3 |
| Water | 30.9 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.2 |
| Ramachandran plot (%) | |
| Favored | 99 |
| Allowed | 1 |
| Outliers | 0 |
a Highest resolution shell is shown in parentheses.
b Rfree was calculated using a test set of 2000 reflections or 2.5% of unique reflections.
Radiolabeled Fak Assay
Assays were carried out in 0.1 m Tris, pH 7.5, 1% Triton X-100, 20 mm MgCl2, 10 mm ATP, 20 μm [1-14C]oleate, and 30 nm FakA. The duplicate 60-μl reactions were initiated with FakB2 or its mutant derivatives and allowed to proceed at 37 °C for 10 min before applying 50 μl to a DE81 filter disk and quenching the reaction in ethanol with 1% acetic acid. Disks were washed three times for 30 min with 20 ml per disk of ethanol and 1% acetic acid before being dried and counted in a PerkinElmer Life Science Tri-Carb 2910TR liquid scintillation analyzer. Data were analyzed, plotted, and fit using GraphPad Prism version 5, and specific activities were determined from the slope by linear regression analysis, and Km values were determined by fitting the data to the Michaelis-Menten equation.
FakB2 Phosphorylation Assay
Assays were carried out in 0.1 m Tris, pH 7.5, 10 mm ATP, 20 mm MgCl2, and either 5 nm, 10 nm, or 2.5 μm FakA in the presence of 5 μm FakB2 or its mutant derivatives previously exchanged with [1-14C]oleate in a final volume of 20 μl. Reactions were started with the addition of FakB2 and proceeded at 37 °C for 10 min before being quenched with 2 μl of 0.25 m EDTA in 50% acetic acid. Samples were quantified using the Bioscan Imaging detector after separating 14C-labeled fatty acid from [14C]acyl-PO4 by thin layer chromatography on Silica Gel G layers developed with chloroform/methanol/acetic acid (90:10:10, v/v). Data were analyzed and plotted in GraphPad Prism version 5.
FakB Thermal Melt Analysis
The thermal stability of FakB2 and its mutant derivatives was measured in 50-μl reactions in triplicate for each protein in a ThermoGrid Optically Clear 96-well PCR plate (Denville Scientific). Each reaction contained 35 μl of Buffer 1 (20 mm HEPES, pH 7.5, 150 mm NaCl), 10 μl of 50 μm protein, and 5 μl of 1:200 SYPRO Orange dye (5000× in DMSO from Invitrogen) in Buffer 1 (2.5× final dye concentration). Plates were centrifuged for 3 min at 1000 × g to remove air bubbles and then subjected to thermal shift analysis in an Applied Biosystems 7500 real time PCR system by monitoring the fluorescence during a temperature ramp from 25 to 94 °C at 1 °C/min. Data were analyzed and plotted in GraphPad Prism version 5 using the Boltzmann sigmoidal equation.
Metabolic Labeling
Experiments were carried out in duplicate as described previously (1) in S. aureus strain SA178R1 and its knock-out and mutant derivatives. Briefly, Luria-Bertani broth containing 10 mg/ml fatty acid-free bovine serum albumin was inoculated with the indicated strain and grown at 37 °C shaking to an A600 = 0.6–1. Cultures were then back diluted to A600 = 0.5 and transferred to a flask containing a final concentration of 20 μm [1-14C]oleate and 180 μm oleate where the ethanol had been previously allowed to evaporate for 1 h. The cultures were then incubated at 37 °C shaking, and 5 ml was removed every 10 min for 30 min. Cells were harvested by centrifugation and washed once with Luria-Bertani broth containing 10 mg/ml fatty acid-free bovine serum albumin and then a second time with H2O. The cell pellet was resuspended in 100 μl of H2O and treated with 360 μl of chloroform/methanol/HCl (1:2:0.02, v/v) to extract the lipids. Phases were separated after the addition of 120 μl of 2 m KCl and 120 μl of chloroform, and the lipid phase was quantified using a combination of scintillation counting and thin layer chromatography analysis. Data were analyzed and plotted in Graph Pad Prism version 5 using linear regression.
Analytical Ultracentrifugation
Experiments were conducted in a ProteomeLab XL-I analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) following standard protocols unless mentioned otherwise (19). All samples in a buffer containing 20 mm Tris, pH 7.5, 200 mm NaCl were loaded into a cell assembly composed of a double sector charcoal-filled centerpiece with a 12-mm path length and either quartz or sapphire windows. The density and viscosity of the ultracentrifugation buffer at 20 °C were measured with a DMA 5000 M density meter and an AMVn viscometer (both Anton Paar, Graz, Austria), respectively. The cell assembly in the sedimentation velocity experiments contained identical sample and reference buffer volumes of 300 μl, was placed in a rotor, and temperature equilibrated at rest at 20 °C for 2 h before it was accelerated from 0 to 50,000 rpm. Rayleigh interference optical data were collected at 1-min intervals for 12 h. The velocity data were modeled with diffusion-deconvoluted sedimentation coefficient distributions c(s) in SEDFIT (20, 21), using algebraic noise decomposition and with signal-average frictional ratio and meniscus position refined with non-linear regression. The s-value was corrected for time, temperature, and radial position, and finite acceleration of the rotor was accounted for in the evaluation of Lamm equation solutions (22, 23). Sedimentation equilibrium was attained at a rotor temperature of 4 °C at increasing speeds of 10,000 revolutions/min (45 h), 15,000 rpm (30 h), and 25,000 rpm (20 h). Protein at concentrations of between 7 and 37 μm (130 μl) were loaded into double-sector centerpieces, and absorbance distributions were recorded at 280 nm in 0.001-cm radial intervals with 20 replicates for each point. In the model, FakA had two sites for FakB2 with two association constants: Ka, 1 for the first binding event and Ka, 2 for the second event. The model was expressed in terms of a macroscopic association constant Ka, 1 for the first binding event and the ratio Ka, 2/Ka, 1 for the second binding event. Global least squares modeling was performed at multiple rotor speeds with the software SEDPHAT (24, 25), using the reversible two-site heterogeneous association model as well as the two discrete species model (19).
Quantitative Real Time PCR
mRNA transcript levels were measured using quantitative real time PCR (qRT-PCR) as described previously (26). Briefly, bacterial cultures were grown to an A600 = 2.0 and then 2-ml cells were removed and pelleted at 4500 × g for 10 min, resuspended in 1 ml of RNA Later (Ambion), and pelleted again. Total RNA was immediately isolated using Ambion RNAqueous kit according to the manufacturer's protocol but with the inclusion of 5 mg/ml lysostaphin to lyse the cells. Genomic DNA was removed using the Turbo DNA-free kit from Ambion, and cDNA was synthesized using SuperScript II reverse transcriptase from Invitrogen according to the manufacturer's instructions. qRT-PCRs were set up in triplicate in a ThermoGrid Optically Clear 96-well PCR plate with SYBR Green PCR master mix (Applied Biosystems), 150 nm forward and reverse primer, and cDNA synthesized from 10 ng of total RNA. Control wells containing nuclease-free water or run without reverse transcriptase were run in duplicate. qRT-PCR was performed in an Applied Biosystems 7500 real time PCR system. Template curves containing a range of 5–50 ng of total RNA were run in duplicate to verify that each primer set had a linear response and same relative efficiency in the PCR as the gmk calibrator. The values were compared using the threshold cycle (CT) method (27), and the amount of cDNA present (2−ΔCT) was reported relative to the gmk calibrator. Primers used were described previously (26) except Sa-SaeP forward 3, 5′-CGGTGAAACTGTTGAAGGTAAAGCTGA, and Sa-SaeP reverse 3, 5′-TTAGCGCCGCCGAAGATGACG.
Results
Conserved Residues That Define the FakB Protein Family
There were three reasons S. aureus why FakB2 was selected as the prototypical member of the FakB protein family for analysis. First, our new 1.2-Å FakB2 structure complexed with oleic acid provided particularly clear insight into the interactions within FakB2 and between FakB2 and the fatty acid (Table 1; Fig. 2A). Second, FakB2 proteins expressed in E. coli could be highly purified using affinity and gel filtration chromatography to examine their biochemical properties in vitro (Fig. 2B). Third, the genetic tools in S. aureus allowed sequences encoding the individual mutant proteins to be knocked into the native fakB2 locus to assess the functions of the altered proteins in vivo.
FIGURE 2.
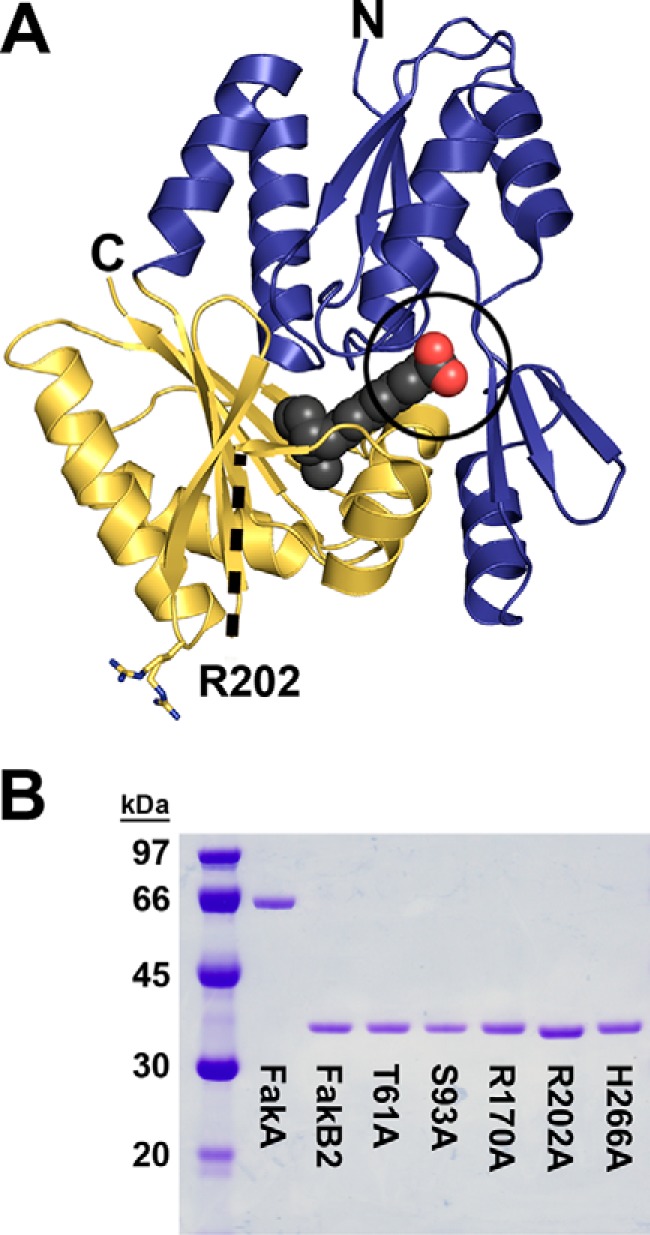
Overall structure of FakB2 and the purity of FakB2 proteins. A, overall structure of FakB2 shown in schematic representation with the bound oleate illustrated as spheres with charcoal-colored carbons and red-colored carbons carboxyl oxygens. The fatty acid carboxyl group is exposed to solvent, and the acyl chain is completely buried within the protein structure. The amino-terminal domain is colored blue, and the carboxyl-terminal domain is colored gold. The section of the protein between residues 171 and 183 is missing from the structure, and its location is denoted by a dashed black line. The location and alternative conformations of Arg-202 (shown as sticks with nitrogens in blue and carbons in gold) in relation to the FakB2 active site containing residues T61A, S93A, R170A, and H266A (black circle) are illustrated. N and C denote the amino and carboxyl termini of the model, respectively. B, gel electrophoresis illustrating the purity of protein reagents used in this study. Coomassie-stained electrophoretic gel analysis of ∼1.5 μg of protein was purified by affinity followed by size-exclusion chromatography. The labeled lanes contained S. aureus FakA, FakB2, and FakB2 mutants T61A, S93A, R170A, R202A and H266A. A molecular mass ladder is shown in lane 1.
The top 6% of evolutionarily conserved residues in the FakB protein family were identified with the program Evolutionary Trace (28) using the T. thermophilus FakB structure (PDB code 2DT8) as the search model to align 288 sequences. Thr-61, Ser-93, Arg-170, Arg-202, and His-266 of FakB2 were 89, 99, 100, 89. and 89% conserved, respectively, and all five residues were changed to alanine to interrogate the function(s) of the side chains. Other residues in the top 6% were not analyzed because they were predicted to have a purely structural role based on their locations within the FakB2 structure. For example, Asp-114 and Gly-193 fixed the position of Ser-93 through hydrogen bonds with the peptide backbone meaning that these residues supported the orientation and function of Ser-93. The conserved Gly-168 was in a tight turn that allowed space for His-266 to form a hydrogen bond with Thr-61. Gly-169 formed another tight β-turn that projected Arg-170 into the active site, and Gly-268 functioned in a similar β-turn that pointed His-266 toward Thr-61. Asp-9, Ser-10, Asp-114, Gly-136, Leu-162, and Leu-165 were predicted to have similar structural roles within the interior of the protein core and were not examined.
Three residues, Thr-61, Ser-93, and His-266, participated in a Ser-93-fatty acid carboxyl-Thr-61-His-266 hydrogen bond network that locked the fatty acid carboxyl group to the protein (Fig. 3A). The hydroxyl group of Ser-93 was 2.6 Å away from one oxygen of the carboxyl headgroup, and on the opposite side of the pocket the hydroxyl group of Thr-61 was 2.7 Å away from the other carboxyl oxygen and 2.9 Å away from the ϵ-nitrogen of His-266 (Fig. 3A). Arg-170 did not contact the hydrogen bond network, but the guanidinium group makes two hydrogen bond interactions with the backbone carbonyl of Leu-92 and the three carbons of Arg-170 pack along the acyl chain in the FakB2-oleate structure. The location of Arg-170 suggests that it would be in position to interact with the dianionic acyl-PO4 product bound to FakB2 and/or participate in the phosphotransfer reaction (Fig. 3B). Arg-170 is 3.2 Å away from water molecule 553 that participates in a hydrogen bonding network of two water molecules and the carboxylate oxygen closest to Thr-61 (Fig. 3B), and one can envision that the phosphate would occupy a similar position in a phosphorylated FakB2 structure. Arg-202 was located on the protein's surface far from the active site (Fig. 2A). Arg-202 adopts two distinct conformations in the FakB2 structure consistent with its flexibility as a surface residue. The conservation of Arg-202 coupled with its surface localization suggests a potential role in protein-protein interactions.
FIGURE 3.

Close-up view of the hydrogen bond network that anchors the fatty acid in FakB2. S. aureus FakB2 crystal structure models are shown in white schematic representation with oleate presented as sticks (dark gray). Selected residues are shown as sticks with oxygen and nitrogen atoms in red and blue, respectively. Waters are depicted as red spheres and hydrogen bonds as black dashed lines along with their distances in Å. A and B, representative simulated annealing 2mFo − DFc composite omit map density (dark gray) contoured at 1.5 σ is rendered around key residues and waters. A, omit map of the fatty acid carboxyl binding pocket highlighting the FakB2 residues Thr-61, Ser-93, and His-266 involved in the hydrogen bond network that fixes the fatty acid carboxyl group. B, omit map illustrating Arg-170 and its interactions with three structured water molecules positioned next to the fatty acid carboxyl group. C, close-up view of the fatty acid binding pocket of FakB2 with the side chains of residues Thr-61, Ser-93, Arg-170, and His-266 superimposed over the FakB2 structures from B. subtilis (PDB code 3FYS; purple), S. pyogenes (PDB code 2G7Z; dark blue), and G. stearothermophilus (PDB code 1PZX; black). D, close-up view of the fatty acid binding pocket of FakB2 with selected residues Thr-61, Ser-93, Arg-170, and His-266 superimposed over the homologous structure from R. gnavus (PDB code 3JR7; purple), which has a tyrosine substituted for histidine in the hydrogen bond network.
The S. aureus FakB2 structure reflects the protein fold characteristic of all crystallized members of the FakB protein family (6, 7). FakB2 crystallized in the C2 space group with one monomer in the asymmetric unit and adopted the same fold as the other bacterial fatty acid-binding proteins (Fig. 2A). The amino-terminal domain consists of an EDD fold (6) and an extension containing a three-stranded anti-parallel β-sheet and one α-helix (Fig. 2A). The carboxyl-terminal domain was constructed of a six-stranded β-sheet flanked by two α-helices on each side that were characteristic of Pfam02645 (7). The oleic acid was bound between the two domains with the fatty acyl tail butting up against the six-stranded mixed β-sheet buried within the carboxyl-terminal domain (Fig. 2A). The carboxyl group was anchored at the protein surface by a hydrogen bond network and the acyl chain was buried within the protein. The three residues participating in the hydrogen bonding network were conserved in homologous FakB proteins. The FakB crystal structures from Bacillus subtilis (PDB code 3FYS) (8), Streptococcus pyogenes (PDB code 2G7Z), and Geobacillus stearothermophilus (PDB code 1PZX) have a range of root mean square deviations compared with FakB2 between 1.5 and 1.9 Å according to PDBefold (29). When these structures are superimposed, the hydrogen bond network and Arg-170 occupied the same position in the active site of the fatty acid-binding proteins (Fig. 3C). The most frequent substitution at the Thr-61 position was the conservative substitution of a serine residue in 9% of the sequences. The most frequent substitution at the His-266 position was a tyrosine residue in 5% of the sequences. The tyrosine substitution was found in FakBs from Ruminococcus gnavus (PDB code 3JR7) and Eubacterium eligens (PDB code 3FDJ). The tyrosine replaces the position of the histidine in both structures to maintain the hydrogen bonding network with Thr-61 (Fig. 3D).
Activity and Stability of FakB2 Mutants
A standardized Fak assay that was linear with time and input FakB2 protein was used to determine the ability of the FakB2 mutants to support kinase activity (Table 2). FakB2(R170A) completely failed to support the Fak reaction. The Fak assay could detect 5 pmol/min/mg, placing an upper limit on the activity of FakB2(R170A). The T61A, S93A, and H266A FakB2 mutants each exhibited severely compromised Fak activity, although the assay was sensitive enough to still be able to detect the thousand-fold defect in catalysis. FakB2(R202A) was also deficient in Fak activity, but much less so than the other mutants. Fak activity assay measures the accumulation of [14C]acyl-PO4 and not only requires FakA phosphorylation of the fatty acid on FakB2, but this event must be followed by the exchange of the acyl-PO4 with a free fatty acid to regenerate another FakB2 substrate for FakA (Fig. 1). Thus, the catalytic defects observed in the Fak assay could arise from the inefficient phosphorylation of FakB2 or from the failure of FakB2 to carry out acyl-PO4-fatty acid exchange. This point was addressed by developing an assay to measure the phosphorylation of FakB2 in the absence of an exchangeable free fatty acid pool (Fig. 4). First, the FakB2 mutant proteins were loaded with [1-14C]oleate overnight, and the radiolabeled proteins were purified from the excess fatty acid by gel filtration chromatography. The fact that we were able to prepare [1-14C]oleate-labeled mutant FakB2s suggested that all of the proteins were competent for the exchange reaction. Next, the labeled FakB2 proteins were phosphorylated by increasing amounts of FakA and the formation of [14C]acyl-PO4 determined by thin layer chromatography. The increase in FakB2 phosphorylation by increasing amounts of FakA established the sensitivity of the assay and the baseline activity of the wild-type protein. The FakB2 T61A, S93A, R202A, and H266A mutant proteins were all clearly defective in FakB2 phosphorylation, but in each case FakB2 phosphorylation did occur at the highest level of input FakA (Fig. 4). FakB2(R170A) was completely defective in FakB2 phosphorylation at all FakA concentrations tested. These data were consistent with the conclusion that the targeted side chains actively participated in the formation of the Fak active site and FakB2 phosphorylation. Arg-170 was clearly the most essential residue involved in Fak catalysis.
TABLE 2.
Specific activities and denaturation temperatures for FakB2 and its mutant derivatives
| FakB2 | Oleoyl-PO4a | Tmb |
|---|---|---|
| nmol/min/mg | °C | |
| Wild type | 54.5 ± 3.1 | 50.3 ± 0.3 |
| T61A | 0.031 ± 0.002 | 38.2 ± 0.7 |
| S93A | 0.034 ± 0.002 | 32.9 ± 0.4 |
| R170A | <0.005 | 45.4 ± 0.3 |
| R202A | 0.497 ± 0.033 | 52.5 ± 0.1 |
| H266A | 0.053 ± 0.002 | 38.0 ± 0.2 |
a The Fak assay contained 0.03 μm FakA, 10 mm ATP, 20 mm MgCl2, 20 μm [1-14C]oleate, and varying concentrations of FakB2, and the specific activities were calculated as described under “Experimental Procedures.”
b The transition temperature for the denaturation of the FakB2 proteins was determined as described under “Experimental Procedures.”
FIGURE 4.
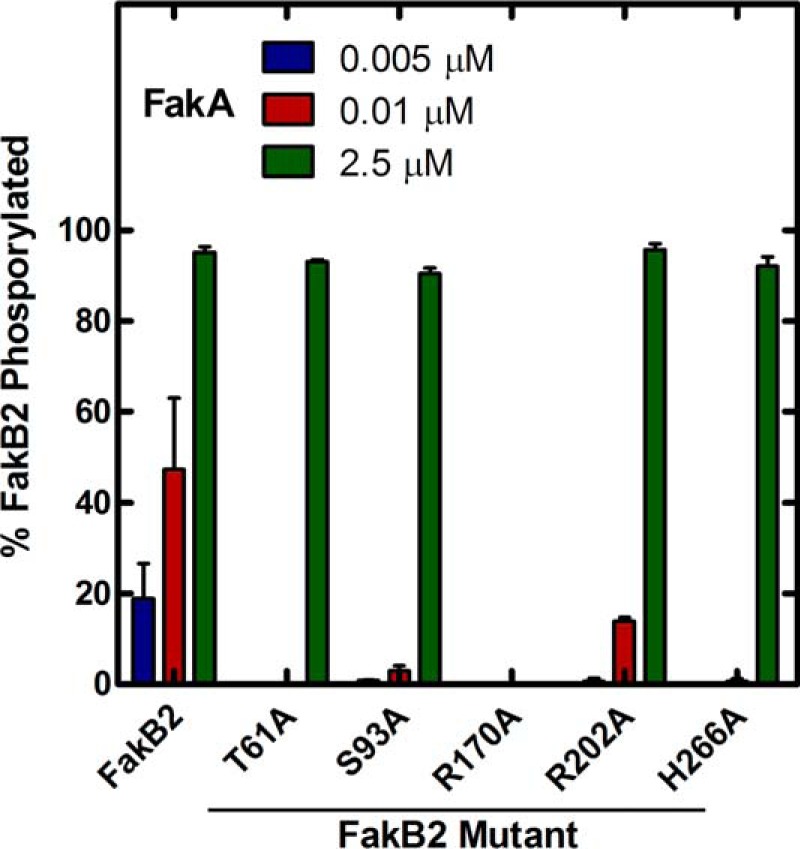
Phosphorylation of the FakB2 mutant panel. [1-14C]Oleate was exchanged onto FakB2 and its mutant derivatives by incubation overnight, and the labeled proteins were purified by gel filtration chromatography to remove all fatty acid that was not bound to the proteins. The labeled proteins were then used in a Fak assay with the indicated concentrations of FakA. The reactions were terminated, and the ratios of [1-14C]oleoyl-PO4 and [1-14C]oleate remaining on FakB2 were determined using a Bioscan Imaging system after separation by thin layer chromatography as described under “Experimental Procedures.”
Thermal denaturation profiles were obtained for each mutant protein to determine whether the mutated residues impacted the overall stability of the FakB2 (Table 2). The disruption of the Ser-93-fatty acid carboxyl-Thr-61–His-266 hydrogen bonding network caused a 12–17 °C decrease in the thermal stabilities of the mutant FakB2s compared with the wild-type protein. These results illustrated the importance of the interactions between the fatty acid carbonyl and the hydrogen bond network to protein stability. In the case of FakB2(S93A), the unfolding temperature was reduced to such an extent (33 °C) that protein denaturation contributed to, or was perhaps the sole reason, for its significantly reduced specific activity in the Fak assay. This behavior was consistent with the difficulties experienced in obtaining high yields of stable FakB2(S93A). Unlike the other FakB2 proteins, there was always a large amount of FakB2(S93A) detected in the insoluble fraction of the expression cell lysate (data not shown), and during storage FakB2(S93A) was the only protein to precipitate from solution. The thermal stability of FakB2(R170A) decreased only 5 °C. The guanidinium of Arg-170 makes two interactions with the peptide backbone carbonyl of Leu-92, and the three-carbon chain of Arg-170 packs along the fatty acyl chain accounting for the decrease in thermal stability in FakB2(R170A). FakB2(R202A) had the same thermal stability as the wild-type protein consistent with the lack of interactions between the protein and the side chain of the Arg-202 surface residue.
FakB2 Binding to FakA
The Fak reaction requires the binding of FakA and FakB, and analytical ultracentrifugation was used to characterize this interaction and to determine whether any of the FakB2 mutants were defective in FakA binding. The first experiments were to determine whether FakA-FakB2 interactions could be detected and to determine the stoichiometry of binding (Fig. 5; Table 3). The sedimentation velocity data of all the protein mixtures were analyzed with the continuous sedimentation coefficient distribution model c(s). FakA alone appeared at 5.59S indicating FakA exists as a dimer with a frictional ratio of 1.62 (1). The FakB2 s-value ranged from 2.5 to 2.76S and corresponded to the theoretical s-value of a FakB2 monomer (2.64S). Mixing an excess of FakB2 with FakA resulted in a new species appearing with a molecular weight consistent with the binding of two FakB2 proteins to each FakA dimer (Fig. 5A). The s-value of the higher molecular weight peak ranged from 6.64 to 6.79S and corresponded to a protein complex containing one FakA dimer and two FakB2 monomers, assuming a frictional ratio value of ∼1.67. The same complex was detected in FakA plus FakB2(R170A) mixture (Fig. 5B) illustrating that the mutant protein with the most severe deficiency in Fak activity bound to FakA. However, the binding of FakB2(R202A) to FakA was not detected under these experimental conditions (Fig. 5C). This result suggested that a binding defect was responsible for the reduced activity in the Fak assay (Table 2). This point was investigated further by determining the apparent Km value for FakB2(R202A) compared with FakB2 in the Fak assay (Fig. 6). FakB2 exhibited an apparent Km of 0.16 ± 0.02 μm, whereas the apparent Km value for FakB2(R202A) was 10-fold higher (1.67 ± 0.17 μm). These data were consistent with Arg-202 being a major determinant of FakB2 binding to FakA. The data obtained from experiments with all five mutant FakB2s are summarized in Table 3. All FakB2 mutant proteins formed the same heterotetrameric complex with FakA, except FakB2(R202A), indicating that FakA binding defects were not responsible for the reduced activity of the mutant FakB2s in the Fak assay.
FIGURE 5.
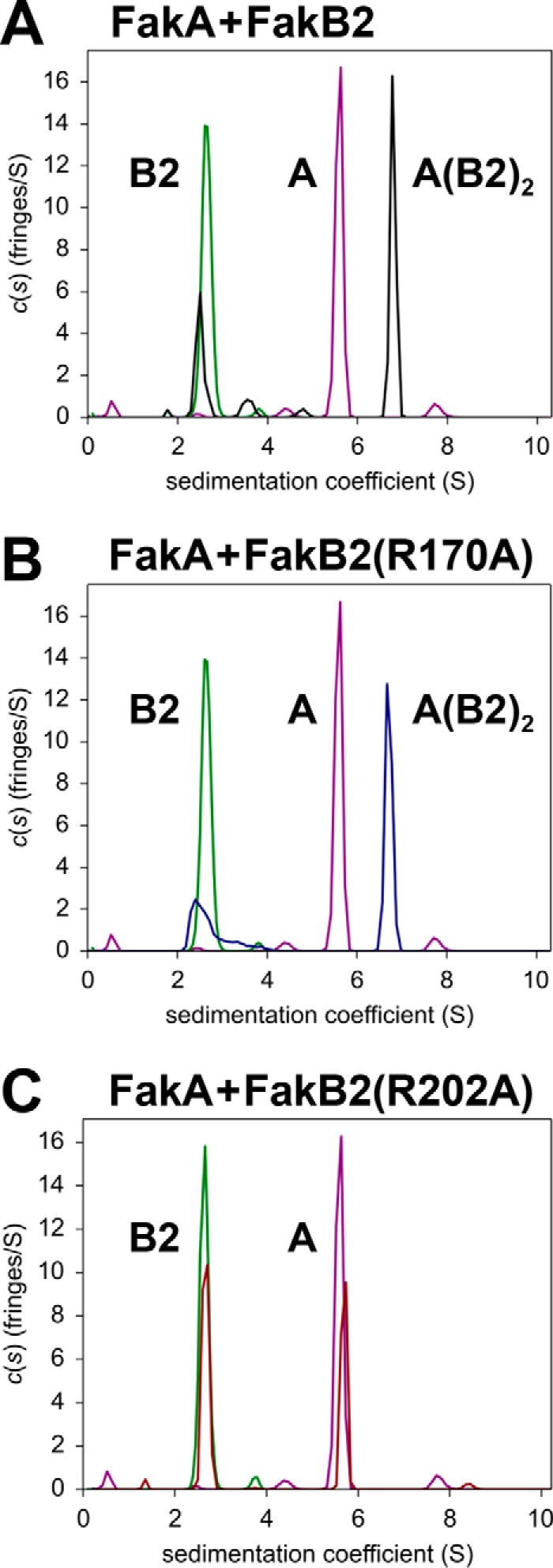
Analysis of FakA-FakB2 binding by analytical ultracentrifugation. A–C, sedimentation velocity profiles (fringe displacement) were fitted to a continuous sedimentation coefficient distribution model c(s). The figure illustrates representative examples of three individual experiments. The s-values for proteins and complexes calculated from experiments with all the mutant proteins are listed in Table 3. All plots show the individual locations of the FakA dimer (purple) and the FakB2 monomer (green) in the experiment. The peaks are labeled to denote a monomer of FakB2 (B2), a dimer of FakA (A) or the complex containing a FakA dimer and two FakB2 molecules (A(B2)2). A, FakA + FakB2 (black). B, FakA + FakB2(R170A) (blue). C, FakA + FakB2(R202A) (red). Additional details are found under “Experimental Procedures.”
TABLE 3.
Sedimentation velocity c(s) analysis of FakA, FakB2, and the mixtures of FakA and FakB2 (and its mutants)
| Sample | μma | mg/mlb | s20 (Svedberg)c | s20, w (Svedberg)d |
|---|---|---|---|---|
| FakA | 122.0 | 1.15 | 5.59 (88%) | 5.86 |
| FakB2 | 231.0 | 1.10 | 2.64 (98%) | 2.79 |
| FakB2 + FakA | 18.91 + 7.85 | 1.23 | 2.50 (29%) | 2.62 |
| 6.79 (61%) | 7.15 | |||
| FakB2(T61A) + FakA | 16.37 + 8.19 | 1.15 | 2.76 (23%) | 2.90 |
| 6.68 (67%) | 7.04 | |||
| FakB2(S93A) + FakA | 14.23 + 7.85 | 1.08 | 2.76 (20%) | 2.90 |
| 6.64 (70%) | 7.07 | |||
| FakB2(R170A) + FakA | 19.68 + 7.91 | 1.17 | 2.60 (33%) | 2.73 |
| 6.70 (65%) | 7.01 | |||
| FakB2(R202A) + FakA | 19.89 + 4.39 | 1.19 | 2.64 (51%) | 2.78 |
| 5.67 (43%) | 5.97 | |||
| FakB2(H226A) + FakA | 19.60 + 8.01 | 1.22 | 2.56 (31%) | 2.70 |
| 6.72 (63%) | 7.08 |
a Total loading concentrations in μm of FakB2 and its mutants (left-hand side) and FakA on the right in 20 mm Tris, pH 7.5, 0.2 m NaCl at 20 °C.
b Total concentration is shown in mg/ml.
c Sedimentation coefficient taken from the ordinate maximum of each peak in the best fit c(s) distribution at 20 °C with percentage protein amount in parentheses. Sedimentation coefficient (s-value) is a measure of the size and shape of a protein in a solution with a specific density and viscosity at a specific temperature.
d Standard sedimentation coefficient (s20, w value) in water at 20 °C.
FIGURE 6.
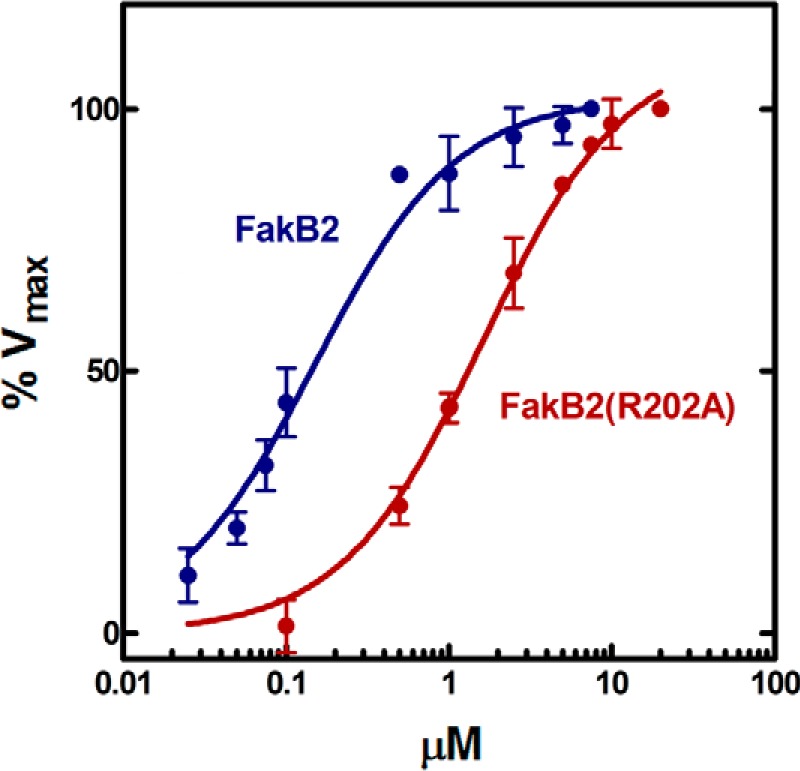
FakB2(R202A) exhibits a lower affinity for FakA. The standard radiochemical Fak assay described under “Experimental Procedures” was used to compare the apparent Km values for FakB2 and FakB2(R202A). The apparent Km value for FakB2 was 0.16 ± 0.02 μm, and the apparent Km value for FakB2(R202A) was 1.67 ± 0.17 μm. The Km values were calculated using non-linear Michaelis-Menten regression analysis using GraphPad software.
The binding of the FakB2 mutant panel to FakA was quantified using sedimentation equilibrium studies. The sedimentation equilibrium data for all the protein mixtures were fitted to a reversible two-site heterogeneous association model that yielded dissociation equilibrium constants Kd, 1 for the first site and Kd, 2 for the second site (Table 4). The association scheme for this model is FakA + FakB2 ↔ FakA-FakB2 + FakB2 ↔ FakA-(FakB2)2. The model posits that FakA dimer has two binding sites for monomeric FakB2 that were defined by Kd, 1 and Kd, 2 (Table 4). The same sedimentation equilibrium data were also fitted to the two discrete species model where the one molecular mass was fixed to the theoretical molecular mass of FakB2, and mutants and the other mass floated in the fit. These best fit molecular masses varied between 179,621 and 191,582 Da and were within 5.9% of the theoretical mass of 190,964 Da of the FakA-(FakB2)2 complex. Based on the values of the root mean square deviations, all the equilibrium data fit both models equally well (Table 4). In all cases, Kd, 2 was greater than Kd, 1, but the differences were small leading to the conclusion that FakA dimer had two independent and nearly equivalent active sites for FakB2 binding and catalysis.
TABLE 4.
Sedimentation equilibrium analysis of the binding of FakB2 and its mutants to FakA
A reversible two-site heterogeneous association model was used for the analysis of the dissociation constants and the two discrete species model was used to determine the molar mass of the complex. CI is confidence interval; r.m.s.d. is root mean square deviation.
| Sample | μma | Kd, 1b (μm) (68.3% CI) | Kd, 2b (μm) (68.3% CI) | r.m.s.d.c | M1d | M2d | r.m.s.d.e |
|---|---|---|---|---|---|---|---|
| Da | Da | ||||||
| FakB2 | 15.75 | 1.71 | 6.84 | 0.0032 | 32,808 | 191,582 | 0.0028 |
| + FakA | + 6.54 | (1.64–11.06) | (6.5–44.26) | ||||
| FakB2(T61A) | 13.64 | 2.63 | 10.54 | 0.0026 | 32,778 | 179,621 | 0.0018 |
| + FakA | + 6.83 | (2.55–3.84) | (10.21–15.38) | ||||
| FakB2(S93A) | 11.86 | 1.46 | 5.87 | 0.0038 | 32,792 | 187,959 | 0.0037 |
| + FakA | + 6.54 | (1.42–3.74) | (5.70–14.96) | ||||
| FakB2(R170A) | 16.41 | 2.25 | 9.01 | 0.0035 | 32,723 | 181,313 | 0.0034 |
| + FakA | + 6.60 | (0.76–3.45) | (3.07–13.81) | ||||
| FakB2(R202A) | 24.80 + 5.39 | Not detected | Not detected | 32,723 | 132,268 | 0.0027 | |
| + FakA | |||||||
| FakB2(H226A) | 16.33 | 1.50 | 6.02 | 0.0033 | 32,742 | 187,964 | 0.0033 |
| + FakA | + 6.67 | (1.47–1.99) | (5.90–7.95) |
a Total loading concentrations in μm of FakB2 and its mutants (left-hand side) and FakA on the right in 20 mm Tris, pH 7.5, 0.2 m NaCl buffer at 4 °C.
b Dissociation equilibrium constants Kd, 1 for first site and Kd, 2 for the second site converted from the association equilibrium constants Ka, 1 and Ka, 2, respectively, for the reversible two-site heterogeneous association model with the association scheme A + B2 ↔ AB2 + B2 ↔ A(B2)2. FakA dimer (125,348 Da) was treated in the analysis as the single species A and FakB2 and mutants as B2.
c Root mean square deviation of the fit, units in absorbance for the reversible complex formations. Errors represent the 68.3% (one standard deviation) confidence interval (CI) using an automated surface projection method (33).
d Fitted molar masses of two discrete species with the first mass (M1) the theoretical molar mass of FakB2 and the mass of the second species (M2) floated in the fit.
e Root mean square deviation of the fit, units in absorbance for the two discrete species model. is shown.
Function of FakB Active Site Residues in Vivo
The function of the mutant proteins in vivo was determined by knocking the mutant alleles into the fakB2 gene by homologous recombination to create a series of isogenic strains expressing mutant proteins from their native promoters. The parent strain for these studies was S. aureus strain SA178R1 (9), a derivative of the commonly used restriction-deficient laboratory strain RN4220 (11). We found during the course of this work that strains RN4220 and SA178R1 both harbored a defective fakB1 allele. The fakB1 gene in strain RN4220 and its derivatives contained an internal 483-bp deletion (t99 to a581) as determined by sequencing of the PCR product derived from amplifying the fakB1 locus. This corresponded to an internal in-frame deletion of residues 33–193 of the translated protein. Thus, a functional FakB1 was not present in these strains, and fakB2 was the only fatty acid-binding protein gene expressed in the cells. There were no differences detected in the growth rates of the wild type, ΔfakB2 knock-out, or mutant-expressing strains when grown in Luria-Bertani medium.
Metabolic labeling experiments were used to determine whether the mutant fakB2 alleles supported incorporation of exogenous [1-14C]oleic acid into membrane phospholipids (Table 5). Phospholipid synthesis required two enzymatic steps as follows: the activation of the fatty acid by Fak, followed by the exchange of acyl-PO4 into the membrane where it is used to initiate phospholipid synthesis by the PlsY acyltransferase. We know that both of these enzymes were absolutely required for phospholipid synthesis from exogenous fatty acids, but we do not know whether Fak or PlsY activity governs the rate of incorporation. We did not detect any incorporation of [1-14C]oleate into phospholipid in the ΔfakB2 strain consistent with the absolute requirement of Fak for fatty acid activation and incorporation (Table 5). Strains expressing FakB2(T61A) or FakB2(H266A) showed 20–40-fold lower incorporation of [1-14C]oleic acid into membrane phospholipids illustrating these proteins had severely impaired Fak activity in vivo (Table 5), as they had in vitro (Table 2). The FakB2(R170A) mutant was indistinguishable from the ΔfakB2 deletion strain with no detectable exogenous fatty acid incorporation into phospholipid (Table 5) corroborating the essential nature of Arg-170 in Fak catalysis (Table 2). FakB2(R202A) mutant was indistinguishable from wild type with respect to fatty acid incorporation, and the expression of FakB2(S93A) resulted in only a 40% decrease in fatty acid incorporation (Table 5). These data suggested that Fak activity may not be the rate-controlling step in the incorporation pathway, although an alternative interpretation of the FakB2(S93A) data may be that the thermal instability of FakB2(S93A) was corrected by its stabilization in the intracellular environment.
TABLE 5.
[1-14C]Oleate incorporation into membrane phospholipids in fakB2 mutant strains
| FakB2 mutanta expressed | [14C]Phospholipidb |
|---|---|
| nmol/min/109 cells | |
| Wild type | 5.11 ± 0.56 |
| ΔfakB2 | <0.005 |
| T61A | 0.13 ± 0.02 |
| S93A | 3.13 ± 0.18 |
| R170A | <0.005 |
| R202A | 4.60 ± 0.37 |
| H266A | 0.23 ± 0.02 |
a All mutant strains were constructed in the SA178R1 background by knocking in alleles with missense mutations that express the altered FakB2 proteins indicated in the column from the native genomic promoter.
b The incorporation of labeled oleate into phospholipids was determined over a 30-min time course as described under “Experimental Procedures.” Linear regression analysis was used to calculate the rates of labeled oleate incorporation into phospholipid.
The second in vivo function of Fak assessed was its ability to support virulence factor transcription. S. aureus expresses a suite of extracellular factors whose transcription was significantly depressed in either ΔfakA or ΔfakB1 ΔfakB2 double mutants (1). The same series of mutant strains used in the fatty acid labeling experiments were grown to mid-log phase and the levels of mRNA for four sentinel virulence factor genes (ehp, efb, hlgC, and saeP) that were regulated by Fak (1) were measured using quantitative real time PCR (Fig. 7). The plsX gene was included in the experiment as a control gene that was not part of the virulence factor regulon. As in strain USA300 (1), the deletion of fakB2 resulted in the significant reduction in virulence factor mRNA levels (Fig. 7). None of the four active site FakB2 mutant alleles were able to restore virulence factor mRNA levels. Virulence gene expression appeared normal in the strain expressing the FakB2(R202A) mutant. These data showed that Fak activity was critical for maintaining the expression of virulence genes. However, understanding why FakB2(R202A) did not affect this process or why FakB2(S93A) was defective in virulence gene expression, but relatively normal in fatty acid incorporation, will require delineating the biochemical steps involved in activating transcription and how FakB2 phosphorylation fits into this putative pathway. Nonetheless, it is clear that fatty acid kinase activity, rather than the proteins themselves, was critical for transcriptional regulation.
FIGURE 7.
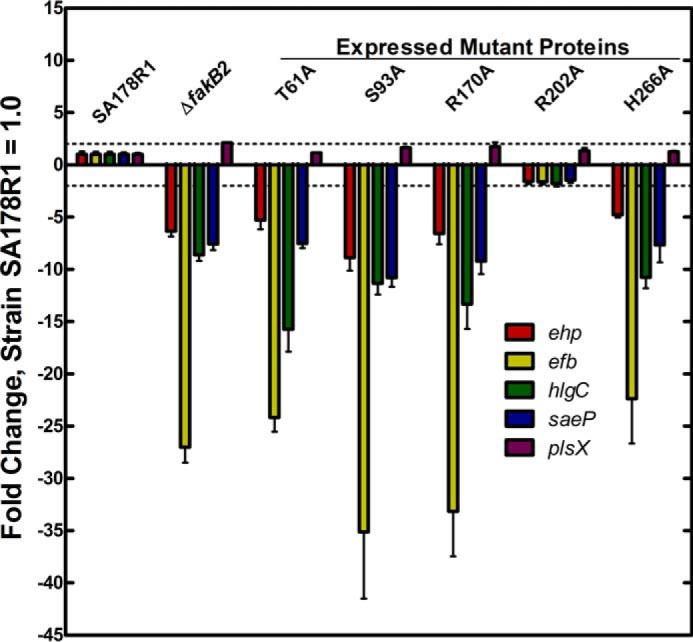
mRNA levels for selected virulence factor genes in strain SA178R1 and its derivatives expressing the panel of fakB2 mutant alleles. A series of strains were constructed by knocking in a fakB2 mutant allele into the fakB2 gene of strain SA178R1 as described under “Experimental Procedures.” The strains were grown to mid-log phase; RNA was extracted, and the levels of mRNA for a panel of five genes were determined by quantitative real time PCR as described under “Experimental Procedures.” Four genes, ehp, efb, hlgC, and saeP, were measured because they belong to the gene set that is known to be regulated by fatty acid kinase (1). The plsX gene was included as a control gene that is not part of the Fak regulon. All mRNAs were quantified by comparing their expression levels to the gmk calibrator, and the levels in the parent strain SA178R1 were set to 1.
Discussion
The biochemical analyses of five conserved residues that define the bacterial fatty acid-binding protein family (Pfam02645) reveal their functions in catalysis and protein stability. Arg-170 is an invariant residue in all members of the FakB protein family. FakB2(R170A) has only a minor defect in protein stability and binds normally to FakA. These data coupled with the complete lack of Fak activity using the FakB2(R170A) mutant points to a key role of Arg-170 in Fak catalysis. Arg-170 interacts with a constellation of three water molecules hovering over the fatty acid carboxyl group that may mimic the positions of the phosphate oxygens in FakB2 bound to acyl-PO4. These observations are consistent with Arg-170 facilitating fatty acid phosphorylation by neutralizing the charge on the acyl-PO4. Three of the conserved residues in the FakB family are involved in a hydrogen bond network that fixes the position of the fatty acid carboxyl moiety. Thr-61 and His-266 play a role in catalysis based on the marked inability of mutations in these residues to support Fak activity in the presence of normal FakA binding. Mutations of Thr-61 and His-266 have the same effect on Fak catalysis, and the 1.2-Å x-ray FakB2 structure shows how these two residues cooperate in polarizing the fatty acid. Thus, we propose that the Fak reaction proceeds by the attack of the fatty acid carboxyl anion activated by the Thr-61–His-266 network on the γ-phosphate of ATP. FakB2(S93A) exhibits marked thermal instability that illustrates the critical importance of this residue in maintaining the overall protein structure. It is difficult to conclude that Ser-93 participates in catalysis because the thermal instability of FakB2(S93A) may underlie its inability to support Fak activity in vitro, and an S. aureus strain expressing FakB2(S93A) is competent for exogenous fatty acid incorporation into phospholipid. The biochemical analysis of the FakB2(R202A) mutant leads to the conclusion that Arg-202 is involved in FakA-FakB2 protein-protein interaction. The FakA protein family has a number of highly conserved Glu and Asp residues that may function as the docking partners for Arg-202, but additional research to determine the structure of FakA will be required to determine whether these conserved acidic residues are involved in FakA-FakB interactions. The high degree of conservation in these residues suggests that they perform the same functions in supporting Fak activity in all members of the bacterial fatty acid-binding protein family.
This study reveals the functional significance of the structural elements that distinguish the bacterial fatty acid-binding protein family (FakB) from the substrate-binding protein/domain of dihydroxyacetone kinases (DhaK). FakB2 is structurally similar to the DhaK of E. coli and Lactococcus lactis dihydroxyacetone kinase (PDB code 3PNK and 3CT4) (30, 31) and the DhaK domain (residues 50–311) of the Citrobacter freundii dihydroxyacetone kinase (PDB code 1UN9) (32). These four structures overlay with root mean square deviations ranging from 2.5 to 2.7 Å. Although the primary sequences of the proteins show little sequence identity (19–22%), the FakB and DhaK proteins share a common amino-terminal EDD domain composed of four β-strands flanked by two α-helices on each side (Fig. 8, blue), and a carboxyl-terminal six-stranded β-sheet flanked by two α-helices (Fig. 8, gold). The two distinguishing structural features of the bacterial fatty acid-binding proteins are an extension of the EDD domain by three β-strands and one α-helix (Fig. 8, dark purple) and the addition of three α-helices to the carboxyl-terminal domain (Fig. 8, pink). One of these helices, helix-8 in the FakB family, is too disordered to be observed in our structure (Fig. 8). This is also the case in the majority of FakB structures in the PDB, but in four cases (PDB codes 2DT8, 3FDJ, 3FYS, and 3LUP) the density for these residues is observed, and an α-helix has been modeled. In these four cases, helix-8 is stabilized by crystal contacts with symmetry mates. The active site Ser-93 is located on the EDD domain; the active site Thr-61 resides on the extension to the EDD domain in FakB2 (Fig. 8, dark purple), and Arg-170 and His-266 are on the structural extension to the carboxyl-terminal domain in FakB2 (Fig. 8, pink). The two structural elements that distinguish the FakB protein family enclose an open area between the amino- and carboxyl-terminal domains of DhaK to form the fatty acid binding pocket. These two structural elements and the presence of the four conserved residues at the active site clearly distinguish the fatty acid-binding protein family from the highly related DhaK proteins.
FIGURE 8.
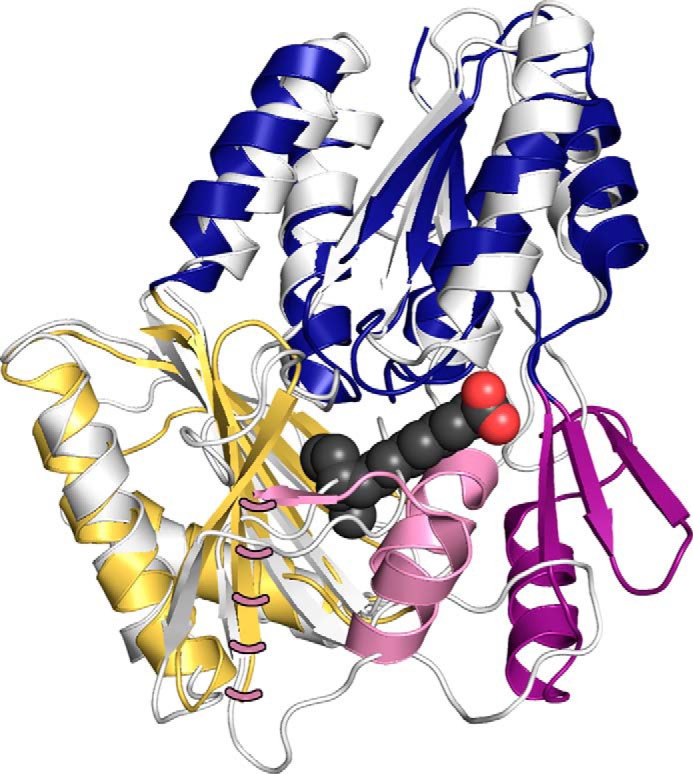
Structures of the bacterial fatty acid-binding protein family (FakB) and the substrate-binding protein/domain of the dihydroxyacetone kinase family (DhaK). The DhaK domain of C. freundii dihydroxyacetone kinase (PDB code 1UN9, residues 50–311) is superimposed on the FaKB2 crystal structure (PDB code 4X9X). The structural elements common to both proteins are the amino-terminal EDD (blue) and carboxyl-terminal domains (gold). The two structural elements the define the FakB family are an extension of the amino-terminal EDD domain by three sheets and one helix (dark purple) and a three-helix extension from the carboxyl-terminal domain (pink). The fatty acid of FakB2 is represented as charcoal-colored carbons and red-colored oxygen spheres. Pink dashes show the position of helix-8 in the FakB2 structure.
Interpreting the functions of FakB2 residues in vivo is more challenging because we do not have specific information about the biochemical pathways that support fatty acid activation/incorporation and virulence factor transcription. Like the ΔfakB2 strain, strains expressing FakB2(R170A), FakB2(T61A), and FakB2(H266A) mutants are defective in both fatty acid incorporation and virulence factor transcription. These data point to the key catalytic roles of these residues and the importance of fatty acid kinase activity to support both of these cellular processes. The strains expressing FakB2(S93A) or FakB2(R202A) are not as deficient in fatty acid incorporation into phospholipid as would be anticipated based on the impact these mutations have on Fak activity in vitro. However, it is likely that the rate-controlling step in phospholipid synthesis is the PlsY acyltransferase and not Fak activity. If Fak is not the controlling step in exogenous fatty acid incorporation, then Fak activity could be significantly diminished in these mutants, but the reduced flux through Fak would have little impact on overall phospholipid synthesis. Alternatively, the temperature-sensitive FakB2(S93A) mutant may be stabilized by the crowded intracellular environment leading to a more functional enzyme in vivo than would be anticipated from the in vitro results. There are even more caveats to interpreting the effect of the mutants on virulence factor transcription. The concomitant loss of Fak activity and virulence factor transcription in strains expressing FakB2(R170A), FakB2(T61A), and FakB2(H266A) clearly shows that Fak kinase activity is critical for its function in both these processes. It is known that Fak activity is a key regulator of virulence factor transcription, but we do not have specific information about the participants in the biochemical phosphorylation cascade that ultimately impacts transcriptional activity. Thus, the inability of FakB2(R202A) expression to effect virulence factor transcription in vivo suggests that the rate-determining step in transcriptional activation is not FakB2 phosphorylation per se, but rather some downstream interaction between FakB2 and the transcriptional machinery. The most interesting mutant was FakB2(S93A) because it is defective in virulence factor transcription but is nearly normal with respect to fatty acid incorporation. It is not clear how to explain this difference at our current level of knowledge. However, the functional contributions of the conserved residues in Pfam02645d delineate their principal biochemical functions in FakB2 catalysis, protein stability, and protein-protein interactions. The next important step will be to define the biochemical steps in the phosphorylation cascade that connects fatty acid kinase to the activation of virulence factor transcription.
Author Contributions
T. C. B. and C. O. R. designed the study. T. C. B., D. J. M., and S. W. W. determined and interpreted the crystal structure. P. J. created and characterized the knock-in strains, and A. N. performed the analytical ultracentrifugation experiments and analyzed the data. All authors contributed to and approved the manuscript.
Acknowledgments
We thank the Protein Production and Hartwell Center DNA Sequencing Shared Resources for protein expression/purification and DNA sequencing, respectively.
This work was supported by National Institutes of Health Grants GM034496 (to C. O. R.) from NIGMS, Training Grant T32 AI106700 from NIAID (to T. C. B.), and Cancer Center Support Grant CA21765 and by the American Lebanese Syrian Associated Charities. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 4X9X) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Fak
- fatty acid kinase
- acyl-PO4
- fatty acylphosphate
- PDB
- Protein Data Bank
- qRT
- quantitative RT.
References
- 1. Parsons J. B., Broussard T. C., Bose J. L., Rosch J. W., Jackson P., Subramanian C., and Rock C. O. (2014) Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 111, 10532–10537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parsons J. B., Frank M. W., Jackson P., Subramanian C., and Rock C. O. (2014) Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol. Microbiol. 92, 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bose J. L., Daly S. M., Hall P. R., and Bayles K. W. (2014) Identification of the vfrAB operon in Staphylococcus aureus: a novel virulence factor regulatory locus. Infect. Immun. 82, 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabirova J. S., Hernalsteens J. P., De Backer S., Xavier B. B., Moons P., Turlej-Rogacka A., De Greve H., Goossens H., and Malhotra-Kumar S. (2015) Fatty acid kinase A is an important determinant of biofilm formation in Staphylococcus aureus USA300. BMC Genomics 16, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 6. Kinch L. N., Cheek S., and Grishin N. V. (2005) EDD, a novel phosphotransferase domain common to mannose transporter EIIA, dihydroxyacetone kinase, and DegV. Protein Sci. 14, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schulze-Gahmen U., Pelaschier J., Yokota H., Kim R., and Kim S. H. (2003) Crystal structure of a hypothetical protein, TM841 of Thermotoga maritima, reveals its function as a fatty acid-binding protein. Proteins 50, 526–530 [DOI] [PubMed] [Google Scholar]
- 8. Nan J., Zhou Y., Yang C., Brostromer E., Kristensen O., and Su X. D. (2009) Structure of a fatty acid-binding protein from Bacillus subtilis determined by sulfur-SAD phasing using in-house chromium radiation. Acta Crystallogr. D Biol. Crystallogr. 65, 440–448 [DOI] [PubMed] [Google Scholar]
- 9. D'Elia M. A., Pereira M. P., Chung Y. S., Zhao W., Chau A., Kenney T. J., Sulavik M. C., Black T. A., and Brown E. D. (2006) Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188, 4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhong J., Karberg M., and Lambowitz A. M. (2003) Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 31, 1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kreiswirth B. N., Löfdahl S., Betley M. J., O'Reilly M., Schlievert P. M., Bergdoll M. S., and Novick R. P. (1983) The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712 [DOI] [PubMed] [Google Scholar]
- 12. Bose J. L., Fey P. D., and Bayles K. W. (2013) Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl. Environ. Microbiol. 79, 2218–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lehman M. K., Bose J. L., and Bayles K. W. (2016) Allelic exchange. Methods Mol. Biol. 1373, 89–96 [DOI] [PubMed] [Google Scholar]
- 14. Bae T., and Schneewind O. (2006) Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63 [DOI] [PubMed] [Google Scholar]
- 15. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., et al. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.6, DeLano Scientific, Palo Alto, CA [Google Scholar]
- 19. Zhao H., Brautigam C. A., Ghirlando R., and Schuck P. (2013) Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. Chapter 20, Unit 20.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown P. H., Balbo A., and Schuck P. (2008) Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr. Protoc. Immunol. 18, 18.15.1–18.15.39 [DOI] [PubMed] [Google Scholar]
- 22. Zhao H., Ghirlando R., Piszczek G., Curth U., Brautigam C. A., and Schuck P. (2013) Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal. Biochem. 437, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghirlando R., Zhao H., Balbo A., Piszczek G., Curth U., Brautigam C. A., and Schuck P. (2014) Measurement of the temperature of the resting rotor in analytical ultracentrifugation. Anal. Biochem. 458, 37–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balbo A., Brown P. H., Braswell E. H., and Schuck P. (2007) Measuring protein-protein interactions by equilibrium sedimentation. Curr. Protoc. Immunol. 18, 18.8.1–18.8.28 [DOI] [PubMed] [Google Scholar]
- 25. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., and Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 [DOI] [PubMed] [Google Scholar]
- 26. Parsons J. B., Kukula M., Jackson P., Pulse M., Simecka J. W., Valtierra D., Weiss W. J., Kaplan N., and Rock C. O. (2013) Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob. Agents Chemother. 57, 2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winer J., Jung C. K., Shackel I., and Williams P. M. (1999) Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal. Biochem. 270, 41–49 [DOI] [PubMed] [Google Scholar]
- 28. Mihalek I., Res I., and Lichtarge O. (2006) Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics 22, 1656–1657 [DOI] [PubMed] [Google Scholar]
- 29. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 30. Shi R., McDonald L., Cui Q., Matte A., Cygler M., and Ekiel I. (2011) Structural and mechanistic insight into covalent substrate binding by Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. U.S.A. 108, 1302–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zurbriggen A., Jeckelmann J. M., Christen S., Bieniossek C., Baumann U., and Erni B. (2008) X-ray structures of the three Lactococcus lactis dihydroxyacetone kinase subunits and of a transient intersubunit complex. J. Biol. Chem. 283, 35789–35796 [DOI] [PubMed] [Google Scholar]
- 32. Siebold C., Arnold I., Garcia-Alles L. F., Baumann U., and Erni B. (2003) Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded α-helical barrel ATP-binding domain. J. Biol. Chem. 278, 48236–48244 [DOI] [PubMed] [Google Scholar]
- 33. Zhao H., and Schuck P. (2012) Global multi-method analysis of affinities and cooperativity in complex systems of macromolecular interactions. Anal. Chem. 84, 9513–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]