

Abstract
A type 3 secretion system is used by many bacterial pathogens to inject proteins into eukaryotic cells. Pathogens insert a translocon complex into the target eukaryotic membrane by secreting two proteins known as translocators. How these translocators form a translocon in the lipid bilayer and why both proteins are required remains elusive. Pseudomonas aeruginosa translocators PopB and PopD insert pores into membranes forming homo- or hetero-complexes of undetermined stoichiometry. Single-molecule fluorescence photobleaching experiments revealed that PopD formed mostly hexameric structures in membranes, whereas PopB displayed a bi-modal distribution with 6 and 12 subunits peaks. However, individually the proteins are not functional for effector translocation. We have found that when added together, the translocators formed distinct hetero-complexes containing 8 PopB and 8 PopD molecules. Thus, the interaction between PopB and PopD guide the assembly of a unique hetero-oligomer in membranes.
Keywords: membrane protein, membrane transport, oligomer, protein translocation, single-molecule biophysics, type III secretion system (T3SS)
Introduction
The transport of proteins across membranes is essential at many stages of pathogen infection and colonization of human cells. This process typically involves the discharge of proteins from the pathogen (secretion) and the introduction of these secreted toxins/effectors into the cytosol of the target cell (translocation). Many pathogens, including the Shigella, Salmonella, Yersinia, and Pseudomonas species, exploit a sophisticated and efficient mechanism of protein secretion and translocation known as type III secretion (T3S)3 system (1, 2). The T3S system is a syringe-like macromolecular machine formed by more than 20 different proteins organized in three major structures to span: (i) the inner bacterial membrane, the periplasmic space, and the outer bacterial membrane (the secreton); (ii) the extracellular space (the needle); and (iii) the host cellular membrane (the translocon) (3–6).
A phylogenetic analysis of bacterial T3S systems based on conservation of their basal body ATPase indicates the presence of at least 7 families of T3S machines. The Pseudomonas aeruginosa genome encodes a single T3S system grouped within the Ysc family, named after the Yersinia spp. T3S system (the archetypical T3S system in this family) (7). The Ysc family includes pathogens like Yersinia pestis, Y. pseudotuberculosis, Y. enterocolitica, Bordetella pertussis, Vibrio parahemeolyticus, and P. aeruginosa, among others. The Ysc family shares structural similarity with the Inv-Mxi-Spa family of T3S systems, which includes the secretion systems used by Salmonella enterica and Shigella spp. (8).
Great progress has been made in the structural characterization of the secreton and the needle for different T3S system families (9). However, little is known about how T3S-secreted proteins are translocated across the plasma membrane of the target cell to alter the normal function of the host (4). Two T3S-secreted proteins, known as the T3S translocators, insert into the target membrane to facilitate effector translocation. P. aeruginosa translocators PopB/PopD and the tip forming protein PcrV are functionally conserved with the Yersinia homologues YopB/YopD and LcrV, suggesting a common translocation mechanism. Placing PopB and PopD in a YopB/YopD-deficient Y. pseudotuberculosis strain rescues full translocation of T3S effectors into HeLa cells as well as a T3S-dependent pore-forming phenotype in erythrocytes, if PcrV is also provided (10). Translocators from the Inv-Mxi-Spa are not able to rescue translocation in Yersinia. The sequence identity between the translocators from the Ysc family and Inv-Mxi-Spa family is poor (less than 20%), and the Shigella and Salmonella translocators are considerably larger than the Yersinia or P. aeruginosa counterparts. These differences suggest that the proteins have evolved to adapt to a different mechanism of invasion and/or to interact with different host cell membranes. Therefore, one needs to be cautious when trying to extrapolate the properties observed for translocators from one family to the other.
Current models for the T3S translocon complex of the Ysc family are quite rudimentary, and they are mostly based on the following observations: (i) the translocators are found associated with cell membranes after incubating the pathogen in close contact with red blood cells (11); (ii) both translocators co-immunoprecipitate after Triton X-100 solubilization of membrane-associated proteins (11); and (iii) ring-like structures are observed using EM when the translocators are incubated with model membranes (12). The proteins are presumed to form a pore in which each PopB/YopB is speculated to have two transmembrane (TM) segments and each PopD/YopD only one TM segment based on sequence analysis (Fig. 1A). Both PopB and PopD can insert individually in model membranes and form discrete and stable pores, yet they interact with each other on the membrane when incubated together (13, 14). Other elements like coiled-coil segments and amphipathic α-helices can be predicted using bioinformatics analysis on PopB and PopD (Fig. 1A), however, how these proteins interact with other proteins or with the membrane is still uncertain.
FIGURE 1.

PopBS164C and PopDF223C derivatives were active for effector translocation. A, scheme of the primary structure of P. aeruoginosa PAO1 translocators PopD (gi 9947683) and PopB (gi 9947682) compared with the homologue Y. enterolcolitica translocators YopD (gi 586795) and YopB (gi 122815801). Location of the single Cys modifications introduced for probe labeling is indicated on PopD and PopB. Predicted hydrophobic segments, amphipathic helices, and coiled coils are shown. The molecular mass for each translocator is shown on the right. The scale at the bottom indicates amino acid number. The activity of the PopB (or PopD) derivative was indistinguishable from the wild-type protein when evaluated for their ability to complement a popB deletion strain (or a popD deletion strain). The activity of the translocators was assessed by the characteristic rounding up of the infected target cells due to actin cytoskeleton disruption caused by effector translocation (15). Strains used in this assay were P. aeruginosa PAK WT (B), PAKΔpopD complemented with pUCP18 (C), PAKΔpopB complemented with pUCPHBS164C (D), and PAKΔpopD complemented with pUCPHDF223C (E). The PAKΔpopB control showed no effect on cell morphology as shown for the PAKΔpopD control in panel C (not shown).
We have established the experimental conditions necessary to maximize association and insertion of the P. aeruginosa translocators PopB and PopD into model membranes (14). Both proteins form pores in model membranes and allow the passage of small proteins and other molecules (13, 14). We sought here to provide specific insights about the stoichiometric arrangement and mechanism of assembly of the PopB and PopD translocators. Steady-state fluorescence spectroscopy of membrane-bound complexes indicated that when added together, PopB and PopD assemble hetero-complexes. Using single-molecule fluorescence photobleaching, we quantified the stoichiometry of membrane-assembled complexes. In the absence of PopD, PopB oligomers presented a bi-modal size distribution with peaks at 6 and 12 subunits. In the absence of PopB, PopD assembled mostly hexameric structures. Strikingly, when PopB and PopD were both present, they assembled into complexes containing 8 PopB and 8 PopD molecules. These findings provide the basis to explain the requirement of both translocators to assemble a functional TM hetero-complex with unique architecture.
Experimental Procedures
Protein Expression, Purification, and Characterization
The purification, structural characterization, and pore forming properties of isolated PopB and PopD were described before in detail (14). The expression and purification of hisPcrH-PopD, hisPcrH-PopB, their derivatives, and the isolation of the translocators from hisPcrH chaperone were done as previously described (14). The protein concentration was estimated using molar absorptivity of 18,910 m−1 cm−1 for hisPcrH, 13,980 m−1 cm−1 for PopD, 6,990 m−1 cm−1 and for PopB as described (14). Single Cys protein derivatives were obtained by site-directed mutagenesis. The functionality of the protein derivatives employed in these studies was evaluated by their ability to form pores in model membranes (14) and their ability to rescue translocation of P. aeruginosa PAK strain effectors into HeLa cells (15). PopBS164C or PopDF223C derivatives were able to rescue effector translocation in P. aeruginosa PAK strains with deletions for PopB (PAKΔpopB) or PopD (PAKΔpopD), respectively (Fig. 1). PAKΔpopB and PAKΔpopD deletion strains were generously provided by Dr. Stephen Lory. A synthetic 1494-bp DNA fragment (named GHD) encoding the P. aeruginosa PAK (Gene Bank AY232997.1) pcrG promoter (400–475), pcrH (1663–2179), and popD (3331–4231) was acquired from Biomatik. The construct was provided in the pUC57-Amp vector with an EcoRI site and a HindIII site at its 5′ and 3′, respectively. The GHD fragment was cut by double digestion with EcoRI and HindIII and cloned into the pUCP18 vector (generously provided by Dr. Dara Frank 16, 17) to generate the pUCPHD vector. The pUCPHB vector was generated by the Gibson assembly method (18) as instructed by the manufacturer (New England Biolabs) (Table 1). The infection assay was adapted from Kaufman et al. (15). Briefly, HeLa cells cultured in DMEM (Caisson) containing 10% FBS were maintained in a 37 °C incubator supplemented with 5% CO2. Before infection, cells were washed once with pre-warmed PBS and incubated in fresh DMEM. Bacteria were grown overnight at 37 °C in Miller lysogeny broth, the next day the cells were diluted to an absorbance at 600 nm of 0.1 in fresh broth and grown until the absorbance reached a value of 1. To establish infection a bacterium/HeLa cell ratio of 30 was used. Bacteria were spun down at 16,000 × g for 2 min and suspended in 100 μl of DMEM, then incubated with HeLa cells grown to confluence in 6-well plates for 4 h at 37 °C (1.2 × 106 cells/well). The pore-forming activity of labeled derivatives was indistinguishable from the one observed for WT translocators (14).
TABLE 1.
PCR primers used to generate the pUCPHD and pUCPHB vectors
The pUCPHB vector was generated using Gibson assembly combining the Cys-less P. aeruginosa PA01 popB gene amplified from plasmid pETDuet1-hispcrHpopB (14) using primers 1 and 2; a fragment from GHD containing the pcrG promoter and pcrH gene amplified using primers 3 and 4; and the pUCP18 vector linearized by double digestion with EcoRI and HindIII. Vectors containing the genes codifying for PopDF223C or PopBS164C were generated similarly using Gibson assembly with the linearized pUCP18 vector and fragments obtained from pUCPHD or pUCPHB with primers 3 and 7, 6 and 5, or 3 and 9, 8 and 2, respectively.
| Primer sequences | |
|---|---|
| 1 | 5′-TCCGATAACGCTTGAACGCGCCGGACTGCCCTA |
| 2 | 5′-GCCAGTGCCAAGCTTTCAGATCGCTGCCGGTCGG |
| 3 | 5′-CTATGACCATGATTACGAATTCTGACGCAGCGAAGCCTGC |
| 4 | 5′-TCAAGCGTTATCGGATTCATATGTTCG |
| 5 | 5′-CGACGGCCAGTGCCAAGCTTCAGACCACTCCGGCCGC |
| 6 | 5′-CAGTCCTGCGTCCAGATGGCCAACGC |
| 7 | 5′-CTGGACGCAGGACTGGATCACCGTGTTG |
| 8 | 5′-CCAGAAGTGCGGTCTGGCAGCCAAAATC |
| 9 | 5′-CAGACCGCACTTCTGGGATTCTTTCGC |
Fluorescent Protein Labeling
PopBS164C and PopDF223C were labeled using the iodoacetamide derivative of BodipyFL (BpyFL C1-IA, Invitrogen) as previously described (14). PopDF223C was labeled using Bodipy® TMR C5-Maleimide (BpyTMR, Invitrogen) using the same described procedure (14). Labeling efficiencies were calculated using the molar absorptivities at 280 nm in urea 6 m for PopB and PopD (see above), at 502 nm for BpyFL (55,000 m−1 cm−1), and at 540 nm for BpyTMR (51,120 m−1 cm−1). Protein concentration was calculated after correcting the absorbance by the contribution of the fluorescent dyes at 280 nm. The absorbance of BpyFL at 280 nm was 4% of its absorbance at 502 nm. The absorbance of BpyTMR at 280 nm was 20% of its absorbance at 540 nm. The percentage of labeling of selected preparations used for single molecule experiments were 96% for PopDF223C-BpyFL, 100% for PopBS164C-BpyFL, and 95% for PopDF223C-BpyTMR (hereafter PopDBpyFL, PopBBpyFL, and PopDBpyTMR, respectively). For single-molecule experiments the labeled proteins were kept in the dark and all incubations were done in the absence of light to avoid photobleaching. To avoid bleaching of protein complexes during focusing of the sample prior to a photobleaching experiment, the focal plane containing single TM complexes was found manually and a Perfect Focus System (PFS®, Nikon) was activated. This system monitors the position of the focal plane using a dedicated near infrared 870-nm light-emitting diode and line charge-coupled device sensor that adjusts focus automatically in milliseconds given a drift event in the Z-axis. After focusing and perfect focus system activation, laser excitation was turned off, the objective lens was moved to a new region never exposed to light, and the single-molecule photobleaching experiment was initiated acquiring the fluorescence emission from time 0 without requiring pre-focusing of the sample.
Liposome Preparation
Liposomes were prepared using identical composition and procedures previously described (14). NBD-PE (N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine) was purchased from Life Technologies.
Liposome Flotation-Membrane Binding Assay
Association of PopB and PopD to liposomes at the indicated pH was assessed by flotation of proteoliposomes through a sucrose gradient, SDS-PAGE, and quantification of the bound proteins by gel densitometry as described previously (14). When added together, PopB and PopD were pre-mixed in 6 m urea at the indicated ratio.
Supported Lipid Bilayers (SLB)
Clean glass coverslips were prepared by boiling coverslips in a 10% (v/v) solution of 7X® detergent (MP Biomedicals) for 20 min. Coverslips were then extensively washed with tap water, followed by extensive wash with pure water, followed by 10 min boiling in pure water. Dried coverslips were heated for 4 h at 400 °C in a kiln oven and stored until use. SLB were formed as follows: a sample containing 400 μl of liposome or proteoliposome (50 mm sodium acetate buffer, pH 4.0, 0.22 mm final lipid concentration, and 5 mm CaCl2) was placed on a Petri dish between two 1-mm thick aluminum spacers. A clean glass coverslip was placed on top of the sample and incubated for 30 min to allow SLB formation. After this, a glass slide was placed next to the coverslip and the Petri dish was filled with buffer. While submerged in buffer, the coverslip was mounted on the glass slide spaced by two strips of double-sided adhesive tape to form a chamber.
Assembly of Protein Complexes for Imaging
For homo-oligomer reaction mixtures (100 μl) containing liposomes (0.1 mm total lipids) and BpyFL-labeled PopB or PopD (30 nm total protein) were incubated in buffer, 50 mm sodium acetate, pH 4.0, at 20–23 °C for 15 min (protein to lipid ratio 1/3,333). For dual labeled hetero-oligomers reaction mixtures were done similarly but total lipid concentrations were 0.3 or 0.6 mm and labeled proteins were 30 nm each (total protein to lipid ratio 1/5,000 or 1/10,000). Before assembly into SLB, proteoliposomes were diluted by addition of 1390 μl of buffer, 50 mm sodium acetate, pH 4.0, plus 10 μl of a solution of liposomes (30 mm total lipids stock concentration). For single-color labeled hetero-oligomers reaction were done similarly as for dual-color labeled ones, maintaining a total protein to lipid ratio of 1/4,550.
Imaging
Single molecule images were acquired using an electron multiplier charge-coupled device Cascade II camera (Photometrics, Tucson, AZ). Total internal reflection fluorescence (TIRF) microscopy used a home-built laser system around a Nikon Eclipse Ti microscope and a high numerical aperture objective (×60, NA 1.49) (Nikon, Melville, NY). TIRF excitation was achieved using a 488-nm argon-ion air laser (Melles-Griot) for PopDBpyFL and PopBBpyFL, and a 532-nm solid state (Crystalaser) laser for PopDBpyTMR. Neutral density filters were employed to attenuate laser light to the desired level and increase the average photobleaching time. The standard exposure and acquisition interval time was 70 ms for BpyFL and 100 ms for BpyTMR probes. Dual-color photobleaching was performed first by constant illumination from a 532-nm laser line to photobleach the red emitting dye BpyTMR, followed by constant illumination with a 488-nm laser line to photobleach the green emitting BpyFL. The consecutive photobleaching of BpyTMR followed by BpyFL was done to minimize any problems associated with potential FRET between the dyes and any direct excitation or photobleaching of BpyTMR when using the 488-nm wavelength. Time point image stacks were independently acquired for PopDBpyTMR and PopBBpyFL, and emission intensity as a function of the illumination time was analyzed for individual protein complexes.
Single-molecule Photobleaching
Photobleaching of labeled translocators was performed on SLB assembled using liposomes with membrane-inserted proteins. Photobleaching data were inspected and analyzed manually. Diffraction-limited spots in image stacks were identified and quantified over time using ImageJ. Time-resolved fluorescence intensity plots of single protein complexes were generated importing the measured intensities into Origin software. The number of fluorescent molecules in each complex was obtained by dividing the background-subtracted initial fluorescence intensity by the intensity contribution of a single dye revealed by single stepwise photobleaching events, most frequently discernible within the last few photobleaching events within each trace (19, 20). Single monomers were not considered in our analysis given the difficulty of distinguish them from background noise. Because ∼100% of the proteins were labeled, and we were able to temporally separate the bleaching of a single fluorophore, the overall photobleaching corresponded to the presence of all the subunits of the complex. This is the ideal situation for photobleaching enabling each subunit to be directly counted without the need to implement discrete statistical distributions to deduce the number of molecules in the complex.
Steady-state Fluorescence Spectroscopy
Steady-state fluorescence measurements and spectra for fluorophore characterization and quantification were collected using a Fluorolog 3–21 spectrofluorometer as described earlier (14). Anisotropy and emission intensity measurements were taken using a Chronos fluorescence lifetime spectrometer (ISS, Champaign, IL) equipped with a 470-nm laser diode and Glan-Thompson prism polarizers (10 × 10 mm aperture for excitation and 14 × 14 mm aperture for emission) (14). Emitted light was collected through a Melles Griot cutoff glass filter GG495 to eliminate scattered light. All measurements were blank subtracted with an equivalent sample lacking the fluorophore. For emission intensity measurements the amount of PopDBpyFL was kept constant (final concentration 10 nm) and the amount of WT PopD (PopDWT) or PopBWT adjusted to render the indicated percentage of labeled protein. The protein:lipid ratio was kept constant at 1/3,000 or 1/10,000 by adjusting the amount of lipids added. For anisotropy measurements total protein concentration was kept constant at 100 nm and total lipid concentration at 1 mm. The percentage of labeling was adjusted by pre-mixing PopDBpyFL with PopDWT (or PopBWT) in 6 m urea. BpyFL was excited with polarized light, and both vertically polarized (‖) and horizontally polarized (⊥) emission intensities were recorded. Anisotropy was calculated using r = (I‖ − I⊥)/(I‖ + 2I⊥), where I is the background-subtracted emission intensity of vertically (or horizontally) polarized light. While working with membrane bilayers it is important to select experimental conditions where the anisotropy (homoFRET) signal originates from the association of proteins, and not as a result of the proximity of randomly distributed proteins. Based on determinations of liposome size by electron microscopy (14), and the protein:lipid ratio used in these studies, we estimated that randomly distributed proteins will be on average more than 200 Å apart in our system.
Time-resolved Fluorescence Spectroscopy
Lifetime measurements were done using a Chronos fluorescence lifetime spectrometer (ISS, Champaign, IL) as described previously (14).
Results
PopB Assists the Association of PopD with Membranes
Both PopB and PopD can efficiently form stable pores at acidic pH on liposomes containing negatively charge phospholipids (12). In addition to their individual ability to insert into membranes, we have shown that the translocators can also interact with each other and form hetero-oligomers that span the bilayer (14). To examine the molecular mechanism of PopB-PopD interactions, we investigated the association of the translocators when simultaneously binding to membranes over a broad pH range, expanding from the optimal acidic pH to neutral conditions.
A single Cys residue was introduced into each translocator at a location predicted to be solvent-exposed (Fig. 1A), and specifically labeled with the fluorescent probe BpyFL. Labeled PopB and PopD were incubated with membranes at the indicated pH, and the association of proteins with lipid bilayers was determined using a liposome flotation assay. The membrane-containing fraction was analyzed by SDS-PAGE and membrane-bound proteins were quantified using gel densitometry (Fig. 2A). In this assay only liposomes and proteoliposomes float to the top of the gradient, whereas free proteins or protein aggregates remain in the bottom fraction. To detect any cooperative interaction between PopB and PopD, their membrane association was first quantified for individual translocators and then when the two translocators were added together. Any difference in the binding properties of PopB or PopD when incubated alone or in combination suggest a potential interaction between the translocators.
FIGURE 2.
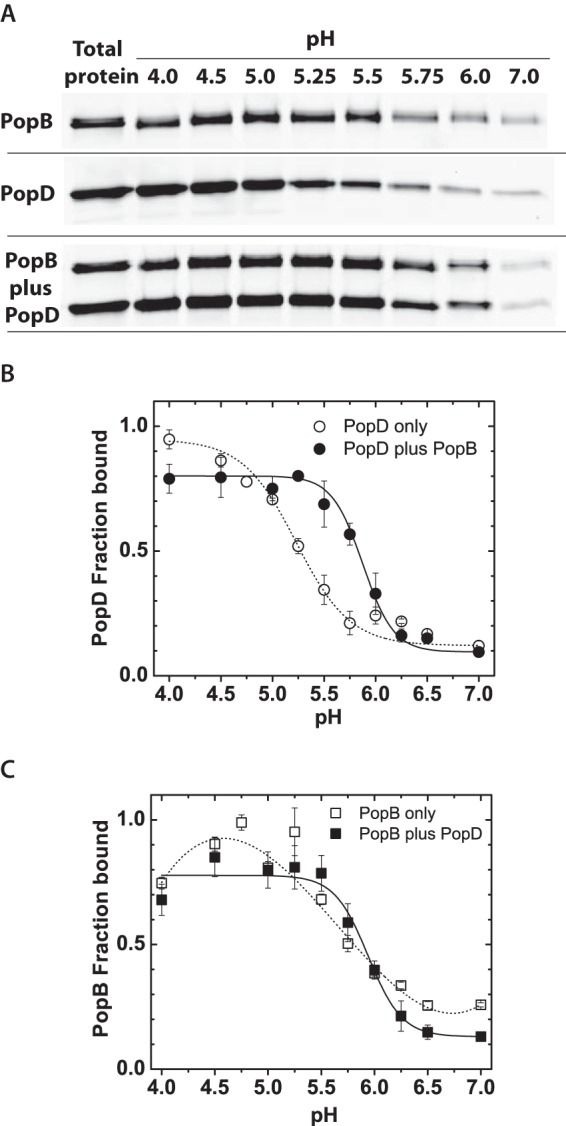
PopB enhanced PopD membrane binding. A, SDS-PAGE gel showing the amount of purified PopDBpyFL and/or PopBBpyFL isolated in the membrane containing fraction after bound and unbound proteins were separated using a liposome flotation membrane-binding assay as described in the text. Total lipids were 2 mm and total protein was 400 nm. Individual proteins or pre-mixed proteins were incubated with membranes at 20–23 °C for 1 h before ultracentrifugation using a sucrose gradient. Proteins were visualized using BpyFL fluorescence. B, PopDBpyFL binding at the indicated pH when the translocator was incubated with membranes alone or when premixed and incubated with an equimolar amount of PopBWT. C, PopBBpyFL binding at the indicated pH when the translocator was incubated with membranes alone or when premixed and incubated with an equimolar amount of PopDWT. BpyFL fluorescence was quantified by gel densitometry and each data point represents the average of at least two independent assays and error bars indicate the data range. Average lines to individual translocators (dashed) and both translocators (solid) are shown only as a guide for the reader.
Membrane association for PopD was higher than 90% at pH 4.0, 50% around pH 5.3, and minimal at neutral pH (Fig. 2B). Interestingly, when PopD and PopB were mixed in equimolar amounts prior to incubation with membranes, the binding of PopD was significantly enhanced in the 5.2–5.8 pH range. PopB membrane association was higher than 90% at pH 4.8, 50% around pH 5.8, and less than 30% at neutral pH. PopB membrane binding was not significantly affected by the presence of PopD (Fig. 2C).
These results suggested that the simultaneous addition of PopB and PopD influences how the proteins interact with membranes, and we hypothesized that this affects their final membrane-inserted arrangement. We therefore focused our analysis on the ability of each translocator to form oligomers on membranes when added individually or combined. We characterize the assembly of membrane complexes at pH 4.5, where binding to membranes reached saturation.
PopD and PopB Form Multimers on Lipid Membranes
FRET or homoFRET (FRET between like molecules), as a function of fractional fluorescent labeling, has been used to determine the oligomerization state of small protein complexes (21). When homoFRET occurs, energy can migrate from dye to dye before a photon is emitted, thereby depolarizing the light and reducing the anisotropy of the sample. How much depolarization occurs depend, among other parameters, on the distance among dyes. Therefore, the steady-state fluorescence anisotropy signal reports on the efficiency of homoFRET (i.e. the distance) between protein molecules labeled with the same fluorophore. In addition to homoFRET, the fluorescence anisotropy can also be influenced by changes in the rotational diffusion of the labeled protein. However, this source of ambiguity is overcome by analyzing anisotropy as a function of the molar fraction for the labeled protein when diluted with unlabeled protein. Under these conditions, changes in fluorescence anisotropy are due to differential separation between the fluorescent probes and reflect on the size of the protein oligomer (21). Both homo- and hetero-oligomerization can be assessed using this approach (22).
In addition to the relatively long range distance information provided by homoFRET measurements (R0 ∼50 Å), BpyFL dyes can form non-fluorescent dimers when positioned a few angstroms apart, which is easily detected by a decrease in the fluorescence intensity, and reports on molecular interactions within a shorter distance range compared with homoFRET (23). BpyFL self-quenching is indicative of adjacent subunits because orbital contact between fluorophores is required (24), and this effect has been exploited in various analytical methods, like biotin-avidin binding detection (25) and single molecule folding studies (26). Moreover, BpyFL is especially useful to study protein-membrane interactions because its fluorescence properties are not sensitive to the pH or polarity of the environment (27, 28).
The ability of PopD to interact with other PopD molecules or with PopB prompted us to investigate the nature of the oligomers formed by these proteins. For PopD-PopD interactions, both the anisotropy (homoFRET) and fluorescence intensity (self-quenching) were measured as a function of the fraction of labeled PopDBpyFL present, which was controlled by addition of PopDWT. Although keeping the total amount of fluorophore constant, an increase in the emission (de-quenching) was observed as the fractional amount of PopDBpyFL was reduced, clearly indicating that PopD formed oligomers on membranes (Fig. 3A and Table 2). At the same time, the homoFRET between subunits decreased (i.e. increase in fluorescence anisotropy) when the fractional amount of PopDBpyFL was reduced.
FIGURE 3.
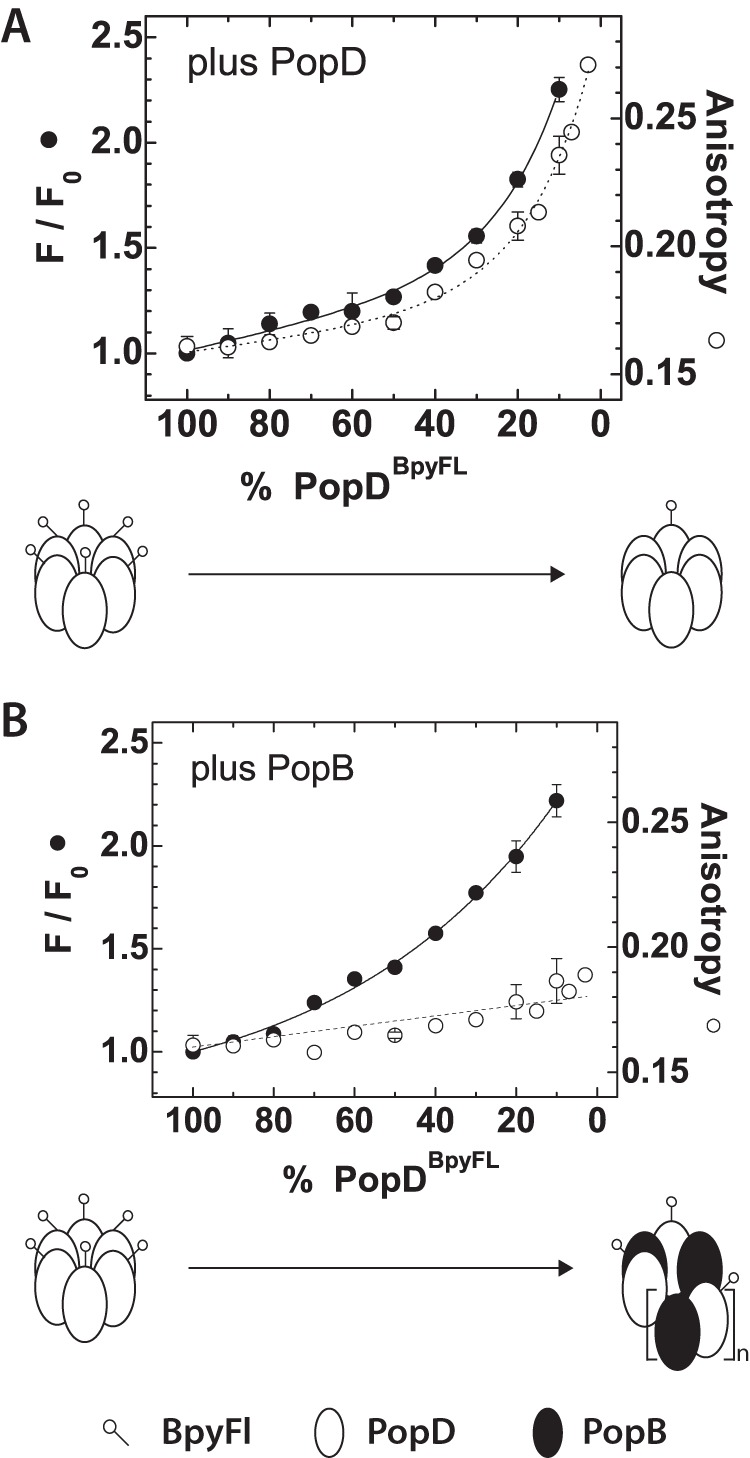
PopD assembled homo- or hetero-oligomeric structures in membranes. Fluorescence intensity and anisotropy changes observed upon the dilution of PopDBpyFL with increasing amounts of PopDWT or PopBWT. A, steady-state fluorescence intensity and anisotropy of PopDBpyFL measured at the indicated percent of labeled protein when diluted with PopDWT. B, steady-state fluorescence intensity and anisotropy of PopDBpyFL when diluted with PopBWT. Scheme indicating the labeled PopDBpyFL distribution expected for complexes containing 100% labeled PopD or a mixture of proteins containing 3% labeled PopDBpyFL, are shown at the bottom of each figure. PopD complexes were drawn as hexamers and hetero-complexes as (PopD-PopB) n-mers for simplicity. Error bars represent the data range of at least two independent assays. Average lines are shown as a guide for the reader.
TABLE 2.
Average fluorescence lifetimes of BpyFL in homo- and hetero-oligomers at varying percent of the fluorescent label
Proteoliposome preparations containing the same protein and lipid concentration, while varying the molar fraction of PopDBpyFL and either PopD or PopB (see Fig. 3), were analyzed using frequency-domain fluorescence spectroscopy as described previously (14). Phase-Delay and Modulation-Ratio data points for each individual sample were fit to a double-exponential decay model. Addition of a third exponential decay component did not significantly improved the fit. The table summarizes the Intensity weighted (〈τ〉I) and Amplitude weighted (〈τ〉A) average fluorescence lifetimes derived from each individual best fit (in nanosecond time units), as well as the reduced chi-squared (χ2) goodness of fit test parameter.
| % PopDBpyFL in sample | 100 | 90 | 80 | 70 | 60 | 50 | 40 | 30 | 20 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Plus PopB | 〈τ〉I | 4.8 | 4.9 | 5.1 | 4.7 | 5.2 | 5.2 | 5.4 | 5.3 | 5.5 | 5.6 |
| 〈τ〉A | 3.2 | 3.2 | 3.6 | 3.2 | 3.9 | 4.0 | 4.5 | 4.7 | 5.1 | 5.3 | |
| χ2 | 5 | 5 | 5 | 5 | 3 | 3 | 2 | 1 | 1 | 1 | |
| Plus PopD | 〈τ〉I | 4.8 | 4.7 | 5.1 | 4.8 | 4.6 | 4.7 | 4.6 | 4.7 | 4.9 | 5.2 |
| 〈τ〉A | 3.3 | 3.2 | 3.6 | 3.3 | 3.2 | 3.4 | 3.4 | 3.7 | 4.3 | 4.9 | |
| χ2 | 3 | 3 | 5 | 3 | 2 | 2 | 2 | 1 | 1 | 1 | |
The interaction of PopD with PopB was explored using a similar approach. While keeping a constant amount ofPopDBpyFL, PopBWT was added in increasing amounts. Emission measurements revealed that PopDBpyFL associated with PopB as efficiently as with PopD, because the same final recovery of the fluorescent intensity was observed when the molar fraction of PopDBpyFL was decreased (Fig. 3B and Table 2). In contrast, very little decrease in homo-FRET (i.e. increase in fluorescence anisotropy) took place when the molar fraction of PopDBpyFL was reduced using PopBWT. These results suggested that PopB was able to intercalate between PopD subunits (as shown by the decrease in the BpyFL self-quenching), but the longer range homoFRET still occurred among subunits in a complex. Given that PopDBpyFL efficiently interacts with PopB, the lack of increase in anisotropy was indicative of the presence of at least two or more PopDBpyFL subunits per PopB/PopD hetero-complex. Taken together, these results showed that PopD assembles homo-oligomeric structures in membranes, but when added together with PopB it forms hetero-oligomeric structures of yet undetermined size and stoichiometry.
Individual Translocators Adopt Mostly Hexameric Arrangements When Assembled into Lipid Bilayers
To effectively determine the stoichiometry of membrane-inserted oligomers and the heterogeneity of the samples, we applied single-molecule fluorescence photobleaching. This technique allows for quantification of the number of protein molecules within a protein complex (19, 29). In practice, fluorescence detection with single-molecule efficiency allows visualization of single photobleaching events as a stepwise decrease in the fluorescence signal. The high sensitivity of this technique combined with the spatial resolution of optical microscopy imaging of single protein complexes, allowed us to quantify the number of photobleaching events occurring in a single membrane inserted oligomer (Fig. 4). The number of photobleaching events is equivalent to the number of fluorophores attached to a protein complex, or in the case of single-labeled polypeptides, to the number of proteins forming part of the complex. We adapted the technique to quantify proteins in their native lipid bilayer environment (i.e. in the absence of detergents) by using SLB (30).
FIGURE 4.

PopD and PopB adopt mostly hexameric arrangements when assembled individually in membranes. A, TIRF microscopy image of PopDBpyFL complexes in SLB. B, example of an intensity time trace obtained from a single membrane-assembled PopDBpyFL complex. Single-molecule photobleaching events were evidenced by the stepwise decrease in the fluorescence intensity. C, quantification of photobleaching counts obtained from 100 time traces of single PopDBpyFL complexes. D, TIRF microscopy image of PopBBpyFL complexes in SLB. E, example of an intensity time trace obtained from a single membrane-assembled PopBBpyFL complex. F, quantification of photobleaching counts obtained from 155 time traces of single PopBBpyFL complexes. Filled dots in C and F represent how much of the total protein represented in the figure was present in each of the bleaching steps groups.
PopBBpyFL or PopDBpyFL were reconstituted into liposomes using previously optimized conditions for membrane association and pore formation (14). The resulting proteoliposomes were diluted with excess liposomes to achieve the optimal low protein density required for single-particle imaging. Diluted samples were used to assemble SLB on glass coverslips by the vesicle fusion method (31). Bilayers were imaged using single-molecule TIRF microscopy as detailed under “Experimental Procedures.”
Assembly of a continuous and fluid lipid bilayer was confirmed in a parallel experiment by including a fluorescently labeled phospholipid in the membranes, NBD-PE, and observing fluorescence recovery after photobleaching of a membrane patch. Fluorescence recovery after photobleaching under our experimental conditions occurred in a time scale consistent with phospholipid diffusion in a fluid lipid bilayer (calculated 10−12-10−11 m2/s lateral diffusion coefficient for NBD-PE, data not shown) (32). In contrast to the freely diffusing lipid molecules, protein complexes remained immobile during the time scale of visualization before complete photobleaching of the attached probes (no significant drifting of the fluorescence complex was observed on membranes). This behavior has been observed for integral membrane proteins inserted into SLB, presumably due to interaction or absorption of the protein to the glass surface caused by protein segments facing the glass side of the membrane (33).
A quantitative analysis of photobleaching steps from 100 individual PopD oligomers and more than 150 individual PopB oligomers was employed to determine the stoichiometry of complexes formed by PopD (or PopB) when assembled into membranes. More than 70% of the total PopD translocators were found in complexes formed by 6 ± 1 subunits, indicating that PopDBpyFL assembled mostly hexameric structures when incubated alone with membranes (Fig. 4C). For PopB, a broader size distribution was observed dominated by hexameric and dodecameric oligomers (Fig. 4F).
PopB and PopD Assemble Hexadecameric Hetero-complexes in Membranes
To effectively determine the stoichiometry of hetero-oligomers we applied a technique that allows for quantification of the number of different protein molecules within a single protein complex (19). Dual-color fluorescence photobleaching was used to simultaneously determine the number of PopB and PopD molecules present in membrane-assembled oligomers. Each translocator was quantified based on the unique properties of the different fluorescence dyes covalently attached to them. PopDF223C was labeled at ∼100% efficiency with the red-emitting fluorescent probe BpyTMR, whereas PopBS164C was labeled with same efficiency using the green emitting fluorescent probe BpyFL. Control experiments showed that no labeling occurred on WT translocators (which lack Cys residues) when they were incubated with labeling reagents (14).
PopBBpyFl and PopDBpyTMR were mixed and reconstituted into liposomes, and SLB were prepared as described above. This technique permitted the analysis of hetero-complexes regardless of the presence of co-existing homo-oligomers of PopB and PopD, as only hetero-complexes display co-localization of green (PopB) and red (PopD) fluorescence (Fig. 5A, yellow spots).
FIGURE 5.

PopB and PopD assemble hexadecameric hetero-complexes in membranes. Protein complexes resulting from the equimolar addition of PopDBpyTMR (red emitting) and PopBBpyFL (green emitting) were imaged using dual color single-molecule TIRF microscopy on SLB. A, merge image of single particles containing PopDBpyTMR (red) and PopBBpyFL (green). Yellow spots indicate co-localization of PopDBpyTMR and PopBBpyFL in individual complexes. B, typical fluorescence intensity time traces for single protein complexes. Photobleaching events appear as stepwise decrease in the intensity. C, quantification of dual-color photobleaching counts obtained from 226 time traces of particles containing both PopDBpyTMR and PopBBpyFL. The heat map for observed PopD:PopB distributions is centered at 8 photobleaching counts for PopD and 8 photobleaching counts for PopB.
A quantitative analysis of the time-dependent photobleaching for more than 220 individual protein complexes showing yellow spots was used to determine the stoichiometry of oligomers formed by both PopD and PopB (Fig. 5B). Remarkably, we found that the distribution of PopB·PopD complexes centered at 8 photobleaching counts for PopDBpyTMR, as well as 8 photobleaching counts for PopBBpyFL (Fig. 5C), constituting an hexadecameric complex. This stoichiometry was independent of the total amount of protein distributed on the vesicles because identical results were obtained when the experiment was done using a 2-fold higher protein:lipid ratio (1/10,000, data not shown).
Individual PopD and PopB homo-oligomers were still observed under these experimental conditions (when the PopB:PopD ratio was 1/1), but it was clear that the proteins arranged into a different oligomeric structure when interacting with each other. The ability of the proteins to form both homo- and hetero-oligomers prompted us to analyze the reversibility of formed complexes. We therefore analyzed if the formation of PopD homo-oligomers was reversible after addition of PopB taking advantage of the PopDBpy self quenching properties. As showed in Fig. 3B, the emission of PopDBpy was low when forming homo-oligomers (Bpy self quenching), but increased when PopDBpy was pre-mixed with an excess of PopB before their addition to the membranes. However, if excess PopB was added after the PopDBpy homo-oligomers were formed, no change in the emission of Bpy was observed (Fig. 6A). This suggested that after assembling a homo-oligomer, the PopD subunits cannot dissociate and form a hetero-oligomer.
FIGURE 6.
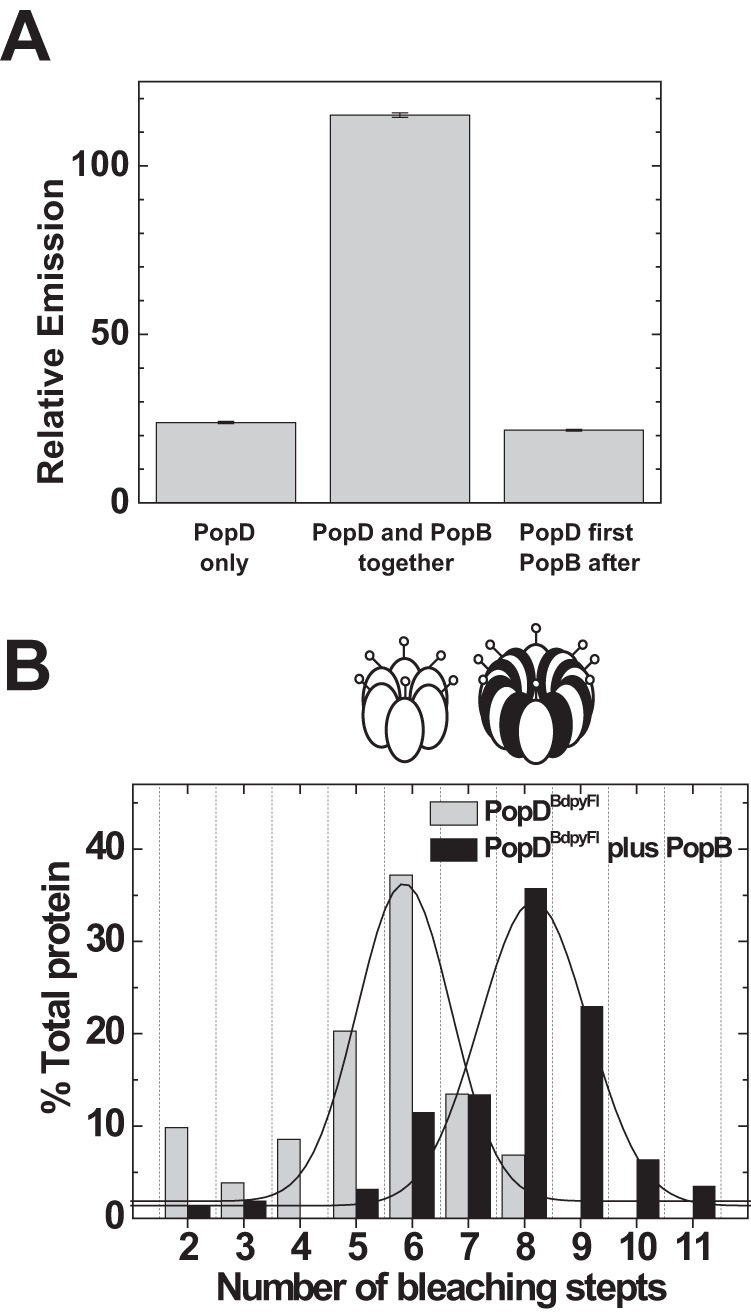
Simultaneous membrane interaction favored hetero-oligomerization over formation of translocator homo-oligomers. A, formation of the PopD homo-oligomer is not reversed by PopB addition to membranes. Left bar, PopDBpyFL was incubated with membranes and the final self-quenched emission determined (PopD only). Center bar, PopDBpyFL was mixed in 6 m urea buffer with a 10-fold excess of PopBWT and the mixture was added to membranes. Excess PopB favored the incorporation of PopD into hetero-oligomers, therefore it de-quenches the emission of the BpyFl probe (see also Fig. 3B). Right bar, PopDBpyFL was incubated with membranes to allow homo-oligomer formation and a 10-fold excess of PopB was subsequently added. No emission de-quenching was observed in this case, indicating that the simultaneous interaction with membranes of both translocators is necessary to form hetero-oligomers. A constant amount of PopDBpyFL (83% labeled with BpyFL) was present in every sample at a final concentration of 10 nm. The protein to lipid ratio was 1/6000 in all samples. Error bars indicate the range between two measurements. B, PopD size distribution shifts from 6 to 8 subunits per complex when membrane incubation is done in the presence of PopB. PopDBpyFL was incubated with membranes in the presence of 10-fold excess PopB whereas maintaining a constant protein:lipid ratio (black bars). Size distributions were obtained using single-molecule photobleaching and compared with the distribution obtained when PopDBpyFL was incubated alone with membranes (Fig. 4C, reproduced in gray bars). Lines indicate Gaussian fits to the histogram data. Oligomers are represented as coded in Fig. 3.
Because the assembly of PopD homo-oligomers is not reversible, how much of each complex would be formed in this model system is dictated by the relative rates of homo- versus hetero-oligomerization. By increasing the PopB/PopD ratio from 1:1 to 10:1 in the reconstitution assay, we were able to optimize the incorporation of PopD into hetero-complexes (Figs. 3B and 6B). Taken together, these data showed that although PopD is able to assemble stable hexamers on membranes, an early interaction event between PopB and PopD along the assembly pathway is required to secure the formation of a hexadecameric hetero-complex.
Discussion
Our spectroscopic analysis of reconstituted membrane protein complexes has provided important insights into the structural organization and assembly of the complex formed by the T3S translocators PopB and PopD. First, PopB facilitated the association of PopD with membranes. Second, in the absence of PopB, PopD formed mostly hexameric membrane complexes. Third, the interaction of PopB with PopD drove the formation of membrane-bound hetero-oligomer with a stoichiometry that differed from the one adopted by isolated proteins in homo-oligomers. Fourth, single molecule dual color photobleaching determinations showed that hetero-oligomers contained 8 PopB and 8 PopD subunits. Therefore, the interaction of PopB with PopD during translocon assembly was critical to chaperone the formation of membrane-inserted hetero-complexes.
Negatively charged lipids and acidic pH have been often used to optimize the interaction of pore-forming toxins and other proteins with membranes in vitro (34–37). In some cases, acidic pH can induce a molten globular state in proteins due to changes on charged amino acids. This relaxed form may facilitate membrane insertion (38). The exposure of hydrophobic surfaces by molten globular states at neutral pH favor protein aggregation in solution (as in the case of PopB and PopD (12, 13, 39)). However, the increase of the net positive charge of the protein at low pH would increase the electrostatic repulsion between proteins monomers and favor the electrostatic interaction with negatively charged membranes. It is therefore not surprising that the association of purified PopB and PopD (i.e. in the absence of the needle complex) to model membranes requires acidic pH and is facilitated by the presence of anionic lipids (13, 14).
The hydrophobic character of PopB and PopD and their propensity to aggregate in aqueous solutions demand a robust, reproducible, and controllable experimental system to study the structural arrangement of the T3S translocon. Unfolding-dependent membrane insertion is a reasonable approach because PopB and PopD are expected to be secreted unfolded through the narrow conduit of the T3S needle.
We therefore optimized the reconstitution of PopB and PopD membrane complexes by spontaneous refolding of urea-solubilized proteins in the presence of membranes (14). This folding-based reconstitution system allowed the study of individual WT translocators and to detect any difference in their properties that may result from the interaction between them. As shown in Fig. 2, when individually incubated with membranes, optimal binding for both PopB and PopD required pH lower than 5, however, PopB assisted the binding of PopD in a narrow pH range (i.e. pH 5–6).
The mechanistic details that govern the interaction between PopD and PopB are far from being understood. Mostly because no high resolution information is available on these proteins. However, the interaction between PopB and PopD was independently confirmed in this work using two spectroscopic properties of the fluorescent dye BpyFL, emission self-quenching, and energy migration. Using these complementary approaches, we found that PopB formed hetero-oligomers with PopDBpyFL (Fig. 3B), and these complexes contained several PopDBpyFL subunits even when a high PopB:PopDBpyFL ratio was used. These observations suggest that the formation of a hetero-dimer may re-direct the assembly and architecture of the TM complexes.
We therefore employed TIRF microscopy and single molecule photobleaching (30, 40, 41) to determine the stoichiometry of homo- and hetero-oligomers formed by PopB and PopD inserted in membranes. These experiments clearly showed that a unique hetero-oligomer is formed when PopB and PopD are added together. These data are in good agreement with results showing that both translocators are required for effector translocation, and that both translocators have to be secreted from the same bacterium for translocation to take place (11). When secreted separately, the encounter between PopB and PopD may take too long and homo-oligomerization dominates under these conditions.
Hetero-oligomers formed by PopB and PopD contained mostly 8 PopB and 8 PopD molecules (Fig. 5C), indicating that the presence of both translocators in a particle was not simply a random distribution of translocators or a co-localization of individual homo-oligomers (which are mostly hexameric, Fig. 4). It is worth noting that a translocon complex containing 8 PopD molecules and 8 PopB molecules will have a molecular mass of 570 kDa (or 601 kDa for a YopD8YopB8, see Fig. 1). Therefore, the molecular mass obtained using this model system is in good agreement with the molecular mass roughly estimated for Yersinia translocons isolated from erythrocyte membranes using blue native gel electrophoresis (600 ± 100 kDa) (42). Taken together, single molecule fluorescence and ensemble experiments suggest a model where the interaction of PopD with PopB, presumably a dimer formation, leads to the assembly of unique hetero-oligomeric structures with defined stoichiometry on lipid bilayers. We can speculate that the sequential secretion of PopB and PopD in the proximity of the target membrane would favor the formation of hetero-oligomers in the proximity of the needle tip. The tip may facilitate the PopB-PopD association and chaperone their insertion into the membrane (4, 43, 44). Unfortunately, the hierarchical mechanism of secretion for the translocators is far from being understood.
Some advances have been made on the assembly of the Shigella counterparts IpaD, IpaB, and IpaC (equivalents to PcrV, PopB, and PopD in P. aeruginosa, respectively (45–47)). However, as mentioned above given the poor sequence identity among the translocators from the Inv-Mxi-Spa and the Ysc families, it is risky to extrapolate results obtained from one family to the other (48). In particular, when these bacteria have evolved to invade different organs and to interact with different cell types. For example, in the Shigella system, binding of bile salts trigger IpaB association with the tip and secretion of IpaC. After secretion, the interaction of IpaC with membranes is required to complete the association of the membrane-associated translocator with IpaD/IpaB at the needle tip (49). In contrast, neither YopB nor PopB are found associated with the tip of the needle (50). Moreover, P. aeruginosa strains where PopD is absent cannot translocate effectors, but PopB is still found associated with the target membrane (11). It is therefore clear that the translocators evolved to adapt to different cell targets, and their functions may differ among different families.
Although homogeneous hetero-complexes were observed on membranes when PopB and PopD are added in equimolar amounts, a great number of homo-complexes were also observed (Fig. 5A). Are there any physiological role(s) for homo-oligomers? It has been claimed that in addition to the essential role in effector translocation, homo-oligomerization of IpaC (or SipC in Salmonella) is essential for events that occurs inside the cytoplasm of the target cell. For example, SipC initiates actin nucleation (51) and IpaC has the ability to induce membrane extensions on macrophages (52, 53). Moreover, IpaB induces Golgi fragmentation and reorganization of the recycling compartment (54) and oligomerization of IpaB form channels that permit potassium influx within endosomal compartments (55). Thus, it is possible that homo-oligomerization of PopD and PopB play other roles in the cytosol of the infected cell, but such abilities remain to be identified for the Ysc family members.
Finally, we would like to emphasize the unique properties of the T3S translocators in the context of protein membrane insertion and folding. Proteins that insert into membranes can be broadly classified in two major groups: (i) those that insert as unfolded polypeptides and require a complex proteinaceous machinery to properly fold and assemble into the lipid bilayer (56, 57), and (ii) those that spontaneous insert but are secreted as well folded proteins, as best exemplified by bacterial pore-forming toxins and complement system proteins (58–60). The T3S translocators do not belong to any of these groups. They are secreted through the needle in an unfolded conformation (61), they do not adopt a well defined folded structure in aqueous solution (39, 62), and no proteinaceous machinery has been identified for insertion and assembly into the target membrane. Therefore, the spontaneous assembly of unfolded proteins into large hetero-complexes with a defined stoichiometry constitutes a novel paradigm among membrane inserted proteins. As shown here, early interactions between PopB and PopD that occur during refolding, membrane binding, and/or insertion direct the spontaneous assembly of a hetero-complex of defined stoichiometry in a membrane bilayer.
Author Contributions
F. B. R. and A. P. H. designed the study; Y. T. performed experiments in Fig. 1 and analyzed Fig. 5C data. F. B. R. performed experiments in Figs. 2–6. K. C. R. performed experiments in Fig. 3; K. R. M. performed experiments in Fig. 6; F. B. R., K. C. R., Y. T., K. R. M., J. R., and A. P. H. analyzed data and wrote the paper.
Acknowledgments
We thank Dr. Leslie Conway and Dr. Daniel Diaz for assistance with single molecule instrumentation and analysis, Dr. Matthew Holden for assistance with the preparation of SLB, Dr. Arjan B. Vermeulen for help on the cloning of pcrHpopD into the PUC18 vector, Dr. Enrico Gratton and Dr. David Jameson for helpful discussion on fluorescence data analysis, and Dr. A. E. Johnson for critically reading the manuscript.
This work was supported, in whole or in part, by National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM097414 (to A. P. H.) and National Science Foundation Grant DMR-1207783 (to J. L. R.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of General Medical Sciences. The authors declare that they have no conflicts of interest with the contents of this article.
- T3S
- type 3 secretion
- TM
- transmembrane
- NBD-PE
- N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine
- SLB
- supported lipid bilayer
- TIRF
- total internal reflection fluorescence
- Bpy
- BODIPY.
References
- 1. Cornelis G. R. (2006) The type III secretion injectisome. Nat. Rev. Microbiol. 4, 811–825 [DOI] [PubMed] [Google Scholar]
- 2. Galán J. E., and Wolf-Watz H. (2006) Protein delivery into eukaryotic cells by type III secretion machines. Nature 444, 567–573 [DOI] [PubMed] [Google Scholar]
- 3. Marlovits T. C., Kubori T., Sukhan A., Thomas D. R., Galán J. E., and Unger V. M. (2004) Structural insights into the assembly of the type III secretion needle complex. Science 306, 1040–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mueller C. A., Broz P., and Cornelis G. R. (2008) The type III secretion system tip complex and translocon. Mol. Microbiol. 68, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 5. Schraidt O., and Marlovits T. C. (2011) Three-dimensional model of Salmonella's needle complex at subnanometer resolution. Science 331, 1192–1195 [DOI] [PubMed] [Google Scholar]
- 6. Matteï P.-J., Faudry E., Job V., Izoré T., Attree I., and Dessen A. (2011) Membrane targeting and pore formation by the type III secretion system translocon. FEBS J. 278, 414–426 [DOI] [PubMed] [Google Scholar]
- 7. Cornelis G. R. (2002) The Yersinia Ysc-Yop “Type III” weaponry. Nat. Rev. Mol. Cell Biol. 3, 742–752 [DOI] [PubMed] [Google Scholar]
- 8. Galán J. E. (2001) Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 17, 53–86 [DOI] [PubMed] [Google Scholar]
- 9. Moraes T. F., Spreter T., and Strynadka N. C. (2008) Piecing together the type III injectisome of bacterial pathogens. Curr. Opin. Struct. Biol. 18, 258–266 [DOI] [PubMed] [Google Scholar]
- 10. Frithz-Lindsten E., Holmström A., Jacobsson L., Soltani M., Olsson J., Rosqvist R., and Forsberg A. (1998) Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis. Mol. Microbiol. 29, 1155–1165 [DOI] [PubMed] [Google Scholar]
- 11. Goure J., Pastor A., Faudry E., Chabert J., Dessen A., and Attree I. (2004) The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 72, 4741–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schoehn G., Di Guilmi A. M., Lemaire D., Attree I., Weissenhorn W., and Dessen A. (2003) Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 22, 4957–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Faudry E., Vernier G., Neumann E., Forge V., and Attree I. (2006) Synergistic pore formation by type III toxin translocators of Pseudomonas aeruginosa. Biochemistry 45, 8117–8123 [DOI] [PubMed] [Google Scholar]
- 14. Romano F. B., Rossi K. C., Savva C. G., Holzenburg A., Clerico E. M., and Heuck A. P. (2011) Efficient isolation of Pseudomonas aeruginosa type III secretion translocators and assembly of heteromeric transmembrane pores in model membranes. Biochemistry 50, 7117–7131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaufman M. R., Jia J., Zeng L., Ha U., Chow M., and Jin S. (2000) Pseudomonas aeruginosa-mediated apoptosis requires the ADP-ribosylating activity of ExoS. Microbiology 146, 2531–2541 [DOI] [PubMed] [Google Scholar]
- 16. West S. E., Romero M. J., Regassa L. B., Zielinski N. A., and Welch R. A. (1995) Construction of Actinobacillus pleuropneumoniae-Escherichia coli shuttle vectors: expression of antibiotic-resistance genes. Gene 160, 81–86 [DOI] [PubMed] [Google Scholar]
- 17. Vallis A. J., Finck-Barbançon V., Yahr T. L., and Frank D. W. (1999) Biological effects of Pseudomonas aeruginosa type III-secreted proteins on CHO cells. Infect. Immun. 67, 2040–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 19. Ross J. L., and Dixit R. (2010) Multiple color single molecule TIRF imaging and tracking of MAPs and motors. Methods Cell Biol. 95, 521–542 [DOI] [PubMed] [Google Scholar]
- 20. Díaz-Valencia J. D., Morelli M. M., Bailey M., Zhang D., Sharp D. J., and Ross J. L. (2011) Drosophila Katanin-60 depolymerizes and severs at microtubule defects. Biophys. J. 100, 2440–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yeow E. K., and Clayton A. H. (2007) Enumeration of oligomerization states of membrane proteins in living cells by homo-FRET spectroscopy and microscopy: theory and application. Biophys. J. 92, 3098–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Runnels L. W., and Scarlata S. F. (1995) Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys. J. 69, 1569–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bergström F., Mikhalyov I., Hägglöf P., Wortmann R., Ny T., and Johansson L. B. (2002) Dimers of dipyrrometheneboron difluoride (BODIPY) with light spectroscopic applications in chemistry and biology. J. Am. Chem. Soc. 124, 196–204 [DOI] [PubMed] [Google Scholar]
- 24. Mikhalyov l., Gretskaya N., Bergström F., and Johansson L. B.-Å. (2002) Electronic ground and excited state properties of dipyrrometheneboron difluoride (BODIPY): dimers with application to biosciences. Phys. Chem. Chem. Phys. 4, 5663–5670 [Google Scholar]
- 25. Song X., and Swanson B. I. (2001) Rapid assay for avidin and biotin based on fluorescence quenching. Anal. Chim. Acta 442, 79–87 [Google Scholar]
- 26. Zhuang X., Ha T., Kim H. D., Centner T., Labeit S., and Chu S. (2000) Fluorescence quenching: a tool for single-molecule protein-folding study. Proc. Natl. Acad. Sci. U.S.A. 97, 14241–14244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karolin J., Johansson L. B.-A., Strandberg L., and Ny T. (1994) Fluorescence and absorption spectroscopic properties of dipyrrometheneboron difluoride (BODIPY) derivatives in liquids, lipid membranes, and proteins. J. Am. Chem. Soc. 116, 7801–7806 [Google Scholar]
- 28. Rosconi M. P., Zhao G., and London E. (2004) Analyzing topography of membrane-inserted diphtheria toxin T domain using BODIPY-streptavidin: at low pH, helices 8 and 9 form a transmembrane hairpin but helices 5–7 form stable nonclassical inserted segments on the cis side of the bilayer. Biochemistry 43, 9127–9139 [DOI] [PubMed] [Google Scholar]
- 29. Kaya S., Abe K., Taniguchi K., Yazawa M., Katoh T., Kikumoto M., Oiwa K., and Hayashi Y. (2003) Oligomeric structure of P-type ATPases observed by single molecule detection technique. Ann. N.Y. Acad. Sci. 986, 278–280 [DOI] [PubMed] [Google Scholar]
- 30. Groulx N., McGuire H., Laprade R., Schwartz J.-L., and Blunck R. (2011) Single molecule fluorescence study of the Bacillus thuringiensis toxin Cry1Aa reveals tetramerization. J. Biol. Chem. 286, 42274–42282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamm L. K., and McConnell H. M. (1985) Supported phospholipid bilayers. Biophys. J. 47, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gawrisch K. (2005) The dynamics of membrane lipids. in The Structure of Biological Membranes (Yeagle P. L., ed) 2nd Ed., pp. 147–171, CRC Press, Boca Raton, FL [Google Scholar]
- 33. Wagner M. L., and Tamm L. K. (2000) Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J. 79, 1400–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y., Malenbaum S. E., Kachel K., Zhan H., Collier R. J., and London E. (1997) Identification of shallow and deep membrane-penetrating forms of diphtheria toxin T domain that are regulated by protein concentration and bilayer width. J. Biol. Chem. 272, 25091–25098 [DOI] [PubMed] [Google Scholar]
- 35. Chenal A., Savarin P., Nizard P., Guillain F., Gillet D., and Forge V. (2002) Membrane protein insertion regulated by bringing electrostatic and hydrophobic interactions into play: a case study with the translocation domain of the diphtheria toxin. J. Biol. Chem. 277, 43425–43432 [DOI] [PubMed] [Google Scholar]
- 36. Thuduppathy G. R., Terrones O., Craig J. W., Basañez G., and Hill R. B. (2006) The N-terminal domain of Bcl-xL reversibly binds membranes in a pH-dependent manner. Biochemistry 45, 14533–14542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vargas-Uribe M., Rodnin M. V., and Ladokhin A. S. (2013) Comparison of membrane insertion pathways of the apoptotic regulator Bcl-xL and the diphtheria toxin translocation domain. Biochemistry 52, 7901–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Goot F. G., González-Mañas J. M., Lakey J. H., and Pattus F. (1991) A “molten-globule” membrane-insertion intermediate of the pore-forming domain of colicin A. Nature 354, 408–410 [DOI] [PubMed] [Google Scholar]
- 39. Faudry E., Job V., Dessen A., Attree I., and Forge V. (2007) Type III secretion system translocator has a molten globule conformation both in its free and chaperone-bound forms. FEBS J. 274, 3601–3610 [DOI] [PubMed] [Google Scholar]
- 40. Das S. K., Darshi M., Cheley S., Wallace M. I., and Bayley H. (2007) Membrane protein stoichiometry determined from the step-wise photobleaching of dye-labelled subunits. ChemBioChem 8, 994–999 [DOI] [PubMed] [Google Scholar]
- 41. Arant R. J., and Ulbrich M. H. (2014) Deciphering the subunit composition of multimeric proteins by counting photobleaching steps. ChemPhysChem 15, 600–605 [DOI] [PubMed] [Google Scholar]
- 42. Montagner C., Arquint C., and Cornelis G. R. (2011) Translocators YopB and YopD from Yersinia enterocolitica form a multimeric integral membrane complex in eukaryotic cell membranes. J. Bacteriol. 193, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goure J., Broz P., Attree O., Cornelis G. R., and Attree I. (2005) Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 192, 218–225 [DOI] [PubMed] [Google Scholar]
- 44. Picking W. L., Nishioka H., Hearn P. D., Baxter M. A., Harrington A. T., Blocker A., and Picking W. D. (2005) IpaD of Shigella flexneri is independently required for regulation of Ipa protein secretion and efficient insertion of IpaB and IpaC into host membranes. Infect. Immun. 73, 1432–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hume P. J., McGhie E. J., Hayward R. D., and Koronakis V. (2003) The purified Shigella IpaB and Salmonella SipB translocators share biochemical properties and membrane topology. Mol. Microbiol. 49, 425–439 [DOI] [PubMed] [Google Scholar]
- 46. Dickenson N. E., Arizmendi O., Patil M. K., Toth R. T. 4th, Middaugh C. R., Picking W. D., and Picking W. L. (2013) N-terminus of IpaB provides a potential anchor to the Shigella type III secretion system tip complex protein IpaD. Biochemistry 52, 8790–8799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adam P. R., Dickenson N. E., Greenwood J. C. 2nd, Picking W. L., and Picking W. D. (2014) Influence of oligomerization state on the structural properties of invasion plasmid antigen B from Shigella flexneri in the presence and absence of phospholipid membranes. Proteins 82, 3013–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veenendaal A. K., Hodgkinson J. L., Schwarzer L., Stabat D., Zenk S. F., and Blocker A. J. (2007) The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol. Microbiol. 63, 1719–1730 [DOI] [PubMed] [Google Scholar]
- 49. Epler C. R., Dickenson N. E., Olive A. J., Picking W. L., and Picking W. D. (2009) Liposomes recruit IpaC to the Shigella flexneri type III secretion apparatus needle as a final step in secretion induction. Infect. Immun. 77, 2754–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mueller C. A., Broz P., Müller S. A., Ringler P., Erne-Brand F., Sorg I., Kuhn M., Engel A., and Cornelis G. R. (2005) The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310, 674–676 [DOI] [PubMed] [Google Scholar]
- 51. Chang J., Myeni S. K., Lin T. L., Wu C. C., Staiger C. J., and Zhou D. (2007) SipC multimerization promotes actin nucleation and contributes to Salmonella-induced inflammation. Mol. Microbiol. 66, 1548–1556 [DOI] [PubMed] [Google Scholar]
- 52. Osiecki J. C., Barker J., Picking W. L., Serfis A. B., Berring E., Shah S., Harrington A., and Picking W. D. (2001) IpaC from Shigella and SipC from Salmonella possess similar biochemical properties but are functionally distinct. Mol. Microbiol. 42, 469–481 [DOI] [PubMed] [Google Scholar]
- 53. Mounier J., Popoff M. R., Enninga J., Frame M. C., Sansonetti P. J., and Van Nhieu G. T. (2009) The IpaC carboxy terminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 5, e1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mounier J., Boncompain G., Senerovic L., Lagache T., Chrétien F., Perez F., Kolbe M., Olivo-Marin J.-C., Sansonetti P. J., and Sauvonnet N. (2012) Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 12, 381–389 [DOI] [PubMed] [Google Scholar]
- 55. Senerovic L., Tsunoda S. P., Goosmann C., Brinkmann V., Zychlinsky A., Meissner F., and Kolbe M. (2012) Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 3, e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cymer F., von Heijne G., and White S. H. (2015) Mechanisms of integral membrane protein insertion and folding. J. Mol. Biol. 427, 999–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Selkrig J., Leyton D. L., Webb C. T., and Lithgow T. (2014) Assembly of β-barrel proteins into bacterial outer membranes. Biochim. Biophys. Acta 1843, 1542–1550 [DOI] [PubMed] [Google Scholar]
- 58. Dunstone M. A., and Tweten R. K. (2012) Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 22, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gilbert R. J., Dalla Serra M., Froelich C. J., Wallace M. I., and Anderluh G. (2014) Membrane pore formation at protein: lipid interfaces. Trends Biochem. Sci. 39, 510–516 [DOI] [PubMed] [Google Scholar]
- 60. Johnson B., and Heuck A. (2014) Perfringolysin O Structure and mechanism of pore formation as a paradigm for cholesterol-dependent cytolysins. in MACPF/CDC Proteins: Agents of Defence, Attack and Invasion (Anderluh G., and Gilbert R., eds) pp. 63–81, Springer, Netherlands: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Radics J., Königsmaier L., and Marlovits T. C. (2014) Structure of a pathogenic type 3 secretion system in action. Nat. Struct. Mol. Biol. 21, 82–87 [DOI] [PubMed] [Google Scholar]
- 62. Dey S., Basu A., and Datta S. (2012) Characterization of molten globule PopB in absence and presence of its chaperone PcrH. Protein J. 31, 401–416 [DOI] [PubMed] [Google Scholar]