

Abstract
A flexible linker (Lnk2) composed of 26 amino acids connects kringle-1 to kringle-2 in the coagulation factor prothrombin. Recent studies point to Lnk2 as a key determinant of the structure and function of this zymogen. Using a combination of mutagenesis, structural biology, and single molecule spectroscopy, we show how Lnk2 influences activation and conformational plasticity of prothrombin. Scrambling the sequence of Lnk2 is inconsequential on activation, and so is extension by as many as 22 residues. On the other hand, below a critical length of 15 residues, the rate of prothrombin activation increases (10-fold) in the absence of cofactor Va and decreases (3-fold) in the presence of cofactor. Furthermore, activation by prothrombinase takes place without preference along the prethrombin-2 (cleavage at Arg271 first) or meizothrombin (cleavage at Arg320 first) pathways. Notably, these transitions in the rate and pathway of activation require the presence of phospholipids, pointing to an important physiological role for Lnk2 when prothrombin is anchored to the membrane. Two new crystal structures of prothrombin lacking 22 (ProTΔ146–167) or 14 (ProTΔ154–167) residues of Lnk2 document striking conformational rearrangements of domains located across this linker. FRET measurements of freely diffusing single molecules prove that these structural transitions are genuine properties of the zymogen in solution. These findings support a molecular model of prothrombin activation where Lnk2 presents the sites of cleavage at Arg271 and Arg320 to factor Xa in different orientations by pivoting the C-terminal kringle-2/protease domain pair on the N-terminal Gla domain/kringle-1 pair anchored to the membrane.
Keywords: coagulation factor, enzyme mechanism, kinetics, protease, structure-function
Introduction
Prothrombin, or coagulation factor II, is a vitamin K-dependent zymogen abundantly present in the blood and comprising a Gla domain (residues 1–46), kringle-1 (residues 65–143), kringle-2 (residues 170–248), and the protease domain (residues 285–579). In the penultimate step of the coagulation cascade, prothrombin is converted to the mature protease thrombin by the prothrombinase complex composed of factor Xa, cofactor Va, Ca2+, and phospholipids (1). Activation is enhanced >2,000-fold by cofactor Va and phospholipids because of increased kcat (1–4), and a significant portion of this effect comes from optimization of the conformation of factor Xa induced by cofactor Va (5, 6). Conversion to thrombin involves cleavage at Arg271 and Arg320 along two alternative pathways, generating prethrombin-2 and meizothrombin, respectively. On synthetic membranes and in the absence of cofactor Va, prothrombin activation proceeds along the prethrombin-2 pathway (1). In the presence of cofactor Va, selection of the pathway is context-dependent. On the surface of platelets (7, 8), prothrombinase activates prothrombin along the prethrombin-2 pathway (8). On the membrane of red blood cells (9) and other prothrombotic surfaces enriched with phosphatydylserine such as circulating microparticles (10) and synthetic liposomes (1, 11–13), prothrombinase activates prothrombin along the alternative meizothrombin pathway. How the pathway is selected at the molecular level remains an intriguing and hotly debated issue (12, 14–17), although its relevance to blood physiology ultimately depends on whether meizothrombin or prethrombin-2 accumulate during prothrombin activation (2, 8, 15).
Notwithstanding decades of investigation, our understanding of the factors that control the rate and pathway of prothrombin activation is largely phenomenological rather than mechanistic and structure-based. For example, perturbations of the Gla domain affect the rate and switch the pathway of activation from meizothrombin to prethrombin-2 (3, 12, 18), but exactly how this domain anchored to the membrane communicates with the sites of cleavage at Arg271 and Arg320 located >80 Å away (19) has not been elucidated. Active site occupancy of prothrombin also switches the pathway of activation from meizothrombin to prethrombin-2 (20, 21), but no structural explanation has been offered for this effect. Previous studies have dissected the role of the Gla domain (3, 12, 18, 22), kringles (23–25), and protease domain (26–28) and have contributed to the formulation of numerous models of the prothrombinase-prothrombin complex (29–31). Unfortunately, these models have not benefited from recent information on the structure of prothrombin and its unanticipated conformational plasticity (19, 32).
Three flexible linkers connect the Gla domain to kringle-1 (Lnk1), the two kringles (Lnk2), and kringle-2 to the protease domain (Lnk3). Among these linkers, Lnk2 (residues 144–169) is unique insofar as it shares exon VII with kringle-2 in the longest coding region of the prothrombin gene (33) and, unlike Lnk1 and Lnk3, connects two adjacent domains of the zymogen not engaged in any intramolecular interaction. This feature presages a key role for Lnk2 in dictating the overall architecture of prothrombin. Movement of Lnk2 could trigger long range perturbation of the conformation of the zymogen, far more pronounced than that produced by changes in Lnk1 and Lnk3 connecting domains in intramolecular contact. Lnk2 is long and nonhelical (34) and should enable different arrangements of the N-terminal Gla domain/kringle-1 pair relative to the C-terminal kringle-2/protease domain pair (32). Indeed, it is easy to envision how prothrombin, anchored to the membrane via its Gla domain, may exploit the flexibility of Lnk2 to position the sites of cleavage at Arg271 in Lnk3 and Arg320 in the protease domain at different heights over the plane of the membrane for presentation to the active site of factor Xa in different order and orientation (see Fig. 6). Through this action, Lnk2 may contribute significantly to the rate and pathway of prothrombin activation.
FIGURE 6.

Conformational flexibility of Lnk2 revealed by x-ray crystallography. A–C, crystal structure of ProTΔ146–167 (A) reported previously (19), along with the new structures presented in this study for ProTΔ154–167 (B) and ProTΔ146–167 in a new conformation (C). The three structures are aligned over the rigid kringle-2/serine protease pair and then visualized separately in the same orientation. Individual domains are labeled and colored as follows: Gla domain (Gla, blue), kringle-1 (K1, red), kringle-2 (K2, green), A chain (Ac, orange), and B chain (Bc, yellow). Linkers are colored in wheat. The flexibility of Lnk2, containing as little as 4 (ProTΔ146–167) or 12 (ProTΔ154–167) residues, causes the Gla domain/kringle-1 pair to move up to 40 Å (B versus A) or to rotate 180° (C versus A) relative to the rigid kringle-2/serine protease domain pair. Differences in the conformation of the Gla domain among the three structures are due to crystallization conditions (Ca2+ or Mg2+), and details are given in Fig. 8 (D–F). D–F, the role of Lnk2 in orienting the two sites of cleavage at Arg271 (not visible and approximated by Thr274 in blue) and Arg320 (red) is readily appreciated when the three structures are docked on a phospholipid surface, with the Gla domain binding almost perpendicularly to the plane and inserting the hydrophobic ω-loop as proposed by Furie and co-workers (57) and Tajkhorshid and co-workers (58). The distance of each site of cleavage from the plane of the membrane is indicated, along with the position of the active site of factor Xa predicted by previous FRET measurements (59–61).
The importance of Lnk2 is clearly established by previous studies (19, 32), but exactly how this domain contributes to prothrombin function has not been elucidated. Here we investigate the role of Lnk2 with deletions, insertions, and scrambling of its sequence. We also present two new structures of prothrombin where Lnk2 lacks 22 (ProTΔ146–167) or 14 (ProTΔ154–167) of its 26 residues. These studies are complemented by measurements of prothrombin conformation in solution at the single molecule level. The combination of these approaches reveals how Lnk2 influences activation and conformational plasticity of prothrombin.
Materials and Methods
Reagents
Prothrombin cDNA wild type (residues 1–579) modified to include an epitope for the HPC4 antibody at the C-terminal was cloned into a pDEST40 expression vector using the Gateway® cloning technology (Life Technologies, Inc.). The constructs ProTΔ145–168, ProTΔ146–167, ProTΔ146–156, ProTΔ154–167, ProTΔ154–159, ProTins11, ProTins22, and ProTscr146–167 carrying deletions, insertions, and scrambling of various portions of Lnk2 (see Fig. 1), and mutants R320A and R271A in the wild type and ProTΔ146–167 backgrounds were generated using the QuikChange Lighting kit (Agilent, Anta Clara, CA) and appropriate primers (Integrated DNA Technologies, Coralville, IA). After sequencing, the proteins were expressed in baby hamster kidney cells and purified by affinity chromatography, ion exchange chromatography, and size exclusion chromatography as described previously (32, 35, 36). SDS-PAGE and N-terminal sequencing verified homogeneity and chemical identity of final preparations. The level of γ-carboxylation was determined by alkaline hydrolysis coupled to amino acid analysis (37, 38). Small unilamellar vesicles composed of phosphatidylcholine and phosphatidylserine in a 3:1 molar ratio were prepared by extrusion using 0.05- or 0.1-μm polycarbonate membranes (Avanti Polar Lipids, Alabaster, AL), and their size was confirmed by DLS. Human purified factor Va, factor Xa, and the thrombin specific inhibitor dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide were purchased from Hematologic Technologies (Essex Junction, VT). Protein concentrations were determined by reading at 280 nm with molar extinction coefficients adjusted based on the amino acid sequence. All other chemicals were purchased from Sigma-Aldrich.
FIGURE 1.

Conservation of Lnk2 and prothrombin constructs. In human prothrombin, Lnk2 is composed of 26 residues, from 144 to 169, and connects kringle-1 (residues 65–143) to kringle-2 (residues 170–248). The site of cleavage for thrombin and factor Xa, Arg155, defines the midpoint of the linker and the partitioning between proximal and distal portions. The result of the sequence alignment is shown as a sequence logo (WebLogo). Residues with frequencies >2 are considered significant and marked with an asterisk. The table lists the sequence and length for the prothrombin constructs of Lnk2 characterized in this study. Deletions of residues 145–168 in ProTΔ145–168 and residues 146–167 in ProTΔ146–167 remove 24 and 22 of the total 26 residues of Lnk2. ProTΔ154–167 lacks the distal portion of Lnk2 and retains most of the proximal one (red). ProTΔ146–156 lacks the proximal portion and retains most of the distal one (green). ProTΔ154–159 eliminates region of Lnk2 around Arg155. Scrambling the entire sequence 146–167 generates ProTscr146–167. The N-terminal half of the scrambled sequence of ProTscr146–167 (purple) was inserted in ProTins11 downstream of Leu167. The entire scrambled sequence of ProTscr146–167 (purple and yellow) was inserted in ProTins22 downstream of Leu167.
Activation of Prothrombin
Prothrombin wild type and mutants were buffer-exchanged into 150 mm NaCl, 20 mm Tris, and 5 mm CaCl2 using a Zeba 7K MWCO desalting column. Prothrombin activation was monitored in two different ways, i.e. using a standard discontinuous assay (12, 16) and a continuous assay analogous to the one developed for the analysis of protein C activation (39, 40). In the discontinuous assay, prothrombin (0–3 μm) was activated by the addition of factor Xa (0.1–5 nm) and phospholipids (20 μm) or factor Xa (10 pm), phospholipids (20 μm), and cofactor Va (10 nm). Aliquots were quenched after 0.5, 1, 2, and 5 min, and active site generation was quantified in terms of the hydrolysis of the chromogenic substrate H-d-Phe-Pro-Arg-p-nitroanilide (FPR) specific for thrombin and meizothrombin. Initial velocities were converted to concentrations using a standard reference curve measured at the time of the experiment. The independent Michaelis-Menten parameters kcat and kcat/Km were obtained by nonlinear fit of the experimental data collected in a SpectraMax i3x multimode detection platform using the software Origin 2015 (OriginLab Corporation, Northampton, MA). In the continuous assay, the real time activation of prothrombin to thrombin was monitored by progress curves of substrate hydrolysis after slight modification of the method developed for the analysis of protein C activation by thrombin (39, 40). Unlike the discontinuous assay, the continuous assay based on progress curves better captures the kinetics of activation, can be analyzed rigorously by integration of the underlying kinetic expressions, and requires small amounts of reagents. The values of kcat/Km or kcat are derived by using prothrombin concentrations below (see Fig. 2A) or above (see Fig. 2, B and C) Km. This feature is particularly useful in the case of reactions that affect mainly kcat, as observed for the effect of cofactor Va on prothrombin activation (Fig. 2, B–E), or when Km is very large and prevents saturation of the enzyme with substrate, as observed when prothrombin activation is studied in the presence of factor Xa only (Fig. 2A). Prothrombin (0.5–1.4 μm) activation was measured in the presence of 20 nm factor Xa (Fig. 2A), 0.1 nm factor Xa and 20 μm phospholipids (Fig. 2B), or 1 pm factor Xa, 20 μm phospholipids, and 10 nm cofactor Va (Fig. 2C), using 24 μm chromogenic substrate H-d-Phe-Pro-Phe-p-nitroanilide (FPF). Factor Xa has no appreciable activity toward FPF, but thrombin and meizothrombin cleave with kcat/Km = 0.13 μm−1 s−1 (41). The lack of product inhibition was the reason for choosing FPF in this assay over the analogous chromogenic substrate FPR. The data were collected on a SpectraMax i3x multimode detection platform and analyzed with Origin 2015 (OriginLab Corporation).
FIGURE 2.

Effect of perturbation of Lnk2 on the rate of prothrombin activation. The various constructs of Lnk2 presented in the figure are listed in the scheme (bottom left). Other constructs listed in Table 2 are not included in the figure for clarity of presentation, but the best fit results are shown below for the continuous assay and in Table 2 for the discontinuous assay. All experiments were carried out in triplicate. A–C, continuous assay of prothrombin activation started by addition of 20 nm factor Xa to 500 μm prothrombin (A); 0.1 nm factor Xa and 20 μm phospholipids to 1 μm prothrombin (B); or 1 pm factor Xa, 20 μm phospholipids, and 10 nm cofactor Va (C) to 1 μm prothrombin. The release of p-nitroaniline upon hydrolysis of FPF was monitored at 405 nm. Analysis of the progress curves yields the value of kcat/Km for the activation of prothrombin when [ProT] ≪ Km (A) and the value of kcat when [ProT] ≫ Km (B and C). The best fit values for kcat/Km in A are 440 ± 10 m−1 s−1 (ProTΔ145–168), 440 ± 10 m−1 s−1 (ProTΔ146–167), 500 ± 20 m−1 s−1 (ProTΔ154–167), 500 ± 10 m−1 s−1 (ProTΔ146–156), 430 ± 10 m−1 s−1 (ProTΔ154–159), 330 ± 10 m−1 s−1 (wild type), 320 ± 10 m−1 s−1 (ProTscr146–167), 300 ± 10 m−1 s−1 (ProTins11), and 300 ± 10 m−1 s−1 (ProTins22). The best fit values for kcat in B are 0.17 ± 0.02 s−1 (ProTΔ145–168), 0.19 ± 0.01 s−1 (ProTΔ146–167), 0.16 ± 0.01 s−1 (ProTΔ154–167), 0.090 ± 0.005 s−1 (ProTΔ146–156), 0.042 ± 0.001 s−1 (ProTΔ154–159), 0.027 ± 0.002 s−1 (wild type), 0.031 ± 0.002 s−1 (ProTscr146–167), 0.025 ± 0.001 s−1 (ProTins11), and 0.019 ± 0.002 s−1 (ProTins22). The best fit values for kcat in C are 8.6 ± 0.2 s−1 (ProTΔ145–168), 8.6 ± 0.3 s−1 (ProTΔ146–167), 11 ± 1 s−1 (ProTΔ154–167), 21 ± 1 s−1 (ProTΔ146–156), 32 ± 1 s−1 (ProTΔ154–159), 33 ± 1 s−1 (wild type), 32 ± 1 s−1 (ProTscr146–167), 36 ± 1 s−1 (ProTins11), and 35 ± 1 s−1 (ProTins22). D and E, discontinuous assay of prothrombin activation in the absence (D) or presence (E) of cofactor Va. Experimental data obey Michaelis-Menten kinetics with best fit parameter values reported in Table 2. Activity is expressed in s−1 to facilitate direct comparison with the results from the continuous assay in B and C. Experimental conditions are 150 mm NaCl, 5 mm CaCl2, 0.1% PEG 8000, 20 mm Tris, pH 7.4, at 25 °C.
Electrophoretic Assays
The pathway of prothrombin activation was monitored by SDS-PAGE. Activation in the presence of 0.2 nm factor Xa, 20 μm phospholipids, and 30 nm cofactor Va was carried out with 1.4 μm prothrombin dissolved in 150 mm NaCl, 20 mm Tris, 5 mm CaCl2 and incubated at 25 °C for 5 min in the presence 60 μm dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide, 20 μm phospholipids, and 10 nm cofactor Va. Reactions involving all prothrombin constructs were performed at the same time to facilitate comparison. Following addition of factor Xa, samples (40 μl) were quenched at different time intervals with 10 μl of NuPAGE LDS buffer containing β-mercaptoethanol as the reducing agent and 20 mm EDTA. The samples were loaded into 12% SDS-polyacrylamide gels or processed by NuPAGE Novex 4–12% Bis-Tris protein gels run with MES buffer. All gels were stained simultaneously with Coomassie Brilliant Blue R-250 and analyzed by quantitative densitometry.
Activated Partial Thromboplastin Time
aPTT2 was measured using the TriniCLOT aPTT kit on a ST4 semiautomated coagulometer (Diagnostica Stago, Gennevilliers, France). Briefly, 50 μl of citrated prothrombin deficient plasma (Hematologic Technologies) was mixed with 50 μl of the desired prothrombin construct (0.1 mg/ml) and 50 μl of reagent S in the appropriate cuvettes. The reaction was started by adding 50 μl of 20 mm CaCl2.
X-ray Studies
Crystallization of the prothrombin mutants ProTΔ146–167 and ProTΔ154–167 lacking 22 or 14 residues of Lnk2, respectively, was achieved at 20 °C by the vapor diffusion technique using an Art Robbins Instruments Phoenix liquid handling robot and mixing equal volumes (0.3 μl) of protein and reservoir solution (Table 1). Optimization of crystal growth was achieved by the hanging drop vapor diffusion method mixing 3 μl of protein (10 mg/ml) with equal volumes of reservoir solution. Crystals of ProTΔ146–167 were grown in less than 2 weeks in the presence of 10 mg/ml protein, 50 mm MgSO4, 0.2 m Na2HPO4, 20% PEG 3350. Crystals of ProTΔ154–167 were also grown in less than 2 weeks in the presence of 10 mg/ml protein, 0.1 m MES, 1.6 m MgSO4,pH 6.5. In both cases, crystals were cryoprotected prior to flash freezing in a solution of 25% glycerol from the original mother liquor. X-ray diffraction data were collected with a home source (Rigaku 1.2 kW MMX007 generator with VHF optics) Rigaku Raxis IV++ detector and were indexed, integrated, and scaled with the HKL2000 software package (42). Initially, the previous structure of Ca2+-free ProTΔ146–167 (19) solved at 2.8 Å resolution (Protein Data Bank code 4NZQ) was used as search model using PHASER from the CCP4 suite (43), but no solution was found because of a different orientation of fragment 1 in the new structures. Then the previous structure of Ca2+-bound ProTΔ146–167 (19) solved at 3.4 Å resolution (Protein Data Bank code 4O03) was partitioned into fragment 1 (1–145) and prethrombin-1 (168–579), and the two separate portions were used as individual search models. Refinement and electron density generation were performed with REFMAC5 from the CCP4 suite, and 5% of the reflections were randomly selected as a test set for cross-validation. Model building and analysis of the structures were conducted with COOT (44). Both structures were subject to a final round of refinement with PDB_REDO (45). TLS tensors modeling rigid body anisotropic temperature factors were calculated and applied to the structure of ProTΔ154–167. Ramachandran plots were calculated using PROCHECK (46). Statistics for data collection and refinement are summarized in Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 5EDK for the new structure of ProTΔ146–167 and 5EDM for ProTΔ154–167).
TABLE 1.
Crystallographic data for prothrombin mutants ProTΔ146–167 and ProTΔ154–167
| ProTΔ146–167 | ProTΔ154–167 | |
|---|---|---|
| Buffer | 200 mm Na2HPO4, 50 mm MgSO4 | 100 mm MES, 1.6 m MgSO4, pH 6.5 |
| PEG | 3350 (20%) | |
| Protein Data Bank code | 5EDK | 5EDM |
| Data collection | ||
| Wavelength (Å) | 1.54 | 1.54 |
| Space group | P41212 | C2221 |
| Unit cell dimensions (Å) | a = 84.2 | a = 109.9 |
| b = 84.2 | b = 168.7 | |
| c = 346.4 | c = 144.3 | |
| Molecules/asymmetric unit | 1 | 1 |
| Resolution range (Å) | 40–3.2 | 40–2.2 |
| Observations | 89,160 | 486,501 |
| Unique observations | 20,053 | 67,055 |
| Completeness (%) | 94.5 (94.0) | 98.0 (97.8) |
| Rsym (%) | 15.5 (44.1) | 8.7 (48.7) |
| I/σ(I) | 7.0 (2.0) | 17.0 (3.0) |
| Refinement | ||
| Resolution (Å) | 40–3.2 | 40–2.2 |
| Rcryst, Rfree | 0.29, 0.32 | 0.20, 0.24 |
| Reflections (working/test) | 18,675/1,009 | 63,362/3,409 |
| Protein atoms | 4,242 | 4,414 |
| Mg2+ ions | 4 | 6 |
| Solvent molecules | 464 | |
| RMSD bond lengths (Å)a | 0.006 | 0.012 |
| RMSD angles (°)a | 1.2 | 1.7 |
| RMSD ΔB (Å2) (mm/ms/ss)b | 3.07/2.39/2.31 | 2.66/2.98/3.29 |
| <B> protein (Å2) | 87.2 | 58.4 |
| <B> Mg2+ ions (Å2) | 59.6 | 57.7 |
| <B> solvent (Å2) | 57.6 | |
| Ramachandran plot | ||
| Most favored (%) | 96.3 | 99.4 |
| Generously allowed (%) | 2.4 | 0.2 |
| Disallowed (%) | 1.3 | 0.4 |
a RMSD from ideal bond lengths and angles and RMSD in B-factors of bonded atoms.
b mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
Single Molecule Measurements
Guided by structural information, residues Ser210 in kringle-2 and Ser101 in kringle-1 were mutated to Cys to generate the double mutant S101C/S210C in the ProTΔ146–167 background. Selective reaction of the unpaired cysteines was achieved by reducing the protein (12–14 μm) in 20 mm Tris, 350 mm NaCl, pH 7.4, at room temperature for 1 h in the dark in the presence of DTT at a molar ratio [SH]:[DTT] = 1:1.4. After removal of excess DTT with G-25 desalting spin column, the solution sat for 1 h at room temperature in the dark before addition of 5-molar excess of Alexa Fluor 555 and Alexa Fluor 647 maleimide dyes (Molecular Probes). The labeling reaction was carried out for 2 h at room temperature. The monomeric protein free of unreacted dyes was purified on an analytical Superdex 200 column, and the efficiency of derivatization (70–90%) was assessed by UV-visible measurements. No incorporation of the fluorescent dyes was observed in the wild type protein under the same conditions.
FRET measurements of freely diffusing single molecules were performed with a confocal microscope MicroTime 200 (PicoQuant, Berlin, Germany). The donor and acceptor dyes were excited with a ps pulsed diode laser at 532 and 638 nm, respectively. To achieve pulsed interleaved excitation (47), the 532-nm laser was electronically delayed 25 ns relative to the 638-nm laser (48, 49). A dual band dichroic mirror reflecting 532 and 638 nm guided the light to a high numerical aperture apochromatic objective (60×, N.A. 1.2, water immersion; Olympus) that focused the light to a confocal volume of 1.0 fl for excitation at 532 nm and detection at 575 nm. Fluorescence from excited molecules was collected with the same objective and focused onto a 50-μm diameter pinhole. The donor and acceptor emission were separated via a dichroic long pass filter with a dividing edge at 620 nm. Suited bandpass filters were inserted to eliminate the respective excitation wavelength and minimize spectral cross-talk. The fluorescence was detected with two avalanche photodiodes using time-correlated single photon counting with the TimeHarp 200 board. The data were stored in the time-tagged time resolved mode. Measurements were performed 25 μm deep in the solution with a total acquisition time of 1 h and repeated fresh up to four times on each protein sample (50 pm) in 20 mm Tris, 145 mm NaCl, 5 mm CaCl2 0.01% Tween 20, pH 7.4. Signals from single molecules were observed as bursts of fluorescence. Appropriate correction for direct excitation of the acceptor at the donor excitation wavelength, leakage of the donor in the acceptor channel, and the instrumental γ factor was calculated using a mixture of double stranded DNA models with known FRET efficiency (E) and stoichiometry (S) labeled with dyes AF555 and AF647 (50). Integration time was set to 0.5 ms, and bursts with more than 35 counts were included in the analysis. Only molecules with a stoichiometry in the range 0.25–0.75 were considered in the final analysis, and their distribution was fit to Gaussian curves using Origin 2015 (OriginLab Corporation). The number of independent Gaussians was determined according to the corrected Akaike information criterion. Data recording and initial data analysis was performed using the SymphoTime Software 6.4 (PicoQuant, Berlin). Further analysis was carried out with Matlab routines kindly provided by Dr. Don C. Lamb (Munich, Germany).
Results
Lnk2 in human prothrombin is composed of 26 amino acids and is particularly rich in Gln, Pro, Ser, and Val residues (Fig. 1) that favor nonhelical and flexible conformations, consistent with recent x-ray studies (32). Sequence alignment among 29 different species reveals high variability in length, from 20 residues in the fugu to 32 residues in dogs. Amino acid composition varies widely among species and only a handful of conserved residues cluster in the proximal portion of Lnk2 (residues 144–154), upstream of the site of cleavage at Arg155 targeted by thrombin and factor Xa in the absence of cofactor Va (19, 51). The distal portion (residues 156–169) contains a single conserved residue, Pro164. The only documented naturally occurring mutation of Lnk2 is the E157K substitution in prothrombin Canberra associated with mild bleeding (52). Taken together, these observations indicate that a viable Lnk2 likely requires a length of 20–30 amino acids but little conservation of sequence. We therefore investigated the role of sequence and length of Lnk2 with substitutions, deletions, and insertions.
To establish the role of any conserved residue within Lnk2, we scrambled the entire sequence 146–167 containing the site of cleavage at Arg155. The scrambled construct ProTscr146–167 shows no appreciable perturbation of the rate of prothrombin activation by factor Xa in the presence of Ca2+ and phospholipids, with or without cofactor Va (Table 2 and Fig. 2, B–E). The specific sequence of residues 146–167 of Lnk2 is largely inconsequential on function. This observation should be considered in the context of our recent analysis of the prothrombin construct ProTΔ146–167 carrying a deletion of residues 146–167 in Lnk2 (19). Notable for ProTΔ146–167 is the 10-fold increase in the rate of activation in the absence of cofactor Va and the decrease (3-fold) in the rate of activation in the presence of cofactor (Table 2). Deletion of a smaller portion of Lnk2, i.e. residues 154–159 in the ProTΔ154–159 construct, was also reported previously (19) and has no significant effect on the rates of activation by factor Xa in the presence of Ca2+ and phospholipids, with or without cofactor Va (Table 2). Given the drastic functional differences between ProTΔ146–167 and ProTΔ154–159, we constructed selective deletions of the proximal (residues 146–156) and distal (residues 154–167) portions of Lnk2 encompassing the site of cleavage at Arg155. Notwithstanding the different degree of conservation between the two portions, much higher for the proximal region (Fig. 1), the two constructs ProTΔ146–156 and ProTΔ154–167 feature rates of activation by factor Xa in the presence of Ca2+ and phospholipids, with or without cofactor Va, that scale with the length of the linker (Table 2 and Fig. 2, B--E). The observed rates are intermediate between those of wild type and ProTΔ146–167. Removal of 24 of the 26 residues of Lnk2 in the construct ProTΔ145–168 produces functional properties very similar to those of ProTΔ146–167 (Table 2 and Fig. 2, B–E). On the other hand, when the length of Lnk2 is increased by partial or full addition of the scrambled sequence of ProTscr146–167 into ProTins11 and ProTins22, the functional properties are practically identical to wild type (Table 2 and Fig. 2, B–E).
TABLE 2.
Kinetic parameters for activation of prothrombin wild type and mutants of Lnk2
Experimental conditions are: 20 μm phospholipids, 150 mm NaCl, 5 mm CaCl2, 0.1% PEG 8000, 20 mm Tris, pH 7.4, at 25 °C. The concentration of factor Xa in the absence of cofactor Va is 0.1–7.5 nmm, depending on the prothrombin construct used, and 10 pm in the presence of cofactor.
| − Cofactor Va |
+ Cofactor Va |
La | |||||
|---|---|---|---|---|---|---|---|
| kcat/Km | kcat | Km | kcat/Km | kcat | Km | ||
| μm−1 s−1 | s−1 | μm | μm−1 s−1 | s−1 | μm | ||
| WT | 0.12 ± 0.01 | 0.016 ± 0.003 | 0.13 ± 0.02 | 270 ± 20 | 32 ± 3 | 0.12 ± 0.01 | 26 |
| ProTΔ145–168 | 0.92 ± 0.08 | 0.11 ± 0.01 | 0.12 ± 0.01 | 100 ± 10 | 11 ± 1 | 0.11 ± 0.01 | 2 |
| ProTΔ146–167 | 1.2 ± 0.1 | 0.13 ± 0.02 | 0.11 ± 0.01 | 110 ± 10 | 11 ± 1 | 0.10 ± 0.01 | 4 |
| ProTΔ154–167 | 0.63 ± 0.06 | 0.095 ± 0.005 | 0.15 ± 0.02 | 120 ± 10 | 10 ± 1 | 0.085 ± 0.008 | 12 |
| ProTΔ146–156 | 0.29 ± 0.02 | 0.041 ± 0.005 | 0.14 ± 0.02 | 190 ± 10 | 21 ± 2 | 0.11 ± 0.01 | 16 |
| ProTΔ154–159 | 0.13 ± 0.01 | 0.018 ± 0.002 | 0.14 ± 0.01 | 270 ± 20 | 25 ± 2 | 0.091 ± 0.008 | 20 |
| ProTscr146–167 | 0.16 ± 0.01 | 0.016 ± 0.003 | 0.10 ± 0.02 | 240 ± 20 | 26 ± 2 | 0.11 ± 0.01 | 26 |
| ProTins11 | 0.12 ± 0.01 | 0.015 ± 0.003 | 0.13 ± 0.02 | 320 ± 20 | 32 ± 2 | 0.10 ± 0.01 | 37 |
| ProTins22 | 0.10 ± 0.01 | 0.011 ± 0.002 | 0.11 ± 0.01 | 280 ± 20 | 31 ± 2 | 0.11 ± 0.01 | 48 |
a Length of the linker (in amino acid residues) connecting the two kringles.
Altogether, these findings indicate that the length of Lnk2 and not its composition influences the rate of prothrombin activation (Fig. 3). As the length of Lnk2 decreases, the value of kcat/Km for prothrombin activation increases up to 10-fold in the absence of cofactor Va and decreases up to 3-fold in the presence of cofactor (Fig. 3). The dependence is sigmoidal in both cases, with a transition centered on a critical length of 15 residues. Interestingly, the documented range of 20–32 residues for Lnk2 found in 29 different species falls in an asymptotic region of the profiles. The length of Lnk2 was optimized during evolution to maximize the effect of cofactor Va on prothrombin activation and the variability in length of Lnk2 observed among species occurs in a range where perturbation of function is minimal. Notably, the dependence of the rate of activation on the length of Lnk2 (Fig. 3) is observed in the presence of phospholipids (Fig. 2, B–E) but not when prothrombin is free in solution (Fig. 2A). This result is consistent with previous mutagenesis studies on prothrombin derivatives lacking the entire fragment 1 comprising residues 1–155 (3, 24, 53).
FIGURE 3.
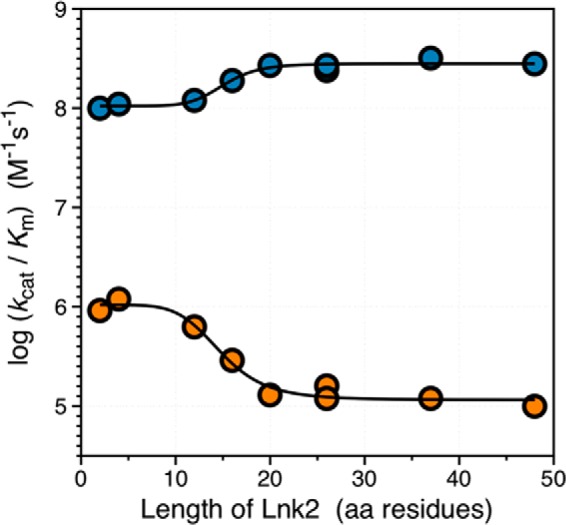
The length of Lnk2 influences the rate of prothrombin activation. The values of the specificity constant kcat/Km for activation of prothrombin by factor Xa and phospholipids in the absence (orange) or presence (blue) of cofactor Va are plotted versus the length of Lnk2. The data are from Table 2 and portray an opposite dependence of the rate of activation that increases (− cofactor Va) or decreases (+ cofactor Va) with the length of Lnk2. In the presence of phospholipids, prothrombin activation in the absence of cofactor Va is significantly enhanced when Lnk2 is <15 residues long. On the other hand, a Lnk2 of <15 residues long slightly compromises activation in the presence of cofactor. The overall effect of cofactor Va is highly dependent on the length of Lnk2 and is optimized when Lnk2 has a length of at least 20 residues, as documented in 29 different species (Fig. 1).
Lnk2 also controls the pathway of prothrombin activation. Activation of prothrombin to thrombin requires cleavage at two sites, Arg271 and Arg320, along two mutually exclusive pathways producing the intermediates prethrombin-2 (cleavage at Arg271 first) or meizothrombin (cleavage at Arg320 first). The prethrombin-2 pathway predominates when activation takes place in the absence of cofactor Va (1, 12, 16) because of a more favorable orientation and accessibility of Arg271 to proteolysis (12). In the presence of cofactor Va in the prothrombinase complex, the choice of the pathway becomes context-dependent. On the surface of platelets (7, 8), activation proceeds along the prethrombin-2 pathway because cleavage at Arg271 is preferred in 95% of the cases over cleavage at Arg320 (7). On nonplatelet surfaces such as red blood cells (9) or synthetic phospholipids (1, 11, 12, 16), prothrombin activation proceeds along the meizothrombin pathway. The relative accessibility of Arg271 and Arg320 is in this case controversial. Measurements carried out under nearly identical solution conditions by three different groups have reported that the sites are either equally accessible (12) or Arg320 is 4-fold (7) or 20-fold (16) more accessible than Arg271. The prothrombinase complex activates prothrombin along the meizothrombin pathway even when the length of Lnk2 is extended by 22 residues (Fig. 4). Two distinguishing features of this pathway are the rapid and transient accumulation of F1.2.A (the fragment of prothrombin containing all domains but the catalytic B chain) and the lack of a significant amount of prethrombin-2. In contrast, reduction of the length of Lnk2 below 15 residues causes activation to proceed without appreciable preference between the meizothrombin and prethrombin-2 pathways, with both intermediates clearly detectable after 10 min (Fig. 4). Densitometric analysis of the intermediates indicates that the integrity of Lnk2 is critical for selecting the meizothrombin pathway and that preference for this pathway is lost when the length of Lnk2 drops below 15 residues.
FIGURE 4.

Effect of perturbation of Lnk2 on the pathway of prothrombin activation. SDS-PAGE analysis of the conversion of prothrombin (1.4 μm) to thrombin by the prothrombinase complex (0.2 nm factor Xa, 20 μm phospholipids, 10 nm cofactor Va). Left panels, progressive deletion of Lnk2 reduces the rate of zymogen consumption and significantly affects accumulation of the intermediates F1.2.A and prethrombin-2 that report cleavage at Arg320 and Arg271, respectively. Meizothrombin accumulates and disappears within the first 10 min in wild type, ProTins22, and ProTΔ146–156. On the other hand, fragment F1.2.A is detected up to 20 min in ProTΔ145–168, ProTΔ146–167, and ProTΔ154–167. Right panels, in ProTΔ146–167, the disappearance of the intact zymogen is linked to the simultaneous appearance of F1.2.A and prethrombin-2, vouching for no preferential cleavage at Arg271 and Arg320 in this prothrombin construct. Introducing the additional R271A or R320A mutation in ProTΔ146–167 generates two constructs cleaved by prothrombinase only at Arg320 (ProTΔ146–167-R271A) or Arg271 (ProTΔ146–167-R320A). All three prothrombin variants ProTΔ146–167, ProTΔ146–167-R271A, and ProTΔ146–167-R320A are processed by prothrombinase with similar rates, indicating no preferential cleavage at Arg320 in the absence of Lnk2.
The change in preferential cleavage at Arg320 versus Arg271 below a critical length of Lnk2 is directly demonstrated by constructs carrying Ala substitutions of Arg271 or Arg320 in the wild type and ProTΔ146–167 backgrounds. The conversion of prothrombin to meizothrombin (cleavage at Arg320 in constructs carrying the R271A mutation) or prethrombin-2 (cleavage at Arg271 in constructs carrying the R320A mutation) was measured directly in view of previous conflicting results (7, 12, 16). Consistent with the findings of Wood et al. (7), cleavage at Arg320 in the construct carrying the R217A mutation is preferred 5-fold over cleavage at Arg271 in the construct carrying the R320A mutation (data not shown). On the other hand, cleavage at Arg320 or Arg271 takes place with comparable rates in constructs carrying the R271A or R320A mutation in the ProTΔ146–167 background (Fig. 4).
The relevance of these findings extends to blood physiology because scrambling, extending, or deleting Lnk2 affects the aPTT in ways consistent with the biochemical data (Fig. 5). Notably, addition of ProTΔ145–168, ProTΔ146–167, and ProTΔ154–167 to prothrombin-deficient plasma prolongs the aPTT 1.5–1.7-fold, indicating significant inhibition of the intrinsic pathway of the coagulation cascade leading to prothrombin activation. The effect is comparable with the defect in thrombin generation documented in patients with hemophilia A or treated with anticoagulants such as heparin. The prolongation of aPTT observed upon deletion of Lnk2 may be due to disruption of preferential activation of prothrombin along the meizothrombin pathway and resulting accumulation of the inactive intermediate prethrombin-2.
FIGURE 5.
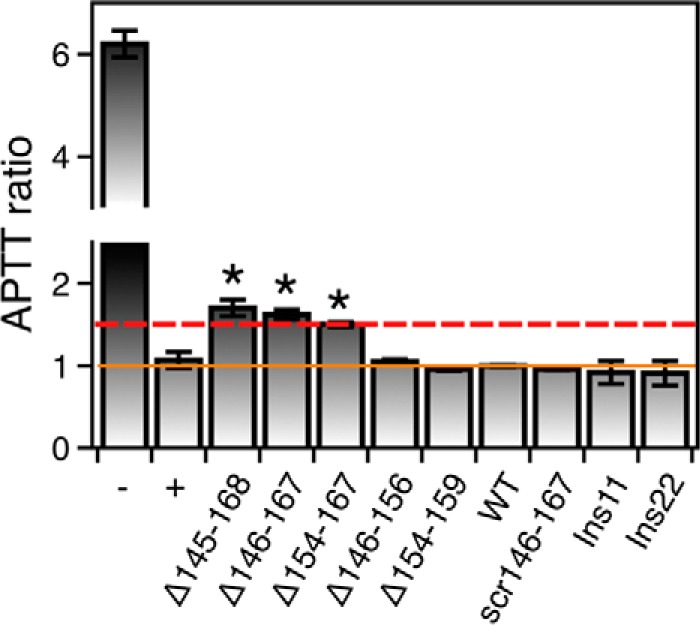
Effect of perturbation of Lnk2 on the aPTT. aPTT values are shown as the ratios of the test to normal, prothrombin wild type (30 ± 2 s). Additional reference values are 32 ± 3 s for plasma collected from a healthy donor (positive control, +) and 185 ± 8 s for prothrombin deficient plasma (negative control, −). The solid orange line identifies the baseline, and the dashed red line highlights the critical value of 1.5. Prothrombin constructs ProTΔ145–168, ProTΔ146–167, and ProTΔ154–167 show significant prolongation of the aPTT (1.50 ± 0.03-, 1.62 ± 0.06-, and 1.72 ± 0.08-fold, respectively). Each measurement is the average of six individual determinations.
Discussion
The factors controlling the rate and pathway of prothrombin activation have been investigated for decades (1, 2, 54–56). Previous studies have pointed out the importance of the context and composition of the membrane in determining the rate and pathway of prothrombin activation (1, 3, 11, 12, 18), implying a long range communication between the Gla domain anchored to the membrane and the sites of cleavage at Arg271 and Arg320 located >80 Å away. The results presented here support Lnk2 as a key structural conduit for this long range communication under conditions relevant to physiology. Scrambling most of Lnk2 is inconsequential on function, and so is its extension by as many as 22 residues. Reducing Lnk2 below a critical length of 15 residues increases the rate of activation (up to 10-fold) in the absence of cofactor Va and decreases it (up to 3-fold) in the presence of cofactor. Furthermore, below the critical length of 15 residues, prothrombinase no longer cleaves prothrombin preferentially at Arg320 over Arg271. All of these effects depend on the presence of phospholipids, which lends support to the physiological relevance of Lnk2 in prothrombin activation.
It is of interest to cast the role of Lnk2 in the context of structural information. The current structure of ProTΔ146–167 (19) has documented a contorted architecture of the zymogen with domains not vertically aligned (Fig. 6A). Overall, prothrombin can be pictured as two rigid bodies, the Gla domain/kringle-1 pair on the N terminus and the kringle-2/protease domain pair on the C terminus, whose relative arrangement is dictated by the flexibility of Lnk2 connecting the two kringles. When the Gla domain is anchored to the membrane as proposed by Furie and co-workers (57) and Tajkhorshid and co-workers (58), the rest of the molecule assumes a bent conformation with the site of cleavage at Arg320 positioned 45 Å over the plane of the phospholipids (Fig. 6D). FRET studies have reported that this plane is 60–70 Å away from a fluorophore conjugated to the active site of factor Xa (59–61). Hence, engagement of the site of cleavage at Arg320 by factor Xa would require a substantial conformational rearrangement of factor Xa and/or prothrombin upon assembly of the enzyme-substrate complex. Furthermore, access to the Arg271 site of cleavage initiating the prethrombin-2 pathway would require an even more drastic rearrangement. Arg271 is in a disordered region of Lnk3. Thr274, the closest residue to Arg271 detected in the density map, sits only 21 Å above the membrane in an orientation predicting nonoptimal cleavage of Arg271 by factor Xa. The lack of preferential cleavage by prothrombinase at Arg271 and Arg320 in the ProTΔ146–167 mutant is difficult to reconcile with the architecture of prothrombin currently documented by x-ray crystallography (19). Two new structures address this issue directly and reveal the importance of Lnk2 in the mechanism of activation.
A new structure of ProTΔ146–167, solved at 3.2 Å resolution, portrays the two rigid portions of the zymogen in a relative arrangement that is flipped almost 180° relative to the original structure (Figs. 6C and 7A). The kringle-2/protease domain pairs in two structures align with an RMSD of only 0.56 Å, but the Gla domain/kringle-1 pair in the new structure of proTΔ146–167 flips on the opposite side of the main vertical axis of the molecule (Fig. 7A) producing an overall architecture that is slightly more elongated (92 versus 85 Å). When this new conformation is docked on a phospholipid surface (Fig. 6F), residues Thr274 and Arg320 flip their position and sit respectively 70 and 60 Å above the plane of the membrane, well within the reach of the active site of factor Xa. Just four of the 26 residues of Lnk2 in the ProTΔ146–167 mutant enable enough flexibility to present Arg271 and Arg320 to the active site of factor Xa in drastically different orientations. The four residues of Lnk2 in ProTΔ146–167 act as a molecular switch between α-helix (Fig. 8A) and β-strand (Fig. 8C) configurations and broker a sharp conformational rearrangement of kringle-1 (Fig. 7A). The remarkable flexibility of Lnk2 documented by x-ray crystallography was investigated further with measurements in solution to rule out any potential artifact caused by crystal packing. Single molecule FRET measurements with probes attached at residues 101 and 210 across Lnk2 in the ProTΔ146-167 background were carried out to complement recent luminescence resonance energy transfer measurements between the same two residues in full-length prothrombin (32). Experiments were performed with pulsed interleaved excitation, which reports the status of both donor and acceptor fluorophores by sorting molecules on the basis of relative donor:acceptor stoichiometry (S) and apparent FRET efficiency (E). The ability of single molecule FRET to detect and resolve dynamic behavior within a single or multiple populations enabled identification of two distinct and comparable populations for ProTΔ146–167 (Fig. 7B), with interprobe distances of 51 Å (low energy transfer) and 36 Å (high energy transfer). These values are consistent with the Cα-Cα distances between residues 101 and 210 in the two conformations of ProTΔ146–167 documented by x-ray crystallography (Fig. 7A) and are also very similar to the distances of 54 and 34 Å measured by luminescence resonance energy transfer between probes attached at residues 101 and 210 in full-length prothrombin (32). Hence, the conformational plasticity of Lnk2 detected by x-ray crystallography in the ProTΔ146–167 mutant is highly relevant to physiology. The x-ray structural data are consistent with solution measurements of the ProTΔ146–167 mutant by single molecule FRET and of full-length prothrombin by luminescence resonance energy transfer (32).
FIGURE 7.

Conformational flexibility of Lnk2 revealed by single molecule FRET. A, structures of ProTΔ146–167 (Protein Data Bank code 4O03, wheat; Protein Data Bank code 5EDK, blue), with the Gla domain removed for clarity. The two structures overlap significantly (RMSD = 0.56 Å) at the level of the kringle-2/protease domain pair but differ sharply in the orientation of kringle-1 (red). The change is due to the conformation of Lnk2 that switches from α-helix (Protein Data Bank code 4O03, position 1) to β-strand (Protein Data Bank code 5EDK, position 2). Residues Ser101 and Ser210 are represented as green and magenta spheres, with arrows indicating Cα-Cα distances. B, FRET histograms for the mutant ProTΔ146–167/S101C/S210C. The lower panel shows a two-dimensional histogram of stoichiometry (S) versus FRET efficiency (E) for each diffusing molecule that contains both AF555 and AF647 fluorophores (i.e. molecules with 0.25 < S < 0.75). The upper panel shows a one-dimensional E histogram of the molecules in the lower panel. The E distribution was fit to a double Gaussian distribution (red). The center and percentage of the population in each Gaussian is indicated. FRET efficiencies were converted to distances using a theoretical R0 = 51 Å and a random orientation factor k = 2/3.
FIGURE 8.

Structural features of prothrombin mutants proTΔ146–167 and proTΔ154–167. A–C, artificial connections between kringle-1 and kringle-2 of proTΔ146–167 (A), proTΔ154–167 (B), and the new conformation of proTΔ146–167 (C) showing the plasticity of Lnk2. The electron density 2Fo − Fc map is countered at 1 σ. Polar interactions are shown as dashed lines, with kringle-1 (red), kringle-2 (green), and Lnk2 (wheat). Lnk2 acts as a molecular switch between α-helix (C) or β-strand (A and B) configurations. Notably, most of the 12 residues of Lnk2 in proTΔ154–167 form an uninterrupted β-sheet with the adjacent 139–143 strand of kringle-1 stabilized by additional contacts between Thr153, Arg81, and Cys170. D–F, structure of the Gla domain of ProTΔ146–167 (19) (D), bovine prothrombin fragment 1 (57) (E) and ProTΔ154–167 (F) bound to Ca2+ (D and E) or Mg2+ (F). Overall, the architecture of the Ca2+ and Mg2+ bound forms are superimposable in the N12-A46 region (RMSD = 0.48 Å) and show a similar globular shape with three α-helices spanning and two connecting loops. The difference between Ca2+ and Mg2+ bound forms is restricted to the first 10 residues from the N termini, in agreement with previous NMR studies on the peptide 1–47 of factor IX (62). Formation of a functional ω-loop is impeded in the presence of Mg2+ (F). Valence calculations (63) identify a total of five binding sites for Mg2+. Mg1, Mg3, Mg4, and Mg5 in ProTΔ154–167 (F) occupy the same sites as Ca1, Ca2, Ca5, and Ca6 in the structure of bovine prothrombin fragment 1, suggesting direct competition in these positions.
Further insight on the role of Lnk2 comes from the structure of ProTΔ154–167, solved at 2.2 Å resolution, that reveals the architecture of prothrombin lacking only 14 residues in unprecedented detail (Fig. 6B). In this structure, the kringle-2/protease domain pair assumes the same conformation seen in the two structures of proTΔ146–167 (Fig. 6, A and C), but the Gla domain/kringle-1 pair features a drastic reorientation. Kringle-1 rotates 3° anticlockwise and moves almost 40 Å downward relative to its position in proTΔ146–167 (Fig. 6A), taking the Gla domain with it (Fig. 8E). As a result, the overall conformation of ProTΔ154–167 stretches up to 100 Å and shows an almost perfect alignment of its two rigid domains (Fig. 6B). The overall shape of ProTΔ154–167 is stabilized by Lnk2 whose 12 residues are all visible in the density map and engage kringle-1 in several H-bonding interactions (Fig. 8B). Lnk2 assumes a β-strand configuration as in the new structure of ProTΔ146–167 (Fig. 8C), supporting a key role for the first two residues of the linker in triggering alternative conformations (Fig. 8, A and C). The trigger likely works in the wild type as well. In fact, the Cα-Cα distance between residues 101 and 210 in the structure of ProTΔ154–167 is 34 Å and practically identical to one of the two distances measured by luminescence resonance energy transfer between probes attached at residues 101 and 210 in full-length prothrombin (32). This makes the high resolution structure of ProTΔ154–167 highly relevant to physiology, just as the structures of ProTΔ146–167. When the conformation of ProTΔ154–167 is docked on a phospholipid surface (Fig. 6E), both Thr274 and Arg320 move up above the plane of the membrane to 55 and 65 Å, respectively, again within the range of the predicted position of the active site of factor Xa (59–61). Unfortunately, the site of cleavage at Arg271 remains too disordered to detect in the structure of ProTΔ154–167, notwithstanding the high resolution.
The structures shown in Fig. 6 offer unprecedented snapshots of the conformational plasticity of prothrombin, with Lnk2 controlling the relative orientation of the two rigid kringle-2/protease domain and Gla domain/kringle-1 pairs. Once anchored to the membrane via the Gla domain, prothrombin utilizes the flexible Lnk2 as a pivot point to orient the kringle-2/protease domain pair containing the sites of cleavage at Arg271 and Arg320. Through this action, Lnk2 adjusts the height of Arg271 and Arg320 over the plane of the membrane and presents them to the active site of factor Xa in the order and orientation necessary to define rate and pathway of activation. This scenario also provides context for the effect of membranes and cofactor Va.
A final comment should be made about the overall conformation of Lnk2 in the three structures of prothrombin discussed in this study (Fig. 6). The arrangement of the N-terminal Gla domain/kringle-1 pair relative to the C-terminal kringle-2/protease pair changes from being distorted when Lnk2 is only 4 residues long in ProTΔ146–167 (Fig. 6, A and C) to almost linear when Lnk2 is 12 residues long in ProTΔ154–167 (Fig. 6B). However, Lnk2 never assumes a conformation promoting full extension of prothrombin along the vertical axis. The rate and pathway of prothrombin activation are perturbed when the length of Lnk2 drops below the critical threshold of 15 residues, and no change is observed when the length is >20 residues (Fig. 3). These structural and functional observations converge to the mechanistically important conclusion that Lnk2 does not function in a fully untethered and extended conformation, contrary to the assumptions of recent models (29–31). In a fully extended Lnk2, the rate of prothrombin activation would decrease both below and above a critical length required for optimal alignment between the sites of cleavage and the active site of factor Xa (Fig. 6), producing a bell-shaped dependence not consistent with the data in Fig. 3. On the other hand, a conformation where Lnk2 is allowed to bulge beyond a critical length would produce a dependence of the rate of activation as shown by the data in Fig. 3.
The conformational plasticity of prothrombin should invite reconsideration of recent musings on the architecture of the zymogen bound to prothrombinase (29–31) or of the long list of alternative mechanisms of prothrombin activation (12, 14–18, 20). The approach presented here shows the benefits, if not the necessity, of combining multiple x-ray structures, site-directed mutagenesis, and single molecule FRET toward a quantitative understanding of prothrombin and its key interactions in the blood coagulation cascade.
Author Contributions
N. P. and E. D. C. designed the research; N. P. and Z. C. carried out the work; N. P., Z. C., and E. D. C. analyzed results; and N. P. and E. D. C. wrote the manuscript.
Acknowledgments
We are grateful to Dr. Alireza Rezaie and Dr. Likui Yang for assistance with aPTT measurements.
This work was supported in part by American Heart Association Grant 15SDG25550094 (to N. P.) and the National Institutes of Health Research Grants HL049413, HL073813, and HL112303 (to E. D. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5EDK and 5EDM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- aPTT
- activated partial thromboplastin time
- RMSD
- root mean square deviation.
References
- 1. Rosing J., Tans G., Govers-Riemslag J. W., Zwaal R. F., and Hemker H. C. (1980) The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 255, 274–283 [PubMed] [Google Scholar]
- 2. Weinreb G. E., Mukhopadhyay K., Majumder R., and Lentz B. R. (2003) Cooperative roles of factor V(a) and phosphatidylserine-containing membranes as cofactors in prothrombin activation. J. Biol. Chem. 278, 5679–5684 [DOI] [PubMed] [Google Scholar]
- 3. Bukys M. A., Orban T., Kim P. Y., Nesheim M. E., and Kalafatis M. (2008) The interaction of fragment 1 of prothrombin with the membrane surface is a prerequisite for optimum expression of factor Va cofactor activity within prothrombinase. Thromb. Haemost. 99, 511–522 [PubMed] [Google Scholar]
- 4. Krishnaswamy S., Church W. R., Nesheim M. E., and Mann K. G. (1987) Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism. J. Biol. Chem. 262, 3291–3299 [PubMed] [Google Scholar]
- 5. Pelc L. A., Chen Z., Gohara D. W., Vogt A. D., Pozzi N., and Di Cera E. (2015) Why Ser and not Thr brokers catalysis in the trypsin fold. Biochemistry 54, 1457–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srivasatava K. R., Majumder R., Kane W. H., Quinn-Allen M. A., and Lentz B. R. (2014) Phosphatidylserine and FVa regulate FXa structure. Biochem. J. 459, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wood J. P., Silveira J. R., Maille N. M., Haynes L. M., and Tracy P. B. (2011) Prothrombin activation on the activated platelet surface optimizes expression of procoagulant activity. Blood 117, 1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haynes L. M., Bouchard B. A., Tracy P. B., and Mann K. G. (2012) Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J. Biol. Chem. 287, 38647–38655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whelihan M. F., Zachary V., Orfeo T., and Mann K. G. (2012) Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood 120, 3837–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nomura S., and Shimizu M. (2015) Clinical significance of procoagulant microparticles. J. Intensive Care 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tans G., Janssen-Claessen T., Hemker H. C., Zwaal R. F., and Rosing J. (1991) Meizothrombin formation during factor Xa-catalyzed prothrombin activation. Formation in a purified system and in plasma. J. Biol. Chem. 266, 21864–21873 [PubMed] [Google Scholar]
- 12. Brufatto N., and Nesheim M. E. (2003) Analysis of the kinetics of prothrombin activation and evidence that two equilibrating forms of prothrombinase are involved in the process. J. Biol. Chem. 278, 6755–6764 [DOI] [PubMed] [Google Scholar]
- 13. Walker R. K., and Krishnaswamy S. (1994) The activation of prothrombin by the prothrombinase complex: the contribution of the substrate-membrane interaction to catalysis. J. Biol. Chem. 269, 27441–27450 [PubMed] [Google Scholar]
- 14. Kim P. Y., and Nesheim M. E. (2007) Further evidence for two functional forms of prothrombinase each specific for either of the two prothrombin activation cleavages. J. Biol. Chem. 282, 32568–32581 [DOI] [PubMed] [Google Scholar]
- 15. Lee C. J., Wu S., Eun C., and Pedersen L. G. (2010) A revisit to the one form kinetic model of prothrombinase. Biophys. Chem. 149, 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orcutt S. J., and Krishnaswamy S. (2004) Binding of substrate in two conformations to human prothrombinase drives consecutive cleavage at two sites in prothrombin. J. Biol. Chem. 279, 54927–54936 [DOI] [PubMed] [Google Scholar]
- 17. Lentz B. R. (2003) Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 42, 423–438 [DOI] [PubMed] [Google Scholar]
- 18. Bradford H. N., Orcutt S. J., and Krishnaswamy S. (2013) Membrane binding by prothrombin mediates its constrained presentation to prothrombinase for cleavage. J. Biol. Chem. 288, 27789–27800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pozzi N., Chen Z., Pelc L. A., Shropshire D. B., and Di Cera E. (2014) The linker connecting the two kringles plays a key role in prothrombin activation. Proc. Natl. Acad. Sci. U.S.A. 111, 7630–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bianchini E. P., Orcutt S. J., Panizzi P., Bock P. E., and Krishnaswamy S. (2005) Ratcheting of the substrate from the zymogen to proteinase conformations directs the sequential cleavage of prothrombin by prothrombinase. Proc. Natl. Acad. Sci. U.S.A. 102, 10099–10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kroh H. K., Panizzi P., Tchaikovski S., Baird T. R., Wei N., Krishnaswamy S., Tans G., Rosing J., Furie B., Furie B. C., and Bock P. E. (2011) Active site-labeled prothrombin inhibits prothrombinase in vitro and thrombosis in vivo. J. Biol. Chem. 286, 23345–23356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harlos K., Holland S. K., Boys C. W., Burgess A. I., Esnouf M. P., and Blake C. C. (1987) Vitamin K-dependent blood coagulation proteins form hetero-dimers. Nature 330, 82–84 [DOI] [PubMed] [Google Scholar]
- 23. Deguchi H., Takeya H., Gabazza E. C., Nishioka J., and Suzuki K. (1997) Prothrombin kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem. J. 321, 729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kotkow K. J., Deitcher S. R., Furie B., and Furie B. C. (1995) The second kringle domain of prothrombin promotes factor Va-mediated prothrombin activation by prothrombinase. J. Biol. Chem. 270, 4551–4557 [DOI] [PubMed] [Google Scholar]
- 25. Taneda H., Andoh K., Nishioka J., Takeya H., and Suzuki K. (1994) Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J Biochem. 116, 589–597 [DOI] [PubMed] [Google Scholar]
- 26. Yegneswaran S., Mesters R. M., Fernández J. A., and Griffin J. H. (2004) Prothrombin residues 473–487 contribute to factor Va binding in the prothrombinase complex. J. Biol. Chem. 279, 49019–49025 [DOI] [PubMed] [Google Scholar]
- 27. Chen L., Yang L., and Rezaie A. R. (2003) Proexosite-1 on prothrombin is a factor Va-dependent recognition site for the prothrombinase complex. J. Biol. Chem. 278, 27564–27569 [DOI] [PubMed] [Google Scholar]
- 28. Yegneswaran S., Mesters R. M., and Griffin J. H. (2003) Identification of distinct sequences in human blood coagulation factor Xa and prothrombin essential for substrate and cofactor recognition in the prothrombinase complex. J. Biol. Chem. 278, 33312–33318 [DOI] [PubMed] [Google Scholar]
- 29. Lee C. J., Wu S., and Pedersen L. G. (2011) A proposed ternary complex model of prothrombinase with prothrombin: protein-protein docking and molecular dynamics simulations. J. Thromb. Haemost. 9, 2123–2126 [DOI] [PubMed] [Google Scholar]
- 30. Lechtenberg B. C., Murray-Rust T. A., Johnson D. J., Adams T. E., Krishnaswamy S., Camire R. M., and Huntington J. A. (2013) Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood 122, 2777–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shim J. Y., Lee C. J., Wu S., and Pedersen L. G. (2015) A model for the unique role of factor Va A2 domain extension in the human ternary thrombin-generating complex. Biophys. Chem. 199, 46–50 [DOI] [PubMed] [Google Scholar]
- 32. Pozzi N., Chen Z., Gohara D. W., Niu W., Heyduk T., and Di Cera E. (2013) Crystal structure of prothrombin reveals conformational flexibility and mechanism of activation. J. Biol. Chem. 288, 22734–22744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Degen S. J., and Davie E. W. (1987) Nucleotide sequence of the gene for human prothrombin. Biochemistry 26, 6165–6177 [DOI] [PubMed] [Google Scholar]
- 34. George R. A., and Heringa J. (2002) An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 15, 871–879 [DOI] [PubMed] [Google Scholar]
- 35. Pozzi N., Chen Z., Zapata F., Niu W., Barranco-Medina S., Pelc L. A., and Di Cera E. (2013) Autoactivation of thrombin precursors. J. Biol. Chem. 288, 11601–11610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pozzi N., Vogt A. D., Gohara D. W., and Di Cera E. (2012) Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 22, 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Price P. A. (1983) Analysis for γ-carboxyglutamic acid. Methods Enzymol. 91, 13–17 [DOI] [PubMed] [Google Scholar]
- 38. Przysiecki C. T., Staggers J. E., Ramjit H. G., Musson D. G., Stern A. M., Bennett C. D., and Friedman P. A. (1987) Occurrence of β-hydroxylated asparagine residues in non-vitamin K-dependent proteins containing epidermal growth factor-like domains. Proc. Natl. Acad. Sci. U.S.A. 84, 7856–7860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dang O. D., Vindigni A., and Di Cera E. (1995) An allosteric switch controls the procoagulant and anticoagulant activities of thrombin. Proc. Natl. Acad. Sci. U.S.A. 92, 5977–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dang Q. D., Guinto E. R., and Di Cera E. (1997) Rational engineering of activity and specificity in a serine protease. Nat. Biotechnol. 15, 146–149 [DOI] [PubMed] [Google Scholar]
- 41. Bush L. A., Nelson R. W., and Di Cera E. (2006) Murine thrombin lacks Na+ activation but retains high catalytic activity. J. Biol. Chem. 281, 7183–7188 [DOI] [PubMed] [Google Scholar]
- 42. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 43. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 44. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 45. Joosten R. P., Joosten K., Cohen S. X., Vriend G., and Perrakis A. (2011) Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank. Bioinformatics 27, 3392–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morris A. L., MacArthur M. W., Hutchinson E. G., and Thornton J. M. (1992) Stereochemical quality of protein structure coordinates. Proteins 12, 345–364 [DOI] [PubMed] [Google Scholar]
- 47. Müller B. K., Zaychikov E., Bräuchle C., and Lamb D. C. (2005) Pulsed interleaved excitation. Biophys J 89, 3508–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gopich I. V., and Szabo A. (2007) Single-molecule FRET with diffusion and conformational dynamics. J. Phys. Chem. B 111, 12925–12932 [DOI] [PubMed] [Google Scholar]
- 49. Kapanidis A. N., Lee N. K., Laurence T. A., Doose S., Margeat E., and Weiss S. (2004) Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. U.S.A. 101, 8936–8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee N. K., Kapanidis A. N., Wang Y., Michalet X., Mukhopadhyay J., Ebright R. H., and Weiss S. (2005) Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 88, 2939–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Petrovan R. J., Govers-Riemslag J. W., Nowak G., Hemker H. C., Tans G., and Rosing J. (1998) Autocatalytic peptide bond cleavages in prothrombin and meizothrombin. Biochemistry 37, 1185–1191 [DOI] [PubMed] [Google Scholar]
- 52. Board P. G., and Shaw D. C. (1983) Determination of the amino acid substitution in human prothrombin type 3 (157 Glu leads to Lys) and the localization of a third thrombin cleavage site. Br. J. Haematol. 54, 245–254 [DOI] [PubMed] [Google Scholar]
- 53. Kotkow K. J., Furie B., and Furie B. C. (1993) The interaction of prothrombin with phospholipid membranes is independent of either kringle domain. J. Biol. Chem. 268, 15633–15639 [PubMed] [Google Scholar]
- 54. Wu J. R., and Lentz B. R. (1994) Phospholipid-specific conformational changes in human prothrombin upon binding to procoagulant acidic lipid membranes. Thromb. Haemost. 71, 596–604 [PubMed] [Google Scholar]
- 55. Wu J. R., Zhou C., Majumder R., Powers D. D., Weinreb G., and Lentz B. R. (2002) Role of procoagulant lipids in human prothrombin activation. 1. Prothrombin activation by factor X(a) in the absence of factor V(a) and in the absence and presence of membranes. Biochemistry 41, 935–949 [DOI] [PubMed] [Google Scholar]
- 56. Nesheim M. E., Taswell J. B., and Mann K. G. (1979) The contribution of bovine factor V and factor Va to the activity of prothrombinase. J. Biol. Chem. 254, 10952–10962 [PubMed] [Google Scholar]
- 57. Huang M., Rigby A. C., Morelli X., Grant M. A., Huang G., Furie B., Seaton B., and Furie B. C. (2003) Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 10, 751–756 [DOI] [PubMed] [Google Scholar]
- 58. Morrissey J. H., Davis-Harrison R. L., Tavoosi N., Ke K., Pureza V., Boettcher J. M., Clay M. C., Rienstra C. M., Ohkubo Y. Z., Pogorelov T. V., and Tajkhorshid E. (2010) Protein-phospholipid interactions in blood clotting. Thromb. Res. 125, S23–S25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koklic T., Chattopadhyay R., Majumder R., and Lentz B. R. (2015) Factor Xa dimerization competes with prothrombinase complex formation on platelet-like membrane surfaces. Biochem. J. 467, 37–46 [DOI] [PubMed] [Google Scholar]
- 60. Husten E. J., Esmon C. T., and Johnson A. E. (1987) The active site of blood coagulation factor Xa. Its distance from the phospholipid surface and its conformational sensitivity to components of the prothrombinase complex. J. Biol. Chem. 262, 12953–12961 [PubMed] [Google Scholar]
- 61. Qureshi S. H., Yang L., Yegneswaran S., and Rezaie A. R. (2007) FRET studies with factor X mutants provide insight into the topography of the membrane-bound factor X/Xa. Biochem. J. 407, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Freedman S. J., Furie B. C., Furie B., and Baleja J. D. (1995) Structure of the metal-free γ-carboxyglutamic acid-rich membrane binding region of factor IX by two-dimensional NMR spectroscopy. J. Biol. Chem. 270, 7980–7987 [DOI] [PubMed] [Google Scholar]
- 63. Nayal M., and Di Cera E. (1994) Predicting Ca2+-binding sites in proteins. Proc. Natl. Acad. Sci. U.S.A. 91, 817–821 [DOI] [PMC free article] [PubMed] [Google Scholar]