

Abstract
Pro-fibrotic mesenchymal cells are known to be the key effector cells of fibroproliferative disease, but the specific matrix signals and the induced cellular responses that drive the fibrogenic phenotype remain to be elucidated. The key mediators of the fibroblast fibrogenic phenotype were characterized using a novel assay system that measures fibroblast behavior in response to actual normal and fibrotic lung tissue. Using this system, we demonstrate that normal lung promotes fibroblast motility and polarization, while fibrotic lung immobilizes the fibroblast and promotes myofibroblast differentiation. These context-specific phenotypes are surprisingly both mediated by myosin II. The role of myosin II is supported by the observation of an increase in myosin phosphorylation and a change in intracellular distribution in fibroblasts on fibrotic lung, as compared with normal lung. Moreover, loss of myosin II activity has opposing effects on protrusive activity in fibroblasts on normal and fibrotic lung. Loss of myosin II also selectively inhibits myofibroblast differentiation in fibroblasts on fibrotic lung. Importantly, these findings are recapitulated by varying the matrix stiffness of polyacrylamide gels in the range of normal and fibrotic lung tissue. Comparison of the effects of myosin inhibition on lung tissue with that of polyacrylamide gels suggests that matrix fiber organization drives the fibroblast phenotype under conditions of normal/soft lung, while matrix stiffness drives the phenotype under conditions of fibrotic/stiff lung. This work defines novel roles for myosin II as a key regulatory effector molecule of the pro-fibrotic phenotype, in response to biophysical properties of the matrix.
Keywords: fibroblast, fibrosis, lung, myofibroblast, myosin, fibroblast migration, myofibroblast differentiation, non-muscle myosin II
Introduction
Fibrotic disorders can occur as a consequence of failure of normal tissue regeneration after either acute or chronic injury, resulting in organ dysfunction of lung, liver, kidney, heart, vasculature, and others (1). As such, fibroproliferative diseases account for nearly half of the deaths worldwide (1, 2). For example, idiopathic pulmonary fibrosis (IPF)2 is an incurable and fatal fibrotic lung disorder. It is characterized by excessive connective tissue accumulation and continuous tissue contraction that creates a cycle of progressive organ deterioration (3). Activated fibroblasts and myofibroblasts are the key “pro-fibrotic” effector cells in this disease and fibroblastic foci, the histological hallmark of IPF, are enriched with these activated fibroblasts and myofibroblasts (4). During pulmonary fibrogenesis, fibroblasts migrate toward and accumulate in fibroblastic foci where they differentiate into myofibroblasts, which produce high levels of pro-fibrotic mediators and have a contractile phenotype (5–7). However, the signals driving this aberrant accumulation and activation of fibroblasts and the intracellular pathways that induce this fibrogenic phenotype have not been fully elucidated.
Enhanced migration and myofibroblast differentiation are characteristic pro-fibrotic phenotypes of mesenchymal cells in IPF (8–11). A number of soluble mediators (e.g. TGF-β, PDGF, LPA) are capable of inducing the pro-fibrotic phenotype in fibroblasts (12–14). However, a growing body of published research demonstrates that fibroblasts can also be activated by specific biophysical properties of their surrounding matrix (15–18). For example, the dimensionality of the matrix (three-dimensional versus two-dimensional) can affect migration rate, and increasing substrate rigidity can induce alpha-smooth muscle actin (α-SMA) expression and its incorporation of into F-actin stress fibers, the hallmarks of myofibroblast differentiation (15, 19, 20). It has recently been shown that fibrotic lung is “stiffer” than normal lung and this difference in rigidity can drive phenotypic changes in fibroblasts (16). Furthermore, lung tissue is 1 × 106 orders of magnitude less rigid than tissue culture plastic (21). As such, traditional cell biological assays using cells plated on plastic may not provide an accurate representation of in vivo fibroblast behavior. Recent advances allowing the seeding of fibroblasts into normal or IPF-cell-derived matrices have demonstrated that IPF-derived ECM promotes the up-regulation of “pro-fibrotic” gene transcription (22) and can induce TGF-β-independent myofibroblast differentiation (23). In this study, we have taken an additional step forward by utilizing a more physiologically-relevant matrix system that more closely mimics the in vivo environment of the lung fibroblast.
Myosins are a family of motor proteins that are involved in many cellular processes that require force. Contractile tension generated within the cytoskeleton by non-muscle myosin II is known to play an important role in both fibroblast migration and differentiation (24–28). Although activation of signals upstream of myosin II has been recently implicated in pulmonary fibrogenesis in an experimental model of human IPF, the direct involvement of myosin II as an effector molecule of fibrogenesis has yet to be documented (20). Furthermore, the mechanisms whereby myosin II contributes to the activation of mesenchymal cells on physiologically relevant matrices, and in vivo, have not been described. Thus, we sought to understand the role of myosin II in mediating the behavior of pro-fibrotic fibroblasts as they interact with physiologically relevant matrices. In doing so, we have uncovered unique and context-specific roles of myosin II in fibrogenesis.
Experimental Procedures
Materials
Normal human lung fibroblasts (HLF, 19Lu, passages 4–9) were purchased from ATCC (Manassas, VA) and maintained/propagated as previously described (29). C57Bl/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). PKH26 Red Fluorescent Cell Linker Kits, monoclonal anti-α-SMA, monoclonal anti-vinculin, and blebbistatin were purchased from Sigma. AF 594 Phalloidin and AF 488 goat anti-mouse antibody were purchased from Life Technologies (Carlsbad, Ca). Antibodies to myosin IIA, myosin IIB, phospho-myosin light chain 2 (p-MLC, Thr-18/Ser-19), and myosin light chain 2 were purchased from Cell Signaling Technology (Beverly, MA). Rabbit IgG was from Jackson Immunoresearch (West Grove, PA). Normal mouse IgG was from R&D Systems (Minneapolis, MN). GAPDH antibody was from Fitzgerald (Acton, MA). MYH9 and MYH10 siRNA were purchased from Thermo Fisher (Waltham, MA). Non-targeting pooled siRNA control was from Dharmacon Inc. (Lafayette, CO). Silentfect lipid transfection reagent was purchased from Bio-Rad Laboratories (Hercules, CA). Glass-bottom plates (12-well) containing activated polyacrylamide gels of 1 kPa, 8 kPa, 25 kPa, and glass were custom-made by Matrigen Life Technologies (Brea, CA).
Lung Tissue Assay System
All animal protocols were performed as approved by the Cleveland Clinic institutional animal care and use committee and using methods in the guidelines for the humane care of animals by the American Physiological Society. Lungs from C57Bl/6 mice (18–20 g, 8–12-week-old females) that had received intratracheal instillation of 4 units/kg bleomycin sulfate (Bristol Laboratories, Syracuse, NY) 2 weeks prior were inflated with OCT as described previously (9, 30, 31). Cryotome-cut, OCT-preserved lung tissue sections (10 μm) were washed in 37 °C PBS to remove the OCT, and blocked with 5% albumin. Normal and fibrotic regions were identified using light microscopy and confirmed with serial H&E-, DAPI- (Fig. 1A), and trichrome- (not shown) stained sections. Samples were mechanically characterized using an atomic force microscope (AFM, MFP-3D; Asylum Research) using methods previously published (16) (Fig. 1B). PKH-labeled normal HLFs were allowed to attach to pre-blocked 10-μm sections (25,000 cells per section) of normal and fibrotic lung as previously published (10, 31), placed in 1% BSA, serum-free medium (5% CO2 at 37 °C) ± blebbistatin, followed by time-lapse video microscopy on an inverted microscope (Leica DM IRBE). Migration tracking was analyzed using ImagePro software (Media Cybernetics, Rockville, MD). Greater than 50 cells from five independent experiments were analyzed. For myofibroblast differentiation, immuno-fluorescence was performed as described below on non-labeled, attached cells ± blebbistatin after 24 h. HLFs were transfected with 100 nm siRNA (MYH9, MYH10, or non-targeting control) using Silentfect lipid and OptiMEM medium (GIBCO/Invitrogen) for 48 h, as previously published (10). Viability of attached cells at 24 h was 91.34 ± 6% on normal lung and 91.25 ± 5% on fibrotic lung (Fig. 1C).
FIGURE 1.

Characterization of lung tissue assay system. Panel A, representative photomicrographs of hematoxylin and eosin (H&E, 20× original magnification, inset 2× magnification) and DAPI (10× original magnification) stained normal and fibrotic lung tissue sections, demonstrating the presence of native nuclear material. Panel B, histogram of stiffness in normal and fibrotic (lesion) lung tissue sections measured using atomic force microscopy. Data indicate median Young's modulus is 3.78 kPa for normal lung and 6 kPa for fibrotic lung. Panel C, comparison of viability of cells seeded onto lung tissue sections over 24 h. % cell viability defined as the number of labeled cells per section actively spreading, migrating, or demonstrating protrusive activity at 24 h, divided by the number of total labeled cells per section at Time 0. n>50 cells from three independent experiments.
Immunofluorescence Staining
Fixed and permeabilized cells were labeled for α-SMA, F-actin stress fibers, and/or p-MLC2 as previously published (31). Immunofluorescence intensity was analyzed using ImageJ Pro software (NIH). Greater than 20 cells for each condition (normal versus fibrotic) were analyzed in triplicate.
Western Blot Analysis
Immunoblotting was performed for the indicated proteins as previously published (10). GAPDH band density was used as a loading control. To selectively harvest cell lysates from normal and fibrotic lung, HLFs were plated inside cloning rings (6.4-mm diameter) onto lung tissue sections, followed by trypsinization to remove the attached cells. To remove any soluble lung proteins, cell suspensions were centrifuged (1000 × g, 5 min). The cell pellet was re-suspended and passed through a 30-micron cell filter. Preliminary experiments demonstrated no significant detection of α-SMA or p-MLC in the absence of added cells.
Polyacrylamide Gel Assays
Custom made 12-well glass-bottom plates containing activated polyacrylamide gels of 1 kPa, 8 kPa, 25 kPa, and glass (Matrigen Life Technologies) were coated with 1 μg/ml fibronectin for 2 h (37 °C). Polyacrylamide gels with stiffnesses of 1 kPa and 25 kPa were chosen to represent the stiffness of normal and fibrotic lung, respectively, based on AFM analysis of our lung tissue sections and on previously published data (16). For migration analyses, HLFs previously labeled with PKH dye were allowed to attach for one hour, and non-adherent cells were washed away. Blebbistatin or vehicle only (DMSO) in 1% BSA in serum-free medium was added, and time-lapse microscopy was performed over 6 h. For differentiation assays, non-labeled HLFs were allowed to attach for one hour, and non-adherent cells were washed away. The gels containing attached cells were incubated for 24 h, followed by permeabilization, fixation, and immuno-fluorescence staining. For vinculin staining, gels with attached cells were incubated in serum-free medium ± TGF-β1 (1 ng/ml, 24 h).
Quantitative Analysis of Cell Phenotype
Time-lapse microscopy was performed at 10× magnification with images captured at 10-min intervals for 6 h. Migration tracking on single cells was performed with ImagePro software (Media Cybernetics). Migration persistence was calculated by dividing the displacement by the total distance. The number of protrusions was manually counted for each cell over the 6 h time period. Polarity was determined by dividing migration persistence by the number of protrusions. Cell area was derived from manual tracings of individual cells using ImageJ Pro software. Elongation (the inverse of circularity) was calculated using the formula 1 − (4π × area/[perimeter]2). Greater than 50 cells for each condition (normal versus fibrotic) were analyzed in five independent experiments.
Statistical Analysis
All data were analyzed using SigmaPlot (SPSS Inc.) with unpaired/paired t test or Mann-Whitney-Wilcoxon test (for non-parametric data), or by ANOVA (for more than 2 groups). Data are expressed as means ± S.E. unless otherwise indicated. A p value < 0.05 was considered statistically significant.
Results
Fibroblasts Have a Different Morphology When Interacting with Normal and Fibrotic Lung
To determine the effect of normal and fibrotic lung on fibroblast phenotype, HLFs were seeded onto murine lung tissue containing both normal and fibrotic areas. Upon attachment, HLFs on both substrates had a rounded appearance and similar area (mean area, normal lung 1162 ± 555 μm2 versus fibrotic lung 1189 ± 560 μm2, Fig. 2, A (green) and B). However, after 6 h, HLFs on fibrotic lung spread less (change in area, fibrotic lung 809 ± 1192 μm2 versus normal lung 1674 ± 1475 μm2, p = 0.007 (Fig. 2, A (red) and B) and continued to maintain a rounded appearance. In contrast, HLFs on normal lung consistently became elongated and spindle-shaped (elongation factor at 6 h, normal lung 0.745 ± 0.12 AU versus fibrotic lung 0.5 ± 0.17 AU, p < 0.001, and these differences persisted through 24 h (Fig. 2C). These data demonstrate that fibroblasts interacting with normal lung assume an elongated, spindle-shaped morphology, while fibroblasts interacting with fibrotic lung assume a rounded morphology.
FIGURE 2.

Fibroblasts spread less and are less elongated on fibrotic lung. Panel A, representative photomicrographs of fibroblast spreading on normal versus fibrotic lung. Green: cell shape immediately following attachment; Red: cell shape 6 h after attachment. Panel B, comparison of fibroblast spreading as characterized by mean cell area (*, p = 0.007 compared with change in area on fibrotic versus normal lung) and shape (*, p < 0.001 compared with change in shape on fibrotic versus normal lung). Elongation: 1 − (4π × (area/[perimeter]2). Panel C, comparison of area and shape over 24-h time period.
Fibroblasts Are Less Motile and Less Polarized on Fibrotic Lung
On fibrotic lung, HLFs consistently migrated over a shorter distance (fibrotic; 63 ± 36 versus normal; 102 ± 48 μm, p < 0.001) and with a slower speed (fibrotic; 10 ± 6 versus normal; 17 ± 8 μm/hr, Fig. 3, A–D) than on normal lung. Migration on fibrotic lung occurred in a significantly more random, less persistent fashion than on normal lung (migration persistence, fibrotic; 0.5 ± 0.18 versus normal; 0.8 ± 0.11 AU, p < 0.001, values closer to 1 indicate more persistent migration, Fig. 3, B and C). There were nearly 3-fold more protrusions per cell in HLFs on fibrotic lung than on normal lung (fibrotic; 13 ± 5 versus normal; 5 ± 2, p < 0.001, Fig. 3C). Overall, HLFs on fibrotic lung were less polarized than those on normal lung, as they exhibited less migration persistence with increased protrusive activity (polarity, fibrotic; 0.05 ± 0.02 versus normal; 0.2 ± 0.07 AU, p < 0.001, higher values indicate greater polarization, Fig. 3C). These differences in migratory behavior persisted over a 24-hour time period (Fig. 3D). Taken together, these data show for the first time that HLFs interacting with fibrotic lung have a less motile, less polarized phenotype than those interacting with normal lung.
FIGURE 3.

Fibroblasts are less motile and less polarized on fibroproliferative lesions. Normal human lung fibroblasts were fluorescently labeled and allowed to migrate on bleomycin-treated lung tissue sections (6 h, 37 °C). Panel A, kymograph representing a single cell migrating on normal lung (top panel) and fibrotic lung (bottom panel). Each image represents a 1-h interval, 10× magnification. Panel B, wind rose plots with 10 representative migration tracks from each condition (6 h). Panel C, comparison of quantitative migration characteristics of HLFs on normal versus fibrotic lung (6 h), *, p < 0.001 compared with normal lung. Panel D, comparison of HLF speed on normal versus fibrotic lung over 24-h time period.
Fibrotic Lung Enhances Myofibroblast Differentiation
As myofibroblast differentiation occurs at longer time points we examined the attached HLFs at 24 h. HLFs on fibrotic lung were more spread with increased dense, thicker stress fibers and increased α-SMA than those on normal lung (Fig. 4A). Fibroblasts on fibrotic lung more frequently possessed phenotypic characteristics of myofibroblasts: HLFs with prominent F-actin stress fibers were 2-fold higher and α-SMA integrated density was >1.5 fold higher on fibrotic lung (*, p < 0.05 versus normal, Fig. 4B). In attached HLFs, α-SMA expression was ∼2.5 fold greater on fibrotic lung than normal lung (*, p < 0.001, Fig. 4C). Together, these results demonstrate that fibrotic lung enhances the myofibroblastic phenotype. Key experiments performed on serial decellularized lung tissue sections (32) revealed similar phenotypes to that on non-decelluarized lung tissue (Fig. 5, A–C).
FIGURE 4.

Fibroproliferative lesions enhance fibroblast transdifferentiation into myofibroblasts. Normal human lung fibroblasts were fluorescently labeled and attached to bleomycin-treated lung tissue sections (24 h, 37 °C). Panel A, representative 10× photomicrographs of fibroblast transdifferentiation on normal and fibrotic lung, labeled for F-actin and α-SMA. Panel B, fibroblasts on fibrotic lesions express α-smooth muscle actin and F-actin stress fibers more frequently, and at higher levels, than fibroblasts on normal lung, *, p < 0.001, compared with fibroblasts on normal lung. Panel C, average α-SMA/GAPDH band density in HLFs from normal (NL) versus fibrotic (FL) lung, from three independent experiments, *, p < 0.001 compared with HLFs on normal lung. Inset shows representative Western blot for α-SMA and GAPDH.
FIGURE 5.

Migration and myofibroblast differentiation response to decellularized matrices. Panel A, representative photomicrographs of H&E (20× original magnification, inset 2× magnification) and DAPI (10× original magnification) stained normal and fibrotic lung tissue sections following decellularization, demonstrating intact alveolar architecture and absence of cellular components in the decellularized lung slice. Panel B, HLF migration characteristics were similar in HLFs on normal lung (white bars) versus decellularized normal lung (gray bars); and similar on fibrotic lung (black bars) versus decellularized fibrotic lung (red bars). Panel C, myofibroblast differentiation (F-actin integrated density) was similar in HLFs on untreated (non-decell) versus decellularized (decell) lung tissue sections. Data indicate means of three independent experiments.
Myosin II Activity Is Increased in Fibroblasts on Fibrotic Lung
As myosin II has been shown to be important for generating intracellular tension necessary for both cell migration and myofibroblast transdifferentiation (24–28), we first analyzed whether phosphorylated myosin light chain (p-MLC) levels, a metric for myosin II activity, was different in HLFs seeded on normal or fibrotic lung. Immunofluorescence for p-MLC in fibroblasts on fibrotic lung demonstrated increased p-MLC oriented along stress fibers diffusely throughout the cell (Fig. 6A, bottom). In contrast, p-MLC was less in HLFs on normal lung, and localized more to the periphery of the cell (Fig. 6A, top). By analyzing either the integrated density signal (Fig. 6B) or protein levels in cells attached to lung tissue (Fig. 6C), we determined that myosin phosphorylation was more than 2-fold greater on fibrotic lung (*, p < 0.05 compared with normal lung,). In summary, p-MLC is increased and more diffusely distributed in HLFs on fibrotic lung.
FIGURE 6.

Phosphorylated myosin II is increased on fibrotic lung. Normal human lung fibroblasts were fluorescently labeled and allowed to differentiate on bleomycin-treated lung tissue sections (24 h at 37 °C). Panel A, representative 10× photomicrographs of fibroblast spreading on normal (top panel) versus fibrotic (bottom panel) lung, immunostained for phosphorylated MLC (p-MLC, 24 h). White arrows denote peripheral distribution, yellow arrow denotes diffuse distribution. Panel B, quantification of p-MLC staining intensity in fibroblasts on normal and fibrotic lung. *, p < 0.05 compared with normal lung. Panel C, average p-MLC/GAPDH band density in HLFs from normal (NL) versus fibrotic (FL) lung, from three independent experiments; *, p = 0.005 compared with HLFs on normal lung. Inset shows representative Western blot for p-MLC and GAPDH.
Inhibition of Myosin II Restores Migration and Polarity on Fibrotic Lung
To determine the effects of myosin II inhibition on HLF motility in our system, the small molecule inhibitor of myosin ATPase activity, blebbistatin (10 μm), was added. In HLFs on normal lung, myosin II inhibition resulted in a decrease in migration distance (bleb; 51 ± 30 versus no bleb; 85 ± 55 μm; *, p < 0.001) and migration speed by 45% (bleb; 8.5 ± 5.1 versus no bleb; 14.2 ± 9.3 μm/hr; *, p < 0.001) (Fig. 7, A, top; B, white bars). Migration persistence in HLFs on normal lung was decreased (bleb; 0.37 ± 0.23 versus no bleb; 0.86 ± 0.08 AU; *, p = <0.001), while the number of protrusions significantly increased (bleb; 13 ± 5 versus no bleb; 6 ± 3; *, p = <0.001), resulting in a 76% decrease in cell polarization (bleb; 0.03 ± 0.02 versus no bleb; 0.16 ± 0.06 AU; *, p = <0.001). On the other hand, HLFs on fibrotic lung in the presence of blebbistatin showed an ∼33% increase in migration distance (bleb; 70 ± 37 versus no bleb; 51 ± 38, *, p = 0.03) and speed (bleb; 11.6 ± 6.3 versus no bleb; 8.5 ± 6.4; *, p = 0.03) (Fig. 7, A, bottom; B, black bars). On fibrotic lung, migration persistence was increased (bleb; 0.8 ± 0.1 versus no bleb; 0.5 ± 0.2 AU, *, p < 0.001) with formation of fewer protrusions (bleb; 11 ± 7 versus no bleb; 14 ± 5; *, p = 0.03), leading to a >2-fold increase in polarity (bleb; 0.1 ± 0.07 versus no bleb; 0.05 ± 0.02 AU; *, p < 0.001). Using a second complementary method of myosin II inhibition, siRNA to myosin heavy chain IIB (MYH10, 100 nm, >70% knockdown efficiency, Fig. 8A), effects similar to those using blebbistatin were observed (Fig. 8, B and C). These data demonstrate that myosin II limits protrusive activity and promotes polarized migration in HLFs on normal lung, while enhancing protrusive activity and limiting polarized migration in HLFs on fibrotic lung.
FIGURE 7.

Pharmacologic inhibition of myosin II restores migration and polarity on fibrotic lung. Normal human lung fibroblasts were fluorescently labeled and allowed to migrate on bleomycin-treated lung tissue sections (6 h at 37 °C, ± 10 μm blebbistatin). Panel A, wind rose plots with 10 representative migration tracks from each indicated condition (6 h). Panel B, comparison of quantitative migration characteristics of HLFs on normal and fibrotic lung (± 10 μm blebbistatin, 6 h); *, p < 0.05 compared with corresponding lung control without blebbistatin.
FIGURE 8.

Myosin heavy chain IIB knockdown restores migration and polarity in fibroblasts on fibrotic lung. Panel A, Western blot demonstrating knockdown of IIB protein in the presence of MHC IIB siRNA for 72 h, compared with non-targeting (NT) control and MHC IIA siRNA. Panel B, wind rose plots with 10 representative migration tracks from each condition (6 h). Panel C, comparison of migration characteristics of HLFs on normal and fibrotic lung (± 100 nm MHC IIB siRNA or NT control, 6 h; *, p < 0.05 compared with corresponding normal or fibrotic lung control with NT control siRNA.
Inhibition of Myosin II Selectively Blocks Myofibroblast Transdifferentiation of Fibroblasts on Fibrotic Lung
To determine the effects of myosin II inhibition on myofibroblast transdifferentiation in our system, HLFs seeded on normal and fibrotic lung were incubated for 24 h in the presence of blebbistatin or serum-free media with vehicle (DMSO). On normal lung, inhibition of myosin II had no effect on F-actin stress fibers or α-SMA expression (Fig. 9). In contrast, inhibition of myosin II in HLFs on fibrotic lung significantly decreased the number of cells expressing F-actin stress fibers and those expressing α-SMA, compared with untreated cells. The mean intensity of F-actin stress fibers was decreased by 68% and α-SMA was reduced by 70%. Inhibition of myosin II using MYH10 siRNA (100 nm) resulted in a similar reduction in stress fiber and α-SMA intensity selectively on fibrotic lung (68% decrease in F-actin, 54% decrease in α-SMA, *, p < 0.001), as compared with HLFs on normal lung (Fig. 10). In summary, myosin II inhibition almost completely abrogated fibrotic lung induction of myofibroblast differentiation, suggesting that myosin II plays a key role in myofibroblast differentiation selectively on fibrotic lung.
FIGURE 9.

Pharmacologic inhibition of myosin II selectively blocks differentiation in fibroblasts on fibrotic lung. Normal human lung fibroblasts were allowed to transdifferentiate on bleomycin-treated lung tissue sections (24 h, 37 °C, ± 10 μm blebbistatin). Panel A, representative 10× photomicrographs of fibroblasts on normal and fibrotic lung stained for F-actin stress fibers. Panel B, representative 10× photomicrographs of HLFs on normal and fibrotic lung stained for α-SMA. Panel C, effect of blebbistatin on myofibroblastic phenotype on normal versus fibrotic lung; *, p < 0.05 relative to fibrotic untreated control.
FIGURE 10.

Myosin heavy chain IIB knockdown selectively blocks differentiation in fibroblasts on fibrotic lung. Normal human lung fibroblasts were allowed to transdifferentiate on bleomycin-treated lung tissue sections (24 h, 37 °C, ± 100 nm MHC IIB siRNA or 100 nm non-targeting (NT) control siRNA). Panel A, representative 10× photomicrographs of fibroblasts on normal and fibrotic lung stained for F-actin stress fibers (see 2× magnification inset). Panel B, effect of MHC IIB siRNA on F-actin density in HLFs on normal versus fibrotic lung; *, p < 0.001 compared with change on normal ± IIB siRNA. Panel C, representative 10× photomicrographs of HLFs on normal and fibrotic lung stained for α-SMA. Panel D, effect of MHC IIB siRNA on α-SMA density in HLFs on normal versus fibrotic lung; *, p < 0.001 compared with change on normal ± IIB siRNA.
Myosin II Inhibition on Substrates of Different Stiffness Recapitulates What Is Seen in Normal and Fibrotic Lung
To determine if differences in tissue stiffness between normal and fibrotic lung were the driving signal for the observed phenotypic changes, HLFs were allowed to attach to and migrate on fibronectin-coated polyacrylamide gels of 1 kPa (representing the stiffness of normal lung) and 25 kPa (representing the stiffness of fibrotic lung) (16) ± blebbistatin. Inhibition of myosin II resulted in a >3-fold increase in the number of protrusions on 1 kPa gels, but caused a 22% decrease in protrusions on 25 kPa gels (Fig. 11A). In addition, blebbistatin resulted in a greater than 75% decrease in polarity on 1 kPa gels, while having the completely opposite effect in polarity on 25 kPa gels (40% increase, Fig. 11B). When HLFs were incubated on 1 kPa gels for 24 h, there was no difference in actin density with TGF-β or blebbistatin (Fig. 11C). The increased stiffness of 25 kPa gels resulted in increased actin density, which was augmented by TGF-β. Blebbistatin reduced actin density by more than half selectively on 25 kPa gels. On 1 kPa gels there was a paradoxical increase in α-SMA with blebbistatin, but α-SMA density was otherwise unaffected by TGF-β (Fig. 11D). Similar to the results with actin density, blebbistatin significantly reduced α-SMA selectively on 25 kPa gels. In the absence of blebbistatin, there were clear differences in baseline motility between HLFs on normal lung and 1 kPa gels, and between HLFs on fibrotic lung and 25 kPa gels. However, the effect of blebbistatin on migration was similar in both systems (Fig. 11E). Further, increased stiffness resulted in the formation of vinculin-containing focal adhesions, which was decreased by myosin II inhibition (Fig. 12). Together, these data demonstrate that both myosin II-mediated polarized migration and myofibroblast differentiation are recapitulated under conditions of matrix stiffness in the pathophysiological range.
FIGURE 11.

Myosin II inhibition on varying stiffness recapitulates what is seen in normal and fibrotic lung. Normal human lung fibroblasts were fluorescently labeled and allowed to migrate on 1 and 25 kPa polyacrylamide gels (A and B; 6 h at 37 °C, ± 10 μm blebbistatin) or allowed to attach for 1 h then incubated in the presence of indicated reagents (C and D; 24 h at 37 °C). Panel A, comparison of the effect of blebbistatin and varying stiffness on number of protrusions (*, p < 0.001 versus 1 kPa vehicle only; +, p = 0.01 versus 25 kPa vehicle only. Panel B, comparison of the effect of blebbistatin and varying stiffness on cell polarity (*, p < 0.001 versus 1 kPa vehicle only; +, p = 0.001 versus 25 kPa vehicle only). 90 cells were analyzed for each condition in three separate experiments. Panel C, comparison of the effects of TGF-β and blebbistatin on actin density (+, p = 0.01 versus 25 kPa control; *, p < 0.001 versus 25 kPa TGF only, p < 0.001 by ANOVA). Panel D, comparison of the effects of TGF-β and blebbistatin on α-SMA density (*, p = 0.01 versus 25 kPa TGF only; +, p < 0.001 versus 1 kPa control by t test; p < 0.001 by ANOVA). 10 cells were analyzed for each condition in three separate experiments. Panel E, wind rose plots illustrating the differences in baseline migration characteristics of HLFs on PA gels versus lung tissue, and similarities in the effect of blebbistatin on migration (6 h, 10 μm blebbistatin).
FIGURE 12.

Myosin II inhibition decreases vinculin-containing focal adhesions on fibrotic lung-range stiffness. Panel A, representative photomicrographs (20× magnification) demonstrating the effects of myosin inhibition on FA size in HLFs on 25 kPa gels (24 h, TGF-β 1 ng/ml, ± blebbistatin 10 μm, inset shows 2× magnification of vinculin-containing FAs). NOTE: No FAs were identified on 1 kPa gels ± TGF-β. Panel B, quantification of mean FA size on 25 kPa gels ± blebbistatin (* denotes p < 0.001 compared with TGF only (no blebbistatin)). Area of individual FAs measured within individual cells from each condition then averaged, >30 cells for each condition from three separate experiments analyzed using ImageJ Pro.
Discussion
In this study, we utilize our unique murine lung tissue model to explore the effect of either normal or fibrotic lung matrix on the activation of myosin II in fibroblasts and its consequences on migration or myofibroblast differentiation. The major findings in this study are that normal lung matrix promotes fibroblast motility and polarization, while fibrotic lung matrix immobilizes the fibroblast and promotes myofibroblast differentiation. These context-specific phenotypes are both mediated through non-muscle myosin II activity, as demonstrated using two independent methods to inhibit myosin II function. Importantly, differences in matrix stiffness alone can mediate this context-specific phenotype through activation of myosin II. These data expand the role of myosin II in fibrosis as a downstream effector of the pro-fibrotic phenotype, and predict that the biophysical properties of the matrix play a critical role in influencing fibroblast pro-fibrotic behavior through myosin II.
We show for the first time that normal and fibrotic lung tissue matrices have opposing effects on polarized migration of fibroblasts, through opposing actions on protrusive activity in the direction of migration. Several findings suggest that the underlying fiber topography is the dominant driver of the polarized migration pattern on normal lung. The highly polarized nature of the migration, myosin-dependence of polarized migration, and myosin-inhibitory effect on lateral protrusive activity in fibroblasts on normal lung all strikingly resemble that seen in prior work using linear pre-patterned substrates (33, 34). Indeed, normal lung exhibits a predominantly unidirectional fiber organization in contrast to the random fiber organization in fibrotic lung and PA gels (34, 35). Moreover, the observed cortical localization of activated myosin in fibroblasts on normal lung further supports the previously described role of cortical myosin in limiting lateral protrusions, and thereby establishing cell polarity (25, 36, 37). Taken together, these data suggest that cortical myosin inhibits lateral protrusions to drive polarized migration when the underlying matrix substrate is linearly organized.
Conversely, under combined conditions of increased stiffness and disorganized fibers (fibrotic lung and stiff PA gels), the high level of protrusive activity did not lead to polarized migration. Moreover, high levels of activated myosin were diffusely distributed throughout the cell. Computer modeling of protrusive force coupling to matrix compliance suggests that increased myosin II-generated intracellular tension, in response to high matrix stiffness, can actually decrease polarized migration through myosin's enhancing effect on randomly-directed focal adhesion maturation (38–44). Extending the work of Hinz (41) and Dugina et al. (45), we demonstrate that fibrotic lung-range stiffness induces myosin II-dependent maturation of vinculin-containing focal adhesions. Moreover, myosin inhibition on fibrotic lung or stiff PA gels decreased lateral protrusive activity and enhanced polarized migration, effects noted previously on infinitely stiff tissue culture plastic (25, 46, 47). Taken together, these data suggest that diffuse high levels of myosin activity immobilize fibroblasts on stiff substrates through inducing lateral focal adhesion maturation.
We observed that that the basal level of myofibroblast differentiation on normal lung and soft gels was independent of myosin. However, enhanced myofibroblast differentiation on fibrotic lung and stiff gels, which share the common features of increased stiffness and disorganized fibers, exhibited strict myosin dependence. Stiff matrices composed of either chemically immobilized fibronectin matrix (48), tissue-derived three-dimensional matrices (49), or collagen (41) all support the development of myofibroblasts. Furthermore, increases in intracellular tension upon application of force to the cell membrane have been shown to initiate myofibroblast differentiation (43). Along with these observations, our data highlight the dominance of increased matrix stiffness above matrix fiber organization in driving myofibroblast differentiation. Interestingly, we observed a significant increase in α-SMA on 1 kPa gels with myosin inhibition (Fig. 11D) and a tendency toward increased α-SMA on normal lung with myosin II inhibition or down-regulation (Fig. 10, C and D). A similar soft substrate-specific effect on actin polymerization has been noted upon myosin inhibition in fibroblasts (50). Further, we have recently shown similar results upon inhibition of the mechanosensing ion channel, TRPV4 in fibroblasts (51). Taken together, these data provide evidence to suggest that the balance between actomyosin intracellular tension and matrix stiffness conditions the cell response to myosin inhibition. We hypothesize that, under conditions of soft matrix, where the intracellular tension is greater relative to matrix stiffness, release of this excess tension by inhibition of myosin II induces compensatory actin polymerization. This actin polymerization results in release of MRTF-A (MLK-1) from actin monomers, and consequent α-SMA transcription (52, 53). As similar results were noted with blebbistatin inhibition of myosin activity as with down-regulation of myosin IIB expression, our findings are unlikely to be a consequence of off-target effects of blebbistatin.
We demonstrate that the consequences of activating myosin II on fibroblast pro-fibrotic behavior appears to be an integrated function of matrix fiber organization and stiffness. Our key findings were replicated using decellularized lung tissue sections, validating our model system and suggesting that matrix components, rather than cellular components, drive the cellular responses. Unmeasured variables such as ECM ligand density, spatial dimensionality, and composition, or integrin signaling and de novo protein synthesis, were not directly examined, and could also play a modifying role in vivo. However, the changes observed as early as 6 h remained stable through 24 h, making it unlikely that new protein synthesis had a significant bearing on the phenotype. We also recognize that our murine lung tissue model system may not fully recapitulate the in vivo environment of human IPF. The data generated using this model is complementary to that generated in decellularized matrix systems and clearly demonstrate novel complexities of interaction between fibroblasts and actual lung tissue matrix, which are lost in traditional assays on protein-coated tissue-culture plastic.
Our laboratory and others have shown that IPF-patient derived fibroblasts migrate faster than normal lung fibroblasts. Much of this work was performed with cells on tissue-culture plastic, which exhibits a stiffness 1 million-fold greater than the physiological range of normal lung in vivo (8, 10, 11, 54, 55). The interpretation of this prior work bears some refinement as our current findings of myosin-dependent migration on actual lung tissue contrast with the myosin-independent migration noted on infinitely stiff tissue-culture plastic. There are clear differences in baseline motility on normal lung versus soft gels and fibrotic lung versus stiff gels suggesting stiffness is not the predominant driver of motility. However, the effects of varying stiffness on lateral protrusions and polarized migration are recapitulated on both PA gels and lung tissue, and similarly reversed with myosin inhibition. This data suggests that myosin II facilitates polarized migration through inhibition of protrusions lateral to the direction of migration in response to mechanically transduced signals. The purpose of this study was to characterize the response of normal human lung fibroblasts to normal and fibrotic lung tissue. Future work will explore and compare migration and differentiation of diseased/fibrotic and normal lung fibroblasts to normal and fibrotic matrix.
Prior work has demonstrated that, on tissue culture plastic, myosin II plays a critical role in establishing cell polarity and limiting lateral protrusions (36, 56, 57). It has also been shown that myosin-actin interactions in stress fibers and enlarging focal adhesions play a role in the increased intracellular tension of fibroblast-to-myofibroblast transition (19, 58, 59). In summary, our data demonstrate for the first time that fibroblast behavior is driven in a context- specific manner through activation of myosin II. In the soft substrate of normal lung, we hypothesize that myosin activation in the periphery of the fibroblast limits lateral protrusions, thereby driving migration along routes dictated by the inherent linear lung fiber architecture. Upon encountering the stiff matrix of fibrotic lung, myosin is diffusely activated in stress fibers extending to focal adhesions, resulting in increased intracellular tension, thereby immobilizing the fibroblast and promoting myofibroblast differentiation. Such a mechanism would lead to progressive accumulation of myofibroblasts in fibrotic lung (Fig. 13). These findings open the door to the possibility that a therapeutic intervention aimed at inhibiting myosin II could impede fibrosis through multiple mechanisms in a matrix context-specific manner.
FIGURE 13.
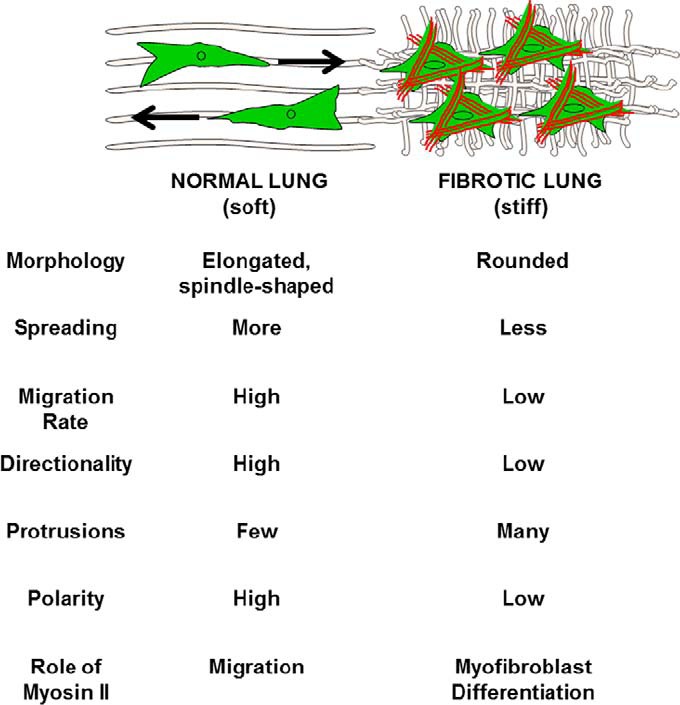
Effect of normal versus fibrotic lung on fibroblast phenotype. The consequence of Myosin II activation differs depending on whether a fibroblast is interacting with normal or fibrotic lung ECM. In normal lung, myosin II activation drives highly directional, polarized migration. In fibrotic lung, myosin II activation results in myofibroblast differentiation. The end result is migration of fibroblasts from normal to fibrotic lung where myofibroblast differentiation is enhanced.
Author Contributions
B. D. S. and M. A. O. conceived and coordinated the study and wrote the paper. L. M. G., S. O. R., S. A., and R. G. S. assisted with experiment design, analysis, and interpretation of the data. K. A. S. performed the animal experiments and maintained the colony. H. S. and E. L. H. assisted with the development of protocols and experiments utilizing decelluarized lung tissue sections. F. L. and D. J. T. assisted with atomic force microscopy data collection and analysis, and with discussion of experimental ideas. T. T. E. and S. R. E. assisted with experiment design and interpretation of data, and provided myosin reagents. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-119792, HL-103553, and HL-085324 (to M. A. O.) and a Genentech Idiopathic Pulmonary Fibrosis Junior Faculty Program Award (to B. D. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- IPF
- idiopathic pulmonary fibrosis
- HLF
- human lung fibroblast
- SMA
- smooth muscle actin
- p-MLC
- phosphorylated myosin light chain.
References
- 1. Wynn T. A. (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bitterman P. B., and Henke C. A. (1991) FIbroproliferative disorders. Chest 99, 81S–84S [DOI] [PubMed] [Google Scholar]
- 3. White E. S., Lazar M. H., and Thannickal V. J. (2003) Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol. 201, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuhn C., and McDonald J. A. (1991) The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 138, 1257–1265 [PMC free article] [PubMed] [Google Scholar]
- 5. Cool C. D., Groshong S. D., Rai P. R., Henson P. M., Stewart J. S., and Brown K. K. (2006) Fibroblast foci are not discrete sites of lung injury or repair. Am. J. Respir. Crit. Care Med. 174, 654–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. King T. E. Jr., Schwarz M. I., Brown K. K., Tooze J. A., Colby T. V., Waldron J. A. Jr., Flint A., Thurlbeck W., and Cherniack R. M. (2001) Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 164, 1025–1032 [DOI] [PubMed] [Google Scholar]
- 7. Phan S. H. (2002) The myofibroblast in pulmonary fibrosis. Chest 122, 286S–289S [DOI] [PubMed] [Google Scholar]
- 8. Cai G. Q., Zheng A., Tang Q., White E. S., Chou C. F., Gladson C. L., Olman M. A., and Ding Q. (2010) Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp. Cell Res. 316, 1600–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ding Q., Gladson C. L., Wu H., Hayasaka H., and Olman M. A. (2008) Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J. Biol. Chem. 283, 26839–26849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grove L. M., Southern B. D., Jin T. H., White K. E., Paruchuri S., Harel E., Wei Y., Rahaman S. O., Gladson C. L., Ding Q., Chapman Jr., Craik C. S., Chapman H. A., and Olman M. A. (2014) Urokinase receptor (u-PA) ligation induces a raft-localized integrin signaling switch that mediates the hypermotile phenotype of fibrotic fibroblasts. J. Biol. Chem. 289, 12791–12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suganuma H., Sato A., Tamura R., and Chida K. (1995) Enhanced migration of fibroblasts derived from lungs with fibrotic lesions. Thorax 50, 984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hiromitsu H., and Sakai T. (2012) Biological significance of local TGF-β activation in liver diseases. Front. Physiol. 3, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tager A. M., LaCamera P., Shea B. S., Campanella G. S., Selman M., Zhao Z., Polosukhin V., Wain J., Karimi-Shah B. A., Kim N. D., Hart W. K., Pardo A., Blackwell T. S., Xu Y., Chun J., and Luster A. D. (2008) The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 [DOI] [PubMed] [Google Scholar]
- 14. Ajayi I. O., Sisson T. H., Higgins P. D. R., Booth A. J., Sagana R. L., Huang S. K., White E. S., King J. E., Moore B. B., and Horowitz J. C. (2013) X-Linked Inhibitor of Apoptosis Regulates Lung Fibroblast Resistance to Fas-Mediated Apoptosis. Am. J. Respir. Cell Mol. Biol. 49, 86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cukierman E., Pankov R., Stevens D. R., and Yamada K. M. (2001) Taking cell-matrix adhesions to the third dimension. Science 23, 1708–1712 [DOI] [PubMed] [Google Scholar]
- 16. Liu F., Mih J. D., Shea B. S., Kho A. T., Sharif A. S., Tager A. M., and Tschumperlin D. J. (2010) Feedback amplification of fibrosis through matrix stiffening and Cox-2 suppression. J. Cell Biol. 190, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mih J. D., Sharif A. S., Liu F., Marinkovic A., Symer M. M., and Tschumperlin D. J. (2011) A multiwell platform for studying stiffness-dependent cell biology. PLoS ONE 6, e19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson L. A., Rodansky E. S., Sauder K. L., Horowitz J. C., Mih J. D., Tschumperlin D. J., and Higgins P. D. (2013) Matrix Stiffness Corresponding to Strictured Bowel Induces a Fibrogenic Response in Human Colonic Fibroblasts. Inflamm. Bowel Dis. 19, 891–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goffin J. M., Pittet P., Csucs G., Lussi J. W., Meister J. J., and Hinz B. (2006) Focal adhesion size controls tension-dependent recruitment of a-smooth muscle actin to stress fibers. J. Cell Biol. 172, 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Y., Huang X., Hecker L., Kurundkar D., Kurundkar A., Liu H., Jin T. H., Desai L., Bernard K., and Thannickal V. J. (2013) Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest. 123, 1096–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Solon J., Levental I., Sengupta K., Georges P. C., and Janmey P. A. (2007) Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 93, 4453–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parker M. W., Rossi D., Peterson M., Smith K., Sikström K., White E. S., Connett J. E., Henke C. A., Larsson O., and Bitterman P. B. (2014) Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 124, 1622–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Booth A. J., Hadley R., Cornett A. M., Dreffs A. A., Matthes S. A., Tsui J. L., Weiss K., Horowitz J. C., Fiore V. F., Barker T. H., Moore B. B., Martinez F. J., Niklason L. E., and White E. S. (2012) Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 186, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bond J. E., Ho T. Q., Selim M. A., Hunter C. L., Bowers E. V., and Levinson H. (2011) Temporal spatial expression and function of non-muscle myosin II isoforms IIA and IIB in scar remodeling. Lab. Invest. 91, 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lo C. M., Buxton D. B., Chua G. C. H., Dembo M., Adelstein R. S., and Wang Y. L. (2004) Nonmuscle myosin IIB is involved in the guidance of fibroblast migration. Mol. Biol. Cell 15, 982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo W. H., and Wang Y. L. (2012) A three-component mechanism for fibroblast migration with a contractile cell body that couples a myosin II-independent propulsive anterior to a myosin II-dependent resistive tail. Mol. Biol. Cell 23, 1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao X. H., Laschinger C., Arora P., Szászi K., Kapus A., and McCulloch C. A. (2007) Force activates smooth muscle a-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 120, 1801–1809 [DOI] [PubMed] [Google Scholar]
- 28. Meyer-ter-Vehn T., Sieprath S., Katzenberger B., Gebhardt S., Grehn F., and Schlunck G. (2006) Contractility as a prerequisite for TGF-b-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest. Ophthalmol. Vis. Sci. 47, 4895–4904 [DOI] [PubMed] [Google Scholar]
- 29. Olman M. A., White K. E., Ware L. B., Simmons W. L., Benveniste E. N., Zhu S., Pugin J., and Matthay M. A. (2004) Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1β-induced IL-6 expression. J. Immunol. 172, 2668–2677 [DOI] [PubMed] [Google Scholar]
- 30. Ding Q., Cai G. Q., Hu M., Yang Y., Zheng A., Tang Q., Gladson C. L., Hayasaka H., Wu H., You Z., Southern B. D., Grove L. M., Rahaman S. O., Fang H., and Olman M. A. (2013) FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am. J. Pathol. 182, 1572–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu S., Gladson C. L., White K. E., Ding Q., Stewart J. Jr., J. E., Jin T. H., Chapman H. A. Jr., and Olman M. A. (2009) Urokinase receptor mediates lung fibroblast attachment and migration towards provisional matrix proteins through interaction with multiple integrins. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L97–L108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun H., Calle E., Chen X., Mathur A., Zhu Y., Mendez J., Zhao L., Niklason L., Peng X., Peng H., and Herzog E. L. (2014) Fibroblast engraftment in the decellularized mouse lung occurs via a β1-integrin-dependent, FAK-dependent pathway that is mediated by ERK and opposed by AKT. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L463–L465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doyle A. D., Wang F. W., Matsumoto K., and Yamada K. M. (2009) One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 184, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toshima M., Ohtani Y., and Ohtani O. (2004) Three-dimensional architecture of elastin and collagen fiber networks in the human and rat lung. Arch. Histol. Cytol. 67, 31–40 [DOI] [PubMed] [Google Scholar]
- 35. Chen Y., Zardi L., and Peters D. M. P. (1997) High-resolution cryo-scanning electron microscopy study of the macromolecular structure of fibronectin fibrils. Scanning 19, 349–355 [DOI] [PubMed] [Google Scholar]
- 36. Vicente-Manzanares M., Ma X., Adelstein R. S., and Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beach J. R., Shao L., Remmert K., Li D., Betzig E., and Hammer III J. A. 3rd (2014) Nonmuscle myosin II isoforms coassemble in living cells. Curr. Biol. 24, 1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bangasser B. L., Rosenfeld S. S., and Odde D. J. (2013) Determinants of maximal force transmission in a motor-clutch model of cell traction in a compliant microenvironment. Biophys. J. 105, 581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chrzanowska-Wodnicka M., and Burridge K. (1996) Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., and Waterman C. M. (2010) Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hinz B. (2006) Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmissions. Eur. J. Cell Biol. 85, 175–181 [DOI] [PubMed] [Google Scholar]
- 42. Huang X., Yang N., Fiore V. F., Barker T. H., Sun Y., Morris S. W., Ding Q., Thannickal V. J., and Zhou Y. (2012) Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 47, 340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hinz B., and Gabbiani G. (2003) Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb. Haemost. 90, 993–1002 [DOI] [PubMed] [Google Scholar]
- 44. Burridge K., Fath K., Kelly T., Nuckolls G., and Turner C. (1988) Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 4, 487–525 [DOI] [PubMed] [Google Scholar]
- 45. Dugina V., Fontao L., Chaponnier C., Vasiliev J., and Gabbiani G. (2001) Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 114, 3285–3296 [DOI] [PubMed] [Google Scholar]
- 46. Even-Ram S., Doyle A. D., Conti M. A., Matsumoto K., Adelstein R. S., and Yamada K. M. (2007) Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat. Cell Biol. 9, 299–309 [DOI] [PubMed] [Google Scholar]
- 47. Liu Z., van Grunsven L. A., Van Rossen E., Schroyen B., Timmermans J. P., Geerts A., and Reynaert H. (2010) Blebbistatin inhibits contraction and accelerates migration in mouse hepatic stellate cells. Br. J. Pharmacol. 159, 304–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Katz B. Z., Zamir E., Bershadsky A., Kam Z., Yamada K. M., and Geiger B. (2000) Physical state of the extracellular matrix regulates the structure and molecular composition of cell-matrix adhesions. Mol. Biol. Cell 11, 1047–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cukierman E., Pankov R., and Yamada K. M. (2002) Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 14, 633–639 [DOI] [PubMed] [Google Scholar]
- 50. Mih J. D., Marinkovic A., Liu F., Sharif A. S., and Tschumperlin D. J. (2012) Matrix stiffness reverses the effect of actomyosin tension on cell proliferation. J. Cell Sci. 125, 5974–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rahaman S. O., Grove L. M., Paruchuri S., Southern B. D., Niese K. A., Abraham S., Scheraga R. G., Ghosh S., Thodeti C.K., Zhang D. X., Moran M. M., Schilling W. P., Tschumperlin D. J., and Olman M. A. (2014) TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Invest. 124, 5225–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sandbo N., Kregel S., Taurin S., Bhorade S., and Dulin N. O. (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-b. Am. J. Respir. Cell Mol. Biol. 41, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Johnson L. A., Rodansky E. S., Haak A. J., Larsen S. D., Neubig R. R., and Higgins P. D. (2014) Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-[β]-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 20, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pierce E. M., Carpenter K., Jakubzick C., Kunkel S. L., Evanoff H., Flaherty K. R., Martinez F. J., Toews G. B., and Hogaboam C. M. (2007) Idiopathic pulmonary fibrosis fibroblasts migrate and proliferate to CC chemokine ligand 21. Eur. Respir. J. 29, 1082–1093 [DOI] [PubMed] [Google Scholar]
- 55. Tang X., Peng R., Phillips J. E., Deguzman J., Ren Y., Apparsundaram S., Luo Q., Bauer C. M., Fuentes M. E., DeMartino J. A., Tyagi G., Garrido R., Hogaboam C. M., Denton C. P., Holmes A. M., Kitson C., Stevenson C. S., and Budd D. C. (2013) Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in-vivo models of lung fibrosis. Am. J. Pathol. 183, 470–479 [DOI] [PubMed] [Google Scholar]
- 56. Totsukawa G., Wu Y., Sasaki Y., Hartshorne D. J., Yamakita Y., Yamashiro S., and Matsumura F. (2004) Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Conti M. A., and Adelstein R. S. (2008) Nonmuscle myosin II moves in new directions. J. Cell Sci. 121, 11–18 [DOI] [PubMed] [Google Scholar]
- 58. Tomasek J. J., Haaksma C. J., Schwartz R. J., and Howard E. W. (2013) Whole animal knockout of smooth muscle α-actin does not alter excisional wound healing or the fibroblast-to-myofibroblast transition. Wound Repair Regen. 21, 166–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Singer I. I., Kawka D. W., Kazazis D. M., and Clark R. A. (1984) In vivo co-distribution of fibronectin and actin fibers in granulation tissue: immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J. Cell Biol. 98, 2091–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]