

Abstract
With clinical trials ongoing, efficient clinical production of adeno-associated virus (AAV) to treat large numbers of patients remains a challenge. We compared distribution of AAV8 packaged with Factor VIII (FVIII) in cell culture media and lysates on days 3, 5, 6, and 7 post-transfection and found increasing viral production through day 6, with the proportion of viral particles in the media increasing from 76% at day 3 to 94% by day 7. Compared to FVIII, AAV8 packaged with Factor IX and Protective Protein/Cathepsin A vectors demonstrated a greater shift from lysate towards media from day 3 to 6, implying that particle distribution is dependent on recombinant vector. Larger-scale productions showed that the ratio of full-to-empty AAV particles is similar in media and lysate, and that AAV harvested on day 6 post-transfection provides equivalent function in mice compared to AAV harvested on day 3. This demonstrates that AAV8 production can be optimized by prolonging the duration of culture post-transfection, and simplified by allowing harvest of media only, with disposal of cells that contain 10% or less of total vector yield. Additionally, the difference in particle distribution with different expression cassettes implies a recombinant vector-dependent processing mechanism which should be taken into account during process development.
Introduction
Adeno-associated virus (AAV) is a powerful gene delivery vehicle capable of safely transducing a variety of tissues to provide long-term expression.1–4 As a result, AAV has long been considered a potential therapeutic vector, with the first clinical trial using AAV starting in 1995 (ref. 5), and the first AAV therapy receiving approval from a regulatory commission for clinical use in 2012 (ref. 6). With a number of clinical trials currently ongoing, the efficient clinical production of sufficient quantities of AAV to treat large numbers of patients remains a significant challenge.
Current cellular systems for producing AAV at scale include the use of adherent 293 cells in cell factories or roller bottles,7,8 suspension 293 cells,9–12 insect cells in suspension using the baculovirus system,13 HeLa cells stably expressing rep/cap genes,14 and coinfection with herpes simplex virus in baby hamster kidney cells.15,16 While each of these systems have unique advantages and disadvantages, none is dramatically more efficient than the rest at generating AAV on a per cell or per culture volume basis. Therefore, in place of a substantially more efficient system, it is necessary to optimize current AAV production methods to the greatest extent possible.
In addition to increasing production of AAV, streamlining the purification process is also essential to clinical production. Typically, cell lysates are harvested alone, though they can be harvested along with cell culture media, with purification of AAV from both fractions. Producing AAV in adherent cell factories involves decanting media from the cell factories and adding ethylenediaminetetraacetic acid to detach the cells, followed by centrifugation and freeze-thawing and/or microfluidization.7 This process, when performed on up to hundreds of cell factories over the course of several weeks, is extremely cumbersome and time-intensive. A suspension system may make cell harvest more efficient, but would nevertheless likely involve either Triton-X100 or large-scale microfluidization.10,13 In addition, any methods which process cell lysates and culture media together have a greater burden in terms of DNA, RNA, and proteins, complicating downstream purification. An ideal method would restrict purification to one fraction (either lysate or, preferably, culture media) and thus minimize downstream processing.
Vandenberghe et al.17 and Lock et al.18 both described serotype-dependent release of AAV particles from cells into cell culture media during production. By using quantitative polymerase chain reaction (qPCR) to measure viral genomes, both papers show that the majority of viral genomes are located in the media at 72 hours with calcium phosphate-based transfection (Vandenberghe et al.17) or at 120 hours with polyethylenimine-based transfection (Lock et al.18). The notable exception was AAV2, which was closely associated with cells, apparently due to its heparin sulfate binding domain.17 Furthermore, Okada et al.19 also showed a sevenfold increase in AAV8 particles in the cell culture media compared to the lysate.
These papers indicate that it should be possible to harvest cell culture media alone during AAV8 production, while losing only a small percentage of product and greatly streamlining the downstream process. To that end, we tested the distribution of AAV8 particles in cell lysates and culture media on days 3, 5, 6, and 7 post-transfection. Using an enzyme-linked immunosorbent assay (ELISA) kit to detect full and empty assembled AAV8 particles,20,21 we found, and show for the first time, a gradual shift in distribution from lysates to media with a daily increase in total AAV production through day 6, allowing for harvest of media only with an overall increase in yield compared to day 3. Furthermore and intriguingly, we found that this shift in distribution is dependent on the recombinant vector. Finally, we found that the full-to-empty particle ratio is equivalent in both lysate and media fractions, and that AAV harvested on day 6 is equivalent in function, after systemic injection in mice, to AAV harvested on day 3.
Results
Distribution of AAV8 particles in cell lysates and culture media changes with time
Four groups (n = 3/group) of HEK 293T/17 cells on 15 cm plates were transfected to produce AAV8 packaged with a 5.1 kb single-stranded genome expressing the human Factor VIII (FVIII) gene.22 Cells and culture media (containing 10% fetal bovine serum in this and all subsequent experiments) were harvested on days 3, 5, 6, and 7 and analyzed by AAV8 capsid ELISA to determine the distribution of assembled viral particles in lysates and media. As shown in Figure 1, AAV-FVIII production increased when culture duration was extended from day 3 to day 5, day 6, and day 7 post-transfection, with day 5 showing a 9% increase over day 3 and day 6 showing a 24% increase over day 3 (all groups significantly different versus day 3 as measured by analysis of variance, P = 0.004). Viral titer decreased from day 6 to 7, but was still 14% higher than on day 3. Seventy-six percent of AAV-FVIII particles were in the culture media at day 3, and this proportion increased to 87% by day 5, 91% by day 6, and 94% by day 7.
Figure 1.
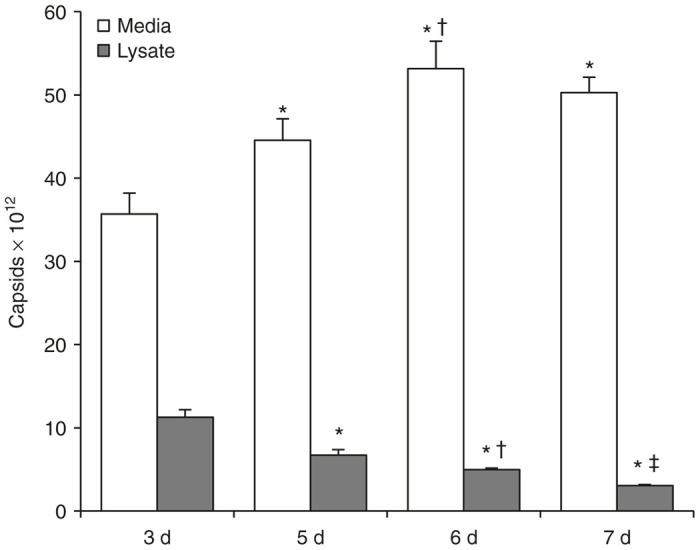
Distribution of adeno-associated virus (AAV)8 particles in cell lysates and culture media changes with time. Four groups (n = 3/group) of HEK 293T/17 cells on 15 cm plates were transfected to produce AAV-FVIII. Cells and culture media were harvested on days 3, 5, 6, and 7 and analyzed by AAV8 capsid enzyme-linked immunosorbent assay to determine the distribution of viral genomes. AAV-FVIII showed an increase in production when culture was extended from day 3 (1.1 × 1013 ± 9.2 × 1011 and 3.6 × 1013 ± 2.5 × 1012 total capsids in the lysate and media, respectively) to day 5 (6.7 × 1012 ± 6.7 × 1011 and 4.5 × 1013 ± 2.6 × 1012 total capsids in the lysate and media, respectively), day 6 (5.0 × 1012 ± 1.9 × 1011 and 5.3 × 1013 ± 3.3 × 1012 total capsids in the lysate and media, respectively), and day 7 (3.0 × 1012 ± 1.3 × 1010 and 5.0 × 1013 ± 1.9 × 1012 total capsids in the lysate and media, respectively) post-transfection, with day 5 showing a 9% increase over day 3 and day 6 showing a 24% increase over day 3 (* denotes significant difference versus day 3; Ɨ denotes significant difference versus day 5; and ǂ denotes significant difference versus day 6 as measured by analysis of variance, P = 0.004). Viral titer decreased from day 6 to 7, but was still 14% higher than on day 3. 76% of AAV-FVIII particles were in the culture media at day 3, and this proportion increased to 87% by day 5, 91% by day 6, and 94% by day 7.
Distribution of AAV8 particles in cell lysates and culture media is dependent on the recombinant vector
Six groups (n = 3/group) of 293T/17 cells on 10 cm plates were transfected to produce AAV-FVIII, or AAV packaged with human Factor IX (FIX),23,24 or AAV packaged with human Protective Protein/Cathepsin A (PPCA; which, along with FIX, is a self-complementary genome of approximately 2.4 kb).25 Cells and culture media were harvested on day 3 or 6 to determine whether AAV distribution was influenced by recombinant vector. As shown in Figure 2, at days 3 and 6 post-transfection, the majority of AAV8 particles were located in the media for all three vectors that were packaged; however, this ratio changed more dramatically for FIX and PPCA than for FVIII. At day 3, 71% of the FVIII particles were in the media, while 65% of the FIX and 59% of the PPCA particles were in the media (the FVIII and PPCA groups were significantly different as measured by analysis of variance, P = 0.008). By day 6, 92% of FVIII, 90% of FIX and 88% of PPCA particles were in the media, indicating a greater shift in AAV particle distribution for FIX and, especially, PPCA. Interestingly, all three vectors showed a 14% increase in total AAV production by day 6 versus day 3.
Figure 2.
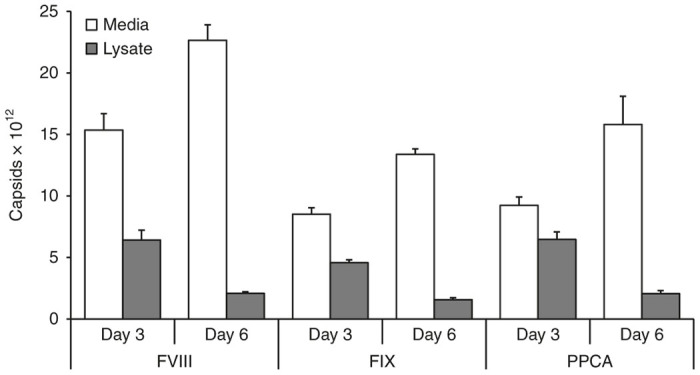
Distribution of adeno-associated virus (AAV)8 particles in cell lysates and culture media is dependent on the recombinant vector. Six groups (n = 3/group) of 293T/17 cells on 10 cm plates were transfected to produce AAV-FVIII, AAV-FIX, or AAV-PPCA. Cells and culture media were harvested on day 3 or 6 to determine whether AAV distribution was influenced by the recombinant vector. At days 3 and 6 post-transfection, the majority of AAV8 particles were located in the media for all three vector genomes, but this ratio changed more dramatically for FIX and PPCA than for FVIII. At day 3, 71% of the FVIII particles were in the media, while 65% of the FIX and 59% of the PPCA particles were in the media (the FVIII and PPCA groups were significantly different as measured by analysis of variance, P = 0.008). By day 6, 92% of FVIII, 90% of FIX and 88% of PPCA particles were in the media, indicating a greater shift in AAV particle distribution for FIX and, especially, PPCA.
Viral genome distribution in cell lysates and culture media tracks with particle distribution
To determine whether AAV viral genome distribution followed the same pattern as AAV8 capsid distribution, three individual 10-stack cell factories containing 293T/17 cells were transfected to produce AAV-FVIII. This experiment was important to verify that empty AAV particles did not preferentially traffic to the culture media, which would result in a high ratio of empty to full particles. Cell lysates and media were harvested 6 days after transfection, and were processed separately for each cell factory through size-exclusion chromatography and anion exchange. As shown in Figure 3, qPCR revealed that an average of 1.6 × 1013 ± 9.6 × 1011 and 2.6 × 1012 ± 3.4 × 1011 total viral genomes were in the media and lysate, respectively. With an average of 85.9 ± 1.8% of FVIII genomes in the media across the three cell factories, the ratio of full-to-empty AAV particles is nearly equivalent in both media and the cell lysates.
Figure 3.
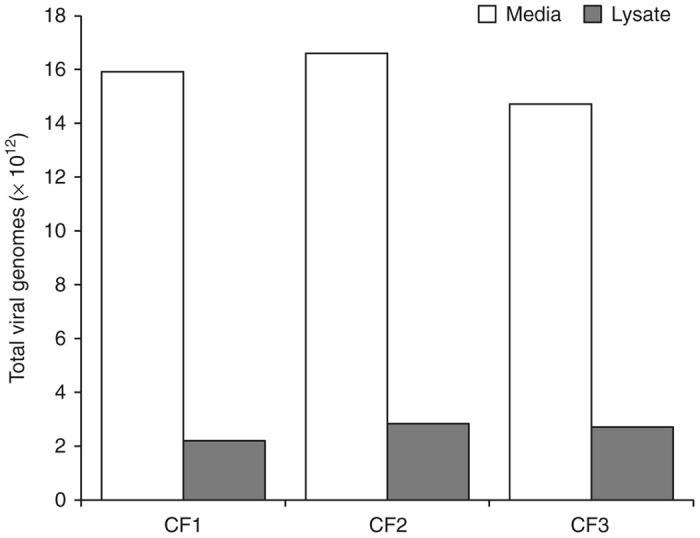
Viral genome distribution in cell lysates and culture media tracks with particle distribution. Three 10-stack cell factories (CF1, CF2, and CF3) containing 293T/17 cells were transfected to produce adeno-associated virus (AAV)-FVIII. Cell lysates and media were harvested 6 days after transfection, and were processed separately for each cell factory. Quantitative polymerase chain reaction revealed that an average of 1.6 × 1013 ± 9.6 × 1011 and 2.6 × 1012 ± 3.4 × 1011 total viral genomes were in the media and lysate, respectively, with an average of 85.9 ± 1.8% of FVIII genomes in the media across the three cell factories.
AAV harvested at D6 provides comparable expression to AAV harvested at D3
To ensure that AAV harvested at later time points was as effective at providing therapeutic expression as AAV harvested at earlier time points, two preparations of three 10-stack cell factories of AAV-FVIII were purified after harvest of cell lysates and culture media at either day 3 or day 6 post-transfection. AAV-FVIII was injected into C57Bl/6 mice at a dose of 5 × 109, 2 × 1010, or 2 × 1011 viral genomes per mouse and plasma FVIII levels were analyzed 7 days postinjection (n = 4–5/group). Figure 4 shows mean FVIII levels in the three dose ranges at both time points, with comparable expression between AAV preparations (21.0 ± 7.6 ng/ml for D3 versus 15.0 ± 6.0 ng/ml for D6 for the low dose; 189.2 ± 83.9 ng/ml for D3 versus 113.7 ± 68.5 ng/ml for D6 for the medium dose; and 1,181.5 ± 371.5 ng/ml for D3 versus 1,019.2 ± 69.9 ng/ml for D6 at the high dose). While AAV-FVIII harvested at day 3 post-transfection provides a slightly higher level of expression at all three time points, these differences were not statistically significant (P = 0.26, P = 0.19, and P = 0.37 at the low, medium, and high doses, respectively).
Figure 4.
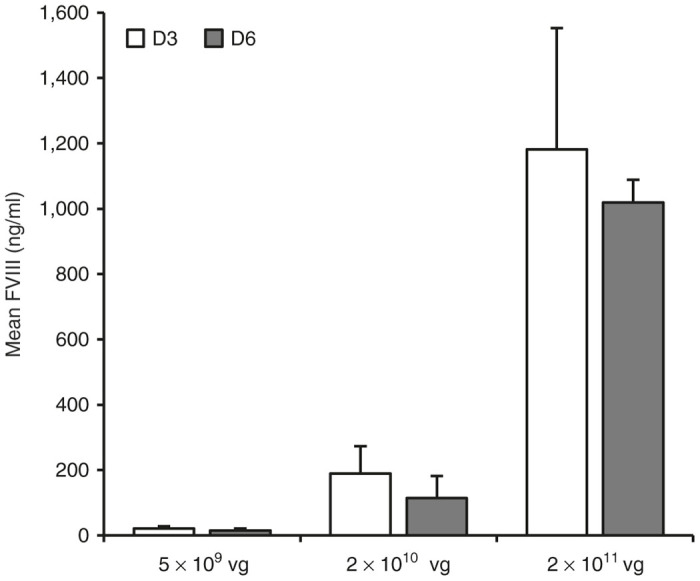
Adeno-associated virus (AAV) harvested at day 6 provides comparable expression to AAV harvested at day 3. Two preparations of three 10-stack cell factories of AAV-FVIII were purified after harvest of cell lysates and culture media at either day 3 or day 6 post-transfection. AAV-FVIII was injected into C57Bl/6 mice at a dose of 5 × 109, 2 × 1010, or 2 × 1011 viral genomes per mouse and plasma FVIII levels were analyzed 7 days postinjection (n = 4–5/group). Expression was comparable between AAV preparations (21.0 ± 7.6 ng/ml for D3 versus 15.0 ± 6.0 ng/ml for D6 for the low dose; 189.2 ± 83.9 ng/ml for D3 versus 113.7 ± 68.5 ng/ml for D6 for the medium dose; and 1,181.5 ± 371.5 ng/ml for D3 versus 1,019.2 ± 69.9 ng/ml for D6 at the high dose). Though AAV-FVIII harvested at day 3 post-transfection provides a slightly higher level of expression at all three time points, these differences were not statistically significant (P = 0.26, P = 0.19, and P = 0.37 at the low, medium, and high doses, respectively).
Discussion
While previous studies have looked at the distribution of various AAV serotypes in the lysate and culture media, this is the first, to our knowledge, to demonstrate a gradual shift from lysate to media with time and a dependence on the recombinant vector. As reported previously,18 this shift takes place without any evident cytopathic effect: adherent 293 cells cultured in 10% fetal bovine serum appear under the microscope on day 6 post-transfection as attached, confluent, and similarly healthy to cells at day 3. Interestingly, however, our experience shows that changing media 24 hours after transfection to serum-free Dulbecco’s Modified Eagle’s Medium and maintaining culture until day 6 leads to a large amount of cell detachment and death, but similar AAV yields in the media (data not shown). Because cells remain healthy in extended culture in the presence of serum, it may be possible to provide further supplements to encourage increased AAV production at later time points.
Importantly, the shift of AAV8 particle distribution toward the media is not primarily an accumulation of empty capsids, as has been speculated.26 Instead, qPCR shows that 86% of viral genomes are located in the media, compared to 90% of capsids. This indicates that the full-to-empty particle ratio may be slightly lower in the media than the lysate, but this difference is likely to be unimportant, especially with downstream procedures that separate full AAV particles from empty particles. Furthermore, in the case of working with adherent 293 cells, the added labor and complications of processing the lysate are not worthwhile for the small increase in yield.
The mechanism behind the dependence on recombinant vector is unknown. AAV-FVIII was more prevalent in the media by day 3 post-transfection, with between 71–76% of total particles in the media, compared to 65 and 59% for FIX and PPCA, respectively. This dependence on recombinant vector could include factors such as size, genome sequence, self-complementarity, vector production levels within the cell, or transgene toxicity, and would be an interesting topic for future studies to address. Nevertheless, this difference indicates that groups should optimize their processes for each recombinant vector that goes into production, rather than assuming a given time point will provide the same AAV particle distribution, even within a given serotype.
While most groups harvest AAV preps at some point between day 2 and day 5 post-transfection, we show here that extended culture to day 6 is optimal for maximizing yields of AAV8 packaged with FVIII, although this finding could well change with a different cellular production system or transfection method. Indeed, as Figure 1 shows, at day 6 there were more AAV-FVIII particles in the media alone than in the media and lysate combined at day 3. In some cases, extension to day 7 could prove worthwhile as well. While we saw a small decrease in production from day 6 to day 7, the difference in total number of capsids was not significant between those two time points. Importantly, there were no statistically significant differences in expression in mice between groups injected with AAV-FVIII harvested at day 3 or day 6 post-transfection, indicating that extended time in culture does not lead to degradation of the AAV8 particle or decreased transduction efficiency in vivo.
In conclusion, increasing the duration of culture post-transfection can significantly improve yields of AAV8 and shift the distribution of particles more greatly toward the culture media. This has important implications for large-scale production, during which the media may be harvested alone, eliminating the more time- and labor-intensive processing of cell lysate, which may contain 10% or less of the total particle yield. Additionally, the differences in particle distribution of different expression cassettes at day 3 post-transfection implies a recombinant vector-dependent processing mechanism which should be taken into consideration when developing upstream production processes.
Materials and Methods
Cell culture and transfection
Adherent HEK 293T/17 cells were cultured in 10 cm plates, 15 cm plates, or 10-stack cell factories (Corning, Corning, NY) using Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum supplemented with 2 mmol/l GlutaMAX (Life Technologies, Grand Island, NY). AAV was produced by two-plasmid transfection using PEIpro (Polyplus-transfection SA, Illkirch, France) 1 day after seeding cells at a density of 7.26 × 104 cells/cm2. Three different viral genomes flanked by AAV2 inverted terminal repeats were packaged into AAV8 capsids: human Factor VIII, human Factor IX, and human Protective Protein/Cathepsin A. While the FVIII genome was single-stranded, FIX and PPCA cassettes included a mutated inverted terminal repeat for packaging of self-complementary genomes. The backbones of genome-containing plasmids also contained the adenoviral helper genes required for AAV production, and the second plasmid in each transfection contained the AAV2 Rep and AAV8 capsid genes. Cell cultures were maintained from between 3 and 7 days post-transfection. For cultures lasting longer than 3 days, additional media (50% of the original transfection volume) was added at day 3 to provide cells with additional nutrients.
AAV harvest
At the time of harvest, culture media were pipetted off plates or decanted from individual cell factories, after which cells were detached using PBS with 5 mmol/l ethylenediaminetetraacetic acid. Cells were pelleted, resuspended in TD buffer, and subjected to five freeze/thaw cycles. Cell lysates were treated with benzonase and centrifuged at high speed to pellet debris. The lysate supernatants containing AAV were removed and pellets were discarded.
Large-scale AAV-FVIII purification
After decanting from cell factories, culture media were filtered through 0.22 micron filters and concentrated by tangential-flow filtration using a 0.005 m2 Pellicon XL Ultrafiltration Module with a 100 kDa cut-off (EMD Millipore, Darmstadt, Germany) for single-cell factories (1.5 l total) or a 0.1 m2 Pellicon 2 membrane for triple-cell factory preparations (3–4.5 l). After concentration, media were treated with benzonase and filtered again prior to column chromatography.
Cell pellets from single-cell factory preparations were processed as described above and filtered through a 0.22 micron filter prior to column chromatography. Pellets from triple-cell factory preparations were lysed using an M-110L Microfluidizer Processor (Microfluidics, Newton, MA), centrifuged to remove debris, treated with benzonase, and filtered through a 0.22 micron filter.
After benzonase treatment and filtration, lysates and media were loaded onto a HiPrep Sephacryl S-300 HR size-exclusion chromatography column equilibrated with 10 mmol/l Tris-Bis Propane, 10 mmol/l Tris, and 200 mmol/l NaCl, at pH 9.0. The flow-through fraction containing AAV was diafiltered to a low-salt buffer and concentrated with a Pellicon XL module with a 100 kDa cut-off. AAV in concentrated diafiltrates was further purified by anion-exchange on a POROS HQ resin equilibrated with 10 mmol/l Tris-Bis Propane, and 10 mmol/l Tris, at pH 9.0. AAV was eluted from the column using a linear NaCl gradient. For the single-cell factory preparations, the lysate and media fractions were purified separately through column chromatography and never combined. For triple-cell factory preparations, lysate and media fractions were combined during post-size-exclusion chromatography concentration and diafiltration.
For triple-cell factory preparations, anion-exchange fractions containing AAV were pooled, concentrated, and underwent cesium-chloride gradient ultracentrifugation to separate viral genome-containing AAV particles from empty particles. CsCl fractions were then dialyzed against PBS and genome-containing fractions were pooled prior to mouse studies.
Supplementary Figure S1 shows a Coomassie Blue-stained SDS-PAGE gel demonstrating the purity of AAV-FVIII through anion exchange. Furthermore, an experiment to determine the activity of benzonase nuclease in concentrated culture media is described in the Supplementary Materials and Methods and demonstrated in Supplementary Figure S1.
AAV8 capsid ELISA
Assembled viral genome-containing and empty AAV8 particles were detected using the AAV8 Titration ELISA Kit (PRAAV8; Progen Biotechnik GmbH, Heidelberg, Germany). Briefly, raw culture media and benzonase-treated lysate samples were diluted in kit-provided sample buffer and incubated on the provided ELISA plate for one hour. Plates were then incubated with biotin-conjugated anti-AAV8, followed by a streptavidin peroxidase conjugate, and a substrate containing tetramethylbenzidine. Absorbance was measured on a Spectramax M2 spectrophotometer (Molecular Devices, LLC, Sunnyvale, CA) at 450 nm.
Quantitative PCR
qPCR was used to determine AAV-FVIII viral genome titers in anion exchange fractions from single-cell factories and in dialyzed CsCl fractions from triple-cell factories. Primers targeted the HLP promoter in the 5′ region of the viral genome, or the middle of the FVIII gene: 5′-CAGGACGCTGTGGTTTCTG-3′ (HLP forward), 5′-TGCCTGAAGCTGAGGAGAC-3′ (HLP reverse), 5′-GGAGATGAAGAAGGAGGACTTTG-3′ (FVIII forward), and 5′-TCCACAGCAGCAATGAAGTAG-3′ (FVIII reverse). Samples were run in duplicates of 25 μl in Bio-Rad iQ Sybr Green Supermix on the Bio-Rad iCycler with attached MyIQ real-time detection system (Bio-Rad Laboratories, Hercules, CA).`
Mouse studies
All animal studies were performed according to protocols approved by the St. Jude Institutional Animal Care and Use Committee. To compare AAV-FVIII preparations purified from harvest on day 3 versus day 6 post-transfection, C57Bl/6 mice were injected via the tail vein with 5 × 109, 2 × 1010, or 2 × 1011 viral genomes of either day 3 AAV-FVIII prep or day 6 AAV-FVIII prep (n = 4–5 mice per group). At 7 days postinjection, blood was collected by retro-orbital bleed with ethylenediaminetetraacetic acid-coated capillary tubes and plasma was isolated. The plasma concentration of human FVIII was determined by ELISA (ASSERACHROM VIII:Ag, DIAGNOSTICA STAGO, France), which does not cross-react with endogenous mouse FVIII.
Statistics
Data are expressed as the average plus or minus the standard deviation, where appropriate. Analysis was performed with Student’s t-test in Excel (Microsoft, Redmond, WA) or one-way analysis of variance with a Tukey comparison in Minitab (Minitab, State College, PA). P < 0.05 was considered significant in all comparisons.
Acknowledgments
The authors would like to thank Rebecca Banks-Spivey for assistance with downstream processing. This research was supported by funding from American Lebanese Syrian Associated Charities (ALSAC) and Hemophilia of Georgia.
Footnotes
There are no institutional or corporate arrangements that would be considered a financial conflict of interest related to this manuscript.
References
- Zincarelli, C, Soltys, S, Rengo, G and Rabinowitz, JE (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16: 1073–1080. [DOI] [PubMed] [Google Scholar]
- Asokan, A, Schaffer, DV and Samulski, RJ (2012). The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki, K, Fuess, S, Storm, TA, Gibson, GA, Mctiernan, CF, Kay, MA et al. (2006). Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 14: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras, BA, O’Connor, DM and French, BA (2013). Systemic delivery of shRNA by AAV9 provides highly efficient knockdown of ubiquitously expressed GFP in mouse heart, but not liver. PLoS One 8: e75894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, BJ (2005). Adeno-associated virus vectors in clinical trials. Hum Gene Ther 16: 541–550. [DOI] [PubMed] [Google Scholar]
- uniQure (2012). uniQure’s Glybera® First Gene Therapy Approved by European Commissionat. http://www.uniqure.com/news/167/182/uniQure-s-Glybera-First-Gene-Therapy-Approved-by-European-Commission.html.
- Allay, JA, Sleep, S, Long, S, Tillman, DM, Clark, R, Carney, G et al. (2011). Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther 22: 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, JF, Wellman, J and High, KA (2010). Manufacturing and regulatory strategies for clinical AAV2-hRPE65. Curr Gene Ther 10: 341–349. [DOI] [PubMed] [Google Scholar]
- Durocher, Y, Pham, PL, St-Laurent, G, Jacob, D, Cass, B, Chahal, P et al. (2007). Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J Virol Methods 144: 32–40. [DOI] [PubMed] [Google Scholar]
- Chahal, PS, Schulze, E, Tran, R, Montes, J and Kamen, AA (2014). Production of adeno-associated virus (AAV) serotypes by transient transfection of HEK293 cell suspension cultures for gene delivery. J Virol Methods 196: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High, KH, Nathwani, A, Spencer, T and Lillicrap, D (2014). Current status of haemophilia gene therapy. Haemophilia 20 Suppl 4: 43–49. [DOI] [PubMed] [Google Scholar]
- Grieger, JC, Soltys, SM and Samulski, RJ (2015). Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol Ther: 1–11. [DOI] [PMC free article] [PubMed]
- Cecchini, S, Virag, T and Kotin, RM (2011). Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum Gene Ther 22: 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin, V, Brument, N, Toublanc, E, Raimbaud, I, Moullier, P and Salvetti, A (2004). Improving rAAV production and purification: towards the definition of a scaleable process. J Gene Med 6 Suppl 1: S223–S228. [DOI] [PubMed] [Google Scholar]
- Thomas, DL, Wang, L, Niamke, J, Liu, J, Kang, W, Scotti, MM et al. (2009). Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum Gene Ther 20: 861–870. [DOI] [PubMed] [Google Scholar]
- Clément, N, Knop, DR and Byrne, BJ (2009). Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther 20: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe, LH, Xiao, R, Lock, M, Lin, J, Korn, M and Wilson, JM (2010). Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum Gene Ther 21: 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock, M, Alvira, M, Vandenberghe, LH, Samanta, A, Toelen, J, Debyser, Z et al. (2010). Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21: 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, T, Nonaka-Sarukawa, M, Uchibori, R, Kinoshita, K, Hayashita-Kinoh, H, Nitahara-Kasahara, Y et al. (2009). Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum Gene Ther 20: 1013–1021. [DOI] [PubMed] [Google Scholar]
- Sonntag, F, Köther, K, Schmidt, K, Weghofer, M, Raupp, C, Nieto, K et al. (2011). The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol 85: 12686–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso, E, Blouin, V, Lock, M, McGorray, S, Leon, X, Alvira, MR et al. (2014). Manufacturing and characterization of a recombinant adeno-associated virus type 8 reference standard material. Hum Gene Ther 25: 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, J, Lenting, PJ, Rosales, C, Lee, D, Rabbanian, S, Raj, D et al. (2013). Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121: 3335–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, Ng, CY, Zhou, J, Spence, Y, Waddington, SN et al. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107: 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, McIntosh, J, Ng, CY, Zhou, J, Spence, Y et al. (2007). Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 109: 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H, Gomero, E, Bonten, E, Gray, JT, Allay, J, Wu, Y et al. (2012). Preclinical dose-finding study with a liver-tropic, recombinant AAV-2/8 vector in the mouse model of galactosialidosis. Mol Ther 20: 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, X (2010). Adeno-associated viral vectors found free in media. Hum Gene Ther 21: 1221–1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.