

Abstract
Low levels of high-density lipoprotein cholesterol (HDL-C) and high triglyceride levels contribute to the excess rate of cardiovascular events seen in subjects with type 2 diabetes. Fenofibrate treatment partially reverses dyslipidemia in these subjects. However, a paradoxical marked reduction in HDL-C and HDL's major protein, apolipoprotein A-I, is a complication of fenofibrate in combination with rosiglitazone, an insulin-sensitizing agent. Risk factors for this condition, termed hypoalphalipoproteinemia, have yet to be identified. Using a case-control study design with subjects enrolled in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial, we tested the hypothesis that alterations in HDL's protein cargo predispose diabetic subjects to fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. HDL was isolated from blood obtained from controls (no decreases or increase in HDL-C while receiving fenofibrate/rosiglitazone therapy) and cases (developed hypoalphalipoproteinemia after fenofibrate/rosiglitazone treatment) participating in the ACCORD study before they began fenofibrate/rosiglitazone treatment. HDL proteins were quantified by targeted parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) with isotope dilution. This approach demonstrated marked increases in the relative concentrations of paraoxonase/arylesterase 1 (PON1), apolipoprotein C-II (APOC2), apolipoprotein C-I, and apolipoprotein H in the HDL of subjects who developed hypoalphalipoproteinemia. The case and control subjects did not differ significantly in baseline HDL-C levels or other traditional lipid risk factors. We used orthogonal biochemical techniques to confirm increased levels of PON1 and APOC2. Our observations suggest that an imbalance in HDL proteins predisposes diabetic subjects to develop hypoalphalipoproteinemia on fenofibrate/rosiglitazone therapy.
Cardiovascular disease (CVD)1 is the primary cause of morbidity and mortality among patients with type 2 diabetes despite effective therapies for treating major risk factors such as elevated blood pressure and cholesterol levels (1, 2). Thus, the risk of cardiovascular events remains markedly elevated in diabetic subjects even during treatment with a statin (an inhibitor of a key enzyme in cholesterol biosynthesis) (3, 4). Subjects with type 2 diabetes and metabolic syndrome suffer from diabetic dyslipidemia, which is characterized by increased blood levels of triglycerides in concert with low levels of high-density lipoprotein-cholesterol (HDL-C) (5–7). This condition strongly associates with an increased risk of atherosclerotic CVD (8, 9). The metabolism of HDL and triglycerides are interrelated, and high levels of triglycerides strongly associate with low levels of HDL-C, even in nondiabetic subjects (10).
The Action to Control Cardiovascular Risk in Diabetes-Lipid (ACCORD-Lipid) trial tested the hypothesis that adding fenofibrate to statin therapy would reduce the risk of cardiovascular events in diabetic subjects (11). In a multivariable model adjusted for a variety of potential confounding factors in this cohort, fenofibrate in combination with statin therapy raised HDL-C by 7.3% and lowered triglycerides by 24%. Subgroup analysis of dyslipidemic participants (subjects with baseline triglyceride levels in the highest tertile and HDL-C levels in the lowest tertile) indicated that addition of fenofibrate to a statin was associated with lowered CVD risk (11).
Hypoalphalipoproteinemia, a striking reduction in HDL-C and HDL's major protein, apolipoprotein A-I (APOA1), is a complication of fenofibrate therapy and typically occurs when fenofibrate is administered in combination with a thiazolidinedione such as rosiglitazone (12–18). However, there are no well-established risk factors for this disorder. Because HDL is a collection of particles that range in size from <7 nm to >14 nm and collectively contain >80 different proteins (19–21), the composition of HDL's protein cargo might predispose subjects to this disorder, perhaps by altering the interplay of triglyceride and HDL metabolism.
Selected reaction monitoring (SRM) has been the mass spectrometry (MS) method of choice for quantitative proteomics due to its sensitivity and precision in complex biological systems (22–25). Recent studies showed that parallel reaction monitoring (PRM) might work as well as SRM for protein quantification (26, 27). PRM does not require prior selection of target peptide transitions (28–30), which facilitates the development and validation of quantitative proteomics analysis. PRM is also less likely to be affected by interfering ions because it monitors product ions with high mass resolution (26). We recently showed that PRM and SRM exhibit comparable linearity, dynamic range, and precision for quantifying HDL proteins by isotope dilution (31).
In the current studies, we used PRM with isotope dilution to test the hypothesis that the protein composition of HDL differ in subjects with fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. HDL was isolated at baseline from plasma obtained from subjects who developed hypoalphalipoproteinemia and matched subjects whose HDL either did not change or increased while receiving fenofibrate/rosiglitazone therapy in the ACCORD-Lipid study. We show that relative levels of certain proteins were markedly elevated in the HDL of subjects that subsequently developed fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. The results were confirmed by SRM analyses and orthogonal biochemical measurements.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Unless otherwise specified, all reagents were obtained from Sigma Aldrich (St. Louis, MO). Water and acetonitrile for MS analyses were Optima LC/MS grade (Fischer Scientific, Pittsburgh, PA). Formic acid was purchased from EMD Millipore (Billerica, MA). 15N-labeled apolipoprotein A-I ([15N]APOA1,15N enrichment >99%) was produced using a bacterial expression system and isolated to greater than 99% purity (32).
Sample Collection
Before subjects in the ACCORD-Lipid trial received fenofibrate/rosiglitazone therapy, fasting blood was collected in EDTA-treated tubes and centrifuged at 4 °C for 10 min at 1,600 g to generate plasma. Plasma was pipetted into cryovials and immediately frozen and stored at −80 °C. The Human Studies Committee at the University of Washington and Columbia University Medical Center approved all studies involving human material. All plasma samples were deidentified, analyzed in a blinded manner, and contained no information that might allow for the identification of individuals.
HDL Isolation
Plasma was quickly thawed at 37 °C, and 50 μl were subjected to sequential ultracentrifugation to isolate HDL (density 1.063–1.210 g/ml) (33). Total protein concentration in HDL was measured using the Bradford assay, with albumin as the standard.
Proteolytic Digestion
HDL (5 μg protein) and [15N]APOA1 (0.25 μg protein, added as an internal standard) (31, 34) were solubilized with 0.2% RapiGest (Waters, Milford, MA) in 100 mm ammonium bicarbonate, reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin (1:20, w/w HDL protein; Promega, Madison, WI) for 4 h at 37 °C. A second aliquot of trypsin (1:20, w/w HDL protein) was added, and the samples were incubated overnight at 37 °C. After acidic hydrolysis of RapiGest with 0.5% trifluoroacetic acid, samples were dried and stored at −20 °C until MS analysis.
Selection of HDL Peptides for Targeted Quantification by PRM
Thirty-eight proteins consistently detected in HDL in previous studies were selected for targeted quantification (19–21, 35). Based on these studies and considering the observed frequency, two to five peptides per protein were selected for PRM quantification. We excluded peptides that are susceptible to ex vivo modification (e.g. containing methionine) and peptides with missing cleavages. Supplemental Table 1 shows a list of proteins and peptides selected for targeted quantification by PRM.
Targeted Analyses by PRM
Digested HDL (250 ng protein) was desalted on a C18 trap column (Waters XBridge BEH C18, 5 μm, 0.075 × 40 mm) for 6.5 min at 3 μl/min in 99% solvent A, and then separated using a C18 column packed in-house (Waters XBridge BEH C18, 3.5 μm, 0.075 × 120 mm) with an integrated electrospray emitter pulled by a laser micropipette puller (Sutter Instrument, Novato, CA). The column was kept at 50 °C and nanoACQUITY UPLC (Waters) was used for the separation, with a linear gradient of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Peptides were eluted from the trap column onto the analytical column at a flow rate of 0.6 μl/min and separated using multistep gradient as follows: 1% to 7% solvent B in 3 min; 7% to 25% solvent B in 16 min; and 25% to 35% solvent B in 3 min. The column was subsequently washed for 3 min at 80% B and re-equilibrated at 99% A for 11 min. Experiments were performed using a Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). The resolution was set at 17,500 (at m/z 200), the automatic gain control target at 5 × 104, the maximum fill time at 30 ms, and the individual isolation window at 2 Th. Normalized collision energy of 25 was employed for fragmentation. A scheduled (3-min window) inclusion list for PRM analyses was generated, using Skyline software (36). The inclusion list consisted of m/z of precursor peptides of interest and corresponding retention times (Supplemental Table 1).
Confirmation of PRM Results by SRM Analyses
The same amount of digested HDL (250 ng protein) and chromatographic conditions used in targeted analyses by PRM were employed by SRM quantification. SRM experiments were performed on a TSQ Vantage Triple Stage Quadrupole Mass Spectrometer (Thermo Scientific). Resolution for Q1 and Q3 were set to 0.7 Da (full width at half-maximum). The collision gas (argon) pressure of Q2 was set to 1.5 mTorr. Each transition had a dwell time of 10 ms, and experiments were performed using a scheduled (3-min window) transition list generated in Skyline (36). This transition list contained each precursor/fragment transition pair along with the collision energy and retention time (Supplemental Table 2 provides transitions and scheduling).
Selection of [15N]APOA1 Peptide to Serve as the Global Internal Standard for Targeted Quantification
Previous studies have shown that [15N]APOA1, the labeled form of APOA1 (HDL's major protein), can serve as the internal standard for quantification of HDL proteins (31, 34). To determine whether [15N]APOA1 peptides corrected for variability in digestion and LC/MS performance equally well, we monitored five labeled tryptic APOA1 peptides by PRM and derived Pearson correlations (r) for each peptide pair for all 60 samples (Supplemental Table 3). The [15N]-labeled peptides DYVSQFEGSALGK and VQPYLDDFQK exhibited the best correlation (r = 0.94). We pre-specified that of the two [15N]APOA1 peptides with the best r, the one with the larger integrated area would be used as internal standard for protein quantification. Therefore, [15N]APOA1 peptide DYVSQFEGSALGK (termed [15N] APOA1 peptide) was used as the internal standard in all quantitative analyses.
Quantification of Targeted Peptides in HDL
Our MS analyses used Skyline (version 2.5.1.6094), an open source software application for quantitative data processing and proteomic analysis (36). All integrated peaks were manually inspected to ensure correct peak detection and integration was adjusted if necessary. The results were expressed as a ratio of the peak area of each peptide and the [15N]APOA1 peptide (DYVSQFEGSALGK).
Among the proteins targeted for HDL quantification, two different protein families containing highly homologous sequences were targeted. Serum amyloid A (SAA) 1, 2, and 4 were quantified for the SAA family; however, only SAA 1 and 2 share significantly homology. Two peptides shared by SAA1 and SAA2 were quantified, and they are termed as SAA1/2 peptides (Table II). In addition, one peptide with a unique sequence for SAA1 and another with a unique sequence for SAA2 were used for quantification (see Table II). For the peptidase S1 family, two peptides shared by the proteins haptoglobin and haptoglobin-related protein were quantified. This joint quantification is termed as haptoglobin/haptoglobin-related protein in Table II. In addition, two unique peptides to the protein haptoglobin were also integrated (represented as HP in Table II).
Table II. Pearson correlations between peptides for each HDL protein.
| Protein | Peptide 1 | Peptide 2 | r |
|---|---|---|---|
| ALB | FQNALLVR | LVAASQAALGL | 0.90 |
| APOA1 | DYVSQFEGSALGK | VQPYLDDFQK | 0.79 |
| APOA2 | EPCVESLVSQYFQTVTDYGK | EQLTPLIK | 0.75 |
| APOA4 | IDQNVEELK | LGEVNTYAGDLQK | 0.97 |
| APOB | SVSLPSLDPASAK | ITENDIQIALDDAK | 0.91 |
| APOC1 | TPDVSSALDK | EFGNTLEDK | 0.76 |
| APOC2 | TYLPAVDEK | ESLSSYWESAK | 0.80 |
| APOC3 | DALSSVQESQVAQQAR | DYWSTVK | 0.78 |
| APOC4 | ELLETVVNR | DGWQWFWSPSTFR | 0.86 |
| APOD | NPNLPPETVDSLK | VLNQELR | 0.86 |
| APOE | SELEEQLTPVAEETR | LEEQAQQIR | 0.83 |
| APOF | SLPTEDCENEK | SGVQQLIQYYQDQK | 0.68 |
| APOH | TCPKPDDLPFSTVVPLK | TFYEPGEEITYSCKPGYVSR | 0.92 |
| APOL1 | VTEPISAESGEQVER | ALADGVQK | 0.90 |
| APOM | SLTSCLDSK | DGLCVPR | 0.79 |
| B2M | VNHVTLSQPK | VEHSDLSFSK | 0.89 |
| C3 | TGLQEVEVK | TIYTPGSTVLYR | 0.95 |
| CLU | LFDSDPITVTVPVEVSR | ASSIIDELFQDR | 0.89 |
| GC | HQPQEFPTYVEPTNDEICEAFR | THLPEVFLSK | 0.91 |
| GPLD1 | NQVVIAAGR | SWITPCPEEK | 0.90 |
| HBB | VNVDEVGGEALGR | LLVVYPWTQR | 0.99 |
| HPX | EVGTPHGIILDSVDAAFICPGSSR | LLQDEFPGIPSPLDAAVECHR | 0.82 |
| HP/HPR | ILGGHLDAK | LPECEAVCGKPK | 0.99 |
| HP | DYAEVGR | VTSIQDWVQK | 0.79 |
| IHH | ATFASHVQPGQYVLVAGVPGLQPAR | AFQVIETQDPPR | 0.87 |
| LBP | SFRPFVPR | VQLYDLGLQIHK | 0.97 |
| LCAT | TYIYDHGFPYTDPVGVLYEDGDDTVATR | LEPGQQEEYYR | 0.81 |
| LPA | TPAYYPNAGLIK | GTLSTTITGR | 0.82 |
| PCYOX1 | IFSQETLTK | LVCSGLLQASK | 0.81 |
| PLTP | AVEPQLQEEER | FLEQELETITIPDLR | 0.92 |
| PON1 | STVELFK | SFNPNSPGK | 0.94 |
| PON3 | LLNYNPEDPPGSEVLR | SVNDIVVLGPEQFYATR | 0.83 |
| PPBP | ICLDPDAPR | NIQSLEVIGK | 0.89 |
| SAA1/2 | DPNHFRPAGLPEKY | SFFSFLGEAFDGAR | 0.99 |
| SAA1 | FFGHGAEDSLADQAANEWGR | - | - |
| SAA2 | GPGGAWAAEVISNAR | - | - |
| SAA4 | GPGGVWAAK | FRPDGLPK | 0.85 |
| SERPINA1 | AVLTIDEK | SASLHLPK | 0.99 |
| VTN | FEDGVLDPDYPR | GQYCYELDEK | 0.91 |
Proteins were identified on the basis of their gene names.
Immunoblotting Analysis for Serum Paraoxonase/Arylesterase 1 (PON1)
Fourteen HDLs that contained a wide range of PON1 concentrations (as quantified by PRM) were selected at random (case or control designation was ignored) for confirmation by immunoblotting. HDL protein (1 μg) was subjected to 4–12% Bis-Tris gel (NuPAGE; Life technology, CA) and then transferred onto a nitrocellulose membrane (Bio-Rad, CA). The membrane was cut into halves and probed with either a PON1 antibody (1:1000, P0123, Sigma Aldrich) or an APOA1 antibody (as loading control, 1:5000, Rabbit pAb 178422, Merck Millipore). After two washes (30 min) with 0.05% Tween 20 in phosphate- buffered saline (137 mm NaCl, 2.7 mm KCl, and 10 mm sodium phosphate, pH 7.4), the membranes were incubated with an anti-rabbit IgG, horseradish peroxidase-linked antibody from goat (1:10,000) (Jackson Immunoresearch) in the same buffer at room temperature for 1 h. The membranes were again washed and then developed with a chemiluminescent detection kit (SuperSignal West Femto Chemiluminescent Substrate, Thermo Scientific). Data were captured with a digital camera.
Immunoassay for Apolipoprotein C-II (APOC2)
The concentration of APOC2 was measured using a sandwich immunoassay (MILLIPLEX MAP, Millipore). The reported intra-assay and interassay CVs were <10% and <20%, respectively, with an accuracy of 92%. Results are expressed as ng/μg protein.
Experimental Design and Statistical Rationale
Hypoalphalipoproteinemia, defined as combination therapy-induced mean fall of HDL-C ≥60% with a mean plasma HDL-C level <10 mg/dl, developed in 130 of the 1,420 subjects randomized to fenofibrate/rosiglitazone therapy in ACCORD-Lipid. We studied a subset of 30 cases (hypoalphalipoproteinemia) and 30 controls (no decreases or increase in HDL-C) that had an adequate volume (50 μl) of plasma available for HDL isolation and MS/MS analysis. Control and case subjects had similar age, gender, and baseline HDL-C levels (Table I).
Table I. Baseline characteristics of subjects.
| Characteristic | Controls | Cases | p value |
|---|---|---|---|
| n | 30 | 30 | |
| Age (years) | 61.6 ± 6.1 | 62.5 ± 5.7 | 0.585 |
| Female (n)a | 4 | 5 | 0.718 |
| HbA1C (%) | 8.1 ± 1.0 | 8.3 ± 1.0 | 0.566 |
| Glucose (mg/dL) | 162 ± 56 | 167 ± 49 | 0.709 |
| LDL-C (mg/dL) | 105 ± 29 | 94 ± 28 | 0.124 |
| HDL-C (mg/dL) | 33.1 ± 5.0 | 31.6 ± 7.8 | 0.376 |
| Triglycerides (mg/dL)b | 187 (129–243)c | 221 (172–319)c | 0.081 |
| Cholesterol (mg/dL) | 177 ± 32 | 174 ± 33 | 0.716 |
| Insulin (n)a | 8 | 6 | 0.542 |
| Smoking status (previous, current)* | 13, 4 | 17, 2 | 0.505 |
a Chi-square test;
b Wilcoxon rank-sum test;
c median and interquartile range.
The relationship between relative levels of peptides for individual proteins quantified by PRM was assessed by Pearson correlation. For each protein, two peptides with the best correlation were selected, and the one with the largest integrated area was chosen to serve as a surrogate for the protein. The results were expressed as a ratio of the area of surrogate peptide and the [15N]APOA1 peptide.
The analytical reliability of the data was validated using the raw area of the [15N]APOA1 peptide. Accessing the variation using the [15N]APOA1 peptide controls for digestion issues, potential losses during the sample preparation, and MS variation. The obtained CVs for PRM (29%) and SRM (12%) are in accordance with the precision recommended for research use assays for quantifying proteins and peptides (20–35%)(37). The areas obtained for the [15N]APOA1 peptide by PRM and SRM are provided in Supplemental Table 4.
Differences in protein expression between controls and cases were initially evaluated by a two-sample t test. To account for multiple comparison testing, p values obtained from the t test were corrected using the method of Benjamini and Hochberg (38–40). This step-up method is a false discovery rate-controlling procedure that assumes a nonnegative correlation. A corrected p value threshold is calculated, and only proteins above the corrected p value are considered significantly different (39, 40). For proteins differing significantly in expression between controls and cases, multiple linear regression analyses controlling for HDL-C and triglycerides (log scale) were performed.
Hierarchical cluster analysis of HDL proteins used average linkage with Pearson correlation as the distance. All statistical analyses were performed with STATA software version 12 (Stata Corp, College Park, TX).
RESULTS
Study Design, Subject Characteristics, and Workflow
We designed a case-control study with subjects selected from the ACCORD-Lipid trial to test the hypothesis that the protein cargo of HDL might identify patients at risk of fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. HDL was isolated from fasting plasma samples obtained while the 60 subjects were on simvastatin therapy (11) but before they were on combination therapy with fenofibrate/rosiglitazone.
Case and control subjects had similar age, percentage of females, and smoking status (Table I). Mean baseline levels of HDL-C for cases and controls were 33 and 31 mg/dL, respectively (p = 0.38), and median triglyceride levels were 187 and 220 mg/dL, respectively, for cases and controls (p = 0.08). All other baseline characteristics were similar in the two groups, with no significant differences in hemoglobin A1c and glucose levels, low density lipoprotein cholesterol (LDL-C), and insulin use (Table I). Full demographics of subjects in ACCORD-Lipid are given elsewhere (11).
An overview of the study's workflow is shown in Fig. 1. HDL was isolated from freshly thawed plasma (stored at −80 °C after collection) of the subjects and digested with trypsin, using [15N]APOA1 as the internal standard. Data from previous studies (20, 31) were used to select 38 HDL protein candidates for relative quantification. Two to five peptides from each targeted protein were then measured by PRM. From the 38 proteins quantified by PRM, 18 were selected for confirmatory analyses using SRM. All statistical analyses were controlled for multiple comparisons. Finally, potential protein candidates found to be differently expressed in cases and controls were confirmed through immunoblotting and immunoassay.
Fig. 1.
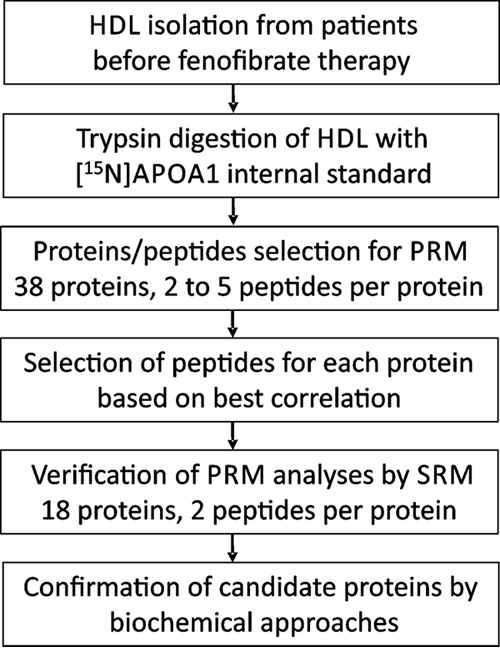
Workflow outlining the study approach.
Selection of HDL Proteins for Quantification
For targeted quantitative analyses by PRM, we selected the 38 most abundant HDL proteins that had been identified previously in at least two different human populations (20, 31, 35)(Table II). Proteins were identified on the basis of their gene names.
Internal Standard Selection
Approximately 70% of HDL's protein mass is composed of APOA1, HDL's major structural protein (41). Previous studies validated the use of [15N]APOA1 as a global internal standard to correct for digestion efficiency and run-to-run variability in LCMS analyses (31, 34).
The same approach was taken in the current study, where we monitored five different [15N]APOA1 peptides to serve as possible internal standards in our PRM analyses. After obtaining Pearson correlations among the peptides in all 60 subjects, the peptides DYVSQFEGSALGK and VQPYLDDFQK were selected because they exhibited the best correlation (r = 0.94, Supplemental Table 3). Before any analyses were performed, we prespecified that the [15N]APOA1 peptide with the larger integrated area would be used as the internal standard. Based on this approach, the [15N]APOA1 peptide DYVSQFEGSALGK ([15N]APOA1 peptide) was chosen as the internal standard.
Targeted Protein Quantification by PRM
Two to five peptides from each targeted protein were selected for PRM analysis. The areas of peptides were integrated using Skyline (36), and the results were expressed as the ratio of the area of each peptide to the area of the [15N]APOA1 peptide (used as an internal standard to correct for variations in sample preparation and instrument response). Correlations among the monitored peptides were calculated for all 38 proteins we quantified in HDL. The purpose of performing such correlations is to determine if using different peptides as surrogates for the same protein produces consistent results. The two peptides from each protein with the highest correlation are presented in Table II. We prespecified that the peptide with the largest integrated area (designated as peptide 1 in Table II) would be used for relative protein quantification with the [15N] APOA1 internal standard peptide.
PRM Analysis Reveals Marked Differences in the Relative Abundances of Multiple HDL Proteins in Diabetic Subjects with Fenofibrate/Rosiglitazone-Induced Hypoalphalipoproteinemia
Using PRM, we performed relative protein quantification, with [15N]APOA1 as the internal standard, of HDLs isolated from plasma of the 60 subjects before they started treatment with fenofibrate/rosiglitazone. Differences in protein expression between the two groups were initially evaluated by a two-sample t test. Then, the Benjamini and Hochberg statistical method (38–40) was applied to account for multiple testing. A corrected p value threshold was calculated by the step-up method, and only proteins above that value were considered significantly different (38–40). Based on the corrected p value, only proteins with p ≤ 0.005 were considered significantly different between cases and controls.
Figure 2A summarizes the results of this analysis, and significantly different proteins are highlighted in Fig. 2B-2E. We did not find any protein significantly depleted in HDL when cases were compared with controls. In contrast, four proteins were elevated in the HDL from subjects who subsequently developed fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia (Fig. 2). PON1 increased the most: the mean level was 47% higher in cases than in controls (p = 0.0001, Fig. 2B). Two members of the apolipoprotein C family were also elevated: The mean level of APOC2 was 45% higher (p < 0.0001, Fig. 2C), and that of apolipoprotein C I (APOC1) was 26% higher (p = 0.0025, Fig. 2D) in cases than in the control subjects. Patients who developed hypoalphalipoproteinemia on fenofibrate/rosiglitazone combination therapy also had a 34% higher level of apolipoprotein H (beta-2-glycoprotein 1, APOH, p = 0.0029, Fig. 2E). These significant changes persisted after controlling for baseline levels of HDL-C and triglycerides (log transformed); in multiple regression analyses that controlled for these two characteristics, subjects that developed hypoalphalipoproteinemia on fenofibrate/rosiglitazone combination therapy had higher levels of PON1 (p < 0.0001), APOC2 (p < 0.0001), APOC1 (p = 0.013), and APOH (p = 0.014) in their HDL. Taken together, these observations indicate that, before the trial began, the relative abundance of certain HDL proteins in subjects who later developed hypoalphalipoproteinemia upon fenofibrate/rosiglitazone treatment differed markedly from that of the control subjects.
Fig. 2.

PRM analysis of HDL proteins in case and control subjects. Proteins measured by PRM (A). For each protein, the p value from the two-sample t test is plotted against the log2 fold change between cases and controls. Proteins overexpressed in cases are displayed to the right of the value 0 on the x axis, while underexpressed proteins are to the left. After we controlled for multiple comparison testing, only proteins situated above the p value of 0.005 on the y axis were considered significantly different between cases and controls. The solid horizontal line shows the corrected overall critical p value threshold (p = 0.005). The dashed horizontal line shows the uncorrected overall critical p value of 0.05. Box plots of PON1 (B), APOC2 (C), APOC1 (D), and APOH (E) in case and control subjects. Peptides were quantified as the integrated peak area relative to that of [15N]APOA1 peptide ([15N]DYVSQFEGSALGK). The box plots show the distribution of the data (median, interquartile ranges), while the dots represent outliers. AU, arbitrary units.
SRM Analyses Confirm PON1, APOC2, APOC1 and APOH Elevation in HDL of Diabetic Subjects with Fenofibrate/Rosiglitazone-Induced Hypoalphalipoproteinemia
From the 38 HDL proteins monitored by PRM, we selected 18 for analysis by SRM. We included all of the proteins that differed significantly in case and control subjects by PRM analyses (PON1, APOC2, APOC1, and APOH), proteins that clustered together with significantly altered proteins (apolipoprotein C-III (APOC3), apolipoprotein C-IV (APOC4), and Lecithin:cholesterol acyltransferase (LCAT)). We also quantified other abundant HDL apolipoproteins (APOA1, A-II (APOA2), A-IV (APOA4), D (APOD), E (APOE), F (APOF), M (APOM), CLU (clusterin, APOJ)) as well as low abundance proteins involved in HDL metabolism (prenylcysteine oxidase, phospholipid transfer protein, and paraoxonase/arylesterase 3).
For each protein, the same peptide selected for PRM quantification was used for SRM analysis. Five transitions (precursor to fragment ion) were selected for SRM quantification of each peptide, based on PRM intensity (Supplemental Table 2). As for PRM analyses, the [15N]APOA1 peptide DYVSQFEGSALGK ([15N]APOA1 peptide) was chosen as the internal standard, and results are reported as the ratio of each peptide area and the [15N]APOA1 peptide. We used the Benjamini and Hochberg method (38–40) to account for multiple testing (38–40). Based on the corrected p value, only proteins with p < 0.014 were considered significantly different between cases and controls.
Figure 3A summarizes the results obtained by SRM quantification. This approach confirmed that PON1, APOC2, APOC1, and APOH were elevated in HDL of subjects that later developed fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. The relative increase in abundance of each protein was similar as assessed by SRM and PRM. As measured by SRM, cases showed a 53% increase in PON1 (Fig. 3B, p < 0.0001), 48% increase in APOC2 (Fig. 3C, p = 0.0005), 26% increase in APOC1 (Fig. 3D, p = 0.002), and 28% increase in APOH (Fig. 3E, p = 0.003). In addition, as measured by SRM, APOC4 of cases showed a 38% increase when compared with controls (p = 0.008). The Pearson correlation between PRM and SRM for APOC4 was 0.93, and PRM showed a 36% increase (p = 0.019) in APOC4 of cases when compared with controls. However, when controlling for multiple comparisons (corrected p value for PRM <0.005), this protein did not reach statistical significance by PRM analyses. This difference between SRM and PRM reflects the more stringent p value required for significance when monitoring more proteins. Thus, the corrected p value threshold for PRM (38 proteins) was more stringent than for SRM (18 proteins).
Fig. 3.

SRM analysis of HDL proteins in case and control subjects. Proteins measured by SRM (A). For each protein, the p value from the two-sample t test is plotted against the log2 fold change between cases and controls. Proteins overexpressed in cases are displayed to the right of the value 0 on the x axis, while underexpressed proteins are to the left. After we controlled for multiple comparison testing, only proteins situated above the p value of 0.014 on the y axis were considered significantly different between cases and controls. The solid horizontal line shows the corrected overall critical p value threshold (p = 0.014). The dashed horizontal line shows the uncorrected overall critical p value of 0.05. Box plots of PON1 (B), APOC2 (C), APOC1 (D), and APOH (E) in case and control subjects as measured by SRM. Peptides were quantified as the integrated peak area relative to that of [15N]APOA1 peptide ([15N]DYVSQFEGSALGK). The box plots show the distribution of the data (median, interquartile ranges), while the dots represent outliers. AU, arbitrary units. Scatter plots showing the correlation of PRM and SRM techniques for PON1 (F), APOC2 (G), APOC1 (H), and APOH (I). r represents the Pearson correlation.
Biochemical Confirmation that PON1 and APOC2 Levels Are Elevated in HDL of Diabetic Subjects with Fenofibrate/Rosiglitazone-Induced Hypoalphalipoproteinemia
We next determined whether orthogonal biochemical approaches would yield similar results for the HDL proteins PON1 and APOC2. We used immunoblot analysis to quantify 14 samples containing different amounts of PON1 as determined by PRM. Samples were selected to cover the full range of values observed by MS/MS. APOA1 was used as a loading control because it accounts for 70% of the protein in HDL. For each sample, the normalized abundance of its MS measurement was compared with the integrated area of PON1 as quantified by immunoblotting (Fig. 4A). Immunoblot quantification of PON1 in HDL correlated well with the PRM data (r = 0.78, Fig. 4B).
Fig. 4.

Quantification of PON1 and APOC2 by immunoblot analysis and ELISA, respectively. (A) Relative quantification of PON1 in 14 subjects. For MS, the relative abundance of PON1 peptide was normalized to [15N]APOA1 peptide. For immunoblot analysis, APOA1 was used as loading control. (B) Pearson correlation between MS and immunoblot measurement for PON1. (C) APOC2 quantification by ELISA in all 60 cases and controls. The box plots show the distribution of the data (median, interquartile ranges), while the dots represent outliers. Concentration is expressed as ηg/μg protein. (D) Pearson correlation between MS and ELISA measurements for APOC2.
We used an ELISA to quantify APOC2 in HDL isolated from all 60 subjects (Fig. 4C). The immunoassay confirmed that APOC2 levels in HDL were higher in the cases than in the controls before the onset of fenofibrate/rosiglitazone therapy (p < 0.001). MS measurements again correlated well with those obtained by immunoassay (r = 0.77, Fig 4D). These observations provide strong evidence that PRM can precisely provide relative quantification of HDL's protein cargo.
Hierarchical Cluster Analysis Demonstrates that Key Proteins Involved in Triglyceride and HDL Metabolism Group Together
We also performed a hierarchical cluster analysis of the 38 HDL proteins, using the normalized peak area ratio (peptide/[15N]APOA1 peptide) obtained for each protein by PRM (Fig. 5). The cluster analysis was conducted using average linkage and Pearson correlation distance. The apolipoprotein C family—APOC1, APOC2, APOC3, and APOC4—formed a tight cluster, suggesting that these family members are coordinately regulated. All of the HDL proteins with elevated levels of expression in the case subjects (APOC2, APOC1, PON1, and APOH) formed a tight cluster. APOC4 clustered strongly with APOC2. LCAT, which was not differentially expressed (p = 0.24, as measured by PRM) but plays a key role in remodeling HDL and apolipoprotein-B-containing lipoproteins, resided in the same cluster (highlighted in Fig. 5).
Fig. 5.
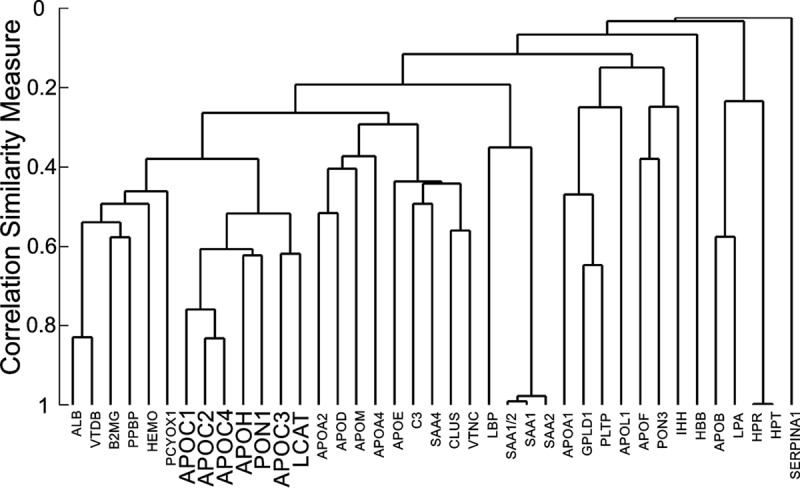
Hierarchical cluster analysis of HDL proteins. Cluster analysis used average linkage with the Pearson correlation (r) as distance. For each protein, the normalized peak area ratio (peptide/internal standard) obtained by PRM was used for relative protein quantification.
DISCUSSION
We tested the hypothesis that variations in HDL's protein cargo can predict diabetic patients who will develop fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. To quantify changes in relative protein abundance, we used PRM in concert with [15N]APOA1 as an internal standard (31, 34).
Previous studies validated the use of a [15N]APOA1 peptide as a global internal standard to quantify multiple proteins, correcting for digestion efficiency and run-to-run variability in the MS analyses (31, 34). Furthermore, the ability of PRM methodology to quantify HDL proteins was compared with the standard SRM methodology and yielded equivalent results in terms of linearity, accuracy, precision, and limits of detection (31). Therefore, we used this approach to investigate levels of HDL protein in diabetic subjects.
After we applied stringent statistical criteria to control for multiple comparisons, our targeted PRM approach identified four HDL proteins—PON1, APOC2, APOC1, and APOH—that were relatively more abundant in HDL isolated from plasma of subjects who subsequently developed fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia. The relative increase in these proteins was confirmed by SRM analyses. In addition, using biochemical approaches, we confirmed our observations for PON1 and APOC2. For PON1, we showed that quantification by PRM correlated well with immunoblot analysis. For APOC2, we obtained a high correlation with ELISA data.
PON1, which has both esterase (42) and lactonase activities (43, 44), associates exclusively with HDL in humans (45). Importantly, hypercholesterolemic mice that are deficient in this enzyme have markedly enhanced atherosclerosis (46). However, human studies have yielded conflicting results (47). Fenofibrate increases PON1 concentration and activity (48, 49), but the plasma samples in our study of diabetic subjects were obtained prior to randomization to fenofibrate therapy. The endogenous substrate(s) of PON1 is unknown, but hydrolysis of lipid peroxides formed during LDL oxidation is one proposed function (50). We tested the possibility that PON1 metabolizes fenofibrate and its metabolites (which contain a lactone moiety) but found no evidence that the enzyme metabolized any of the compounds under a wide range of concentrations and pHs (data not shown).
Interestingly, three (APOC2, APOC1, and APOH) out of four proteins that were more abundant in the HDL of diabetic subjects who developed hypoalphalipoproteinemia on fenofibrate/rosiglitazone therapy are involved in the metabolism of HDL and triglyceride-rich lipoproteins. In hierarchical cluster analysis, they clustered with other proteins that play key roles in remodeling triglyceride-rich lipoproteins and HDL, such as APOC3, and LCAT.
APOC3 resides on the surface of HDL and triglyceride-rich lipoproteins (51), and it is an important inhibitory cofactor for lipoprotein lipase-mediated hydrolysis of triglycerides (52). Recently, mutations that disrupt APOC3 function were shown to be associated with lower levels of triglycerides and a reduced risk of CVD (53). APOC1 is an inhibitor of lipoprotein lipase-mediated triglyceride lipolysis (54), and overexpression of this protein is associated with decreased clearance of very low density lipoprotein, triglycerides, and lipoprotein remnants (55). APOC1 may also activate LCAT (56). APOC2 is an activator of lipoprotein lipase, and its presence is required for triglyceride-rich lipoproteins lipolysis (57).
Dynamic interaction between triglyceride-rich lipoproteins and HDL in the circulation leads to the remodeling of HDL particles (10). Patients with diabetes can have dysfunctional intravascular lipolysis of triglycerides, causing hypertriglyceridemia (58, 59). Because a significant proportion of the surface constituents of HDL is derived from hydrolysis of triglyceride-rich lipoproteins, reduced lipolysis of these particles could significantly impact the formation of nascent and mature HDL particles by reducing the availability of the necessary surface constituents (60). However, there was no significant difference between the cases and controls in baseline levels of triglycerides in our study, suggesting that HDL's altered protein composition affects HDL-cholesterol levels in fenofibrate/rosiglitazone-treated subjects by a mechanism independent of the plasma levels of triglyceride-rich lipoproteins.
How elevated levels of APOH might affect HDL metabolism is unclear because the protein's major role is thought to be regulation of coagulation (61). However, about 35% of circulating APOH is bound to lipoprotein particles, which are distributed mainly between triglyceride-rich lipoproteins and HDL (62, 63). In the presence of APOC2, APOH significantly increases the activity of the lipoprotein lipase (62). APOH gene variations associate with measures of lipid metabolism, such as triglyceride and apolipoprotein E levels (64).
The impact of fenofibrate/rosiglitazone-induced hypoalphalipoproteinemia on cardiovascular disease risk is unknown. However, many lines of evidence show that low levels of HDL-C are an independent risk factor for CVD (8). Moreover, a decrease in HDL-C was associated with an increased risk of hospitalization for coronary artery disease and stroke in a retrospective study of diabetic patients (65), suggesting that therapy-induced lowering of HDL-C might increase the risk of cardiovascular disease.
Our study has potential limitations. Imbalance in HDL proteins was detected in diabetic patients; it remains unclear whether our findings may be generalized to other populations. The number of patients in the current study is relatively small; given the importance of this condition, the results need to be validated in additional cohorts.
Our observations demonstrate the power of using targeted proteomics in concert with isotope dilution to quantify HDL's protein cargo. Importantly, we found marked differences in proteins that play key roles in HDL and triglyceride-rich lipoproteins metabolism in diabetic subjects before starting fenofibrate/rosiglitazone therapy. This finding strongly suggests that these proteins contribute to the risk of developing paradoxical hypoalphalipoproteinemia. In future studies, it will be of great interest to determine prospectively whether proteins associated with HDL alter lipid metabolism in diabetic subjects treated with fenofibrate and rosiglitazone and to test the hypothesis that HDL's protein cargo predicts CVD risk, the leading cause of death in people with diabetes.
Supplementary Material
Acknowledgments
We thank Dr. Priska D. von Haller (University of Washington) for technical assistance and helpful discussions, Ms. Marianna Pavlyha for laboratory assistance, and Laura Lovato for her assistance with identifying and characterizing our ACCORD cohort for this study. Mass spectrometry experiments were performed in the University of Washington's Proteomics Resource (UWPR95794) and the Quantitative and the Functional Proteomics Core of the Diabetes Research Center, University of Washington.
Footnotes
Author contributions: G.E.R., G.R., H.G., and J.W.H. designed the research; G.E.R., Y.H., and J.W.H. performed the research; G.R., Y.H., M.O., and H.G. contributed new reagents or analytic tools; G.E.R. and J.W.H. analyzed data; and G.E.R. and J.W.H. wrote the paper.
* This work was supported by grants from the National Institutes of Health (R01HL110418–03, R01HL108897, R01HL112625, P01HL092969, and DP3 DK108209), the American Heart Association (14POST18620020), the University of Washington's Diabetes Research Center (P30DK017047), the Columbia University Center for Clinical and Translational Research (UL1 RR024156), and the California Tobacco-Related Disease Research Program (TRDRP - 22RT-0095). None of the sponsors had any role in study design, data analysis, or reporting of the results. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material Supplemental Tables 1–4.
This article contains supplemental material Supplemental Tables 1–4.
1 The abbreviations used are:
- CVD
- cardiovascular disease
- HDL-C
- high-density lipoprotein-cholesterol
- ACCORD
- Action to Control Cardiovascular Risk in Diabetes
- APOA1
- apolipoprotein A-I
- PRM
- parallel reaction monitoring
- MS
- mass spectrometry
- SRM
- selected reaction monitoring
- [15N]APOA1
- 15N-labeled apolipoprotein A-I
- PON1
- paraoxonase/arylesterase 1
- APOC2
- apolipoprotein C-II
- APOC1
- apolipoprotein C-I
- APOH
- apolipoprotein H
- APOC3
- apolipoprotein C-III
- APOC4
- apolipoprotein C-IV
- LDL-C
- low density lipoprotein cholesterol
- SAA1
- serum amyloid A-1 protein
- SAA2
- serum amyloid A-2 protein
- LCAT
- Lecithin: cholesterol acyltransferase.
REFERENCES
- 1. Stamler J., Vaccaro O., Neaton J. D., and Wentworth D. (1993) Diabetes, other risk-factors, and 12-yr cardiovascular mortality for men screened in the multiple risk factor intervention trial. Diabetes Care 16, 434–444 [DOI] [PubMed] [Google Scholar]
- 2. Haffner S. M., Lehto S., Rönnemaa T., Pyörälä K., and Laakso M. (1998) Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New Engl. J. Med. 339, 229–234 [DOI] [PubMed] [Google Scholar]
- 3. Collins R., Armitage J., Parish S., Sleigh P., and Peto R. (2003) MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 361, 2005–2016 [DOI] [PubMed] [Google Scholar]
- 4. Colhoun H. M., Betteridge D. J., Durrington P. N., Hitman G. A., Neil H. A., Livingstone S. J., Thomason M. J., Mackness M. I., Charlton-Menys V., and Fuller J. H. (2004) Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 364, 685–696 [DOI] [PubMed] [Google Scholar]
- 5. Taskinen M. R. (2003) Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 46, 733–749 [DOI] [PubMed] [Google Scholar]
- 6. Grant R. W., and Meigs J. B. (2007) Prevalence and treatment of low HDL cholesterol among primary care patients with type 2 diabetes: An unmet challenge for cardiovascular risk reduction. Diabetes Care 30, 479–484 [DOI] [PubMed] [Google Scholar]
- 7. Reyes-Soffer G., and Ginsberg H. (2014) Special populations: Diabetes and metabolic syndrome. In: Ballantyne C. M., ed. Clinical Lipidology, A Companion to Braunwald's Heart Disease, 2nd ed., pp. 401–417, Elsevier, Philadelphia, PA [Google Scholar]
- 8. Gordon D. J., and Rifkind B. M. (1989) High-density lipoprotein—The clinical implications of recent studies. New Engl. J. Med. 321, 1311–1316 [DOI] [PubMed] [Google Scholar]
- 9. Chapman M. J., Ginsberg H. N., Amarenco P., Andreotti F., Borén J., Catapano A. L., Descamps O. S., Fisher E., Kovanen P. T., Kuivenhoven J. A., Lesnik P., Masana L., Nordestgaard B. G., Ray K. K., Reiner Z., Taskinen M. R., Tokgozoglu L., Tybjaerg-Hansen A., and Watts G. F. (2011) Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: Evidence and guidance for management. Eur Heart J 32, 1345–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carr M. C., and Brunzell J. D. (2004) Abdominal obesity and dyslipidemia in the metabolic syndrome: Importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J. Clin. Endocrinol. Metab. 89, 2601–2607 [DOI] [PubMed] [Google Scholar]
- 11. Ginsberg H. N., Elam M. B., Lovato L. C., Crouse J. R. 3rd, Leiter L. A., Linz P., Friedewald W. T., Buse J. B., Gerstein H. C., Probstfield J., Grimm R. H., Ismail-Beigi F., Bigger J. T., Goff D. C. Jr., Cushman W. C., Simons-Morton D. G., and Byington R. P. (2010) Effects of combination lipid therapy in type 2 diabetes mellitus. New Engl. J. Med. 362, 1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beghin L., Capps N., Duhal N., Davies J., Staels B., and Luc G. (1999) Metabolism of apolipoproteins AI and AII in a patient with paradoxical reduction in high-density lipoprotein due to ciprofibrate. Ann. Clin. Biochem. 36, 523–525 [DOI] [PubMed] [Google Scholar]
- 13. Keidar S., Guttmann H., Stam T., Fishman I., and Shapira C. (2007) High incidence of reduced plasma HDL cholesterol in diabetic patients treated with rosiglitazone and fibrate. Pharmacoepidemiol. Drug. Saf. 16, 1192–1194 [DOI] [PubMed] [Google Scholar]
- 14. Mymin D., Dembinski T., and Friesen M. H. (2009) Iatrogenic severe depression of high-density lipoprotein cholesterol. J. Clin. Pharmacol. 49, 865–871 [DOI] [PubMed] [Google Scholar]
- 15. Normén L., Frohlich J., Montaner J., Harris M., Elliott T., and Bondy G. (2004) Combination therapy with fenofibrate and rosiglitazone paradoxically lowers serum HDL cholesterol. Diabetes Care 27, 2241–2242 [DOI] [PubMed] [Google Scholar]
- 16. Schwing W., Hustak L., and Taylor H. C. (2010) Paradoxical severe decrease in high-density lipoprotein cholesterol due to rosiglitazone-fenofibrate interaction. Endocr. Pract. 16, 382–388 [DOI] [PubMed] [Google Scholar]
- 17. Shetty C., Balasubramani M., Capps N., Milles J., and Ramachandran S. (2007) Paradoxical HDL-C reduction during rosiglitazone and fibrate treatment. Diabet. Med. 24, 94–97 [DOI] [PubMed] [Google Scholar]
- 18. Linz P. E., Lovato L. C., Byington R. P., O'Connor P. J., Leiter L. A., Weiss D., Force R. W., Crouse J. R., Ismail-Beigi F., Simmons D. L., Papademetriou V., Ginsberg H. N., and Elam M. B. (2014) Paradoxical reduction in HDL-C with fenofibrate and thiazolidinedione therapy in type 2 diabetes: The ACCORD Lipid trial. Diabetes Care 37, 686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah A. S., Tan L., Long J. L., and Davidson W. S. (2013) Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 54, 2575–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaisar T., Pennathur S., Green P. S., Gharib S. A., Hoofnagle A. N., Cheung M. C., Byun J., Vuletic S., Kassim S., Singh P., Chea H., Knopp R. H., Brunzell J., Geary R., Chait A., Zhao X. Q., Elkon K., Marcovina S., Ridker P., Oram J. F., and Heinecke J. W. (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Invest. 117, 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davidson W. S., Silva R. A., Chantepie S., Lagor W. R., Chapman M. J., and Kontush A. (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 29, 870–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gillette M. A., and Carr S. A. (2013) Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 10, 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whiteaker J. R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., Yan P., Schoenherr R. M., Zhao L., Voytovich U. J., Kelly-Spratt K. S., Krasnoselsky A., Gafken P. R., Hogan J. M., Jones L. A., Wang P., Amon L., Chodosh L. A., Nelson P. S., McIntosh M. W., Kemp C. J., and Paulovich A. G. (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., and Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Picotti P., and Aebersold R. (2012) Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat Methods 9, 555–566 [DOI] [PubMed] [Google Scholar]
- 26. Gallien S., Bourmaud A., Kim S. Y., and Domon B. (2014) Technical considerations for large-scale parallel reaction monitoring analysis. J. Proteomics 100, 147–159 [DOI] [PubMed] [Google Scholar]
- 27. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Domon B., and Gallien S. (2015) Recent advances in targeted proteomics for clinical applications. Proteomics Clin. Appl. 9, 423–431 [DOI] [PubMed] [Google Scholar]
- 29. Gallien S., Duriez E., Demeure K., and Domon B. (2013) Selectivity of LC-MS/MS analysis: Implication for proteomics experiments. J. Proteomics 81, 148–158 [DOI] [PubMed] [Google Scholar]
- 30. Gallien S., Duriez E., Crone C., Kellmann M., Moehring T., and Domon B. (2012) Targeted proteomic quantification on quadrupole-Orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ronsein G. E., Pamir N., von Haller P. D., Kim D. S., Oda M. N., Jarvik G. P., Vaisar T., and Heinecke J. W. (2015) Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J. Proteomics 113, 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryan R. O., Forte T. M., and Oda M. N. (2003) Optimized bacterial expression of human apolipoprotein A-I. Protein Expr. Purif. 27, 98–103 [DOI] [PubMed] [Google Scholar]
- 33. Mendez A. J., Oram J. F., and Bierman E. L. (1991) Protein kinase C as a mediator of high density lipoprotein receptor-dependent efflux of intracellular cholesterol. J. Biol. Chem. 266, 10104–10111 [PubMed] [Google Scholar]
- 34. Hoofnagle A. N., Becker J. O., Oda M. N., Cavigiolio G., Mayer P., and Vaisar T. (2012) Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin. Chem. 58, 777–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon S. M., Deng J., Tomann A. B., Shah A. S., Lu L. J., and Davidson W. S. (2013) Multi-dimensional co-separation analysis reveals protein:protein interactions defining plasma lipoprotein subspecies. Mol. Cell. Proteomics 23, 3123–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Hüttenhain R., Koomen J. M., Liebler D. C., Liu T., Maclean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J. S., Lee S. W., Moritz R., Oses-Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., and Weintraub S. (2014) Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simes R. J. (1986) An improved Bonferroni procedure for multiple tests of significance. Biometrika 73, 751–754 [Google Scholar]
- 39. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57, 289–300 [Google Scholar]
- 40. Benjamini Y., and Yekutieli D. (2001) The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 29, 1165–1188 [Google Scholar]
- 41. Huang R., Silva R. A., Jerome W. G., Kontush A., Chapman M. J., Curtiss L. K., Hodges T. J., and Davidson W. S. (2011) Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat. Struct. Mol. Biol. 18, 416–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mackness M. I. (1989) ‘A’-esterases: Enzymes looking for a role? Biochem. Pharmacol. 38, 385–390 [DOI] [PubMed] [Google Scholar]
- 43. Khersonsky O., and Tawfik D. S. (2005) Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 44, 6371–6382 [DOI] [PubMed] [Google Scholar]
- 44. Draganov D. I., Teiber J. F., Speelman A., Osawa Y., Sunahara R., and La Du B. N. (2005) Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 46, 1239–1247 [DOI] [PubMed] [Google Scholar]
- 45. Mackness M. I., Hallam S. D., Peard T., Warner S., and Walker C. H. (1985) The separation of sheep and human serum “A”-esterase activity into the lipoprotein fraction by ultracentrifugation. Comp. Biochem. Physiol. B 82, 675–677 [DOI] [PubMed] [Google Scholar]
- 46. Shih D. M., Gu L., Xia Y. R., Navab M., Li W. F., Hama S., Castellani L. W., Furlong C. E., Costa L. G., Fogelman A. M., and Lusis A. J. (1998) Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 394, 284–287 [DOI] [PubMed] [Google Scholar]
- 47. Costa L. G., Cole T. B., Jarvik G. P., and Furlong C. E. (2003) Functional genomic of the paraoxonase (PON1) polymorphisms: effects on pesticide sensitivity, cardiovascular disease, and drug metabolism. Annu. Rev. Med. 54, 371–392 [DOI] [PubMed] [Google Scholar]
- 48. Phuntuwate W., Suthisisang C., Koanantakul B., Chaloeiphap P., Mackness B., and Mackness M. (2008) Effect of fenofibrate therapy on paraoxonase1 status in patients with low HDL-C levels. Atherosclerosis 196, 122–128 [DOI] [PubMed] [Google Scholar]
- 49. Paragh G., Seres I., Harangi M., Balogh Z., Illyés L., Boda J., Szilvássy Z., and Kovács P. (2003) The effect of micronised fenofibrate on paraoxonase activity in patients with coronary heart disease. Diabetes Metab. 29, 613–618 [DOI] [PubMed] [Google Scholar]
- 50. Mackness M. I., Arrol S., Abbott C., and Durrington P. N. (1993) Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis 104, 129–135 [DOI] [PubMed] [Google Scholar]
- 51. Brown W. V., and Baginsky M. L. (1972) Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem. Biophys. Res. Commun. 46, 375–382 [DOI] [PubMed] [Google Scholar]
- 52. Ginsberg H. N., Le N. A., Goldberg I. J., Gibson J. C., Rubinstein A., Wang-Iverson P., Norum R., and Brown W. V. (1986) Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J. Clin. Invest. 78, 1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crosby J., Peloso G. M., Auer P. L., Crosslin D. R., Stitziel N. O., Lange L. A., Lu Y., Tang Z. Z., Zhang H., Hindy G., Masca N., Stirrups K., Kanoni S., Do R., Jun G., Hu Y., Kang H. M., Xue C., Goel A., Farrall M., Duga S., Merlini P. A., Asselta R., Girelli D., Olivieri O., Martinelli N., Yin W., Reilly D., Speliotes E., Fox C. S., Hveem K., Holmen O. L., Nikpay M., Farlow D. N., Assimes T. L., Franceschini N., Robinson J., North K. E., Martin L. W., DePristo M., Gupta N., Escher S. A., Jansson J. H., Van Zuydam N., Palmer C. N., Wareham N., Koch W., Meitinger T., Peters A., Lieb W., Erbel R., Konig I. R., Kruppa J., Degenhardt F., Gottesman O., Bottinger E. P., O'Donnell C. J., Psaty B. M., Ballantyne C. M., Abecasis G., Ordovas J. M., Melander O., Watkins H., Orho-Melander M., Ardissino D., Loos R. J., McPherson R., Willer C. J., Erdmann J., Hall A. S., Samani N. J., Deloukas P., Schunkert H., Wilson J. G., Kooperberg C., Rich S. S., Tracy R. P., Lin D. Y., Altshuler D., Gabriel S., Nickerson D. A., Jarvik G. P., Cupples L. A., Reiner A. P., Boerwinkle E., and Kathiresan S. (2014) Loss-of-function mutations in APOC3, triglycerides, and coronary disease. New Engl. J. Med. 371, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berbée J. F., van der Hoogt C. C., Sundararaman D., Havekes L. M., and Rensen P. C. (2005) Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J. Lipid Res. 46, 297–306 [DOI] [PubMed] [Google Scholar]
- 55. Shachter N. S., Ebara T., Ramakrishnan R., Steiner G., Breslow J. L., Ginsberg H. N., and Smith J. D. (1996) Combined hyperlipidemia in transgenic mice overexpressing human apolipoprotein Cl. J. Clin. Invest. 98, 846–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soutar A. K., Garner C. W., Baker H. N., Sparrow J. T., Jackson R. L., Gotto A. M., and Smith L. C. (1975) Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: Cholesterol acyltransferase. Biochemistry 14, 3057–3064 [DOI] [PubMed] [Google Scholar]
- 57. LaRosa J. C., Levy R. I., Herbert P., Lux S. E., and Fredrickson D. S. (1970) A specific apoprotein activator for lipoprotein lipase. Biochem. Biophys. Res. Commun. 41, 57–62 [DOI] [PubMed] [Google Scholar]
- 58. Panarotto D., Remillard P., Bouffard L., and Maheux P. (2002) Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur. J. Clin. Invest. 32, 84–92 [DOI] [PubMed] [Google Scholar]
- 59. Taskinen M. R., Beltz W. F., Harper I., Fields R. M., Schonfeld G., Grundy S. M., and Howard B. V. (1986) Effects of NIDDM on very-low-density lipoprotein triglyceride and apolipoprotein B metabolism. Studies before and after sulfonylurea therapy. Diabetes 35, 1268–1277 [DOI] [PubMed] [Google Scholar]
- 60. Lamarche B., Rashid S., and Lewis G. F. (1999) HDL metabolism in hypertriglyceridemic states: An overview. Clin. Chim. Acta 286, 145–161 [DOI] [PubMed] [Google Scholar]
- 61. Schousboe I. (1985) beta 2-Glycoprotein I: A plasma inhibitor of the contact activation of the intrinsic blood coagulation pathway. Blood 66, 1086–1091 [PubMed] [Google Scholar]
- 62. Polz E., and Kostner G. M. (1979) The binding of beta 2-glycoprotein-I to human serum lipoproteins: Distribution among density fractions. FEBS Lett. 102, 183–186 [DOI] [PubMed] [Google Scholar]
- 63. Polz E., Wurm H., and Kostner G. M. (1980) Investigations on β2-glycoprotein-I in the rat: Isolation from serum and demonstration in lipoprotein density fractions. Int. J. Biochem. 11, 265–270 [DOI] [PubMed] [Google Scholar]
- 64. Leduc M. S., Shimmin L. C., Klos K. L., Hanis C., Boerwinkle E., and Hixson J. E. (2008) Comprehensive evaluation of apolipoprotein H gene (APOH) variation identifies novel associations with measures of lipid metabolism in GENOA. J. Lipid Res. 49, 2648–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nichols G. A., Vupputuri S., and Rosales A. G. (2011) Change in high-density lipoprotein cholesterol and risk of subsequent hospitalization for coronary artery disease or stroke among patients with type 2 diabetes mellitus. Am. J. Cardiol. 108, 1124–1128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.