

Abstract
Deleted in breast cancer 1 (DBC1) has emerged as an important regulator of multiple cellular processes, ranging from gene expression to cell cycle progression. DBC1 has been linked to tumorigenesis both as an inhibitor of histone deacetylases, HDAC3 and sirtuin 1, and as a transcriptional cofactor for nuclear hormone receptors. However, despite mounting interest in DBC1, relatively little is known about the range of its interacting partners and the scope of its functions. Here, we carried out a functional proteomics-based investigation of DBC1 interactions in two relevant cell types, T cells and kidney cells. Microscopy, molecular biology, biochemistry, and mass spectrometry studies allowed us to assess DBC1 mRNA and protein levels, localization, phosphorylation status, and protein interaction networks. The comparison of DBC1 interactions in these cell types revealed conserved regulatory roles for DBC1 in gene expression, chromatin organization and modification, and cell cycle progression. Interestingly, we observe previously unrecognized DBC1 interactions with proteins encoded by cancer-associated genes. Among these interactions are five components of the SWI/SNF complex, the most frequently mutated chromatin remodeling complex in human cancers. Additionally, we identified a DBC1 interaction with TBL1XR1, a component of the NCoR complex, which we validated by reciprocal isolation. Strikingly, we discovered that DBC1 associates with proteins that regulate the circadian cycle, including DDX5, DHX9, and SFPQ. We validated this interaction by colocalization and reciprocal isolation. Functional assessment of this association demonstrated that DBC1 protein levels are important for regulating CLOCK and BMAL1 protein oscillations in synchronized T cells. Our results suggest that DBC1 is integral to the maintenance of the circadian molecular clock. Furthermore, the identified interactions provide a valuable resource for the exploration of pathways involved in DBC1-associated tumorigenesis.
Deleted in breast cancer 1 (DBC1)1 was first identified by cloning a human chromosomal region observed to be homozygously deleted in multiple breast cancers (1). Having gained prominence as an important regulator of gene expression, DBC1 is now known to have additional functions in chromatin remodeling, transcriptional regulation, and modulation of the cell cycle through its interactions with epigenetic modifiers, nuclear hormone receptors, and proteins implicated in RNA processing (2–5). DBC1 possesses several functional domains, in particular an N-terminal nuclear localization signal, a coiled-coil region, a leucine zipper (LZ), an inactive EF hand, an inactive Nudix hydrolase domain, and a S1-like RNA-binding domain (Fig. 1A) that contribute to its formation of multiple protein interactions (2, 4). Intriguingly, these known interactions of DBC1 link its function to both the suppression and promotion of tumorigenesis (6–9). A number of studies have classified DBC1 as a tumor suppressor: DBC1 was found to be down-regulated in pancreatic ductal adenocarcinoma cell lines and in patients with squamous cell lung carcinoma (10, 11). Furthermore, DBC1-knock-out mice are more susceptible to tumor formation than wild-type mice, and DBC1 was shown to directly promote p53 stability (9). Critically connected to many of its functions, DBC1 was demonstrated to negatively regulate the human deacetylases sirtuin 1 (SIRT1) and histone deacetylase 3 (HDAC3) (Fig. 1A) (3, 12). Comprehensive analysis of the interactions of all 11 members of the HDAC family revealed that DBC1 associates with additional deacetylases, including HDAC9 (13), and other deacetylase studies found DBC1 associated to HDAC5 and SIRT7 (13, 14). DBC1 is further linked to regulation of histone modifying enzymes through its inhibition of the histone methyltransferase Suv39H1 (Fig. 1A), which implicates DBC1 in the control of cell proliferation (15). Together, these reports indicate that DBC1 may act as an important global regulator of deacetylase activity, chromatin organization, and gene expression. DBC1-mediated inhibition of SIRT1 and HDAC3 further indicates a mechanism by which DBC1 may act as a tumor suppressor. SIRT1 is overexpressed in a range of cancers, including prostate (16), breast (17), colorectal (18), and acute myeloid leukemia (19). Increased HDAC3 deacetylation activity has also been strongly linked to the development of colon (20), renal (21), and classical Hodgkin's lymphoma tumors (22).
Fig. 1.
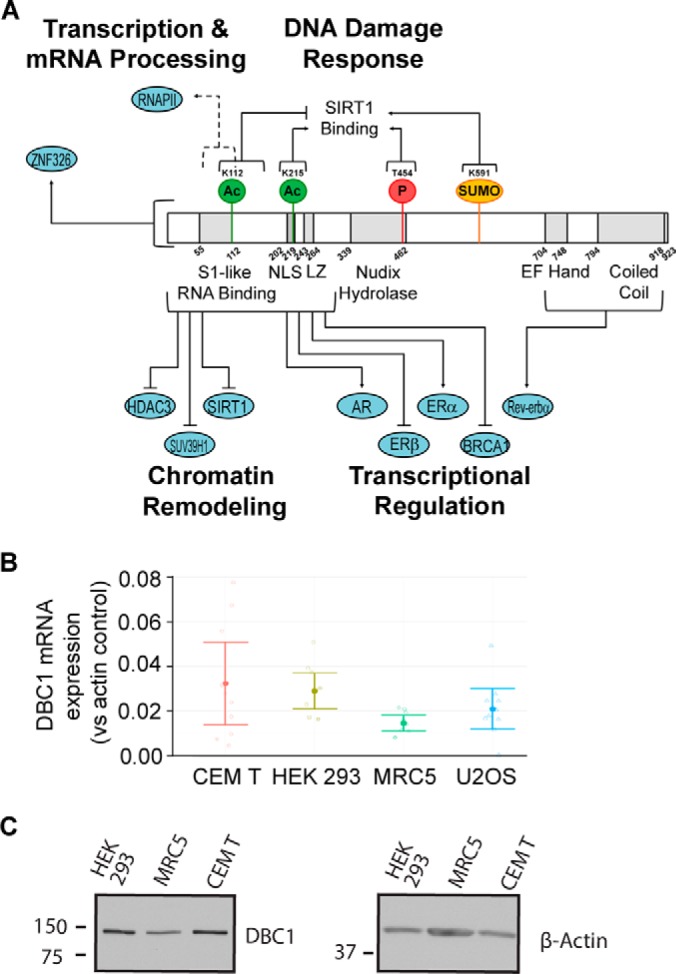
DBC1 domains, regulatory functions, and relative mRNA levels in different cell types. A, Model of DBC1 structure with predicted domain boundaries and direct interacting partners. Numbers indicate amino acid positions. Functionally characterized post-translational modifications (PTMs) are indicated. B, Endogenous mRNA expression of DBC1 in wild-type HEK 293, CEM T, U2OS, and MRC5 cells determined by qRT-PCR, was normalized to endogenous β-actin (ΔCt) for each cell type. Fold change (2−ΔCt) is plotted for each cell type. Mean fold change (solid circle) and 95% confidence intervals (error bars) are indicated. n = 10 biological replicates and n = 3 technical replicates for each biological replicate. C, Endogenous DBC1 protein levels in wild-type HEK 293, MRC5, and CEM T cells assessed by Western blotting. Loading was normalized by equal cell number (75,000 cells/sample).
Intriguingly, additional evidence points to a role for DBC1 in tumor promotion. DBC1 was observed to be up-regulated in colorectal cancer and human hepatocellular carcinoma, as well as in certain breast cancers (23, 24). In esophageal squamous cell carcinoma patients, overexpression of DBC1 correlated with poor prognosis, and knockdown of DBC1 reduced tumor cell migration and invasion (25). Similarly, increased expression of DBC1 has been associated with a greater likelihood of tumor metastasis (26). The associations between DBC1 and nuclear hormone receptors provide various pathways by which DBC1 may act as a tumor promoter (Fig. 1A). In particular, DBC1 interactions with nuclear receptors, including the estrogen receptor α (ERα) have been associated with cell proliferation (6). Additionally, DBC1 may act as a tumor promoter through its inhibition of the tumor suppressor BRCA1, particularly in the presence of apoptosis-inducing conditions (7).
DBC1 is connected to additional intracellular functions, for example through the formation of the DBIRD complex (DBC1, ZNF326/ZIRD), which is important for regulation of transcription and RNA processing (Fig. 1A) (5). The DBIRD complex integrates RNA polymerase II (RNAPII)-dependent transcription and mRNP particle formation with alternative mRNA splicing (5). Altogether, these observations indicate that the roles of DBC1 extend beyond its functions as a direct mediator of chromatin remodeling enzymes and transcriptional activation, suggesting its involvement in a wide variety of cellular processes through the formation of a diverse range of protein interactions. However, the prior knowledge accumulated with regard to DBC1 interactions was primarily derived from targeted studies, in most part focused on the interaction partner rather than on DBC1. Currently no study has focused on characterizing DBC1 interactions in several cell types, which would provide important insights into both house-keeping and cell type-specific functions of DBC1.
Here, we perform the first functional proteomics study of DBC1. To obtain insight into the activity and regulatory mechanisms of DBC1, we characterize DBC1 functional interaction networks in two relevant human cell types, T cells and HEK293 kidney cells. These cell types were selected based on our observation that DBC1 is well expressed in these cells, as well as on prior knowledge. The initial studies on DBC1 and SIRT1 were performed in HEK293. Importantly, there is a need for a better understanding of HDAC regulatory proteins, such as DBC1, in T cells considering their promise as therapeutic targets for treatment of T-cell lymphomas. Using microscopy and immunoaffinity purification (IP)-mass spectrometry, we characterized the DBC1 localization and protein interaction networks in these two cell types. Our results discovered a core set of interactions conserved in these cell types, likely representative of house-keeping DBC1 functions. The most prominent DBC1 associations were with proteins involved in the regulation of gene expression, chromatin structure, and cell cycle progression. We went on to validate a subset of these interactions using reciprocal isolations. Furthermore, we observed that proteins encoded by cancer-associated genes were enriched within the DBC1 interactome relative to the human proteome, pointing to pathways through which DBC1 could be functionally linked to tumorigenesis. Interestingly, we identified proteins that implicate DBC1 in a variety of previously unrecognized functions, including the regulation of mitotic chromosome condensation and segregation. Among the newly discovered DBC1 interactions were proteins with known functions in regulating the circadian cycle, including DDX5, DHX9, and SFPQ. We validated this association using microscopy and reciprocal isolations. Importantly, we performed functional assays to demonstrate that DBC1 protein levels are important for the regulation of CLOCK protein oscillations in synchronized T cells. Altogether, our results reveal a role for DBC1 in regulating the circadian molecular clock, as well as provide a resource for future studies of DBC1 roles in cancer progression and transcriptional regulation.
EXPERIMENTAL PROCEDURES
Generation of DBC1-EGFP Cell Lines
DBC1-EGFP cell lines were constructed by retroviral transduction using previously described methods (13) and a pLXSN-DBC1-Flag-EGFP plasmid. Briefly, that PhoenixTM retroviral transduction system (Orbigen, San Diego, CA) was used to generate stable CEM T, HEK293, MRC5, and U2OS cell lines expressing the pLXSN-DBC1-Flag-EGFP construct. Cells expressing the plasmid were selected using 300 mg/L G418 (EMD, Gibbstown, NJ) for 14 days, followed by FACS analysis (Vantage S.E. with Turbosort II, Beckton Dickinson, Franklin Lakes, NJ).
Cell Culture
Wild-type CEM T, CEM T EGFP, and CEM T DBC1-EGFP cells were cultured in Gibco® RPMI media supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin. Wild-type HEK293, HEK293 EGFP, HEK293 DBC1-EGFP, and wild-type U2OS, and MRC5 cells were cultured in Gibco® DMEM media supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin.
RNA Isolation and Quantitative RT-PCR
Total cellular RNA was isolated from wild-type HEK293, CEM T, MRC5, and U2OS, or HEK293 and CEM T cells expressing DBC1-EGFP or EGFP, using an RNeasy kit (Qiagen). cDNA was synthesized from RNA samples (1–1.5 μg) following the protocol described in the RETROScript kit (ThermoFisher Scientific, Inc.). RT-qPCR was carried out in reactions containing cDNA, DBC1 or actin primer pairs (supplemental Table S1), and SYBR Green PCR Master Mix (Applied Biosystems), on an ABI 7900HT (Applied Biosystems; Microarray Core Facility, Department of Molecular Biology, Princeton University). Relative quantification of DBC1 PCR products was performed by calculating fold change relative to endogenous β-actin. Data were analyzed by the comparative ΔCt method or ΔΔCt method (27).
DBC1 mRNA expression in wild-type cells was compared using 2−ΔCt values. For each biological replicate, the Ct values of three technical replicates were averaged, and average DBC1 Ct values were normalized by the average β-actin Ct values from the same replicate, to give the ΔCt. Statistical tests were run on the transformed values (2−ΔCt) in R-3.1.3 (28). To evaluate statistical significance of the differences in mean fold change across cell types, we built a linear model using cell type and replicate as variables, and compared the mean fold change using ANOVA (28). We assumed normal distribution of residuals. DBC1 mRNA expression in transformed cells was evaluated using the comparative 2−ΔΔCt method (27).
Immunofluorescence Microscopy
WT HEK293, HEK293-EGFP, and HEK293-DBC1-EGFP cells were cultured on chambered slides and fixed with 4% paraformaldehyde (v/v) in phosphate-buffered saline (PBS) for 15 min at 4 °C. At room temperature, cells were washed 3× with 0.1 m Glycine in PBS for 5 min, permeabilized with 0.1% Triton-X 100 in PBS for 15 min, washed 3× with 0.2% Tween in PBS (PBS-T) for 5 min, and blocked with 2% BSA and 0.2% Tween in PBS for 60 min. WT HEK293 cells were incubated in the dark for 1 h with 1:1000 rabbit polyclonal α-DBC1 primary antibody (Cell Signaling #5693) and incubated in the dark with goat α-mouse antibody conjugated to Alexa-488 (ThermoFisher Scientific, Inc.) for 60 min. Room temperature cells were incubated in the dark for 1 h with primary antibody then with secondary antibodies conjugated to either AlexaFuor-488 or -568 in PBS-T. Cells were stained with DAPI solution (1:1000 in PBS-T) in the dark for 30 min. After each incubation with antibodies and DAPI solution, cells were washed for 15 min in the dark with PBS-T. Cover slips were mounted on the slides with Aqua-PolyMount (Polysciences, Inc.) antifade solution added to each chamber, topped with a coverslip, sealed with nail polish, and stored at 4 °C in the dark. Cells were visualized with a Nikon A1 confocal microscope using (Nikon Instruments, Inc.; Confocal Microscopy Core Facility, Department of Molecular Biology, Princeton University) using a 60× immersion oil objective.
Analysis for DBC1 colocalization in the nucleus by microscopy was performed with ImageJ 1.50d. Each image contained data from three channels: DAPI, DBC1-EGFP, and either SFPQ or HCFC1. Background was subtracted for each image, and the nucleus was manually selected as the region of interest. Pearson's correlation coefficients were calculated from pixel intensities between each paired combination of the three channels using the “Coloc 2” plugin. A total of 20 nuclei were selected from each experiment. To statistically determine whether there is a difference in the correlation of one channel with either of the other two, a paired t test was used by pairing correlation coefficients from the same nucleus.
Immunoaffinity Purification (IP) of EGFP-tagged DBC1
DBC1-EGFP was isolated via the EGFP tag from either CEM T or HEK293 cells. Parallel isolations of GFP were performed to assess the specificity of interactions. The IP experiments were performed in biological replicates (n = 3 for HEK293 IPs and n = 4 for CEM T IPs) to assess the reproducibility of the observed associations. For these studies, CEM T EGFP, CEM T DBC1-EGFP, HEK293 EGFP, and HEK293 DBC1-EGFP cells were harvested by centrifugation for 5 min at 250 rcf; the cell pellet was twice washed with PBS and spun down for 5 min at 250 rcf, then resuspended in 1 ml/g of cells of 20 mm Na-HEPES (pH 7.5) + 1.2% polyvinylpyrolidone (w/v) + 1:100 protease inhibitor mixture (v/v), and frozen as pellets in liquid nitrogen. Frozen cells were cryogenically lysed at 30 Hz for 8 cycles of 2.5 min, using a Retsch MM 301 Mixer Mill (Retsch, Newtown, PA), as described (13).
Magnetic beads (M-270 Epoxy Dynabeads, ThermoFisher Scientific Inc.) were conjugated with in-house generated polyclonal rabbit anti-GFP antibodies, as described (29), washed 3× in wash buffer (1× TBT with salts, 0.5% Triton-X 100, and 200 mm NaCl), and resuspended in 100 μl wash buffer. Ground cell powder was resuspended in 10 ml/g of cells of optimized lysis buffer (1× TBT with salts, 0.5% Triton-X 100, 200 mm NaCl, 1/100 (v/v) protease inhibitor mixture (Sigma-Aldrich), and 10 μg/ml DNase (Sigma-Aldrich)), vortexed for 30 s, incubated at room temperature for 5 min with agitation, then homogenized with a Polytron PT 10–35 GT (Kinematica) for 2 cycles of 30 s. Cell debris was precipitated by centrifugation for 10 min at 8000 rcf, at 4 °C. The lysate supernatant was incubated with antibody-conjugated magnetic beads for 90 mins with rotation, at 4 °C. After immunoaffinity isolation, magnetic beads were washed 4x with 1 ml wash buffer and twice with 1 ml PBS. Isolated proteins were eluted in 30 μl 1x NuPAGE LDS sample buffer (ThermoFisher Scientific, Inc.) by shaking for 10 min at room temperature and incubation for 10 min at 70 °C. Elution was performed twice.
Eluates were stored at −20 °C until further use. To test the efficiency of DBC1 isolation, 5% or 10% of the whole cell lysate, cell pellet (insoluble fraction after incubation with lysis buffer), flow-through (unbound proteins after incubation with magnetic beads), and both eluates were preserved for Western blot analysis. Proteins in the whole cell lysate and flow-through were precipitated with 4 volumes of ice-cold acetone for 20 min at −20 °C, collected by centrifugation at 5000 rcf for 5 min, dried, and resuspended in 20 μl 1× LDS-Sample Buffer.
Mass Spectrometry-based Analysis of IP-isolated Proteins
Samples were prepared for mass spectrometry analysis with protocols modified from (30). Primary eluates from immunoaffinity solutions were reduced with 1× NuPAGE Reducing Agent (Thermo FisherScientific, Inc.) at 70 °C for 10 min and incubated in the dark at room temperature with 100 mm iodoacetamide, for 20 min. Samples were partially resolved (1/3-gel length) on a 4–12% NuPAGE Novex Bis-Tris gels (ThermoFisher Scientific, Inc.) with a PrecisionPlus Dual Color Standards (Bio-Rad Laboratories), in 1× MOPS-SDS running buffer (ThermoFisher Scientific, Inc.); proteins were visualized using SimplyBlueTM SafeStain (ThermoFisher Scientific, Inc.) and gels were destained with Milli-Q water. Whole gel lanes were cut into 1-mm slices. For additional destaining, slices were agitated at 4 °C for 10 min in 50% acetonitrile (ACN) and 50 mm ammonium bicarbonate, then dehydrated in 100% ACN at room temperature. Destaining/dehydration steps were performed three times. In-gel trypsin digestion was carried out overnight at 37 °C, with 12.5 ng/μl trypsin (Promega) in 50 mm ammonium bicarbonate (trypsin cleaves at the C terminus of arginine and lysine residues). Digested peptides were first extracted from gel with equal volume 1% formic acid (FA) for 4 h at room temperature, in the dark, then with 0.5% FA/50% ACN for 2 h at room temperature in the dark. Both extractions were combined and concentrated to ∼50 μl using vacuum centrifugation, and adjusted to 0.1% trifluoroacetic acid (TFA). To desalt peptides, samples were loaded onto StageTips (p200 tips containing 3 m SDB-RPS Empore Discs, Sigma-Aldrich), (31) and centrifuged at 1000 rcf to collect ∼8–10 μg peptides per tip. StageTip samples were washed with 100 μl 0.2% TFA and peptides manually eluted by air-pressure with 50 μl 5% ammonium hydroxide/80% ACN. Peptide samples were concentrated to ∼1 μl by vacuum centrifugation and diluted to a final volume of 9 μl with 1% FA/4% ACN.
Four microliters of each peptide sample were analyzed on a Dionex UltiMate 3000 RSLCnano System coupled to an ESI-equipped linear trap quadrupole (LTQ)-Orbitrap Velos mass spectrometer (ThermoFisher Scientific, San Jose, CA). Peptides were separated by reverse phase chromatography (Acclaim PepMap RSLC, 1.8 μm × 75 μm × 25 cm) at a flow rate of 250 nL/min on a 90 min gradient (4–40% ACN). The mass spectrometer was operated in a data-dependent acquisition mode; each acquisition cycle consisted of a single full-scan mass spectrum (m/z = 350–1700) in the Orbitrap (r = 30,000 at m/z = 400) followed by MS/MS acquisition (collision induced dissociation fragmentation) of the top 20 most abundant ions per cycle.
Raw MS/MS spectra files from each biological sample (1 gel lane with n = 8–10 fractions) were extracted and analyzed by Proteome Discoverer (PD1.4)/SEQUEST (v1.3, ThermoFisher Scientific), as described in (30); peptide spectrum matches were generated by searching against the forward and reverse sequences in the UniProt SwissProt database (22,630 entries containing human, herpesvirus, and common contaminant sequences; downloaded in August 2013), as described in (31). Search parameters were: full trypsin specificity, maximum of two missed cleavages, ion precursor mass tolerance of 10 ppm, fragment ion mass tolerance of 0.5 Da, fixed modification including cysteine carbamidomethylation, variable modifications including methionine oxidation, serine, threonine, and tyrosine phosphorylation. Search results were analyzed by Scaffold (v3.6.5 and 4.4.1; Proteome Software) and, using the X!Tandem algorithm (Beavis Informatics), refined for detection of the following additional modifications: deamidation of glutamine and asparagine, acetylation of lysine, and oxidation of tryptophan. Protein and peptide confidences were set to 99 and 95%, respectively, to achieve a protein and peptide global false discovery rate of < 2 and 1%, respectively, estimated based on searches against the reverse database. Sample contaminants entries were removed and unweighted spectrum counts were exported to Excel for additional analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (32) with the data set identifier PXD002733.
To evaluate the specificity of identified interactions, unweighted spectrum counts from DBC1-EGFP IPs were compared with EGFP control IPs from the corresponding cell type (4 replicates for CEM T cells or 3 replicates for HEK293 cells) with the SAINT algorithm, as described by (33). The SAINT algorithm assigns a probability score (ranging from 0 to 1) or specificity based on spectrum counts for bait-prey pairs, while also taking into account protein length and the total number of spectra identified in a single experiment. Proteins with a SAINT score ≥0.9 were chosen for construction of cell-specific interaction networks using the STRING database (34). The STRING network was exported to xml files and visualized with Cytoscape (35). The ratio of each protein's normalized spectral abundance factor (NSAF) to protein abundance (PAX) was calculated to account for the impact of length, relative spectral abundance among co-isolated proteins, and total cellular abundance, as reported in (14). NSAF values were calculated as in (36). Enrichment of bait proteins was calculated using the following equation: log2 (105 × NSAF/PAX).
Quantification of DBC1 by Selected Reaction Monitoring (SRM) Mass Spectrometry
Total cellular protein was extracted from wild-type CEM T, HEK 293, and cells expressing EGFP alone or DBC1-EGFP (n = 3, 105 cells per replicate) in 20 μl of pre-heated (70 °C) lysis buffer (0.1 m ammonium bicarbonate containing 5% sodium deoxycholate, 50 mm TCEP (pH-neutral), and 50 mm chloroacetamide). Extraction and subsequent processing was performed in triplicate. Samples were heated at 95 °C for 5 min and bath sonicated, then cysteine was added (50 mm final concentration), incubating for 10 min at 37 °C to quench free chloroacetamide. Samples were diluted with 80 μl of water containing 0.8 μg of MS-grade trypsin and incubated at 37 °C for 7 h. To remove deoxycholate, a phase transfer approach was used, as previously described (37). Briefly, samples were diluted with an equal volume of ethyl acetate and then acidified to 0.5% TFA. Samples were vortexed briefly, shaken for 1 min, and centrifuged at 16,000 × g for 2 min. The aqueous phase was recovered and desalted over SDB-RPS StageTips by washing sequentially with 50:50 ethyl acetate/0.5% TFA and 0.2% TFA, then eluting in 40 μl of 80:15:5 (v:v:v) ACN/water/ammonium hydroxide (38). Samples were immediately concentrated to near-dryness and suspended in 100 μl of 1% FA/2% ACN, except for MRC5 cells, which were suspended in 20 μl volume.
Candidate DBC1 peptides and histone peptides for targeted SRM quantitative analysis were selected from a spectral library of tandem mass spectra from HEK 293 whole cell tryptic digests collected by data-dependent acquisition. Based on a previous study using histone proteins as normalization standards for label-free quantification (39), six candidate tryptic peptides were screened from three histones (H2A1A, H2B1A, and H4). The tryptic peptide sequences were selected to largely avoid known sites of modifications, and in particular, the sequences from the N-terminal “tail” regions were excluded, where the most prominent post-translational modification are present. For DBC1, five candidate peptides were selected from the spectral library. Skyline (40) (ver. 3.1.1.9007) was used to calculate and export theoretical masses and retention time windows (4 min) for candidate peptides into the Xcalibur method editor.
Desalted peptides (2 μl) were analyzed by nanoliquid chromatography-selected reaction monitoring mass spectrometry on a Dionex Ultimate 3000 RSLC coupled directly to an Orbitrap Velos ETD mass spectrometer equipped with an EASY-Spray ion source (Thermofisher Scientific). Peptides were injected directly on to the analytical column (EASY-Spray 2.0 μm, 75 μm ID x 50 cm) and separated over a 60 min acetonitrile gradient from 4 to 40% B (A: 0.1% FA, B: 97% ACN in 0.1% FA). Full scan tandem mass spectra were acquired in the dual-pressure linear ion trap (isolation width = 2.0, normalized collision energy = 35, maximum ion time = 150 ms, and maximum AGC target value = 1E4) from a scheduled MS/MS mass inclusion list. After each cycle of MS/MS, a single MS spectrum was collected in the orbitrap analyzer (resolution = 7500, m/z = 350 - 1700).
Skyline was used to extract fragment ion intensities (QIT resolution = 0.7 m/z) from the ThermoRAW files, and the reproducibility of fragment ion detection and integration boundaries were manually inspected. After refinement, two histone peptides (AGLQFPVGR from H2A1A and LLLPGELAK from H2B1A) and three DBC1 peptides (VQTLSNQPLLK, SPAPPLLHVAALGQK, VLLLSSPGLEELYR) were selected (3–5 transitions with dotp > 0.90) to represent total protein abundance as these candidates had the highest chromatographic (ΔRT < 20 s) and integrated area reproducibility across biological replicates and cell types. The total fragment ion area measurements were exported to Excel, and for each biological replicate, DBC1 fragment ion areas were summarized at the protein level and normalized to the respective sum of histone H2 fragment ion areas. For DBC1-EGFP and EGFP control cell lines, the relative increase in DBC1 abundance was expressed versus the respective wild-type cells. Skyline data was published to the Panorama Public server accessible at https://panoramaweb.org/labkey/DBC1.url
Reciprocal Co-immunopurifications and Western Blot Analysis
A 0.2 g sample of cryogenically lysed HEK293 or CEM T DBC1-EGFP or EGFP cell powder was resuspended in 2 ml of cold-optimized lysis buffer (1× TBT with salts, 0.5% Triton-X 100, 200 mm NaCl, 1/100 (v/v) protease inhibitor mixture (Sigma-Aldrich), and 10 μg/ml DNase (Sigma-Aldrich)). Ten percent of lysate was saved for input analysis and the remainder was divided into two separate isolations (IgG and target protein), and incubated with rotation for 1.5 h at 4 °C with 2 μg primary antibody: mouse α-TBL1XR1 (Santa Cruz Biotechnology), mouse α-DNAPKCS (Santa Cruz Biotechnology), rabbit α-DDX5 (Abcam), and either rabbit or mouse IgG (Santa Cruz Biotechnology). Samples were incubated with 20 μl Protein A/G beads per isolation at 4 °C with gentle rotation for 2.5 h. Beads were washed twice with lysis buffer and twice with PBS, then resuspended in 25 μl LDS sample buffer and eluted by heating at 95 °C for 5 min. Co-isolated proteins were analyzed by Western blot.
To evaluate protein expression and IP efficiency, samples were analyzed by Western blotting. To measure IP efficiency, 5% or 10% of flow-through (unbound proteins), cell pellet (insoluble fraction after cell lysis), beads (following elution of bound protein), and primary elution from immunoaffinity solutions were reduced with 1× NuPAGE Reducing Agent (ThermoFisher Scientific, Inc.) at 70 °C for 10 min and incubated in the dark at room temperature with 100 mm iodoacetemide, for 20 min; to measure protein expression, whole cell lysates were prepared in the same way. Samples underwent SDS-page electrophoresis (5%-acrylamide stacking gel, 10%-acrylamide resolving gels) with PrecisionPlus Dual Color Standard ladder (Bio-Rad Laboratories) and transferred to a PVDF membrane. Proteins were visualized by antibody staining and exposure with Amersham Biosciences ECL Western blotting Detection Reagents (GE Healthcare).
Circadian Synchronization of CEM T Cells
CEM T cells expressing DBC1-EGFP or EGFP alone were synchronized by treatment with dexamethasone (Sigma Aldrich). Cells were incubated for 20 min with standard culture media supplemented with 300 nm dexamethasone. Dexamethasone-containing media was aspirated and replaced with standard culture media to release cells from treatment, and cells were plated into separate wells to allow for collection over a time-course. Cells were harvested and snap frozen at 12 h intervals, beginning at 0 h post treatment (t0) to 24 h. Cell lysates were analyzed by Western blotting to detect expression of circadian cycle-associated proteins using anti-CLOCK antibodies (Abcam).
Next, a more comprehensive analysis of circadian rhythm was performed. CEM T cells were incubated with gentle shaking at 37°C for 2 h with 300 nm dexamethasone, followed by a PBS wash and incubation in standard culture media on separate culture plates. Cells were harvested and snap frozen at 4 h intervals starting at 2 h post treatment to 50 h post treatment. Cells were lysed and analyzed by Western blotting to determine the expression of another core clock component, BMAL1, using rabbit anti-BMAL1 antibodies (Abcam #ab140646) and β-Actin as loading control. In our hands, the anti-BMAL1 antibody gave better signal-to-noise ratio than the anti-CLOCK antibody, and was preferred for more accurate quantification. BMAL1 abundance was calculated by densitometry using β-actin for normalization. A sine wave function was fit to normalized BMAL1 abundance as a function of time:
Parameters were estimated by minimizing the residual sum of squares while penalizing for large frequency (f) parameters to avoid overfitting. Periodicity was obtained from the estimated parameter f.
Experimental Design and Statistical Rationale
IP-MS experiments of DBC1-EGFP from HEK 293 and CEM T cells were performed in three and four biological replicates, respectively. For each replicate, a control IP-MS experiment was performed with an EGFP-expressing cell line. In total 18 biological samples were subjected to GeLC-MS/MS analysis. EGFP and DBC1-EGFP samples from the same replicate were analyzed sequentially by mass spectrometry. For DBC1 SRM experiments, three biological replicates and no more than three cell types/lines were processed in parallel. For quantification of DBC1 levels in DBC1-EGFP overexpression cell lines, EGFP control cell lines and wild-type cells were used as controls. Samples were analyzed in a block design, where each block was a different biological replicate that consisted of three condition/samples analyzed sequentially (wild type, EGFP, and DBC1-EGFP). Sample blank and instrument QC standards were analyzed between blocks. Statistical significance of DBC1 protein abundance was assessed by one-way ANOVA with a Bonferroni post-test (p < 0.05). For the protein colocalization experiments by microscopy, 20 nuclei were visualized for each sample. Pearson's correlation coefficients were calculated from pixel intensities, and paired t-tests were used to statistically determine differences between correlations.
RESULTS AND DISCUSSION
DBC1 mRNA Expression and Protein Abundance Varies Across Cell Types
Given the anticipated range of DBC1 functions and its absence in certain cancer tissues, understanding DBC1 expression patterns in different cell types is a necessary starting point for determining relevant cell types for assessing DBC1 interactions and tissue-specific regulatory roles. First, we used quantitative RT-PCR to determine the mRNA levels of endogenous DBC1 in T (CEM T), embryonic kidney (HEK293), fibroblast (MRC5), and osteosarcoma (U2OS) cells (Fig. 1B). These cells were selected to represent cell types previously used in DBC1 studies (HEK293) (3, 41, 42), cells in which the lysine deacetylases targeted by DBC1 (SIRT1 and HDAC3) have been functionally investigated (CEM T and MRC5) (13, 43), and an example of a cancer cell line (U2OS). We used R-3.1.3 to build a linear model using cell type and biological replicates as variables, and determined that 2−ΔCt values varied significantly across cell types (p = 0.0427). Using the Tukey Honest Significant Differences test (44), we found that MRC5 and CEM T 2−ΔCt values were significantly different (p = 0.0297). Overall, as the highly abundant β-actin mRNA was present at ∼30- to ∼70-fold greater levels than DBC1 in the sampled cells, our results indicate that DBC1 mRNA is relatively abundant within these cell types, particularly in CEM T and HEK293 cells. We next assessed the relative protein levels of endogenous DBC1 using Western blotting (Fig. 1C). Actin levels are shown as a comparison, as this control was used in previous studies. However, these cells have different amounts of cytoplasm and cytoskeleton; therefore the normalization was performed by loading equal number of cells for each sample. The protein abundance of DBC1 displayed similar relative levels to those observed for its mRNA levels, illustrating higher levels of DBC1 in CEM T and HEK293 cells when compared with MRC5 cells. Because both DBC1 mRNA and protein levels varied across the cell types investigated, its expression may be regulated on a cell type-specific basis.
DBC1-EGFP is Nuclear, Functionally Modified by Phosphorylation, and retains Its Interaction with SIRT1
Attempts to study DBC1 interactions using commercially available anti-DBC1 antibodies has resulted in inefficient isolation of the DBC1 bait protein. Therefore, in order to investigate DBC1 interactions we had to adopt a tagging approach. We opted to generate a DBC1 construct with a C-terminal EGFP tag, which we previously used for effective α-EGFP IP-MS studies and for characterization of novel protein functions (13, 45). Although tagging can significantly improve the efficiency of protein isolation, the addition of the tag can interfere with the localization, interactions, and function of the protein of interest. Therefore, first, to avoid an excessive overexpression of DBC1, we used retroviral transduction to generate CEM T and HEK293 cell lines stably expressing DBC1-EGFP or an EGFP-only control. We selected these two cell types for our interaction studies based on the functional relevance of these cell types and on our observation that DBC1 is well expressed in these cells (Fig. 1B and 1C). Specifically, HEK293 cells were selected because of their use in the initial functional studies implicating DBC1 in the regulation of SIRT1 activity (3, 41, 42). Additionally, given the ability of DBC1 to negatively regulate HDAC3, we further selected CEM T cells as a relevant cell type for investigating DBC1 functions. Small molecule pan-HDAC inhibitors are currently in use for the treatment of T-cell lymphomas, and we have previously characterized the functions and interactions of HDACs in T cells (13). We confirmed the stable expression of DBC1-EGFP in HEK293 and CEM T cells at the mRNA level using qRT-PCR (Fig. 2A). Next, we assessed the protein levels using both Western blotting (Fig. 2B) and selected reaction monitoring mass spectrometry (SRM-MS) (Fig. 2C). The Western blotting using an anti-DBC1 antibody allowed distinguishing between the endogenous DBC1 and the tagged DBC1-EGFP in HEK293 and CEM T cells (Fig. 2B). The SRM-MS analysis was performed using three unique DBC1 tryptic peptides (see Methods), and afforded a more accurate quantification of the total DBC1 protein levels in these cells (Fig. 2C). Our results showed that the overexpression of DBC1-EGFP increased the total cellular abundance of DBC1 by only ∼20 to 40% in HEK293 and CEM T cells, respectively. These results are consistent with the retroviral LTR-driven expression of DBC1-EGFP, which limits profound overexpression often induced with other promoters (e.g. CMV). We further assessed the GFP protein levels in these cells, either as free GFP or as DBC1-EGFP. DBC1 retained the GFP tag, as no prominent GFP cleavage was evident (Fig. 2D).
Fig. 2.

Expression and localization of DBC1-EGFP in T cells and kidney cells. A, DBC1 mRNA expression was measured by qRT-PCR in HEK 293 (left) or CEM T (right) cells stably expressing DBC1-EGFP or EGFP alone relative to wild-type (WT). B, Relative protein levels of endogenous DBC1 versus DBC1-EGFP assessed by Western blotting in HEK 293 (left) or CEM T (right) cells; β-actin levels are used as loading controls. C, DBC1 protein abundance was measured by selected reaction monitoring (SRM) mass spectrometry using three unique DBC1 tryptic peptides (see Materials and Methods). Experiments were performed in HEK 293 (left) and CEM T (right) cells stably expressing DBC1-EGFP or EGFP alone and in their respective WT cells. The abundance of total DBC1 (endogenous + overexpressed) was increased DBC1-EGFP cell lines, but not in the GFP control, compared with wild type cells (one-way ANOVA, HEK293: p = 0.011, CEM: p < 0.0001, mean ± S.E., n = 3). D, DBC1-EGFP and EGFP protein expression in HEK 293 or CEM T cells by Western blotting. E, DBC1 protein localization shown for the endogenous (DBC1 in WT cells) or the tagged protein (DBC1-EGFP) in HEK293 or CEM T cells. The localization of free GFP is illustrated in the control HEK293 and CEM T cell lines (EGFP, green); Nucleus, (DAPI, blue); Scale bar, 10 μm; 60x oil immersion lens.
Next, we verified the subcellular localization of EGFP-tagged DBC1 using confocal immunofluorescence (IF) microscopy. DBC1-EGFP localized primarily to the nucleus in both HEK293 and CEM T cell lines (Fig. 2E). This phenotype is consistent with previous reports on DBC1 subcellular localization (46) and with our observations of endogenous DBC1 localization in wild-type HEK293 and CEM T cells (Fig. 2E, DBC1 in WT cells). Conversely, cells expressing EGFP alone displayed free EGFP localization to both the nuclear and cytoplasmic compartments (Fig. 2E, EGFP). Additionally, using small-scale IP-Western blotting studies, we confirmed that the tagged DBC1-EGFP can still form its known functional interactions with SIRT1 and HDAC3 under the lysis buffer conditions selected for subsequent experiments (supplemental Fig. S1A). Lastly, recent studies have indicated that DBC1 is phosphorylated at numerous residues throughout its amino acid sequence (47–50). One critical modification is the phosphorylation at T454 within its Nudix hydrolase domain (Fig. 1A), a modification shown to be important for its binding to SIRT1 (50). Therefore, to further verify that DBC1-EGFP is functional, we tested whether it retains these functional phosphorylations. Mass spectrometry analysis of immunoaffinity purified DBC1-EGFP (Fig. 3A) led to the identification of 11 phosphorylation sites in both HEK293 cells and CEM T cells (red nodes, Fig. 3D). As all these sites have been previously observed in other cell types, these results indicate that the tagged DBC1-EGFP can be regulated at the post-translational level by phosphorylation similarly to endogenous DBC1 (51). Importantly, the functional T454 phosphorylation was identified in our study of DBC1-EGFP. Overall, our assessment of DBC1-EGFP shows that the tagged protein retains its nuclear localization, the interaction with SIRT1 and HDAC3, and the functional phosphorylation sites previously observed, providing a suitable model for studying DBC1 protein interactions.
Fig. 3.

Workflow for investigating DBC1 protein interactions and phosphorylation sites. A, Proteomic workflow for immunoaffinity purifications (IP) of DBC1-EGFP and EGFP from stable CEM T and HEK293 cell lines. Immunoisolates were subjected to label-free IP-MS analysis with interaction specificity determined by SAINT. DBC1 protein interaction networks were constructed and specific DBC1 interactions were functionally clustered (using GO annotations, the UniProt database, and literature searches). B–C, Representative Coomassie-stained gel lanes of DBC1-EGFP isolations, and assessment of the isolation efficiency by Western blotting (10% of each fraction was loaded). B, CEM T DBC1-EGFP cell line and C, HEK 293 DBC1-EGFP cell line. *, nonspecific band. D, DBC1 phoshorylation sites identified in this study following DBC1-EGFP isolation from both CEM T and HEK293 cells.
Identification of DBC1 Protein Interactions
Having validated the localization, known functional interactions, and modification state of DBC1 in our generated T cells and HEK293 cells, we next performed immunoaffinity purifications of DBC1-EGFP from these two cell lines using magnetic beads conjugated to in-house generated anti-GFP antibodies, and analyzed the interacting proteins by mass spectrometry (Fig. 3A). To ensure an efficient isolation of DBC1 with its functional interactions, we first performed a series of optimization experiments (supplemental Figs. S1, S2). We aimed to find a lysis buffer condition that provided sufficient stringency for effective isolation of DBC1, while still being mild enough to retain its interactions. We used DBC1's known association to SIRT1 as a test for maintenance of interactions (supplemental Fig. S1). Furthermore, we confirmed the efficient recovery of DBC1-EGFP by verifying that the majority of the protein is in the immunoaffinity purified fraction (elution) when compared with the remaining cell pellet, the flow through, and the material left on the beads after elution (Fig. 3B, 3C). The proteins co-isolated with DBC1 were next analyzed by nLC-MS/MS.
To assess the specificity of the observed DBC1 interactions, we used the Significance Analysis of Interactome (SAINT) algorithm (33) to compare proteins identified in the biological replicates of DBC1-EGFP isolations with those found in EGFP control isolations. We selected a stringent SAINT score cutoff (SAINT p ≥ 0.9) for inclusion of a prey protein in the DBC1 interaction network. This score is more stringent than the scores commonly used in previous functional protein interaction network studies (13, 30). However, given the limited knowledge regarding DBC1 interactions, we selected this SAINT cutoff to focus only on the high-confidence interactions of DBC1. From this set of DBC1 protein associations predicted as highly specific, we constructed interaction networks using information regarding the localization and cellular functions of these proteins (curated by UniProtKB and STRING-db) (34, 52). To further determine which proteins are most enriched in the DBC1 interaction network, we calculated the ratio between the normalized spectrum abundance factor (NSAF) and proteome abundance (PAX) for each interacting protein. We have previously shown that this analysis aids in the selection of the most prominent interactions for further validation and characterization (14); PAX values (http://www.pax-db.org) describe the total estimated cellular abundance of proteins, whereas NSAF values (53) reflect the relative abundance of each protein within the IP. The log2(105*NSAF/PAX) values are depicted in the interaction networks by the relative sizes of the nodes (Figs. 4, 5). Protein subcellular localization (UniProtKB Database) is denoted using node color, and proteins that have been causally linked to cancer (COSMIC database, Genome Research Ltd.) are depicted as black-outlined nodes.
Fig. 4.

Functional DBC1 interaction network in HEK 293 cells. A, DBC1 interaction network of 95 SAINT-filtered (SAINT p > 0.90) protein interactions identified in HEK293 cells (n = 3 biological replicates). Nodes indicate individual proteins; edges, known protein interactions (curated by STRING-db); bold edge, previously known DBC1 interactions). Node color depicts subcellular localization. Node size indicates NSAF/PAX values. Nodes with black borders indicate proteins causally associated with cancer (COSMIC database). B, Reciprocal isolation of DBC1-EGFP with DNAPKcs and TBL1XR1. IgG is shown as negative control.
Fig. 5.

Functional DBC1 interaction network in CEM T cells. A, DBC1 interaction network of 82 SAINT-filtered (SAINT p > 0.90) protein interactions identified in CEM T cells (n = 4 biological replicates). Nodes indicate individual proteins; edges, known protein interactions (curated by STRING-db); bold edge, previously known DBC1 interactions). Node color indicates subcellular localization. Node size indicates NSAF/PAX values. Nodes with black borders indicate proteins causally associated with cancer (COSMIC database). B, Representative immunofluorescence microscopy images of fixed DBC1-EGFP HEK293 cells stained for HCFC1 (red), DBC1-EGFP (EGFP, green), and DAPI (nucleus, blue). Scale bar, 10 μm. C, Additional assessment of HCFC1 and DBC1-EGFP colocalization within subnuclear regions. Pearson's correlation coefficients were calculated from pixel intensities between each paired combination of the three channels (DAPI, DBC1-EGFP, and HCFC1). n = 20 cells for each experiment. The points in each box represent the correlation coefficients between one of the channels (base channel) and two other channels individually. The coefficients obtained from the same cell are connected by a line to represent the difference in correlation from the base channel to the other two other channels. Paired t test p values assess whether there is a significant difference in correlation between the base channel and the other two channels.
DBC1-EGFP Interacts Primarily with Proteins Implicated in Gene Expression and Cell Cycle Progression in HEK293 Cells
We first investigated the DBC1-EGFP interactions in HEK293 cells. The co-isolation of SIRT1 and additional reported DBC1 interactions (CDK9, HNRPLL, CEP170, Fig. 4A, thick edges) (54–56) demonstrated successful isolation of functional DBC1 protein complexes. HDAC3 was also present specifically in our DBC1 isolations (and not in control isolations), but with low spectral counts. This is in agreement with our previous observation that HDAC3 is not always well detected by mass spectrometry, possibly because of the low ionization efficiency of HDAC3 peptides or the high abundance of proteins present in the same ∼50 kDa region of the gel. These low spectral counts (covering up to 12% of its amino acid sequence) precluded HDAC3 from passing our stringent SAINT filter, even if it was not detected in the control isolations. Given that the DBC1-HDAC3 is a known functional interaction and that we confirmed it by both Western blotting (supplemental Fig. S1A) and mass spectrometry, we manually included this association in our interaction network. Consistent with the predominately nuclear localization of DBC1 (Fig. 2E), a majority of the observed DBC1 interactions are reported to localize to the nucleus or have dual nucleo-cytoplasmic localization (Fig. 4A, blue and green nodes).
One dominant functional group among DBC1 interactions was formed by proteins involved in cell cycle regulation. For example, several previously unrecognized DBC1 associations function in chromosome condensation and segregation (Fig. 4A). These included members of the condensin complex (NCAPG2 and SMC4), which is responsible for establishing mitotic chromosome architecture and regulating chromosome segregation (57). The microtubule organizer, CEP170, is known to associate with the centrosome during interphase and with the spindle apparatus in mitosis (58). NUMA1 helps tether spindle microtubules to the centrosomes (59). CHTF18, PDS5A, PDS5B, POGZ, and STAG2 are all implicated in sister chromatid cohesion, which is critical in chromosome segregation and DNA repair (60, 61). Further strengthening the functional association of DBC1 with proteins involved in DNA repair, we validated the observed interaction with DNAPKcs by reciprocal isolation of the endogenous protein (Fig. 4B). Interestingly, both HDAC8 and HDAC9 have been shown to associate with members of the cohesin complex. Although HDAC8 and HDAC9 were not co-isolated with DBC1 in this study, possibly because of the low abundances of these proteins, DBC1 has been shown to interact with HDAC9 upon immunoaffinity isolation of HDAC9-EGFP (13). Furthermore, HDAC8 is known to regulate acetylation levels of cohesin complex members, promoting turnover of the cohesin complex (62). Cohesin removal from chromatin is essential for sister chromatid segregation, and deacetylation of cohesin complexes facilitates release (63, 64). DBC1 may coordinate its activity with HDAC8 and HDAC9 to regulate acetylation levels of these proteins, cohesin complex turnover, and chromosome segregation. To our knowledge, DBC1 has not previously been linked to sister chromatid cohesion or chromosome segregation.
Maybe not surprisingly, proteins that function in the regulation of gene expression represented the largest contingent within DBC1 associations. Within this group were proteins with roles in mRNA processing and splicing, including the TREX complex proteins THOC2 and THOC5. The TREX complex is thought to couple transcription with RNA processing and export (65), and deletion of THOC2 is associated with reduced transcription elongation in yeast (66). It is possible that DBC1's transcriptional regulatory and mRNA processing activities are coupled through this association with the TREX complex. In agreement with the known role of DBC1 in regulating gene expression as a cofactor for nuclear hormone receptors (Fig. 1A), our interaction study revealed its interaction with multiple nuclear hormone receptor transcription factors. These factors included SAFB2 (estrogen receptor corepressor), PELP1 (estrogen receptor co-activator), and PSPC1 (mediator of androgen receptor transcriptional activity). The presence of additional transcriptional regulatory elements, TLE1 and TLE3, further implicates DBC1 in multiple signal transduction pathways, including the Wnt/β-catenin and mitogen-activated signaling pathways.
Of particular interest was the identified DBC1 interaction with TBL1XR1 (Fig. 4A–4B), a component of the HDAC3-containing NCoR complex (67). In view of the known inhibitory function of DBC1 on HDAC3, we further investigated this association. Indeed, we validated this interaction by reciprocal immunoaffinity isolation of endogenous TBL1XR1 (Fig. 4B). It is possible that TBL1XR1 facilitates recruitment of DBC1 to the NCoR complex to promote inhibition of HDAC3. As the NCoR complex is known to repress ERα and AR-dependent transcription (68, 69), TBLX1R1-mediated recruitment could provide a mechanism for DBC1-associated co-activation of estrogen and/or androgen receptors (6, 70) .
The Functional Associations of DBC1 with Gene Expression and Cell Cycle Progression are Retained in T Cells
Given the relevance of T cells in HDAC biology and cancer development, we next investigated DBC1 interactions in T cells. Similar to our studies in HEK293 cells, we constructed an interaction network for predicted specific DBC1 interaction in CEM T cells (82 proteins with SAINT p ≥ 0.90) (Fig. 5A). As expected, the group of proteins that passed this stringent criteria included SIRT1 and other known DBC1 interactions—p53, ZNF326, and PRMT5 (5, 9, 71), and most identified proteins had nuclear or nucleo-cytoplasmic annotations. Among the DBC1 interactions that appeared to be unique or enriched in T cells when compared with HEK293 cells were proteins involved in RNA processing and splicing, as well as in the control of the circadian cycle. However, despite these cell type-dependent differences in interactions, one striking feature was again the prominent association of DBC1 with proteins involved in the regulation of gene expression, chromatin structure, and cell cycle progression (Fig. 5A). To determine the subset of proteins that maintained their interactions with DBC1 within these broad functional categories, we constructed an interaction network from proteins found in both T cells and HEK293 cells (supplemental Fig. S3). In performing this comparison between cell types, in addition to our initial analysis at a SAINT probability cutoff of 0.9 (Figs. 4, 5), we also performed an analysis with a lower filtering cutoff (≥ 0.75) to try to partially compensate for the likely differences in protein abundances in these two cell types (supplemental Fig. S3). The identified overlap in interactions with proteins regulating gene expression and the cell cycle aligns well with the previously reported functions of DBC1, and points to core interactions that may be associated with the house-keeping roles of DBC1 in various cell types. Therefore, future investigation of these previously unrecognized interactions can help to shed light on mechanisms modulating DBC1 functions. For example, the interaction with SPIN1 links DBC1 to chromatin binding, Wnt signaling, and cancer cell proliferation (72). Similarly, the identified interaction with host-cell factor HCFC1 strengthens the link between the roles of DBC1 in regulating the cell cycle and chromatin remodeling. HCFC1 is required for cell cycle progression, as well as an important regulator of transcription through its functional association with chromatin modifying enzymes, including deacetylases (Sin3-HDAC complex), acetyltransferases (p300), and methyltransferases (Set1/Ash acting on H3K4) (73–75). Indeed, we observed the colocalization of DBC1 and HCFC1 within the nucleus (Fig. 5B). However, both of these proteins display disperse subnuclear localizations, making it challenging to qualitatively determine the level of colocalization. Therefore, we next assessed their subnuclear colocalization using Pearson's correlation coefficient. At the subnuclear level, DBC1-EGFP and HCFC1 show similar distribution, as evident by the intensity pattern of these proteins in the nucleus (Fig. 5C, top panel). In contrast, the nuclear marker DAPI, which is not expected to localize with these proteins, shows a distinct and more diffuse distribution. This observation was quantified using Pearson's correlation of pixel intensities in 20 cell nuclei. The average correlation coefficients of DBC1-EGFP and HCFC1 (0.87) were higher to those of DAPI to either DBC1-EGFP (0.64) or HCFC1 (0.59). The statistical significance was assessed using paired t-tests (Fig. 5C, bottom panel), showing that DBC1-EGFP correlation is significantly higher to HCFC1 than to DAPI (p = 2.7 × 10–7) and HCFC1 correlation is significantly higher to DBC1-EGFP than to DAPI (p = 6.7 × 10–7). In contrast, the coefficients of DAPI correlation to either DBC1-EGFP or HCFC1 are not significantly different (p = 0.22). This analysis suggests that these proteins have the opportunity to interact within the nucleus, further supporting DBC1 coordinated regulation of multiple types of chromatin modifying enzymes and remodeling events.
The DBC1 Interactome is Enriched in Proteins Causally Associated with Cancer
To further explore the connection between cancer development and functional DBC1 interactions, we used the COSMIC database (Wellcome Trust Sanger Institute, Genome Research Ltd.) to identify proteins within the interaction networks that are causally associated with cancer (black outlined nodes in Figs. 4, 5 and supplemental Fig. S3). Currently, according to the COSMIC database, ∼1% of the genome (572 human genes) has been causally implicated in cancer. These cancer-associated genes were enriched within the DBC1 interactome relative to the human proteome (Table I): 9.4% of specific interactions in HEK293 cells, 9.8% of specific interactions in CEM T cells, and 11.4% of shared interactions in HEK293 and CEM T cells are causally linked to cancer. In agreement with prior reports that DBC1 can both inhibit and promote tumorigenesis, the observed interacting proteins are characterized as both tumor promoters and tumor suppressors (Table I). The observed variation in DBC1-EGFP interactions and function across cell types highlights the complex and multifaceted roles for DBC1 in tumorigenesis, as it implies that DBC1 has multiple cell-specific functions and behaviors.
Table I. DBC1 interactions with proteins encoded by genes causally associated with human cancers. TP = tumor promoter; TS = tumor suppressor; D = dominant; HI = haploinsufficient; r = recessive.
| Protein | Link | Mutation type | Cancer types | |
|---|---|---|---|---|
| CEM T& HEK293 | FIP1L1 | Kinase fusion (with PDGFRA) | Cryptic chromosomal deletion; D | Hypereosinophilic syndrome |
| SF3B1 | TS; potential oncogenic mutations | Missense; D, HI | Myelodysplastic syndrome | |
| STAG2 | TS; inactivation may be linked to aneuploidy | Various | Bladder carcinoma, glioblastoma, melanoma, Ewing's sarcoma, myeloid neoplasms, pancreatic ductal adenocarcinoma | |
| ZCCHC8 | Kinase fusion (with ROS1) | Translocation, D | Spitzoid tumor | |
| CEM T | DDX5 | TP | Missense, translocation; D | Prostate, breast |
| IKZF1 | TS; dominant-negative isoform is TP | Translocation, deletion; D, HI | Acute lymphocytic leukemia, B-cell lymphoma | |
| PPP2R1A | TS | Missense | Clear cell ovarian carcinoma | |
| p53 | TS | Missense; multiple types | Multiple tumor types | |
| SFPQ | Fusion protein (with TFE3); disruption of normal activity | Translocation; D | Papillary renal | |
| HEK293 | ARID1A | TS | Insertions & deletions (frame-shifts, nonsense); sometimes HI | Clear cell ovarian carcinoma, breast, renal cell |
| LCP1 | Fusion (with LAZ3) | Translocation (with BCL6); D | B-cell non-Hodgkin lymphoma | |
| MSH6 | TS | Substitution, multiple types; R | Colorectal, endometrial, ovarian | |
| NUMA1 | Fusion (with RARA); TP | Translocation; D | Acute promyelocytic leukemia | |
| SMARCA4 | TS | Multiple | Lung, ovarian | |
| TBL1XR1 | TS; TP | Frame-shift, substitution; R | Multiple | |
| TRIM27 | Fusion (with RET); TP | Translocation; D | Papillary thyroid |
One particularly relevant DBC1 interaction identified in our study was that with the SWI/SNF (SWItch/Sucrose Nonfermentable) complex (Fig. 4). Five SWI/SNF components were identified in DBC1 isolations in HEK293 cells: ADNP, ARID1A, SMARCA2, SMARCA4, and SMARCC2. Strikingly, the SWI/SNF is the most frequently (∼20%) mutated chromatin remodeling complex in human cancers (76, 77). Moreover, SMARCA4 has been shown to be a tumor suppressor through its role in regulating nuclear receptor-dependent gene expression (78). As DBC1 is already known to modulate gene expression associated with nuclear receptors (6, 70, 79), its interaction with SMARCA4 may indicate a regulatory role for DBC1 in SMARCA4-mediated tumor suppression.
A DBC1 interacting partner observed in both HEK293 and T cells (supplemental Fig. S3), STAG2, was reported in a study of 21 human cancer types as one of 12 genes frequently mutated in more than four tumor types (80). Mutations of proteins associated with sister chromatid cohesion have been found within multiple cancer genomes, and may be linked with aneuploidy in some cancers (81). Through its interaction with STAG2, it is possible that the role of DBC1 in regulating sister chromatid cohesion and segregation can be associated with tumorigenesis.
Aberrant DBC1 expression has been associated with multiple cancer types, and consequently, it is not surprising that the DBC1 interactomes are enriched in cancer-associated genes. However, the identification of specific DBC1 interactions that are already linked to tumorigenesis or tumor suppression (Table I) provides promising targets for future studies in a therapeutic context and further deciphering of the role of DBC1 in cancer progression.
DBC1 Interactions in T Cells Point to a Role in Regulating the Circadian Cycle and Alternative Splicing
Despite the overlap in DBC1 interactions in T cells and kidney cells, numerous SAINT-filtered proteins were only observed to associate with DBC1 in a single cell type (Figs. 4, 5, supplemental Fig. S3). The relatively small degree of overlap between these interaction networks implies that DBC1 participates in a range of cellular pathways through interactions that vary on a cell-by-cell basis.
An intriguing DBC1 interaction enriched in T cells was that with proteins known to regulate RNA processing (in particular splicing) and the circadian cycle (Fig. 5A). Nine of the isolated proteins belong to the spliceosomal C and the U4/U5/U6 tri-snRNP complexes: PNN, DDX5, SF3B3, SNRNP200, SF3B1, ZCCHC8, PRPF8, SFPQ, and CDC5L (82, 83). SF1, UPF1, and ADAR also belong to the spliceosome or supraspliceosome machinery (84, 85). These interactions with members of the spliceosome complex are likely linked to DBC1's role as a component of the DBIRD complex, which integrates transcript elongation and mRNA splicing (5). Indeed, ZNF326, the other known component of the DBIRD complex, was also present in our interaction network in T cells. However, our finding that DBC1 interacts with multiple components of the spliceosome complex suggests that DBC1 may have a role in the integration of transcription with alternative splicing beyond the promotion of exon exclusion. RNAPII-dependent transcription is functionally coupled to the assembly of the spliceosome (86). The spliceosome is a large and dynamic ribonucleoprotein (RNP) complex, consisting of the U1, U2, U4/U6, and U5 small nuclear (sn) RNPs, as well as over a hundred non-snRNP proteins (87). Notably, protein acetylation and deacetylation plays a role in cotranscriptional spliceosome assembly (88). For example, histone post-translational modifications create binding sites for proteins that promote spliceosome assembly (89). Moreover, one study has found that the deletion of HDACs limits the recruitment and rearrangement of snRNPs downstream of U2, inhibiting the formation of a fully functional spliceosomal complex (90). HDAC inhibitors, both of classical HDACs and of sirtuins, as well as HAT inhibitors, lead to the formation of stalled complexes that represent intermediates of spliceosomal assembly and block pre-mRNA splicing (88). DBC1 may modulate spliceosomal assembly through the regulation of chromatin remodeling and protein acetylation levels.
Sharing oscillatory features with cell cycle progression, the circadian cycle emerged as another interesting functional category for DBC1 interactions in T cells. We identified several proteins belonging to the human circadian clock PERIOD (PER) complex (SFPQ, DDX5, and DHX9), which suggests a regulatory role for DBC1 at the interface of chromatin remodeling and the circadian cycle. The PER complex mediates repression of circadian gene targets, while SFPQ recruits the SIN3-HDAC complex, promoting histone deacetylation and transcriptional repression (91). DDX5 (p68) has been found within spliceosomal B and C complexes, is necessary for the transition between the prespliceosomal and the spliceosomal complex, and co-immunoprecipitates with the PER complex component, PER2 (83, 91). Given the potential significance of this DBC1 association, we performed further studies to validate it. Using reciprocal isolation of the endogenous RNA helicase DDX5 from T cells, we indeed confirmed this interaction by observing the co-isolation of DBC1 only in the DDX5 IP and not in the parallel IgG control isolation (Fig. 6A). Furthermore, we also confirmed the pan-nuclear colocalization of DBC1 with DDX5 (Fig. 6C) by immunofluorescence microscopy. We observed a similar colocalization of DBC1 with SFPQ (Fig. 6D), but were not able to obtain an antibody suitable for additional validation by reciprocal isolations. Therefore, we further assessed the subnuclear colocalization of these proteins. DBC1-EGFP and SFPQ displayed good colocalization as shown by zoomed images of the nucleus (Fig. 6E, top panel) and averaged Pearson's correlation coefficient of 20 nuclei (0.79). This correlation is significantly higher than the averaged coefficients of DAPI correlation to either DBC1-EGFP (0.47) or SFPQ (0.45), as shown by paired t-tests (Fig. 6E, bottom panel). DBC1-EGFP and SFPQ correlation is significantly higher than the correlation of either of these proteins to DAPI (p = 8.8 × 10−9 and p = 1.2 × 10−7). In contrast, the correlation of DAPI to either of these proteins is not significantly different (p = 0.58). Altogether, these analyses indicate that DBC1 has the potential to interact with members of the circadian clock PER complex within the nuclear compartment.
Fig. 6.

DBC1 interacts with regulatory components of the circadian cycle. A, Isolation of DBC1-EGFP in immunoaffinity purifications of endogenous DDX5 using rabbit anti-DDX5 antibodies or rabbit IgG (IgG); loading represents 10% of each sample (IgG IP, DDX5 IP, and Input). B, Confirmation of bait (DDX5) isolation using rabbit anti-DDX5 antibodies, when compared with control isolation; loading represents 1% of input, 10% for flow through (FT), and 10% for IP elution. C–D, Representative immunofluorescence images of fixed DBC1-EGFP CEM T cells stained for PER complex components DDX5 (panel C, red) and SFPQ (panel D, red) and, DBC1-EGFP (green), and DAPI (nucleus, blue). Scale bar, 10 μm. E, Additional assessment of the subnuclear colocalization of DBC1-EGFP with SFPQ, as in Fig. 5C. Pearson's correlation coefficients were calculated from pixel intensities between each paired combination of the three channels (DAPI, DBC1-EGFP, and SFPQ). n = 20 cells for each experiment. F, CLOCK protein levels by immunoblotting (anti-Clock) following circadian cycle synchronization by dexamethasone treatment (300 nm) at 0, 12, and 24 h post-treatment in control EGFP overexpression (EGFP Ctrl) or DBC1-EGFP overexpression (DBC1 OE) CEM T cell line backgrounds. Values represent relative levels as assessed by densitometry. G, BMAL1 relative protein levels as quantified by Western blot densitometry after circadian cycle synchronization. BMAL1 levels were measured every 4 h from 2 to 50 h after dexamethasone treatment in EGFP control (EGFP Ctrl) and DBC1-EGFP overexpression (DBC1 OE). BMAL1 levels normalized to actin are shown as points in the graph, and a fitted sine curve is displayed as a line. The periodicity was calculated from the parameters of the fitted sine curve (representative of two biological replicates). H, Model for an expanded role for DBC1 in the regulation of the circadian cycle. Protein interactions known to have direct roles in regulation of the circadian cycle are shown as solid gray lines. Known impact of DBC1 interactions that have not been previously linked to the circadian cycle are shown as solid orange lines. Proposed regulatory roles for DBC1 are shown as dashed orange lines. HDAC corepressor complexes are shown in red.
By integrating our findings with previous reports, we propose a model for DBC1 functions in the regulation of the circadian cycle (Fig. 6H). DBC1 has been previously shown to directly regulate the circadian receptor Rev-erbα by promoting Rev-erbα stability and transcriptional repression of BMAL1 expression (92), which is mediated by NCoR-HDAC3 (93). Conversely, the CLOCK-BMAL1 complex promotes transcription of circadian genes, in part through HAT-mediated acetylation of CLOCK (94) and through physical association with SIRT1 (95). Cyclic SIRT1-dependent deacetylation of BMAL1 promotes BMAL1 stability and CLOCK-BMAL1 activity, whereas SIRT1-mediated histone deacetylation likely represses expression of CLOCK-BMAL1 target genes (95, 96). The DBC1-mediated regulation of SIRT1 activity further links DBC1 function to the circadian cycle (3, 4). Furthermore, if DBC1-mediated inhibition of HDAC activity extends to SIN3-HDAC complexes, DBC1 may also control repression of PER complex activity and promotion of CLOCK-BMAL1 target gene expression.
Given the potential for DBC1 to regulate the expression of proteins associated with the circadian cycle and our identification of a specific association with PER complex components, we next tested whether DBC1 levels can impact CLOCK functions. Toward this goal, we synchronized CEM T cells with dexamethasone, a glucocorticoid agonist, and assessed the impact of DBC1-EGFP overexpression on the oscillation of CLOCK protein expression relative to EGFP control cells (Fig. 6F). The EGFP cell line was selected as the control in this experiment to both account for any potential impact of the EGFP tag on circadian cycling, as well as to provide a matched cell line that underwent the same transformations (e.g. retrovirus transduction) as the DBC1-EGFP cell line. As expected, upon circadian clock synchronization in EGFP control cells, CLOCK protein levels were decreased at 0 h, increased at 12 h, and then decreased again at 24 h. In contrast, we observed loss of CLOCK protein oscillation in cells expressing DBC1-EGFP (Fig. 6F). Our results suggest that DBC1 protein expression is important for the temporal regulation of CLOCK protein levels.
To further support this proposed role of DBC1 in the regulation of circadian rhythm, we performed a more temporally comprehensive analysis by looking at the regulation of another core clock component, BMAL1. A 50-h time course with 4 h intervals was collected from dexamethasone-synchronized CEM T EGFP control and DBC1-EGFP overexpressing cells. BMAL1 abundance was measured throughout this time course (Fig. 6G). In EGFP control cells, BMAL1 oscillated with a periodicity of 20.6 h, as expected for the circadian clock. Interestingly, DBC1-EGFP overexpressing cells showed BMAL1 oscillations with an apparently slowed clock of 53.9 h periods. The oscillations of this slowed clock could only be observed by looking at this longer time-course, whereas a shorter time-course (<24 h) only showed an apparent loss of circadian oscillations (as observed in Fig. 6F). Our results suggest that DBC1 protein abundance is important for the temporal regulation of two core clock components, BMAL1 and CLOCK.
DBC1 may also regulate PER complex activity as a component of the DBIRD complex. Whereas the PER complex (DDX5 and DHX9) acts to inhibit transcript termination and to reduce the rate of transcription, the DBIRD complex promotes elongation (5, 91). As both the PER complex and DBIRD complex are known to interact with RNAPII and are found in our study to be DBC1 interactions, DBC1 may counteract the PER-dependent inhibition of RNAPII transcription. Altogether, by revealing these DBC1 interactions with components of the PER complex, the DBIRD complex, chromatin remodeling proteins, and transcriptional regulatory factors, our results point to several mechanisms through which DBC1 can contribute to regulating the human circadian cycle.
CONCLUSIONS
Here we present the first proteomics study of DBC1 interactions in HEK293 and CEM T cells. These interactomes point to both shared and unique DBC1 functional protein associations in these diverse cell types. We demonstrate conserved regulatory roles for DBC1 in gene expression, chromatin organization and modification, and cell cycle progression. We further observed that numerous newly identified interactions of DBC1 are causally associated with cancer progression. Among these are the interactions with five components of the SWI/SNF complex, which is the most frequently mutated chromatin remodeling complex in human cancers. Another important example is our identified DBC1 interaction with STAG2, a protein associated with sister chromatid cohesion and found to be mutated in multiple cancer genomes. Of particular interest was the identified interaction with TBL1XR1, a component of the NCoR complex, which we validated by reciprocal isolations. Our results implicate DBC1 activity in transcriptional regulation through its association with HDAC-containing corepressor complexes and in maintenance of DNA strand integrity via specific interaction with the DNA repair protein DNAPKcs.
Interestingly, we discover that DBC1 associates with multiple proteins involved in the circadian cycle. We show that DBC1 interacts with multiple components of the PER complex (DDX5, DHX9, and SFPQ), a critical regulator of the circadian cycle. We validate this interaction by colocalization and reciprocal isolation. Importantly, we demonstrate that DBC1 protein levels are important in the regulation of CLOCK and BMAL1 protein oscillations in T cells during circadian cycling.
Altogether, these interactions support and further expand our knowledge of DBC1 as both a specific regulator of histone deacetylation and a broad regulator of chromatin remodeling. Our results lead us to propose that DBC1 functions are integral to the maintenance of coordinate circadian cycle progression. It is possible that DBC1-mediated transcriptional events and epigenetic modifications are regulated in a temporal manner through its participation in circadian clock-associated complexes. In addition to our functional insights into the roles of DBC1, we expect that our broad interaction networks will provide a resource for future investigations of DBC1 roles in cancer progression and transcriptional regulation.
Supplementary Material
Acknowledgments
We appreciate the technical help provided by Gary Laevsky and C. DeCoste (Microscopy and Flow Cytometry Core Facilities, Princeton University).
Footnotes
Author contributions: A.J.G., P.M. Jean Beltran, P.M. Joshi, T.M.G., and I.M.C. designed research; S.S.G., A.J.G., P.M. Jean Beltran, P.M. Joshi, T.M.G., O.L.Q., and J.K. performed research; S.S.G., A.J.G., P.M. Jean Beltran, P.M. Joshi, T.M.G., O.L.Q., and I.M.C. analyzed data; S.S.G., A.J.G., and I.M.C. wrote the paper.
* This work was supported by NIH grants R01 GM114141, R33 AI102187, and R01 HL127640 to IMC, by a postdoctoral fellowship from the New Jersey Commission on Cancer Research (NJCCR) to PMJ and TMG, and by an NSF GRFP to AJG. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental Figs. S1 to S3 and Tables S1 to S5.
This article contains supplemental Figs. S1 to S3 and Tables S1 to S5.
1 The abbreviations used are:
- DBC1
- deleted in breast cancer 1
- ACN
- acetonitrile
- CEM T cells
- human T-cell lymphoblasts
- CID
- collision induced dissociation
- ETD
- electron transfer dissociation
- FA
- formic acid
- HDAC
- histone deacetylase
- HEK293
- human embryonic kidney cells
- NSAF
- Normalized Spectral Abundance Factor
- SAINT
- Significance Analysis of Interactome computational tool
- SIRT
- sirtuin
- WT
- wild type.
REFERENCES
- 1. Hamaguchi M., Meth J. L., von Klitzing C., Wei W., Esposito D., Rodgers L., Walsh T., Welcsh P., King M. C., and Wigler M. H. (2002) DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 99, 13647–13652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sundararajan R., Chen G. H., Mukherjee C., and White E. (2005) Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-alpha-mediated death signaling. Oncogene 24, 4908–4920 [DOI] [PubMed] [Google Scholar]
- 3. Zhao W., Kruse J.-P., Tang Y., Jung S. Y., Qin J., and Gu W. (2008) Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–U511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim J.-E., Chen J., and Lou Z. (2008) DBC1 is a negative regulator of SIRT1. Nature 451, 583–U510 [DOI] [PubMed] [Google Scholar]
- 5. Close P., East P., Dirac-Svejstrup A. B., Hartmann H., Heron M., Maslen S., Chariot A., Soeding J., Skehel M., and Svejstrup J. Q. (2012) DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 484, 386–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trauernicht A. M., Kim S. J., Kim N. H., and Boyer T. G. (2007) Modulation of estrogen receptor alpha protein level and survival function by DBC-1. Mol. Endocrinol. 21, 1526–1536 [DOI] [PubMed] [Google Scholar]
- 7. Hiraike H., Wada-Hiraike O., Nakagawa S., Koyama S., Miyamoto Y., Sone K., Tanikawa M., Tsuruga T., Nagasaka K., Matsumoto Y., Oda K., Shoji K., Fukuhara H., Saji S., Nakagawa K., Kato S., Yano T., and Taketani Y. (2010) Identification of DBC1 as a transcriptional repressor for BRCA1. Br. J. Cancer 102, 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim H. J., Kim S. H., Yu E. J., Seo W. Y., and Kim J. H. (2014) A positive role of DBC1 in PEA3-mediated progression of estrogen receptor-negative breast cancer. Oncogene [DOI] [PubMed] [Google Scholar]
- 9. Qin B., Minter-Dykhouse K., Yu J., Zhang J., Liu T., Zhang H., Lee S., Kim J., Wang L., and Lou Z. (2015) DBC1 Functions as a tumor suppressor by regulating p53 stability. Cell Rep. 10, 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wauters E., Sanchez-Arevalo Lobo V. J., Pinho A. V., Mawson A., Herranz D., Wu J., Cowley M. J., Colvin E. K., Njicop E. N., Sutherland R. L., Liu T., Serrano M., Bouwens L., Real F. X., Biankin A. V., and Rooman I. (2013) Sirtuin-1 regulates acinar-to-ductal metaplasia and supports cancer cell viability in pancreatic cancer. Cancer Res. 73, 2357–2367 [DOI] [PubMed] [Google Scholar]
- 11. Tseng R.-C., Lee C.-C., Hsu H.-S., Tzao C., and Wang Y.-C. (2009) Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients. Neoplasia 11, 763–U765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chini C. C. S., Escande C., Nin V., and Chini E. N. (2010) HDAC3 is negatively regulated by the nuclear protein DBC1. J. Biol. Chem. 285, 40830–40837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., and Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Sys. Biol. 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsai Y.-C., Greco T. M., Boonmee A., Miteva Y., and Cristea I. M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell Proteomics 11, 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Z., Chen L., Kabra N., Wang C., Fang J., and Chen J. (2009) Inhibition of SUV39H1 methyltransferase activity by DBC1. J. Biol. Chem. 284, 10361–10366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huffman D. M., Grizzle W. E., Bamman M. M., Kim J. S., Eltoum I. A., Elgavish A., and Nagy T. R. (2007) SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 67, 6612–6618 [DOI] [PubMed] [Google Scholar]
- 17. Elangovan S., Ramachandran S., Venkatesan N., Ananth S., Gnana-Prakasam J. P., Martin P. M., Browning D. D., Schoenlein P. V., Prasad P. D., Ganapathy V., and Thangaraju M. (2011) SIRT1 is essential for oncogenic signaling by estrogen/estrogen receptor alpha in breast cancer. Cancer Res. 71, 6654–6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen X., Sun K., Jiao S., Cai N., Zhao X., Zou H., Xie Y., Wang Z., Zhong M., and Wei L. (2014) High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Scientific Rep. 4, 7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bradbury C. A., Khanim F. L., Hayden R., Bunce C. M., White D. A., Drayson M. T., Craddock C., and Turner B. M. (2005) Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19, 1751–1759 [DOI] [PubMed] [Google Scholar]
- 20. Spurling C. C., Godman C. A., Noonan E. J., Rasmussen T. P., Rosenberg D. W., and Giardina C. (2008) HDAC3 overexpression and colon cancer cell proliferation and differentiation. Molecular Carcinog. 47, 137–147 [DOI] [PubMed] [Google Scholar]
- 21. Fritzsche F. R., Weichert W., Roske A., Gekeler V., Beckers T., Stephan C., Jung K., Scholman K., Denkert C., Dietel M., and Kristiansen G. (2008) Class I histone deacetylases 1, 2, and 3 are highly expressed in renal cell cancer. BMC Cancer 8, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams H., Fritzsche F. R., Dirnhofer S., Kristiansen G., and Tzankov A. (2010) Class I histone deacetylases 1, 2, and 3 are highly expressed in classical Hodgkin's lymphoma. Expert Opin. Therap. Targets 14, 577–584 [DOI] [PubMed] [Google Scholar]
- 23. Kim J. E., Chen J., and Lou Z. (2009) p30 DBC is a potential regulator of tumorigenesis. Cell Cycle 8, 2932–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y., Gu Y., Sha S., Kong X., Zhu H., Xu B., Li Y., and Wu K. (2014) DBC1 is overexpressed and associated with poor prognosis in colorectal cancer. Int. J. Clin. Oncol. 19, 106–112 [DOI] [PubMed] [Google Scholar]
- 25. Kim S. H., Kim J. H., Yu E. J., Lee K. W., and Park C. K. (2012) The overexpression of DBC1 in esophageal squamous cell carcinoma correlates with poor prognosis. Histol. Histopathol. 27, 49–58 [DOI] [PubMed] [Google Scholar]
- 26. Kim J. R., Moon Y. J., Kwon K. S., Bae J. S., Wagle S., Yu T. K., Kim K. M., Park H. S., Lee J. H., Moon W. S., Lee H., Chung M. J., and Jang K. Y. (2013) Expression of SIRT1 and DBC1 is associated with poor prognosis of soft tissue sarcomas. PloS One 8, e74738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmittgen T. D., and Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 28. Team R. C. (2015) R: a language and environment for statistical computing. [Google Scholar]
- 29. Cristea I. M., Williams R., Chait B. T., and Rout M. P. (2005) Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941 [DOI] [PubMed] [Google Scholar]
- 30. Diner B. A., Li T., Greco T. M., Crow M. S., Fuesler J. A., Wang J., and Cristea I. M. (2015) The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol. Syst. Biol. 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greco T. M., Miteva Y., Conlon F. L., and Cristea I. M. (2012) Complementary proteomic analysis of protein complexes. Methods Mol. Biol. 917, 391–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., and Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 37. Masuda T., Tomita M., and Ishihama Y. (2008) Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 7, 731–740 [DOI] [PubMed] [Google Scholar]
- 38. Kulak N. A., Pichler G., Paron I., Nagaraj N., and Mann M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 [DOI] [PubMed] [Google Scholar]
- 39. Wisniewski J. R., Hein M. Y., Cox J., and Mann M. (2014) A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics 13, 3497–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu E. J., Kim S. H., Heo K., Ou C. Y., Stallcup M. R., and Kim J. H. (2011) Reciprocal roles of DBC1 and SIRT1 in regulating estrogen receptor alpha activity and coactivator synergy. Nucleic Acids Res. 39, 6932–6943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nin V., Escande C., Chini C. C., Giri S., Camacho-Pereira J., Matalonga J., Lou Z., and Chini E. N. (2012) Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J. Biol. Chem. 287, 23489–23501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koyuncu E., Budayeva H. G., Miteva Y. V., Ricci D. P., Silhavy T. J., Shenk T., and Cristea I. M. (2014) Sirtuins are evolutionarily conserved viral restriction factors. mBio 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tukey J. (1949) Comparing individual means in the analysis of variance. Biometrics 5, 99–114 [PubMed] [Google Scholar]
- 45. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., and Cristea I. M. (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garapaty S., Xu C.-F., Trojer P., Mahajan M. A., Neubert T. A., and Samuels H. H. (2009) Identification and characterization of a novel nuclear protein complex involved in nuclear hormone receptor-mediated gene regulation. J. Biol. Chem. 284, 7542–7552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villen J., Li J., Cohn M. A., Cantley L. C., and Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 49. Molina H., Horn D. M., Tang N., Mathivanan S., and Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yuan J., Luo K., Liu T., and Lou Z. (2012) Regulation of SIRT1 activity by genotoxic stress. Genes Dev. 26, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cohen P. (2000) The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 52. (2015) UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paoletti A. C., Parmely T. J., Tomomori-Sato C., Sato S., Zhu D., Conaway R. C., Conaway J. W., Florens L., and Washburn M. P. (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. U.S.A. 103, 18928–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jeronimo C., Forget D., Bouchard A., Li Q., Chua G., Poitras C., Therien C., Bergeron D., Bourassa S., Greenblatt J., Chabot B., Poirier G. G., Hughes T. R., Blanchette M., Price D. H., and Coulombe B. (2007) Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell 27, 262–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kristensen A. R., Gsponer J., and Foster L. J. (2012) A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9, 907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chatr-Aryamontri A., Breitkreutz B. J., Oughtred R., Boucher L., Heinicke S., Chen D., Stark C., Breitkreutz A., Kolas N., O'Donnell L., Reguly T., Nixon J., Ramage L., Winter A., Sellam A., Chang C., Hirschman J., Theesfeld C., Rust J., Livstone M. S., Dolinski K., and Tyers M. (2015) The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmiesing J. A., Ball A. R. Jr., Gregson H. C., Alderton J. M., Zhou S., and Yokomori K. (1998) Identification of two distinct human SMC protein complexes involved in mitotic chromosome dynamics. Proc. Natl. Acad. Sci. U.S.A. 95, 12906–12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guarguaglini G., Duncan P. I., Stierhof Y. D., Holmstrom T., Duensing S., and Nigg E. A. (2005) The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol. Biol. Cell 16, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Silk A. D., Holland A. J., and Cleveland D. W. (2009) Requirements for NuMA in maintenance and establishment of mammalian spindle poles. J. Cell Biol. 184, 677–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prieto I., Pezzi N., Buesa J. M., Kremer L., Barthelemy I., Carreiro C., Roncal F., Martinez A., Gomez L., Fernandez R., Martinez A. C., and Barbero J. L. (2002) STAG2 and Rad21 mammalian mitotic cohesins are implicated in meiosis. EMBO Rep. 3, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Losada A., Yokochi T., and Hirano T. (2005) Functional contribution of Pds5 to cohesin-mediated cohesion in human cells and Xenopus egg extracts. J. Cell Sci. 118, 2133–2141 [DOI] [PubMed] [Google Scholar]
- 62. Deardorff M. A., Bando M., Nakato R., Watrin E., Itoh T., Minamino M., Saitoh K., Komata M., Katou Y., Clark D., Cole K. E., De Baere E., Decroos C., Di Donato N., Ernst S., Francey L. J., Gyftodimou Y., Hirashima K., Hullings M., Ishikawa Y., Jaulin C., Kaur M., Kiyono T., Lombardi P. M., Magnaghi-Jaulin L., Mortier G. R., Nozaki N., Petersen M. B., Seimiya H., Siu V. M., Suzuki Y., Takagaki K., Wilde J. J., Willems P. J., Prigent C., Gillessen-Kaesbach G., Christianson D. W., Kaiser F. J., Jackson L. G., Hirota T., Krantz I. D., and Shirahige K. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nasmyth K., and Haering C. H. (2005) The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74, 595–648 [DOI] [PubMed] [Google Scholar]
- 64. Chan K. L., Roig M. B., Hu B., Beckouet F., Metson J., and Nasmyth K. (2012) Cohesin's DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 150, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Strasser K., Masuda S., Mason P., Pfannstiel J., Oppizzi M., Rodriguez-Navarro S., Rondon A. G., Aguilera A., Struhl K., Reed R., and Hurt E. (2002) TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417, 304–308 [DOI] [PubMed] [Google Scholar]
- 66. Piruat J. I., and Aguilera A. (1998) A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J. 17, 4859–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang J., Kalkum M., Chait B. T., and Roeder R. G. (2002) The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 9, 611–623 [DOI] [PubMed] [Google Scholar]
- 68. Cheng S., Brzostek S., Lee S. R., Hollenberg A. N., and Balk S. P. (2002) Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol. Endocrinol. 16, 1492–1501 [DOI] [PubMed] [Google Scholar]
- 69. Stossi F., Likhite V. S., Katzenellenbogen J. A., and Katzenellenbogen B. S. (2006) Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J. Biol. Chem. 281, 16272–16278 [DOI] [PubMed] [Google Scholar]
- 70. Fu J., Jiang J., Li J., Wang S., Shi G., Feng Q., White E., Qin J., and Wong J. (2009) Deleted in breast cancer 1, a novel androgen receptor (AR) coactivator that promotes AR DNA-binding activity. J. Biol. Chem. 284, 6832–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C. L., Pena V., Luhrmann R., and Stelzl U. (2012) Dynamic protein–protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580 [DOI] [PubMed] [Google Scholar]
- 72. Wang J. X., Zeng Q., Chen L., Du J. C., Yan X. L., Yuan H. F., Zhai C., Zhou J. N., Jia Y. L., Yue W., and Pei X. T. (2012) SPINDLIN1 promotes cancer cell proliferation through activation of WNT/TCF-4 signaling. Mol. Cancer Res. 10, 326–335 [DOI] [PubMed] [Google Scholar]
- 73. Mahajan S. S., and Wilson A. C. (2000) Mutations in host cell factor 1 separate its role in cell proliferation from recruitment of VP16 and LZIP. Mol. Cell. Biol. 20, 919–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wysocka J., Myers M. P., Laherty C. D., Eisenman R. N., and Herr W. (2003) Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 17, 896–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cai Y., Jin J., Swanson S. K., Cole M. D., Choi S. H., Florens L., Washburn M. P., Conaway J. W., and Conaway R. C. (2010) Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem. 285, 4268–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., and Crabtree G. R. (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shain A. H., and Pollack J. R. (2013) The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS One 8, e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Romero O. A., Setien F., John S., Gimenez-Xavier P., Gomez-Lopez G., Pisano D., Condom E., Villanueva A., Hager G. L., and Sanchez-Cespedes M. (2012) The tumor suppressor and chromatin-remodeling factor BRG1 antagonizes Myc activity and promotes cell differentiation in human cancer. EMBO Mol. Med. 4, 603–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Koyama S., Wada-Hiraike O., Nakagawa S., Tanikawa M., Hiraike H., Miyamoto Y., Sone K., Oda K., Fukuhara H., Nakagawa K., Kato S., Yano T., and Taketani Y. (2010) Repression of estrogen receptor beta function by putative tumor suppressor DBC1. Biochem. Biophys. Res. Commun. 392, 357–362 [DOI] [PubMed] [Google Scholar]
- 80. Lawrence M. S., Stojanov P., Mermel C. H., Robinson J. T., Garraway L. A., Golub T. R., Meyerson M., Gabriel S. B., Lander E. S., and Getz G. (2014) Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 505, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guo G., Sun X., Chen C., Wu S., Huang P., Li Z., Dean M., Huang Y., Jia W., Zhou Q., Tang A., Yang Z., Li X., Song P., Zhao X., Ye R., Zhang S., Lin Z., Qi M., Wan S., Xie L., Fan F., Nickerson M. L., Zou X., Hu X., Xing L., Lv Z., Mei H., Gao S., Liang C., Gao Z., Lu J., Yu Y., Liu C., Li L., Fang X., Jiang Z., Yang J., Li C., Chen J., Zhang F., Lai Y., Zhou F., Chen H., Chan H. C., Tsang S., Theodorescu D., Li Y., Zhang X., Wang J., Yang H., Gui Y., and Cai Z. (2013) Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 45, 1459–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gozani O., Patton J. G., and Reed R. (1994) A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J. 13, 3356–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jurica M. S., Licklider L. J., Gygi S. R., Grigorieff N., and Moore M. J. (2002) Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8, 426–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang X., Bruderer S., Rafi Z., Xue J., Milburn P. J., Kramer A., and Robinson P. J. (1999) Phosphorylation of splicing factor SF1 on Ser20 by cGMP-dependent protein kinase regulates spliceosome assembly. EMBO J. 18, 4549–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Agranat L., Raitskin O., Sperling J., and Sperling R. (2008) The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc. Natl. Acad. Sci. U.S.A. 105, 5028–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Das R., Dufu K., Romney B., Feldt M., Elenko M., and Reed R. (2006) Functional coupling of RNAP II transcription to spliceosome assembly. Genes Dev. 20, 1100–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wahl M. C., Will C. L., and Luhrmann R. (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 [DOI] [PubMed] [Google Scholar]
- 88. Kuhn A. N., van Santen M. A., Schwienhorst A., Urlaub H., and Luhrmann R. (2009) Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. RNA 15, 153–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sims R. J. 3rd, Millhouse S., Chen C. F., Lewis B. A., Erdjument-Bromage H., Tempst P., Manley J. L., and Reinberg D. (2007) Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 28, 665–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gunderson F. Q., Merkhofer E. C., and Johnson T. L. (2011) Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. U.S.A. 108, 2004–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Padmanabhan K., Robles M. S., Westerling T., and Weitz C. J. (2012) Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science 337, 599–602 [DOI] [PubMed] [Google Scholar]
- 92. Chini C. C., Escande C., Nin V., and Chini E. N. (2013) DBC1 (Deleted in Breast Cancer 1) modulates the stability and function of the nuclear receptor Rev-erbalpha. Biochem. J. 451, 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yin L., and Lazar M. A. (2005) The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 19, 1452–1459 [DOI] [PubMed] [Google Scholar]
- 94. Doi M., Hirayama J., and Sassone-Corsi P. (2006) Circadian regulator CLOCK is a histone acetyltransferase. Cell 125, 497–508 [DOI] [PubMed] [Google Scholar]
- 95. Nakahata Y., Kaluzova M., Grimaldi B., Sahar S., Hirayama J., Chen D., Guarente L. P., and Sassone-Corsi P. (2008) The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hirayama J., Sahar S., Grimaldi B., Tamaru T., Takamatsu K., Nakahata Y., and Sassone-Corsi P. (2007) CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450, 1086–1090 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.