

Abstract
Defective copper excretion from hepatocytes in Wilson's disease causes accumulation of copper ions with increased generation of reactive oxygen species via the Fenton-type reaction. Here we developed a nanoflow liquid chromatography-nanoelectrospray ionization-tandem mass spectrometry coupled with the isotope-dilution method for the simultaneous quantification of oxidatively induced DNA modifications. This method enabled measurement, in microgram quantities of DNA, of four oxidative stress-induced lesions, including direct ROS-induced purine cyclonucleosides (cPus) and two exocyclic adducts induced by byproducts of lipid peroxidation, i.e. 1,N6-etheno-2′-deoxyadenosine (εdA) and 1,N2-etheno-2′-deoxyguanosine (εdG). Analysis of liver tissues of Long-Evans Cinnamon rats, which constitute an animal model of human Wilson's disease, and their healthy counterparts [i.e. Long-Evans Agouti rats] showed significantly higher levels of all four DNA lesions in Long-Evans Cinnamon than Long-Evans Agouti rats. Moreover, cPus were present at much higher levels than εdA and εdG lesions. In contrast, the level of 5-hydroxymethyl-2′-deoxycytidine (5-HmdC), an oxidation product of 5-methyl-2′-deoxycytidine (5-mdC), was markedly lower in the liver tissues of Long-Evans Cinnamon than Long-Evans Agouti rats, though no differences were observed for the levels of 5-mdC. In vitro biochemical assay showed that Cu2+ ions could directly inhibit the activity of Tet enzymes. Together, these results suggest that aberrant copper accumulation may perturb genomic stability by elevating oxidatively induced DNA lesions, and by altering epigenetic pathways of gene regulation.
Many endogenous and exogenous chemical events may produce DNA damage, including assault from reactive oxygen species (ROS)1 (1, 2). ROS are routinely generated in cells as a consequence of metabolic activity and exposure to various environmental agents. Under physiological conditions, transition metal ion-mediated Fenton reaction constitutes a major endogenous source of ROS, and aberrant accumulation of transition metal ions may elicit deleterious effects on cells. Elevated ROS could lead to DNA damage directly, or indirectly via byproducts of lipid peroxidation (LPO). In this vein, hydroxyl radical can directly react with DNA to yield a myriad of DNA lesions, including the bulky 8,5′-cyclopurine-2′-deoxynucleosides (cPu) (3, 4). Byproducts of LPO could react with DNA to generate exocyclic adducts, including 1,N6-etheno-2′-deoxyadenosine (εdA) and 1,N2-etheno-2′-deoxyguanosine (εdG) (5, 6). Multiple lines of evidence showed that these DNA lesions in mammalian cells, tissues and blood may serve as potential biomarkers of oxidative stress (7–14).
Some oxidatively induced DNA modifications are also exploited by mammalian cells to regulate gene expression. In this context, Fe2+- and 2-oxoglutarate (2-OG)-dependent ten-eleven translocation (TET) family dioxygenases were recently found to be involved in sequential oxidation of 5-methyl-2′-deoxycytidine (5-mdC) to yield 5-hydroxymethyl-2′-deoxycytidine (5-HmdC), 5-formyl-2′-deoxycytidine (5-FodC), and 5-carboxyl-2′-deoxycytidine (5-CadC) (15–20). Subsequent excision of 5-FodC and 5-CadC from DNA by thymine DNA glycosylase (TDG), coupled with restoration of an unmethylated 2′-deoxycytidine by base-excision repair (BER) pathway, may facilitate active DNA cytosine demethylation in mammals (16, 21). In addition, 5-hmdC could be recognized by some specific cellular proteins (22–25), suggesting that the oxidized derivatives of 5-mdC may also serve as epigenetic marks on their own.
Transition metal ion homeostasis is essential for normal cellular function and excess accumulation of metal ions, such as iron or copper, may result in diseases. In this vein, Wilson's disease (WD) is an autosomal recessive genetic disorder arising from mutations in the P-type ATPase gene, ATP7B, and patients suffering from this disease are manifested by defective excretion of copper ions into bile (26, 27). This leads to accumulation of copper in the body with progressive damage in liver, brain and kidneys (26, 27). The Long-Evans Cinnamon (LEC) rat, which was found in a colony of healthy Long-Evans Agouti (LEA) rat, harbors a partial deletion at the 3′ end of the Atp7b gene and shares many clinical attributes of Wilson's disease in humans, including excess copper accumulation in the liver along with spontaneous acute liver failure and progressive liver damage with extensive hepatic cholangiofibrosis (28). Therefore, the LEC rat serves as an excellent animal model for studying the pathophysiology of human Wilson's disease.
Increased copper content, and elevated levels of ROS and oxidative DNA damage have been observed in liver tissues of LEC rats (10, 29). However, it remains unclear whether abnormal copper accumulation perturbs epigenetic signaling by modulating the formation of 5-HmdC. In light of our previous finding that 5-HmdC could be induced from oxidation of 5-mdC in synthetic duplex DNA by Fenton-type reagent (Cu2+/H2O2/ascorbate) in vitro (30), we reasoned that increased copper accumulation might also elicit higher levels of 5-HmdC in vivo. On the other hand, the Jumonji C domain-containing histone demethylases (JHDMs), which are also Fe2+- and 2-OG-dependent dioxygenases, were previously shown to be perturbed by exposure to carcinogenic heavy metals, including nickel, chromium, and possibly arsenic (31–33). The replacement of iron in the active site of JHDMs by nickel, and depletion of ascorbic acid, a crucial cofactor of these enzymes, from chromium or arsenic exposure, could attenuate the demethylase activity of JHDMs in cells (31–33). Likewise, other Fe2+- and 2-OG-dependent dioxygenases, including hypoxia inducible factor (HIF)-prolyl hydroxylase PHD2 and DNA repair enzymes ALKBH2/3, could also be inhibited by nickel ion (31, 34). Therefore, accumulation of copper in Wilson's disease may compromise enzymatic activities of Tet proteins in vivo, thereby resulting in lower 5-HmdC levels.
Herein, we comprehensively assessed the effect of abnormal copper accumulation on oxidatively induced DNA modifications in liver and brain tissues of LEC and LEA rats by LC-MS/MS combined with the stable isotope-dilution technique. The goal was to gain a better understanding of genetic and epigenetic alterations arising from aberrant copper accumulation.
MATERIALS AND METHODS
Materials and Animals
All chemicals and enzymes, unless otherwise specified, were purchased from Sigma-Aldrich (St. Louis, MO) and New England Biolabs (Ipswich, WA), respectively. Stable isotope-labeled compounds were obtained from Cambridge Isotope Laboratories (Cambridge, MA). Erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) hydrochloride was from Tocris Bioscience (Ellisville, MO). LEA and LEC rats (14–16 week old) were bred in a Core Facility at Einstein as described previously (10). The stable isotope-labeled S-cdA, S-cdG and 5-HmdC were synthesized previously (10, 30). The εdA and εdG as well as their stable isotope-labeled counterparts were synthesized following procedures published previously (35, 36).
Extraction and Enzymatic Digestion of DNA
Genomic DNA was extracted from rat tissues by using a high-salt method (10). The liver and brain tissues of LEA/LEC rats were grounded into fine powders under liquid nitrogen in a mortar. A 100-μl lysis buffer, containing 20 mm Tris (pH 8.1), 20 mm EDTA, 400 mm NaCl, 1% SDS (w/v) and 10–15 μl of proteinase K (20 mg/ml), was added to the powdered tissue with incubation in a water bath at 55 °C overnight. Saturated NaCl solution (0.5 volume) was subsequently added to the mixture, which was vortexed for 1 min and incubated at 55 °C for another 15 min. The resulting mixture was centrifuged at 13,000 rpm for 30 min. Nucleic acids were precipitated from the supernatant by ethanol. The RNA in the nucleic acid mixture was digested with 3 μl RNase A (10 mg/ml) and 2 μl RNase T1 (25 units/μl) at 37 °C overnight, followed by extraction with an equal volume of chloroform/isoamyl alcohol (24:1, v/v). The DNA was precipitated from the aqueous layer by ethanol, re-dissolved in water and quantified by UV spectrophotometry.
The DNA sample (10 μg) was digested with 1 unit of nuclease P1 and 0.00125 unit of phosphodiesterase II in a 15-μl buffer containing 300 mm sodium acetate (pH 5.6), 10 mm ZnCl2 and 2.5 nmol of EHNA, which served as an inhibitor of adenine deaminase to minimize deamination of dA to 2′-deoxyinosine (10). After incubation at 37 °C for 48 h, to the mixture were added 1.0 unit of alkaline phosphatase, 0.0025 unit of phosphodiesterase I, and 20 μl of buffer containing 500 mm Tris-HCl (pH 8.9). After a 2-hr incubation at 37 °C, the digestion mixture was neutralized with 1 m formic acid, and uniformly 15N-labeled S-cdA, S-cdG, εdA and εdG internal standards were added to the mixture. The enzymes in the digestion mixture were removed by extraction with chloroform/isoamyl alcohol (24:1, v/v), and the resulting aqueous layer was subjected to off-line HPLC enrichment.
For quantification of 5-HmdC and 5-mdC, [1,3-15N2-2′-D]-5-HmdC and [1′,2′,3′,4′,5′-13C5]-5-mdC were added to the enzymatic digestion mixture of 50 ng of genomic DNA. The enzymes in digestion mixture were again removed by extraction with chloroform/isoamyl alcohol (24:1, v/v), and the resulting aqueous layer was subjected directly to LC-MS/MS analysis on an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific).
HPLC Enrichment
The HPLC separation was performed on a Beckman HPLC system with pump module 125 and UV detector module 126. An Alltima HP-C18 column (4.6 × 250 mm, 5 μm in particle size, 300 Å in pore size, Grace Davison, Deerfield, IL) was used to enrich oxidatively induced DNA lesions from the aforementioned nucleoside mixture. The mobile phases were 10 mm ammonium formate (Solution A) and methanol (Solution B). A gradient of 42 min 0% B, 1 min 0–2% B, 17 min 2% B, 1 min 2–5% B, 9 min 5% B, 10 min 5–13% B, 20 min 13% B, and 50 min 13–60% B was used, and the flow rate was 1 ml/min. The HPLC fractions containing S-cdG (33.5–38.0 min), S-cdA (66.0–70.0 min), and εdA + εdG (81.5–87.5 min) were pooled individually and dried in a Speed-vac. The dried samples were then reconstituted in water for NanoLC-NSI/MS2 analysis.
NanoLC-NSI/MS2 Analysis
The NanoLC-NSI-MS/MS analysis was conducted on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific) equipped with a nano-electrospray ionization source and coupled with an EASY nLC II (Thermo Fisher Scientific). HPLC separation was carried out using a homemade trapping column (150 μm × 40 mm) and an analytical column (75 μm × 200 mm), both packed with Magic C18 AQ (200 Å, 5 μm, Michrom BioResources, Auburn, CA). Mobile phase A was 0.1% formic acid in H2O and mobile phase B was 0.1% formic acid in acetonitrile. Initially, the sample was loaded onto the trapping column with mobile phase A at a flow rate of 2.5 μl/min. The analytes were eluted using a 40-min linear gradient of 0–40% mobile phase B at a flow rate of 300 nL/min. The TSQ Vantage mass spectrometer was operated in the positive-ion mode, where the spray voltage was 1.8 kV and the temperature for ion-transport tube was 275 °C. The instrument was set up in multiple-reaction monitoring (MRM) mode, with MRM transitions being listed in supplemental Table S1. The sensitivities for detecting all modified nucleosides were optimized by varying the collision energy (supplemental Table S1). The amounts of oxidative stress-induced DNA modifications (in moles) in nucleoside mixtures were calculated from area ratios of peaks found in selected-ion chromatograms (SICs) for the analytes over their corresponding isotope-labeled standards, the amounts of the labeled standards added (in moles) and the calibration curves (Fig. S2). The final levels of the modified nucleosides, in terms of the numbers of modified nucleosides per 107 nucleosides, were calculated by dividing the amounts of individual modified nucleosides with the total amount of nucleosides (in moles) in the digested DNA.
Capillary LC-tandem MS Analyses of 5-HmdC and 5-mdC
The LC-MS3 quantification of 5-HmdC was conducted on an LTQ linear ion trap mass spectrometer coupled with an Agilent 1200 capillary HPLC pump, as described previously (37). The LC-MS2 measurement of 5-mdC was performed on the same instrument. The HPLC separation was conducted using a 0.5 × 250 mm Zorbax SB-C18 column (5 μm in particle size, Agilent Technologies). A solution of 2 mm ammonium bicarbonate in water (pH 7.0, solution A) and methanol (solution B) were employed as mobile phases. A gradient of 5 min 0–20% B and 25 min 20–70% B was used for separating analytes, and the flow rate was 8.0 μl/min. The voltage for electrospray was 5.0 kV, and the ion-transport tube was maintained at a temperature of 275 °C. The isolation width for precursor ion selection was 3 m/z units, the normalized collision energy was 35, the activation Q was 0.25, and the activation time was 30 ms. The levels of 5-HmdC and 5-mdC were calculated with the method described above for the oxidative stress-induced DNA lesions.
Biochemical Assay of Tet1-mediated Oxidation of 5-mdC in Duplex DNA
The Tet1-mediated 5-mdC oxidation assay was conducted with the use of the 5mC Tet1 Oxidation Kit (Wisegene, Chicago, IL) and a 5-mdC-containing duplex DNA, d(AGCTC(5-mdC)GGTCA)/d(GTGACCGGAGCTG), following procedures reported previously (38) with slight modifications. Briefly, 20 pmol of the aforementioned duplex DNA was incubated with 0.4 μl of mouse Tet1 protein, along with a reaction buffer containing 50 mm HEPES (pH 8.0), 100 mm sodium chloride, 2 mm ascorbic acid, 1 mm 2-oxoglutarate, 1 mm ATP, 1 mm DTT, along with 75 μm ammonium iron(II) sulfate and various concentrations of copper sulfate (at Cu2+/Fe2+ molar ratios of 0, 0.25, 0.50, 1.0, 2.0 and 5.0) in a total volume of 50 μl. After incubation at 37 °C for 10 min, the reaction mixtures were immediately frozen on dry ice. The enzyme in the mixture was removed using chloroform extraction. The resulting aqueous layer was subjected directly to LC-MS and MS2 on an LTQ linear ion trap mass spectrometer to identify reaction products and to quantify conversions of 5-mdC to its oxidation products. A 0.5 × 250 mm Zorbax SB-C18 column was employed for the separation and the flow rate was 8.0 μl/min. 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP, pH adjusted to 7.0 with triethylamine, solution A) and methanol (solution B) were employed as mobile phases, and a gradient of 5 min of 0–20% B and 35 min of 20–40% B was used. The voltage for electrospray was 4.0 kV and the ion-transport tube of the mass spectrometer was set at 300 °C. The MS2 and higher-resolution “ultra-zoom-scan” MS were acquired for the [M - 3H]3− ions of the initial 11mer 5-mdC-containing ODN and corresponding oxidized 5-mdC products. The intensities of the monoisotopic and M + 1 isotopic peaks were employed for calculating fractions of 5-mdC oxidation product in the reaction mixtures.
RESULTS
NanoLC-NSI/MS2 Method for Quantifying Oxidative Stress-induced DNA Lesions in Rat Tissues
Relatively large amounts of unmodified nucleosides and buffer salts are present in the enzymatic digestion mixture of tissue DNA, which can significantly interfere with the detection of low levels of DNA lesions. To overcome this, we employed off-line HPLC separation to enrich DNA lesions of interest from the digestion mixtures prior to NanoLC-NSI/MS2 analysis. Relative to previously reported capillary LC-MS3 methods (10), the low flow rate used for nanoflow LC (at 300 nL/min) further improved the sensitivity of analyte detection by improving the ionization efficiencies and transmission of ions from the ionization interface to the mass analyzer. Consequently, the amount of tissue DNA consumed in sample preparation was reduced from 80–100 μg to 10–20 μg.
Upon collisional activation, the [M + H]+ ions of unlabeled εdA and εdG can eliminate a 2-deoxyribose moiety to yield the protonated nucleobase portion as the dominant fragment ions in MS/MS, i.e. the ions of m/z 160 and 176 for εdA and εdG, respectively (Fig. 1A and1B and supplemental Fig. S1A). The collisional activation of [M + H]+ ions of unlabeled S-cdA and S-cdG, on the other hand, gave fragment ions of m/z 164 and 180, respectively, which arise from cleavages of both the N-glycosidic bond and the C4′-C5′ bond in the 2-deoxyribose moiety (39) (Fig. 1C and 1D and supplemental Fig. S1B). The nearly identical elution time for the analytes and their stable isotope-labeled counterparts, along with their specific MRM transitions, allowed for unambiguous identification and reliable quantification of oxidative stress-induced lesions in rat tissue DNA (calibration curves shown in supplemental Fig. S2).
Fig. 1.
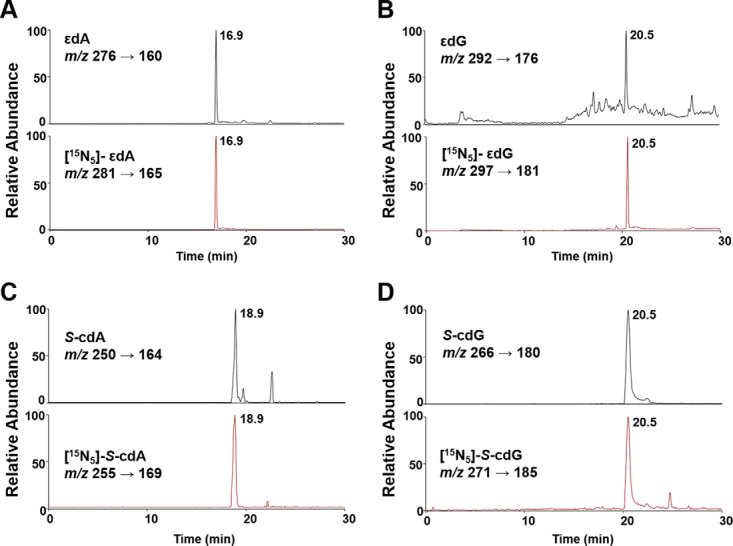
Representative NanoLC-NSI/MS2 results for the quantifications of εdA (A), εdG (B), S-cdA (C), and S-cdG (D) in DNA of rat tissues. Shown are the selected-ion chromatograms (SICs) for monitoring the indicated MRM transitions for the analytes (top trace) and the isotope-labeled standards (bottom trace).
Levels of Oxidative Stress-induced DNA Lesions in Liver and Brain Tissues of LEA and LEC Rats
With the NanoLC-NSI/MS2 method, we were able to measure the levels of cPus and LPO-induced exocyclic DNA lesions in rat liver and brain tissues with low-μg quantities of DNA. Our quantification results revealed that S-cdA, S-cdG, εdA, and εdG levels in LEA rat liver tissues were 12.0, 20.2, 2.3, and 1.1 lesions per 107 nucleosides, respectively (Fig. 2 and supplemental Table S2). The corresponding levels of these lesions were 26.8, 44.5, 3.0, and 1.6 adducts per 107 nucleosides, respectively, in DNA from LEC rat liver tissues (Fig. 2 and supplemental Table S2). Thus, the levels of all four oxidative stress-induced DNA lesions were significantly higher in liver tissues of LEC than LEA rats, which is in agreement with markedly increased liver copper content and liver injury in LEC rats (28). Consistent with the absence of gross neurological defects in LEC rats (40), the levels of oxidative stress-induced DNA lesions in brain tissues of LEC and LEA rats were similar.
Fig. 2.
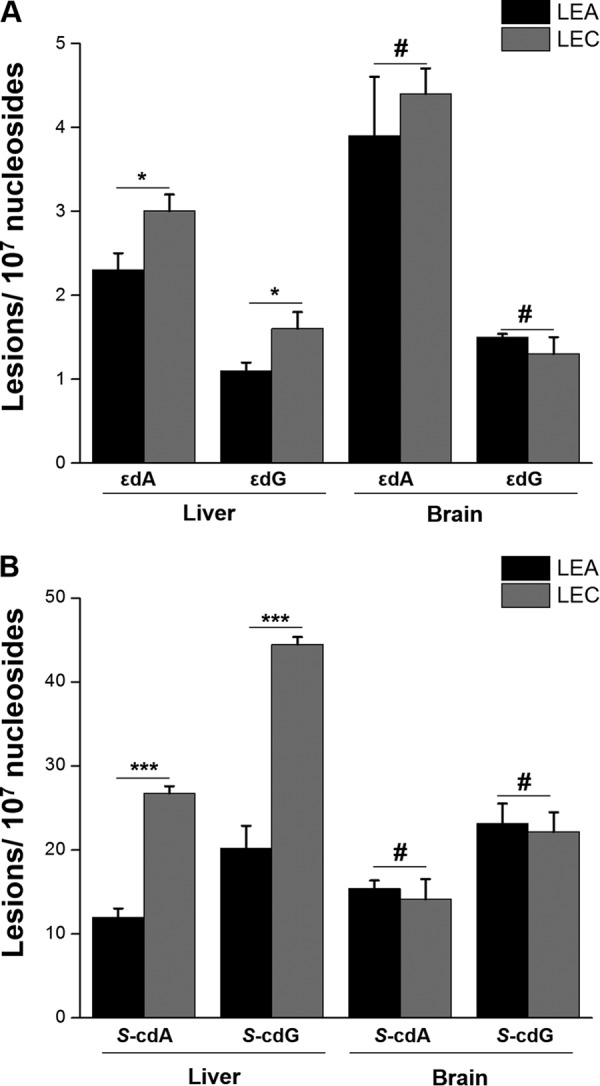
Quantification results for the levels of εdA and εdG (A), as well as S-cdA and S-cdG (B) in liver tissues of LEA (n = 6) and LEC (n = 5) rats as well as in brain tissues of LEA (n = 3) and LEC (n = 3) rats. The data represent the mean and standard deviation of the measurement results. The p values were calculated using unpaired two-tailed Student's t test. #, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Our quantification results also showed that S-cdA and S-cdG (12.0–44.5 lesions per 107 nucleosides) are present endogenously in rat liver tissues at much higher levels than εdA and εdG (1.1–4.4 lesions per 107 nucleosides, Fig. 2 and supplemental Table S2). Similarly, the magnitudes of increases in the levels of S-cdA and S-cdG (∼2.2-fold), arising from aberrant copper accumulation in liver tissues of LEC versus LEA rats, were more pronounced than those for εdA and εdG (∼1.3-fold, Fig. 2 and supplemental Table S2). These results suggested more efficient formation and/or less facile repair of direct ROS-induced cPu lesions than εdA and εdG, which arise from byproducts of LPO.
The Levels of 5-HmdC and 5-mdC in Liver and Brain Tissues of LEA and LEC Rats
5-HmdC is considered as a key nexus in DNA cytosine demethylation in mammalian cells that can either be passively diluted through DNA replication or actively converted to dC through further oxidation (i.e. to 5-FodC and 5-CadC) and action by TDG-mediated BER process (16, 21, 41). On the basis of our previous finding that Cu2+/H2O2/ascorbate could induce oxidation of 5-mdC to 5-HmdC in synthetic duplex DNA in vitro, we initially hypothesized that copper accumulation may also stimulate 5-HmdC formation in liver tissues of LEC rats. However, our quantification data revealed significantly lower levels of 5-HmdC in liver tissues of LEC rats than LEA rats (at 176 and 339 modifications per 106 nucleosides, respectively. Fig. 3A and supplemental Fig. S3). The LC-MS/MS quantification results showed similar levels of 5-mdC in liver of LEA and LEC rats (Fig. 3B and supplemental Fig. S3); thus, the lower level of 5-HmdC in liver of LEC rats was not attributed to alterations in 5-mdC levels. In line with the lack of difference in the prevalence of cPu and etheno DNA lesions in brain tissues of LEA and LEC rats, we found no appreciable difference in 5-HmdC levels in DNA extracted from brain of LEA and LEC rats (Fig. 3A and supplemental Table S2).
Fig. 3.
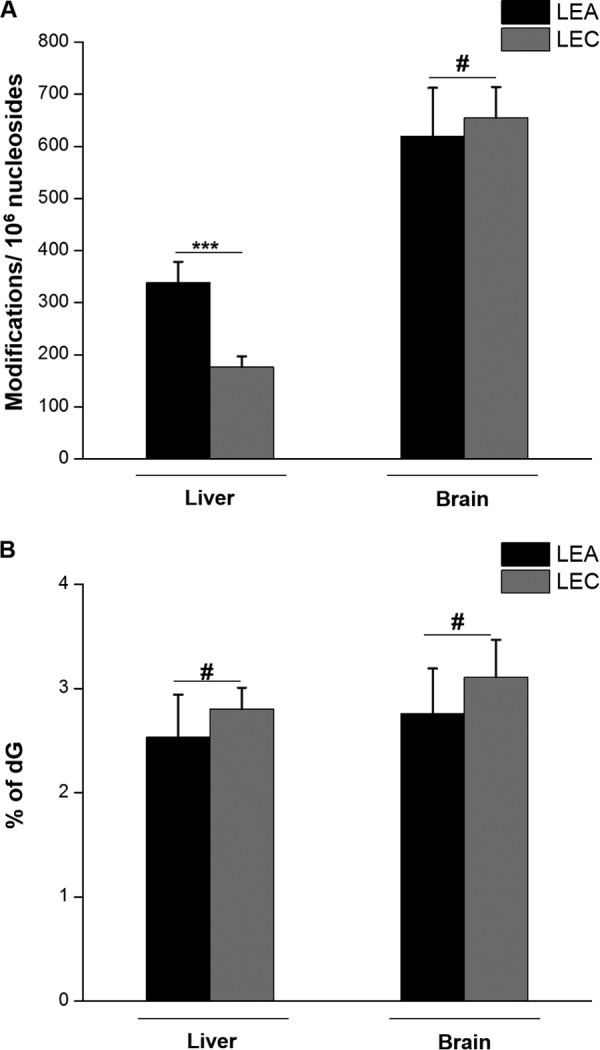
Quantification results for the levels of 5-HmdC (A) and 5-mdC (B) in LEA rat liver tissues (n = 6), LEC rat liver tissues (n = 5), LEA rat brain tissues (n = 3) and LEC rat brain tissues (n = 3). The data represent the mean and standard deviation of the measurement results. The p values were calculated using unpaired two-tailed Student's t test. #, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Biochemical Assay for Tet1-mediated Oxidation of 5-mdC in Duplex DNA
The above results prompted us to reason that decreased levels of 5-HmdC in liver tissues of LEC rats might result from inhibition of Tet enzymes by elevated levels of copper ions. To explore this, we examined how the presence of Cu2+ ion affects the TET1-mediated oxidation of 5-mdC in duplex DNA in vitro (Fig. 4A and supplemental Fig. S4)(38). “Ultra-zoom-scan” MS-based quantification results revealed 5-HmdC and 5-FodC as the major Tet1 oxidation products of 5-mdC in double-stranded DNA. Addition of Cu2+ ion to the reaction mixture significantly reduced the overall catalytic efficiencies of Tet1 in all samples with Cu2+ treatment compared with the reaction without Cu2+ (Fig. 4B). In particular, we observed that the inhibition of Tet1-mediated formation of 5-FodC is dependent on the concentration of Cu2+ (Fig. 4B). Moreover, the remaining TET1 activity in Cu2+-treated samples was largely similar to the control samples without addition of Fe2+ or Cu2+, suggesting that only a portion of the Tet1 protein was initially charged with Fe2+, and Cu2+ may not displace Fe2+ already incorporated in the active center of the Tet1 enzyme. In this context, it is worth noting the difference between the Tet1 protein used in the in vitro biochemical assay and the Tet proteins in liver tissues of LEC rats. The Tet1 protein used for the in vitro assay was purified from host cells that were not exposed to high concentrations of copper. Therefore, the catalytic center of the protein is expected to be occupied, in part, by Fe2+ which, as noted above, may not be readily replaced with Cu2+ during the in vitro reaction. In the hepatocytes of LEC rats, the Tet proteins are folded in an environment with elevated concentrations of Cu2+, and the resulting Cu2+-bound Tet proteins are not expected to be active (vide infra).
Fig. 4.

Cu2+ inhibits Tet1-mediated oxidation of 5-mdC in duplex DNA in vitro. A, Representative LC-MS data for monitoring the Tet1-mediated oxidation of 5-mdC in a duplex DNA, d(AGCTC(5-mdC)GGTCA)/d(GTGACCGGAGCTG). Shown are the higher resolution “ultra-zoom-scan” MS results for monitoring the [M - 3H]3− ions of the initial 5-mdC-bearing DNA and its oxidation products. B, Quantification results for the formation of 5-HmdC and 5-FodC in Tet-mediated oxidation of 5-mdC in the presence of a reaction buffer containing 75 μm Fe2+ and various concentrations of Cu2+ (at Cu2+/Fe2+ molar ratios of 0, 0.25, 0.50, 1.0, 2.0 and 5.0). A control reaction was also conducted in the presence of 75 μm Cu2+ alone (1× Cu).
DISCUSSION
In the present study, we employed a novel NanoLC-NSI/MS2 coupled with the stable isotope-dilution method for the first comprehensive assessment of oxidatively induced modifications of DNA. These included nucleoside modifications induced directly by ROS (cdA and cdG), indirectly by ROS (εdA and εdG from byproducts of LPO), and enzymatically from Tet-catalyzed 5-mdC oxidation. This method offers unambiguous chemical specificity for analyte identification based on the co-elution of the analyte with its stable isotope-labeled standard and the characteristic fragment ions observed in the MS/MS or MS/MS/MS (Fig. 1). Although the ROS-induced cyclopurine lesions were quantified in our previous work (10), in the present study we were able to rigorously compare the levels of direct ROS-induced cyclopurine lesions with the LPO-induced εdA and εdG lesions in DNA isolated from the tissues of the same animals and based on measurements made on the same analytical platform. Our results revealed for the first time that, relative to LPO-induced εdA and εdG lesions, the direct ROS-induced S-cdA and S-cdG were present at much higher levels in liver and brain tissues of LEA and LEC rats, and were stimulated to a greater extent by aberrant accumulation of copper ions in liver tissues of LEC rats (Fig. 2).
We found significantly lower levels of 5-HmdC in DNA isolated from the liver tissues of LEC than LEA rats (Fig. 3A), though no appreciable differences were observed for the levels of 5-mdC (Fig. 3B). This result is seemingly incongruent with our previous in vitro data concerning the dose-dependent induction of 5-HmdC by Fenton-type reagent (30). However, results from our in vitro biochemical assay suggested that aberrant accumulation of copper may directly perturb the TET-mediated oxidation of 5-mdC to 5-HmdC. In this vein, Ni2+ ion is known to be able to replace Fe2+ from the catalytic center of other Fe2+- and 2-OG-dependent dioxygenases (31, 34). The stability of bivalent transition metal ion complexes follows the Irving-Williams order, i.e. Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+, irrespective of the nature of the ligand (42). In addition, Cu2+ was previously shown to be capable of substituting Fe2+ from the DNA repair enzyme, AlkB, another Fe2+/2-OG-dependent dioxygenase (43). Thus, Cu2+ may occupy the catalytic center of TET1 in lieu of Fe2+. The catalytic cycle mediated by the Fe2+/2-OG-dependent dioxygenases, including TET proteins, involves Fe4+ (44), an oxidation state that cannot be adopted by Cu2+. Thus, Cu2+ may occupy the Fe2+-binding site of TET enzymes and disrupt their catalytic activities because of the incapability of Cu2+ to undergo further oxidization. Viewing that aberrant epigenetic regulation of gene expression is associated with cancer development (45), our results also suggest that abnormal copper accumulation-induced dysfunction of TET enzymes may contribute, in part, to progressive hepatic injury in Wilson's disease.
Aside from TET enzymes, other members of the Fe(II)- and 2-OG-dependent dioxygenase family may also act as potential molecular targets for copper toxicity. Escherichia coli AlkB protein, which is an ortholog of human ALKBH2 and ALKBH3, were shown to be capable of repairing etheno DNA lesions (46, 47). Thus, compromised ALKBH repair activity arising from elevated copper content may also account, in part, for greater accumulation of etheno DNA lesions in liver tissues of LEC rats relative to LEA rats. As observed for Ni2+ ion (31, 33), elevated copper accumulation may also inhibit the Fe2+/2-OG-dependent dioxygenases involved in histone demethylation, which may play roles in hepatic damage in human Wilson's disease.
In summary, we employed a sensitive LC-MS/MS and MS3 coupled with the stable isotope-dilution method for simultaneous quantification of oxidative stress-induced cPus and etheno DNA lesions, as well as 5-HmdC and 5-mdC epigenetic marks in tissue DNA. Our results illustrated, for the first time, relative levels of ROS- and LPO-induced DNA adducts in vivo, which supported the implications of oxidative DNA damage in pathogenesis of Wilson's disease and revealed the relative contributions of these two subgroups of oxidatively induced DNA lesions (i.e. the direct ROS-induced cdA and cdG versus LPO-induced εdA and εdG) to this process. Moreover, assessment of 5-HmdC and 5-mdC levels in DNA, in combination with results of TET1-mediated 5-mdC oxidation in vitro, suggest a role of copper-induced inhibition of TET enzymes and resultant perturbation of 5-HmdC epigenetic mark in the pathophysiology of Wilson's disease. Taken together, our data provide mechanistic insights into copper toxicity in vivo, where accumulation of copper may compromise genomic integrity by oxidative DNA damage and perturbation of epigenetic pathways of gene regulation.
Supplementary Material
Footnotes
Author contributions: Y.Y., S.G., and Y.W. designed research; Y.Y. performed research; C.R.G., S.L., N.J.A., and Y.S. contributed new reagents or analytic tools; Y.Y. and Y.W. analyzed data; Y.Y., S.G., and Y.W. wrote the paper.
* This work was supported in part by the National Institutes of Health (R01 CA 101864 to Y.W. and R01 DK 071111, R01 DK088561 and P30 DK41296 to S.G.), and N.J.A. was supported by an NRSA Institutional Training Grant (T32 ES018827).
 This article contains supplemental Figs. S1 to S4 and Tables S1, S2.
This article contains supplemental Figs. S1 to S4 and Tables S1, S2.
1 The abbreviations used are:
- ROS
- reactive oxygen species
- cPu
- 8,5′-cyclopurine-2′-deoxynucleoside
- εdA
- 1,N6-etheno-2′-deoxyadenosine
- εdG
- 1,N2-etheno-2′-deoxyguanosine
- LPO
- lipid peroxidation
- 2-OG
- 2-oxoglutarate
- TET
- ten-eleven translocation
- SIC
- selected ion chromatogram.
REFERENCES
- 1. Finkel T., and Holbrook N. J. (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247 [DOI] [PubMed] [Google Scholar]
- 2. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2006) DNA Repair and Mutagenesis, ASM Press, Washington, D.C. [Google Scholar]
- 3. Brooks P. J. (2007) The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 145, 1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y. (2008) Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 21, 276–281 [DOI] [PubMed] [Google Scholar]
- 5. Chung F. L., Chen H. J., and Nath R. G. (1996) Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 17, 2105–2111 [DOI] [PubMed] [Google Scholar]
- 6. Marnett L. J. (2000) Oxyradicals and DNA damage. Carcinogenesis 21, 361–370 [DOI] [PubMed] [Google Scholar]
- 7. Chen H. J., Lin G. J., and Lin W. P. (2010) Simultaneous quantification of three lipid peroxidation-derived etheno adducts in human DNA by stable isotope dilution nanoflow liquid chromatography nanospray ionization tandem mass spectrometry. Anal. Chem. 82, 4486–4493 [DOI] [PubMed] [Google Scholar]
- 8. Chen H. J., and Lin W. P. (2009) Simultaneous quantification of 1,N2-propano-2′-deoxyguanosine adducts derived from acrolein and crotonaldehyde in human placenta and leukocytes by isotope dilution nanoflow LC nanospray ionization tandem mass spectrometry. Anal. Chem. 81, 9812–9818 [DOI] [PubMed] [Google Scholar]
- 9. Wang J., Clauson C. L., Robbins P. D., Niedernhofer L. J., and Wang Y. (2012) The oxidative DNA lesions 8,5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 11, 714–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang J., Yuan B., Guerrero C., Bahde R., Gupta S., and Wang Y. (2011) Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal. Chem. 83, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang S., Villalta P. W., Wang M., and Hecht S. S. (2006) Analysis of crotonaldehyde- and acetaldehyde-derived 1,N2-propanodeoxyguanosine adducts in DNA from human tissues using liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 19, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitra D., Luo X., Morgan A., Wang J., Hoang M. P., Lo J., Guerrero C. R., Lennerz J. K., Mihm M. C., Wargo J. A., Robinson K. C., Devi S. P., Vanover J. C., D'Orazio J. A., McMahon M., Bosenberg M. W., Haigis K. M., Haber D. A., Wang Y., and Fisher D. E. (2012) An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 491, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shaked H., Hofseth L. J., Chumanevich A., Chumanevich A. A., Wang J., Wang Y., Taniguchi K., Guma M., Shenouda S., Clevers H., Harris C. C., and Karin M. (2012) Chronic epithelial NF-kB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. U.S.A. 109, 14007–14012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tilstra J. S., Robinson A. R., Wang J., Gregg S. Q., Clauson C. L., Reay D. P., Nasto L. A., St Croix C. M., Usas A., Vo N., Huard J., Clemens P. R., Stolz D. B., Guttridge D. C., Watkins S. C., Garinis G. A., Wang Y., Niedernhofer L. J., and Robbins P. D. (2012) NF-kB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfaffeneder T., Hackner B., Truss M., Muenzel M., Mueller M., Deiml C. A., Hagemeier C., and Carell T. (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. 50, 7008–7012 [DOI] [PubMed] [Google Scholar]
- 16. He Y. F., Li B. Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C. X., Zhang K., He C., and Xu G. L. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ito S., D'Alessio A. C., Taranova O. V., Hong K., Sowers L. C., and Zhang Y. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kriaucionis S., and Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., and Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maiti A., and Drohat A. C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mellén M., Ayata P., Dewell S., Kriaucionis S., and Heintz N. (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yildirim O., Li R., Hung J. H., Chen P. B., Dong X., Ee L. S., Weng Z., Rando O. J., and Fazzio T. G. (2011) Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spruijt C. G., Gnerlich F., Smits A. H., Pfaffeneder T., Jansen P. W., Bauer C., Münzel M., Wagner M., Müller M., Khan F., Eberl H. C., Mensinga A., Brinkman A. B., Lephikov K., Müller U., Walter J., Boelens R., van Ingen H., Leonhardt H., Carell T., and Vermeulen M. (2013) Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159 [DOI] [PubMed] [Google Scholar]
- 25. Frauer C., Hoffmann T., Bultmann S., Casa V., Cardoso M. C., Antes I., and Leonhardt H. (2011) Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE 6, e21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ala A., Walker A. P., Ashkan K., Dooley J. S., and Schilsky M. L. (2007) Wilson's disease. Lancet 369, 397–408 [DOI] [PubMed] [Google Scholar]
- 27. Roberts E. A., and Schilsky M. L. (2008) Diagnosis and treatment of Wilson disease: An update. Hepatology 47, 2089–2111 [DOI] [PubMed] [Google Scholar]
- 28. Li Y., Togashi Y., Sato S., Emoto T., Kang J. H., Takeichi N., Kobayashi H., Kojima Y., Une Y., and Uchino J. (1991) Spontaneous hepatic copper accumulation in Long-Evans Cinnamon rats with hereditary hepatitis. A model of Wilson's disease. J. Clin. Invest. 87, 1858–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nair J., Strand S., Frank N., Knauft J., Wesch H., Galle P. R., and Bartsch H. (2005) Apoptosis and age-dependant induction of nuclear and mitochondrial etheno-DNA adducts in Long-Evans Cinnamon (LEC) rats: enhanced DNA damage by dietary curcumin upon copper accumulation. Carcinogenesis 26, 1307–1315 [DOI] [PubMed] [Google Scholar]
- 30. Cao H., and Wang Y. (2007) Quantification of oxidative single-base and intrastrand cross-link lesions in unmethylated and CpG-methylated DNA induced by Fenton-type reagents. Nucleic Acids Res. 35, 4833–4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen H., Giri N. C., Zhang R., Yamane K., Zhang Y., Maroney M., and Costa M. (2010) Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem. 285, 7374–7383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chervona Y., Arita A., and Costa M. (2012) Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics 4, 619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chervona Y., and Costa M. (2012) The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med. 53, 1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen H., and Costa M. (2009) Iron- and 2-oxoglutarate-dependent Dioxygenases: an emerging group of molecular targets for nickel toxicity and carcinogenicity. Biometals 22, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H. J., Chiang L. C., Tseng M. C., Zhang L. L., Ni J., and Chung F. L. (1999) Detection and quantification of 1,N6-ethenoadenine in human placental DNA by mass spectrometry. Chem. Res. Toxicol. 12, 1119–1126 [DOI] [PubMed] [Google Scholar]
- 36. Garcia C. C., Freitas F. P., Di Mascio P., and Medeiros M. H. (2010) Ultrasensitive simultaneous quantification of 1,N2-etheno-2′-deoxyguanosine and 1,N2-propano-2′-deoxyguanosine in DNA by an online liquid chromatography-electrospray tandem mass spectrometry assay. Chem. Res. Toxicol. 23, 1245–1255 [DOI] [PubMed] [Google Scholar]
- 37. Liu S., Wang J., Su Y., Guerrero C., Zeng Y., Mitra D., Brooks P. J., Fisher D. E., Song H., and Wang Y. (2013) Quantitative assessment of Tet-induced oxidation products of 5-methylcytosine in cellular and tissue DNA. Nucleic Acids Res. 41, 6421–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fu L., Guerrero C. R., Zhong N., Amato N. J., Liu Y., Liu S., Cai Q., Ji D., Jin S. G., Niedernhofer L. J., Pfeifer G. P., Xu G. L., and Wang Y. (2014) Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaruga P., Birincioglu M., Rodriguez H., and Dizdaroglu M. (2002) Mass spectrometric assays for the tandem lesion 8,5′-cyclo-2′-deoxyguanosine in mammalian DNA. Biochemistry 41, 3703–3711 [DOI] [PubMed] [Google Scholar]
- 40. Ahn T. B., Cho S. S., Kim D. W., and Jeon B. S. (2005) Absence of nigrostriatal degeneration in LEC rats up to 20 weeks of age. Neurol. Res. 27, 409–411 [DOI] [PubMed] [Google Scholar]
- 41. Kohli R. M., and Zhang Y. (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Irving H., and Williams R. J. P. (1953) The stability of transition-metal complexes. J. Chem. Soc., 3192–3210 [Google Scholar]
- 43. Bleijlevens B., Shivarattan T., Sedgwick B., Rigby S. E., and Matthews S. J. (2007) Replacement of non-heme Fe(II) with Cu(II) in the a-ketoglutarate dependent DNA repair enzyme AlkB: spectroscopic characterization of the active site. J. Inorg. Biochem. 101, 1043–1048 [DOI] [PubMed] [Google Scholar]
- 44. Lu X., Zhao B. S., and He C. (2015) TET family proteins: Oxidation activity, interacting molecules, and functions in diseases. Chem. Rev. 115, 2225–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lund A. H., and van Lohuizen M. (2004) Epigenetics and cancer. Genes Dev. 18, 2315–2335 [DOI] [PubMed] [Google Scholar]
- 46. Delaney J. C., Smeester L., Wong C., Frick L. E., Taghizadeh K., Wishnok J. S., Drennan C. L., Samson L. D., and Essigmann J. M. (2005) AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat. Struct. Mol. Biol. 12, 855–860 [DOI] [PubMed] [Google Scholar]
- 47. Frick L. E., Delaney J. C., Wong C., Drennan C. L., and Essigmann J. M. (2007) Alleviation of 1,N6-ethanoadenine genotoxicity by the Escherichia coli adaptive response protein AlkB. Proc. Natl. Acad. Sci. U.S.A. 104, 755–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.