

Abstract
Protein phosphorylation, one of the most common and important modifications of acute and reversible regulation of protein function, plays a dominant role in almost all cellular processes. These signaling events regulate cellular responses, including proliferation, differentiation, metabolism, survival, and apoptosis. Several studies have been successfully used to identify phosphorylated proteins and dynamic changes in phosphorylation status after stimulation. Nevertheless, it is still rather difficult to elucidate precise complex phosphorylation signaling pathways. In particular, how signal transduction pathways directly communicate from the outer cell surface through cytoplasmic space and then directly into chromatin networks to change the transcriptional and epigenetic landscape remains poorly understood. Here, we describe the optimization and comparison of methods based on thiophosphorylation affinity enrichment, which can be utilized to monitor phosphorylation signaling into chromatin by isolation of phosphoprotein containing nucleosomes, a method we term phosphorylation-specific chromatin affinity purification (PS-ChAP). We utilized this PS-ChAP1 approach in combination with quantitative proteomics to identify changes in the phosphorylation status of chromatin-bound proteins on nucleosomes following perturbation of transcriptional processes. We also demonstrate that this method can be employed to map phosphoprotein signaling into chromatin containing nucleosomes through identifying the genes those phosphorylated proteins are found on via thiophosphate PS-ChAP-qPCR. Thus, our results showed that PS-ChAP offers a new strategy for studying cellular signaling and chromatin biology, allowing us to directly and comprehensively investigate phosphorylation signaling into chromatin to investigate if these pathways are involved in altering gene expression. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD002436.
Protein phosphorylation is a reversible modification of proteins in which a covalently bound phosphate group can be added to a serine, a threonine, or a tyrosine residue by a protein kinase or can be removed by a phosphatase (1, 2). This reversible modification activates many enzymes and receptors that play important roles in protein function, subcellular localization, cell cycle control, receptor-mediated signal transduction, degradation of proteins, and cell signaling networks (3–5). It is estimated that 30–50% of proteins are phosphorylated at some point during their lifetime. As phosphorylation is an important regulatory mechanism that occurs in cells, understanding the “state” of a cell requires knowing the phosphorylation status of the proteome (6).
A variety of technologies such as one-dimensional and two-dimensional gels in combination with 32P labeling or Western blotting with phosphosite-specific antibodies have been developed for determining phosphorylation sites of proteins (7, 8). Protein phosphorylation analysis can also be performed in a large scale and in a somewhat high-throughput fashion due to the great improvements in the speed and sensitivity of mass spectrometers and higher selectivity of phosphopeptide enrichment strategies (9–11). This powerful technology has been utilized to identify and quantify dynamic changes in phosphorylated proteins (i.e. phosphoproteomics) for a systems-wide global look at phosphorylated proteins. Combined with labeling technologies, quantitative phosphoproteomics allows researchers to investigate aberrantly activated signal transduction pathways under different biological conditions (12, 13). Accordingly, phosphoproteomics has been successfully used for large-scale identification of phosphorylation sites and dynamic changes in the phosphorylation status after stimulation.
Various phosphoproteomic enrichment strategies have been developed, such as immunoaffinity chromatography, immobilized metal affinity chromatography, strong cation exchange chromatography, calcium phosphate precipitation, and titanium dioxide (TiO2) chromatography (14–17). The enrichment techniques themselves also have specific advantages and disadvantages. For example, anti-phosphotyrosine antibodies are very useful in immunoblotting for identification of phosphotyrosine containing proteins while there are still some disadvantages such as they do not distinguish between isolating tyrosine-phosphorylated proteins and proteins associated with tyrosine-phosphorylated proteins. Also, antibodies are not as robust as metal-based affinity interaction approaches. Another example is that TiO2 chromatography enriches monophosphorylated peptides more efficiently, while immobilized metal affinity chromatography has a preference for multiply phosphorylated peptides. No method is perfect for phosphoproteomic enrichment, but one can decide which to choose from depending on the sample type and research aims.
Other approaches are being developed for the enrichment of phosphorylated peptides and proteins. These include approaches to directly label protein phosphorylation sites for detection by mass spectrometry using chemical approaches or ATP analogs such as ATP-γ-S (for thiophosphorylation) (18–20). ATP-γ-S can be utilized by a majority of kinases as a phospho-donor since the substrate-binding pocket is separated from the ATP-binding pocket in kinases, so a subtle structural change in ATP is unlikely to change the absolute substrate specificities of most kinases (21). Although some kinases use ATP-γ-S less efficiently than physiological ATP, thiophosphorylation does have several advantages for the analysis of phosphorylation. First, thiophosphorylation is enzymatically irreversible and therefore has been widely used in studies where the phosphorylation site must be maintained. Second, thiophosphorylation strategies can uniquely mark phosphorylation sites because this modification is not endogenous to cells (e.g. distinguishes newly phosphorylated events from previously old phosphorylation). Finally, thiophosphorylation is amenable to chemical modifications since the thiophosphate group has similar chemical properties as the sulfhydryl group that can be exploited for highly specific affinity purifications.
Although these methods have greatly expanded knowledge about the numbers and types of phosphoproteins along with their role in signaling networks, it is still challenging to elucidate the timing or order or interactions within a complex signaling pathway. One long-standing unresolved question in the chromatin biology field is how signal transduction pathways directly communicate into chromatin to change the transcriptional or epigenetic landscape and influence gene expression (22–24). It has been shown that chromatin itself can be the direct target of upstream signaling, and chromatin landscape alteration is emerging as a major consequence of signaling events. Additionally, a signal can be relayed directly onto transcription factors and enzymes that modify the molecular interactions in epigenetic network pathways. For example, chromatin-modifying enzymes will add or remove chemical modifications to and from histones, transcription factors, and DNA, and several have been implicated to be targets of various kinases (25). Additionally, kinase cascades can also communicate with nucleosome remodelers and histone chaperones to alter chromatin modifications, chromatin remodeling, and the deposition of histone variants. The significance to fully understand the interaction between cell signaling and epigenetic regulation is at an all-time high, as comprehensive efforts have not reported in this area, despite the fact that over ten years ago it was postulated that signaling might have direct effects on chromatin.
To better understand how phosphorylation signaling dynamically interacts with chromatin and how chromatin phosphorylation modifications change under different stimuli, we evaluated several thiophosphorylation affinity approaches for isolating phosphorylated proteins. The best-performing thiophosphorylation enrichment strategy was further developed to be used to monitor phosphorylation signaling directly on chromatin-associated nucleosomes, a methodology we term phosphorylation-specific chromatin affinity purification (PS-ChAP). We demonstrate the utility of this method by executing quantitative proteomics studies of chromatin-associated protein phosphorylation changes following activation or inhibition of global transcriptional patterns. Additionally, we show that the PS-ChAP method can isolate DNA that contains phosphorylated proteins for further PCR or genomic sequencing experiments to identify the genes associated with particular phosphorylated protein states. Overall, we feel this method will allow us to monitor signaling into chromatin that changes gene expression and can be a useful tool to interrogate the relationship between kinases, cellular signaling, and chromatin biology/epigenetics on a holistic level.
EXPERIMENTAL PROCEDURES
Standard Cell Culture
HeLa S3 cells were maintained in Joklik's modified Eagle's medium supplemented with 10% newborn calf serum (Gibco) and penicillin-streptomycin solution diluted 1:100 (10,000 units penicillin G and 10 mg streptomycin/ml) (Fisher). Cells were considered to have an optimal seeding density at 2 × 105 cells/ml and harvested at 8 × 105 cells/ml.
Stable Isotope Labeling of Amino Acids in Cell Culture (SILAC)
SILAC media was made in-house using the formulation of Eagle's minimum essential medium (Sigma; catalogue number M 8028) as a template. Arginine and lysine were excluded from the standard formulations, and the pH was adjusted to 7.4. For SILAC labeling, l-lysine and l-arginine were prepared as 1,000× stock solutions in either light (Lys0; Arg0) or heavy (Lys8; Arg10) forms to give a final concentration of 72.5 g/ml for lysine and 126 g/ml for arginine. SILAC medium was filtered (0.22 μm) into equal volumes in autoclaved containers. Proteins were tested for >99% incorporation of the label after six cell passages by mass spectrometry.
HeLa cells were cultured in SILAC media supplemented with 10% dialyzed fetal bovine serum (FBS) (Invitrogen) (10 kDa cut-off) and 1X penicillin/streptomycin in a humidified atmosphere containing 5% CO2. Cell lines were grown for six cell divisions in labeling media containing either light amino acid (Lys0; Arg0) or heavy amino acid (Lys8; Arg10) before introducing stimuli. Heavy-labeled HeLa cells (2 × 107) were serum starved with 0.2% dialyzed FBS for 72 h and then fed with 10% dialyzed FBS for 4 h while light-labeled HeLa cells were starved with 0.2% dialyzed FBS for 72 h. In another experiment, heavy-labeled HeLa cells (2 × 107) were stimulated with 1 μg/ml α-amanitin for 24 h while light-labeled HeLa cells were grown with 10% dialyzed FBS without any treatment. In EGF stimulation experiment, light-labeled HeLa cells and heavy-labeled HeLa cells were serum starved without FBS for 24 h and then stimulated with 100 ng/ml EGF for 10 min or 120 min before harvest.
In Nucleo Kinase Assay
Harvested HeLa cells were washed three times in cold phosphate buffered saline (PBS) and were then lysed in ice-cold hypotonic lysis buffer (10 mm KCl, 1.5 mm MgCl2, 10 mm HEPES-KOH (pH 7.5), 1 × HALT protease and phosphatase inhibitor mixture that was freshly added). The extent of lysis was monitored using trypan blue staining. Intact nuclei were collected by centrifugation and resuspended in 37 °C ATP reaction buffer (35 mm NaCl, 10 mm KCl, 5 mm MgCl2, 2 μm CaCl2, 10 mm Tris-HCL (pH 7.5)) with 1 × HALT and 10 mm adenosine 5-[γ-thio]triphosphate tetralithium salt (ATP-γ-S; ≥75% purity; Sigma-Aldrich). The nuclei were incubated in a 37 °C water bath for 4 h, and ATP-γ-S was subsequently removed by centrifugation and washes. The aliquots were either sonicated to break apart the chromatin to obtain thiophosphorylated proteins or digested by micrococcal nuclease to obtain thiophosphorylated mononucleosomes. The same amount of thiophosphorylated protein (1 mg) was used for the thiophosphoproteins enrichment or was digested by trypsin to generate the thiophosphopeptides for affinity enrichment.
Anti-Thiophosphate Ester Antibody
The nuclei were sonicated to break apart the chromatin and release thiophosphorylated proteins then treated with p-nitrobenzyl mesylate (PNBM) at room temperature for 2 h, and finally analyzed by Western blotting. Proteins (30 μg) were loaded on a 15% SDS gel subsequently transferred to a PVDF membrane in a wet chamber at 300 mA for 1.5 h. Afterward, the membrane was blocked with phosphate buffered saline (PBST) + 5% milk for 1 h at room temperature and subsequently incubated with anti-thiophosphate ester antibody (1:5,000) (Abcam) overnight. The membrane was washed with PBST three times and subsequently incubated with HRP-anti-rabbit (pierce) (1:10,000) in PBST + 5% milk for 1 h at room temperature. Then the membrane was washed with PBST three times prior to detection with the ECL Western blotting detection reagent (Amersham Biosciences) according to the manufacturer's instruction.
Thiophosphorylated Peptides/Proteins Enrichment
Thiophosphorylated peptides/proteins (1 mg) were enriched using titanium dioxide (TiO2), UltraLink iodoacetyl resin (iodoacetyl-beads) or anti-thiophosphate ester antibody. Thiophosphorylated peptides were obtained by trypsin digestion (overnight, substrate:enzyme ratio 100:1) after the sonication of the nuclei. After that, the peptides were desalted using SepPak C18 columns (Waters, MA).
Titanium beads (GL Sciences, Tokyo, Japan) were washed twice using loading buffer (50% acetonitrile, 2 m lactic acid) before being mixed with the peptides at a ratio of 4 mg beads to 1 mg peptide and rotated at room temperature for 30 min. After mixing, the beads were rinsed once with loading buffer, twice with wash buffer (50% ACN, 0.1% TFA), then eluted in a basic elution buffer (50% ACN, pH to 10 with ammonium hydroxide). The elution solution was dried and then desalted using reversed phase C18 tips (made in-house) followed by mass spectrometric analysis (26).
UltraLink iodoacetyl resin (iodoacetyl-beads) were washed twice using 50 mm Tris buffer before being mixed with the peptides/proteins at a ratio of 200 μl beads to 1 mg peptide/proteins and rotated at room temperature overnight (protected from light). For the peptide enrichment, the beads were washed by water, 5 m NaCl, water, 50% acetonitrile, and 5% formic acid. After that, the beads were treated with 0.5 ml oxone buffer (1 mg/ml in H2O) for 0.5 h. Then the supernatant was dried and then desalted using reversed phase C18 tips (made in-house) followed by mass spectrometric analysis as described above. For the protein enrichment, the beads were washed by 50 mm Tris buffer and HEPES 100 (20 mm HEPES, 5 mm MgCl2, 100 mm NaCl) then followed by trypsin digestion. After digestion, the beads were washed by water, 5 m NaCl, water, 50% ACN and 5% formic acid and then eluted using oxone as described above.
Immunoaffinity Purification
The PNBM-treated nuclei were sonicated to obtain the protein for affinity purification, some of which was digested by trypsin to obtain peptides for affinity purification. Anti-thiophosphate ester antibody (10 μl) was first incubated with agarose A/G beads (100 μl), followed by addition of proteins or peptides (3 mg) in immunoaffinity purification (IAP) buffer (50 mm MOPS, pH 7.2; 10 mm sodium phosphate, 50 mm NaCl) and rotated overnight. The beads were washed extensively and the antigens were eluted by using 0.15% TFA. Then the eluted proteins were digested by trypsin (overnight, substrate:enzyme ratio 100:1) and desalted for mass spectrometric analysis while the eluted peptides were desalted followed by mass analysis.
Western Blot Analysis to Confirm the MS Results
Normal cells and α-amanitin treated cells were washed three times in cold phosphate buffered saline (PBS) and were then lysed in ice-cold hypotonic lysis buffer (10 mm KCl, 1.5 mm MgCl2, 10 mm HEPES-KOH (pH 7.5), 1 × HALT protease and phosphatase inhibitor mixture that was freshly added). The lysis was sonicated to break apart the chromatin and the supernatant was used for Western blotting. Proteins (30 μg) were loaded on a 15% SDS gel and then subsequently transferred to a 0.45 μm PVDF membrane in a wet chamber at 300 mA for 1.5 h or 1 h for histone (0.22 μm PVDF). After that, the membrane was blocked with PBST + 5% BSA for 1–2 h at room temperature and subsequently incubated with anti-phospho MCM2 (S27) (1:10,000) (One World Lab) or anti-histone H3 (pT45) (1:5,000) (Abcam or active motif) overnight. The membrane was washed with PBST three times and subsequently incubated with HRP-anti-rabbit (Pierce) (1:10,000) in PBST + 5% BSA for 1 h at room temperature. Then the membrane was washed with PBST three times prior to detection with the ECL Western blotting detection reagent (Amersham Biosciences) according to manufacturer's instruction.
Quantitative Polymerase Chain Reaction (qPCR)
Nonstimulated cells and EGF-stimulated cells were harvested and crosslinked with 1% formaldehyde. Then cells were washed three times. Following PS-ChAP, DNA was quantified by qPCR using standard procedures on a 7900HT Fast-Real-Time PCR platform (ABI). Primers were designed by primer express 2.0 of Applied Biosystems at the promoter region.
GAPDH forward: TCTGTCCCTCAATATGGTCCT reverse: TCCACGACGTACTCAGCG H2B forward: AGGTGCTGAAACAGGTCCAT reverse: GGTCGAGCGCTTGTTGTAAT 18sRNA forward: GTAACCCGTTGAACCCCATT reverse: CCATCCAATCGGTAGTAGCG IL8forward: TGATGACTCAGGTTTGCCCTG reverse: CCACGATTTGCAACTGATGG EGR1 forward: ACCCCTCACCACAAGGACC reverse: AGGCCTGATTCTTGTTCTCACC FOS forward: CATCCCGAACTGACCACCC reverse: GGTAGGGAGTGCGAGGTGTG EGR2 forward: ATAGCAGCAGGTTCTGGCTTG reverse: CGCACTGGGAGGTAGAAGCTT JUNB forward: AACCCTCCCGATTTACAGTGC reverse: GAGACCCCAAAAAGCAGAAATG.
Nano Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (nano-LC-ESI-MS/MS)
We analyzed the thiophosphopeptide/protein-enriched samples and the flow-through samples on an LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific) attached to an Eksigent AS2 autosampler and an Eksigent nano-LC ultra two-dimensional plus system run at 250 nl/min. The samples were loaded on a pulled-tip fused silica column with a 100 μm inner diameter packed in-house with 12 cm of 3 μm C18 resin (Reprosil-Pur C18-AQ) that served both as a resolving column and as a nanospray ionization emitter. Peptide elution used solvents comprised of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). The gradient for most experiments are from 2% solvent B to 35% solvent B over 95 min, then increasing to 100% solvent B in the next 10 min and staying at 100% solvent B for 3 min and finally decreasing to 2% solvent B. The gradient for SILAC experiments is from 2% solvent B to 45% solvent B over 95 min then increasing to 100% solvent B in the next 35 min and staying at 100% solvent B for 10 min and finally decreasing to 2% solvent B.
The mass spectrometers were operated in positive ion mode and in the data-dependent mode with dynamic exclusion enabled (repeat count: 1, exclusion duration: 0.5 min). For the LTQ-Orbitrap Elite MS, every cycle, one full MS scan (m/z 350 to 1,650) was collected at a resolution of 60,000 at an Automatic Gain Control (AGC) target value of 1 × 106, followed by fifteen MS2 scans of the most intense peptide ions using Collision-induced dissociation (CID), also known as collisionally activated dissociation (CAD) (normalized collision energy = 35%, isolation width = 2 m/z) at an AGC target value of 1 × 104 or higher-energy collisional dissociation (HCD) (normalized collision energy = 36%, isolation width = 2 m/z, resolution = 15,000) at an AGC target value of 5 × 104. Ions with a charge state of one and a rejection list of common contaminant ions (exclusion width = 10 ppm) were excluded from the analysis.
Data Analysis
pFind Studio 2.8 was used for data analysis, including pFind for database search (27, 28) and pBuild for result validation. The data were searched using pFind against the human International Protein Index (IPI) database (version 3.87, 91,464 sequences), using a mass tolerance of 20 ppm for precursor ions and 0.5 Da for fragment ions. Serine, threonine and tyrosine phosphorylation, methionine oxidation, lysine, and arginine SILAC label were set as variable modifications, and up to two missed cleavages were allowed for trypsin digestion. The target and reversed database were used for searching, and pBuild used the target-decoy approach to filter search results with false discovery rate less than 1% at the MS/MS level (3) and then obtained MS/MS based peptide and protein identifications.
After the identification, pQuant was used for peptide and protein quantification (29). From the identified MS/MS, the light (unlabeled) and heavy (SILAC labeled) peptide pair was found in MS scans, and then the chromatography profiles for each peptide isotopic peak were reconstructed. The least interfered peaks from light and heavy isotopic peaks were selected, respectively, and then they were used to calculate more-accurate heavy-to-light (H/L) peptide ratio. Protein ratio was calculated based on the distribution of peptide ratios.
Bioinformatics Analysis
Enrichment analysis in the gene ontology (GO) cellular component, molecular function categories, and interactive analysis were performed for all phosphoproteins identified by large-scale thiophosphoproteomic screening of HeLa nuclei. This analysis was performed using the GOrilla annotation website, a web-based tool for identifying and visualizing enriched GO terms in ranked lists of genes (30). GOrilla is publicly available at: http://cbl-gorilla.cs.technion.ac.il. Panther was also used for GO analysis to determine enrichment in protein classes (http://www.pantherdb.org/).
Motif-X is a powerful algorithm that was developed at Harvard Medical School for the analysis of sequences that surround identified phosphorylation sites by mass-spectrometry-based proteomics (31). It was used here to predict the specificity of kinases according to identified phosphosites. The phosphopeptide sequences for those phosphorylation sites were prealigned by a custom Perl script, and their lengths were adjusted to ± 6 amino acids from the central position and submitted to the Motif-X algorithm as foreground. Parameters were set to peptide length = 13, occurrence = 10, and significance p value less than .00001.
RESULTS
Enrichment of Thiophosphorylated Peptides via Different Affinity Purification Approaches
Techniques to directly label protein phosphorylation sites for mass spectrometry detection using chemical approaches or other ATP analogs, such as ATP-γ-S, have been previously reported (19, 20, 32). The Shokat lab has developed individual mutant kinases that can utilize N6-(benzyl) ATP-γ-S to tag the direct substrates of specific kinases. This method allows for direct affinity purification of the substrates of any of these engineered kinases in the genome by capturing the thiophosphate-containing proteins transferred by the respective kinase. This chemical approach allows mapping of any specific kinase pathway in a cell and contributes toward understanding complex kinase signaling networks. However, to date, a global large-scale characterization of thiophosphosite-labeled proteins utilizing ATP-γ-S that can be utilized by most kinases has not been performed. With this approach, a general mapping of all the thiophosphorylated sites in the cellular compartments can be achieved. Furthermore, our overall goals include capturing phospho-signaling directly on the chromatin-associated nucleosomes, which at the current time cannot be performed in a large-scale manner using normal phospho-enrichment strategies that target phosphate-containing peptides (i.e. TiO2) but possibly can be performed by the thiophospho covalent bond enrichment protocols described here.
A few methods have been developed for the enrichment of thiophosphopeptides, but a comprehensive comparison of different methods to isolate thio-phosphorylated peptides or proteins has not been systematically studied as we have performed in these studies. As shown Fig. 1, we chose to use titanium dioxide (TiO2) beads, UltraLink iodoacetyl resin (iodoacetyl-beads) and the anti-thiophosphate ester antibody to perform the enrichments at either the peptide (Fig. 1A) or protein level (Fig. 1B). We chose these methods over others for several reasons. First, it has been shown that titanium dioxide (TiO2) is a highly efficient strategy to selectively enrich and purify phosphopeptides. Since thiophosphopeptides mimic phosphopeptides, titanium dioxide (TiO2) was chosen as a method for thiophosphopeptide enrichment. Additionally, ultraLink iodoacetyl resin (iodoacetyl-beads) is a newly developed method that has been successfully used for thiophosphopeptide enrichment, as thiophosphopeptides have a unique thio-group that can be chemically distinguished from other functional groups. This strategy is based on a chemical reaction between iodoacetamide beads and thiol groups that occurs at pH 8. However, previous researchers performed the reaction at pH 4 to give more specific binding to the beads by excluding cysteine from the reaction (21). Here, we characterize thiophosphopeptide and thiophosphoprotein enrichment using iodoacetyl enrichment using both pH 4 and pH 8. Finally, we performed the enrichment using anti-thiophosphate ester antibodies, which were designed to purify the substrates of the analog-specific (AS) kinase (33). AS kinases can utilize N6-alkylated ATP-γ-S to produce thiophosphorylated substrates. Alkylation with PNBM then yields a thiophosphate ester that can be specifically recognized by the anti-thiophosphate ester antibody, as shown in Fig. 1C.
Fig. 1.

Overview of enrichment methods for thiophosphorylated peptides and proteins. The orange box displays the work flow to prepare the peptides and protein mixtures before the enrichment. (A) Schematic of different methods to enrich thiophosphopeptides, which include titanium dioxide (TiO2) beads, UltraLink iodoacetyl resin (iodoacetyl-beads), and an anti-thiophosphate ester antibody. After all the steps, peptides are eluted and analyzed by nanoLC-MS/MS. (B) Schematic of different methods used for thiophosphoprotein enrichment. The methods include UltraLink iodoacetyl resin (iodoacetyl-beads) and anti-thiophosphate ester antibody. After all the steps, peptides are eluted and analyzed by nanoLC-MS/MS. (C) PNBM derivatized thiophosphorylated proteins detected by Western blotting using the anti-thiophosphate ester antibody. The first lane shows that HeLa nuclei only using alkylation with PNBM (but without ATP-γ-S), and the second lane shows HeLa nuclei incubated with ATP-γ-S followed by alkylation with PNBM.
Evaluation of the Efficiency of the Different Thiophosphopeptide Enrichment Methods
For all three methods described above, we compared the identified numbers of total peptides, total protein, target peptides (thiophosphopeptides or phosphopeptides), and the ratio of target peptides/total peptides after analysis of the eluted peptides by nanoflow liquid chromatography coupled to an LTQ-Orbitrap Elite mass spectrometer. As shown in Fig. 2A (Method 4), the titanium dioxide (TiO2) method recovered a total of 1,177 peptides, only 15 of which were the target thiophosphopeptides (1.18%) in the first replicate, with similar numbers in a second biological replicate. Among all those peptides detected, 1,013 peptides were phosphopeptides, which suggests that phosphorylated peptides bind much more strongly to the TiO2 beads than thiophosphorylated peptides. Presumably, this is due to changes in the binding affinity caused by electron cloud density differences between O and S atoms.
Fig. 2.

Comparison of the enrichment efficiency for thiophosphopeptide and thiophosphoprotein isolation. (A) Table showing the results of two biological replicates for the various enrichment strategies (Method 1: iodoacetyl resin, pH 4; Method 2: iodoacetyl resin, pH 8; Method 3: anti-thiophosphate ester antibody; and Method 4: TiO2 beads). For this comparison, the identified numbers of total peptides, total protein, target peptides (thiophosphopeptides or phosphopeptides), and the ratio of target peptides/total peptides are displayed. (B) Histogram showing the enrichment efficiency for thiophosphopeptide isolation. The X-axis represents different methods as listed in A, while the Y-axis shows the amount of peptides identified. The numbers of nontargeted peptides identified are shown in light blue and targeted peptides are shown in pink. (C) Histogram showing shows the enrichment efficiency toward thiophosphoprotein isolations. The X-axis represents different methods while the Y-axis shows the amount of target peptides identified. Light blue indicates the amount of nontargeted peptides identified, while pink indicates the amount of targeted peptides identified.
We also tested the iodoacetyl resin method under two pH conditions: pH 4.0 and pH 8.0 (Methods 1 and 2) (21, 32). At pH 8, thiophosphorylated peptides can covalently bind the iodoacetyl resin. This strategy relies on the nucleophilicity of the thiophosphate group, indicating that cysteine thiol groups can also react with the iodoacetyl resin. However, by reacting and eluting with oxone, the two groups can be distinguished, and thiophosphate groups can be specifically isolated and eluted even in the presence of excess peptide cysteine groups as shown in Fig. 1A. Elution with oxone from the iodoacetyl resin results in the release of phosphorylated peptides, as the thiophosphoryl sulfur atom is replaced with oxygen after the oxidation-promoted hydrolysis of the sulfur-phosphorus bond. With the oxone elution, cysteine containing peptides remain on the beads as shown in the Fig. 1. Using iodoacetyl-beads, we identified 466 phosphorylated peptides mapping to 321 proteins, and the ratio of target phosphorylated peptides/total peptides was 93.5% in the first analysis, with similar numbers in a second biological replicate (Fig. 2A). All of the proteins identified from MS analysis using iodoacetyl-beads at pH 8 are listed in Supplemental Table S1, and all the MS/MS spectra are also listed in Supplemental data. The identified phosphorylated proteins included various nucleic-acid-binding proteins, cytoskeletal proteins, and transcription factors. At pH 4, only thiophosphorylated peptides can bind the iodoacetyl resin by presumably excluding cysteines from the reaction at a more acidic pH. Using this pH 4 condition, however, we were only able to identify 13 and 16 phosphorylated peptides, respectively, in two biological experiments after enrichment with the iodoacetyl beads. The efficiency of the reaction between thiophosphorylated peptides and the iodoacetyl resin decreased dramatically under acidic pH, although we are not sure of the exact cause of the efficiency drop.
The Shokat lab also developed an anti-thiophosphate ester antibody for specific kinase substrate purification (33). For this method to be effective, thiophosphopeptides are first alkylated using p-nitrobenzylmesylate (PNBM), producing a phosphoryl-conjugate that is then recognized by the antibody. Interestingly, sulfhydryl groups on cysteines can also be alkylated with PNBM. Therefore, a key feature of this strategy is the discrimination of closely related reaction products by the antibody. Following the PNBM alkylation, the derivatized substrates of thiophosphopeptides were detected by the anti-thiophosphate ester antibody, which can specifically recognize thiophosphate esters over thioethers (cysteines). Western blotting results from cell lysate (shown in Fig. 1C) showed that several thiophosphoproteins in the whole cell nucleus isolation were detected only in the nuclei treated with ATP-γ-S. We next tested the enrichment efficiency of the thiophosphate ester-specific antibody for thiophosphopeptide enrichment (Method 3, Fig. 2A). A total of 1,819 peptides were detected by this strategy, while among these only 29 peptides were derivatized substrates of thiophosphopeptides (2.43%) in the first replicate, with similar values in a second biological replicate. These data suggest that the antibody has a rather weak affinity for derivatized substrates of thiophosphopeptides, and indeed as far as we know, there no publications using this approach for comprehensive and global thiophosphorylated peptide enrichment. This could be that the antibody is not suited for peptide immunoprecipitations but, rather, only for Western blotting or that the ionization efficiency of the thiophosphopeptides is severely compromised after reaction with PNBM.
Experiments were repeated in biological replicate, and the peptide enrichment results are listed in Fig. 2A and visually shown in Fig. 2B. More than 1,000 peptides were identified by using TiO2 beads and the anti-thiophosphate ester antibody, but the overwhelming majority of these were nontargeted phopeptides. In contrast, we can clearly see that the oxone elution from iodoacetyl resin (at pH 8) results in a more complete characterization of the phosphorylated peptides with a high number of phosphopeptides detected (target peptides). Consistent within both biological replicates, we identified the largest number of phosphopeptides by using iodoacetyl-beads (pH 8) while only very few targeted peptides were found by using other methods (Fig. 2B).
Evaluation of the Efficiency of the Different Thiophosphoprotein Enrichment Methods
For thiophosphoprotein enrichment, we did not attempt to use the titanium dioxide beads since the results for the thiophosphopeptides enrichment showed little promise, and these approaches have never had much success at the protein level. However, we tested all of the other methods used in the thiophosphopeptide enrichment for thiophosphoprotein enrichment. For the thiophosphoprotein enrichment, we incubated the iodoacetyl beads with thiophosphorylated proteins overnight followed by a trypsin digest. Only covalent binding peptides (thiophosphopeptides) should be still left on the beads after several stringent washes, including using high concentration salts (Fig. 1B). The results showed that we identified 240 phosphorylated peptides by using iodoacetyl-beads at pH 8 (Fig. 2A; 57.14%), While at pH 4, we only identified only nine phosphorylated peptides out of 11 total identified peptides after enriching thiophosphorylated proteins using iodoacetyl beads. This result was essentially reproducible in a second biological replicate (Fig. 2A) All of the proteins identified from MS analysis using iodoacetyl beads at pH 8 are listed in Supplemental Table S2, and all the MS/MS spectra are also listed in Supplemental data. We also tested the enrichment efficiency of the anti-thiophosphate ester antibody to enrich thiophosphorylated proteins. As shown in Fig. 1B, alkylation with PNBM yields thiophosphate esters and thioethers. The anti-thiophosphate ester antibody can specifically bind thiophosphate esters that can be monitored with Western blotting (Fig. 1B). Bound proteins were eluted from the antibody and then digested with trypsin, followed by mass spectrometry analysis. The results showed that we detected a total of 1,187 peptides; however, only four peptides were actually derivatized substrates of thiophosphopeptides (<1%). This result is not surprising because the abundance of peptides containing thiophosphate esters should be very low compared with nonmodified peptides from other regions of the eluted thiophosphorylated proteins. Furthermore, the ionization efficiency of the thiophosphopeptides might be limited after reacted with PNBM and lower compared with non-thiophosphorylated peptides. These experiments were also repeated twice, and the results are listed in Fig. 2A. We consistently identified the largest number of phosphopeptides using iodoacetyl beads at pH 8, while only very few targeted peptides were found by using other methods via protein level enrichment. Nevertheless, as expected, peptide-level enrichment for thiophosphorylation analysis far exceeded any protein-level approaches.
Bioinformatics Analysis of the HeLa Nuclear Thiophosphoproteome
All the phosphorylated peptides/proteins identified from whole HeLa nuclei lysis by using UltraLink iodoacetyl resin (iodoacetyl-beads) at pH 8 for thiopeptide enrichment are listed in the supporting information. To our knowledge, this constitutes the first large-scale screening of the HeLa cell nuclear thiophosphoproteome. We therefore investigated the overall properties of our high-confidence HeLa nuclear thiophosphoproteome using gene ontology (GO), kinase motif analysis, cellular component, and interactive analysis.
The identified phosphorylated proteins were functionally grouped into three biological processes by GO-term analysis: catalytic activity, binding, and protein-binding transcription factor activity as shown in Supplemental Fig. S1A. A p value of ≤ 10−6 was considered significant. Significantly overrepresented biological processes included: DNA binding, chromatin binding, transcription factor binding function, and transcription cofactor activity, all of which are tightly connected with the regulation of gene transcription. The pie chart (Supplemental Fig. S1B) represents protein class analysis of all phosphorylated proteins identified by using panther website (http://www.pantherdb.org/), which has comprehensive function information about genes and was designed to also facilitate analysis of large numbers of genes. According to Supplemental Figs. S1A and S1B, the identified proteins were classified into several protein type classes and the most represented protein class was binding such as nucleic acid binding, chromatin binding, and transcription binding, which showed similar results as described above of molecular function.
Identified phosphorylated proteins were annotated with several GO terms. To identify the cellular component of the hundreds of identified phosphoproteins, we performed a cellular component annotation by using the GOrilla annotation website (Supplemental Fig. S1C). Again, a p value of ≤ 10−6 was considered highly identified. GO terms annotated in the “cellular component” category revealed a clustered distribution of thiophosphoproteins in nuclear and chromosomal compartments. We further performed GO enrichment analysis of gene expression networks by using the GOrilla annotation again and found that thiophosphorylated proteins generally tend to interact physically or function in related processes. It was shown that regulation of transcription was the most overrepresented process in our data (Supplemental Fig. S1D).
Motif-based kinase-substrate predictions have been widely used to better understand kinase-substrate interactions. To determine the residue composition surrounding the phosphorylation sites, phosphopeptide sequences for those sites were prealigned by a custom Perl script that keeps the peptide length to ± 6 amino acids from the central position (the position of the thiophosphate). These sequences were submitted to Motif-X, and the resulting enriched sequence motifs for thio-phosphorylation sites are shown in Supplemental Fig. S2. Most of the phosphorylation motifs are classified into PKC kinase substrate motif, rsp5 (WW) domain binding motif, and 14-3-3 domain binding motif. Since some protein kinases share the same or similar recognition motifs, it was not as feasible to match a phosphorylation site to a specific protein kinase at this time using this data.
Isolation of Nucleosomes Containing Thiophosphorylated Proteins Utilizing Phosphorylation-Specific Chromatin Affinity Purification (PS-ChAP)
To further develop this method for monitoring signaling to chromatin, we adapted this chemistry-based purification method to isolate thiophosphorylated mononucleosomes, in an approach we term phosphorylation-specific chromatin affinity purification (PS-ChAP). The strategy is depicted in Fig. 3A. First, a nuclear pellet was obtained after a dounce homogenization procedure. Next, the pellet was treated with ATP-γ-S followed by micrococcal nuclease (MNase) digestion. The digested solution was then incubated with the iodoacetyl-beads at pH 8. After several stringent washes, we performed on-bead trypsin digestion, which leaves only thiophosphorylated peptides covalently bound to the resin. Lastly, we used oxone to elute the binding thiophosphorylated peptides before nanoLC-MS/MS analysis.
Fig. 3.

Phosphorylation specific chromatin affinity purification (PS-ChAP) for mononucleosome enrichment. (A) Illustration of the steps in the PS-ChAP strategy performed on a mononucleosome mixture after MNase digestion. By using iodoacetyl-beads, thiophosphorylated mononucleosomes can be isolated. After trypsin digestion, washes, and elution, thiophosphorylated sites on the mononucleosomes can be specifically detected using nanoLC-MS/MS. (B) The left figure shows the electrophoretic gel analysis of micrococcal nuclease-digested chromatin, and the right two figures show protein gels using Coomassie blue and silver staining. 0.05% of the total input protein and flow-through protein were loaded while the entire elution solution was loaded on the gel. (C) Three biological replicates were performed, and the figure shows the efficiency of this method for mononucleosome enrichment. The X-axis represents different biological replicates while the Y-axis shows the amount of peptides identified. The amount of nonphosphopeptides identified is shown in light blue and phosphopeptides are shown in pink.
To make sure that we can isolate mononucleosomes using the PS-ChAP approach, DNA fragments pulled down after PS-ChAP enrichment were isolated and analyzed on a 2% agarose gel. As we expected, most of the DNA fragments correspond to the size of mononucleosomes, around 146bp (Fig. 3B, left). We also performed protein separation by gel electrophoresis using the eluted solution before trypsin digestion to confirm the results of MS analysis (Fig. 3B). Staining by Coomassie blue demonstrated that the protein amount in the flow through is significantly decreased compared with the input, while only a few light bands can be observed from the elution (especially some faint bands in the histone region <15 kDa). Given the low amount of protein observed after elution and Coomassie staining, we performed silver staining to improve protein detection. After silver staining, it was easier to detect more proteins, and bands corresponding to the molecular weight of histones were more readily detected on the gel. Mass spectrometry results of the mononucleosome isolation show that we detected a total of 183 peptides, 156 peptides of which were phosphorylated (85.2%). Three biological replicates were performed, and the results are listed in Fig. 3C. The results show that the efficiency of this method to enrich mononucleosomes containing thiophosphorylated proteins is very high, with one of the replicates showing 97% efficiency. According to the MS analysis, more than 100 proteins were identified in the elution, including many variants of histones H2A, H2B, and H3. All of the proteins identified from MS analysis are listed in Supplemental Table S3, and all the MS/MS spectra are also listed in Supplemental data.
Alterations in the Chromatin-Associated Phosphorylation-Specific Chromatin Affinity Purification (PS-ChAP) Captured Thiophosphoproteome Following Inhibition of Transcription
As we mentioned earlier, the overall goal of this research is to develop a method to monitor phosphorylation signaling down to the gene level. Given the success of thiophosphorylated mononucleosome enrichment, we decided to combine stable isotope labeling by amino acids in cell culture (SILAC) and this isolation enrichment strategy to improve our capacity for relative quantification of chromatin-associated protein phosphorylation changes. This quantitative thiophosphoproteomics method would be an ideal tool for in-depth characterization of thiophosphoproteome-wide chromatin changes related to gene activation upon different stimuli that affect gene transcription. As shown in Fig. 4A, cells are differentially labeled by growing them in light medium with standard amino acids (Lys0; Arg0) or medium with heavy amino acids (Lys8; Arg10). These two samples were treated with or without stimuli and mixed in a 1:1 ratio. The whole cells were lysed, the nuclei were digested by MNase, and thiophosphorylated mononucleosome enrichment was performed. Then after several washes, the bound proteins were digested with trypsin and the thiophosphorylated peptides eluted (as shown in Fig. 3A) were subjected to mass spectrometry interrogation.
Fig. 4.

Analysis of PS-ChAP SILAC results under amanitin treatment for identifying phosphorylation events related to gene transcription. (A) Light amino acids (Lys0; Arg0) or heavy amino acids (Lys8; Arg10) were used for the SILAC experiment. Untreated cells were grown in the light medium while the stimulated cells were grown in the heavy medium. Cells were mixed in a 1:1 ratio, and whole cell lysis was performed before nuclei isolation and Mnase digestion. As described in Fig. 3, thiophosphorylated sites on the mononucleosomes can be specifically detected from nanoLC-MS/MS after thiophosphorylated mononucleosome enrichment. Light-labeled HeLa cells served as a control while heavy-labeled HeLa cells were treated with α-amanitin for 24h. (B) Histogram of log2-transformed normalized protein ratios for all quantified proteins identified in the experiment. (C) Plot of the normalized ratios of all quantified proteins plotted against their summed heavy and light peptide intensities. (D) MS-based quantification analysis of a phosphopeptide on MCM2 (GDPLTSpSPR) that displays a decreased abundance during inhibition of transcription. The SILAC peptide pair shows a heavy to light ratio (H/L) of 0.489 corresponding to a more than twofold reduction of the protein level. (E) MS-based quantification analysis of another peptide on histone H3 (YRPGpTVALR). The SILAC peptide pair shows a heavy to light ratio (H/L) of 0.491 also corresponding to about twofold reduction of the protein level.
Our hypothesis is that certain protein phosphorylations are linked to gene transcription (34), and thus, perturbing transcriptional events could cause changes in chromatin-associated protein phosphorylation. This precedent has been previously observed, as one example would be the highly regulated phosphorylation events that govern RNA polymerase II function (35, 36). We decided to inhibit genome-wide transcription in two ways using a drug designed to stop transcription and also by serum starvation, which has a similar effect. In the first experiment, we SILAC-labeled HeLa cells and treated the heavy cell population with α-amanitin (an RNA polymerase II inhibitor) for 24 h. The heavy- and light-labeled cells were combined in equal amounts and lysed, nuclei digested using Mnase, followed by our PS-ChAP thiophosphonucleosome enrichment (Fig. 4A). Thiophosphopeptides were eluted and detected by nanoLC-MS/MS. To minimize random effects and increase fidelity, two independent biological replicates of enrichment fractions were isolated and prepared for nanoLC-MS/MS identification, and pQuant was used for quantitative MS analysis of the data. In the first biological replicate, 181 unique proteins were identified, most of which had quantifiable ratios. For the second biological replicate, 240 unique proteins were identified; still most of the proteins were quantified (Supplemental Table S4). In total, 145 unique proteins were identified from between both the first and the second datasets.
The quantitative MS data of the chromatin-associated thiophosphoproteome analysis following α-amanitin treatment were analyzed and the results are shown in Fig. 4. Prior to in-depth analysis, we plotted the histogram of the normalized H/L SILAC ratios that were transformed using the binary logarithm (log2) as shown in Fig. 4B. Log2-fold changes ranged from -2 to 2.5, and the standard deviation was 0.25. As shown in the Fig. 4B, there were a few more down-regulated phosphorylation events than up-regulated after the treatment, consistent with protein phosphorylation being associated with gene transcription. In Fig. 4C, the same ratios are plotted as a function of peptide abundance. Next, we wanted to further analyze the data and identify protein candidates that significantly change in abundance on chromatin upon α-amanitin treatment. We highlight two SILAC peptide pairs as shown in Fig. 4D and Fig. 4E, which demonstrated significantly changing abundance from both transcriptional states. One of these peptides is GDPLTSSPR from MCM2 in which the second serine is thiophosphorylated, while the other peptide is YRPGTVALR from histone H3 containing a threonine phosphorylation at position 45 (H3T45ph).
The pQuant program quantifies SILAC pairs by first reconstructing a chromatogram for each isotopic peak and pairing one set of isotopic chromatograms for the light peptide with another set for the heavy peptide. On the basis of these isotopic pairs, pQuant then calculates the peptide ratio and the associated confidence interval. Finally, a protein ratio is calculated from peptide ratios and their confidence intervals by kernel density estimation. The result shows that the H/L ratios of the two selected peptides were both around 0.5, which indicates there was about twofold decrease in the level of phosphorylation of both MCM2-Ser27 and H3-Thr45 after α-amanitin treatment (RNA II polymerase inhibition). Fig. 5A and Fig. 5B show the MS and MS/MS spectra of the GNDPLTSpSPR identified from the eluted fragment, respectively. Validation of this specific protein phosphorylation site (MCM2-Ser27) by immunoprecipitation-Western blot analysis is shown in Fig. 5C and shows that there was a visual decrease of MCM2-S27 after drug treatment, similar to the quantitative profile suggested by our SILAC proteomics analysis. The MS and MS/MS spectra of YRPGpTVALR (histone H3Thr45ph) identified from the SILAC data are shown in Fig. 6A and Fig. 6B. This mark was found to decrease twofold upon transcriptional inhibition with α-amanitin. While this phosphorylation site in histone H3 at threonine 45 (H3-Thr45) has been detected previously, it still remains poorly understood with regards to its function, as several lines of research show it is associated with diverse areas of chromatin biology (37–39). H3Thr45ph lies in a structurally important region in the nucleosome, being positioned at the extreme N terminus of the first helix of H3 (50) (Fig. 6C) and making important contacts points with other chromatin factors (Fig. 6D). Previous research has shown that H3Thr45 is actually subject to phosphorylation during DNA replication, and as H3Thr45ph was decreased after inhibition of RNA polymerase II, we believe this mark may play a role in transcriptional processes during S-phase (40).
Fig. 5.

Quantitative SILAC-based proteomics and PS-ChAP reveal phosphorylation events related to gene transcription. (A) Pairs of chemically identical peptides (GNDPLTSpSPGR) with a different stable-isotope composition can be differentiated by mass spectrometry due to the mass difference between the pair (Δm = 5 m/z). The relative protein abundance was reflected by comparing the differences in the intensities of the peaks. (B) MS/MS spectra obtained for GNDPLTSpSPGR ([M+2H]2+ = 595.758). The sequence, modification, and peptide spectrum match score are listed on the top of the figure. The y-type fragment ions are shown in yellow while the b-type fragment ions are shown in green. (C) MS quantification result of GNDPLTSpSPGR from combined control and α-amanitin treated cells were obtained by using pQuant. Western blotting results are shown in the second part of the panel and are consistent with the MS observations.
Fig. 6.

Histone H3T45 phosphorylation identified as potentially associated to transcription. (A) Pairs of chemically identical peptides (YRPGpTVALR) with a different stable-isotope composition can be differentiated by mass spectrometry due to the mass difference between the pair as shown in the image. The relative protein abundance was reflected by comparing the differences in the intensities of the peaks (Δm = 10 m/z). (B) MS/MS spectra obtained for YRPGpTVALR. The sequence, modification, and score are listed on the top of the figure. The y-type fragment ions are showing in yellow while the b-type fragment ions are showing in green. (C) Structural positions of H3Thr45 site in the nucleosome (PDB ID: 1KX5) (Davey et al. 2002). The position of H3Thr45 (red) is indicated by the arrows; H3Thr45 is located at the N terminus of the first helix of H3 (the αN1-helix), a position close to where the DNA both enters and exits the nucleosome. Images were constructed using the PyMOL software. (D) According to recent literature, RNA polymerase II and histone methyltransferase Set2 interact with the structured nucleosome core surface formed by the listed residues in the figure, including H3Thr45.
Another method of transcriptional inhibition we used is serum starvation. This approach will lead to transcriptional inactivation and decreased cellular proliferation during starvation but then will result in robust transcriptional and kinase activation leading to increased global phosphorylation following serum replenishing. Heavy-labeled HeLa cells were serum starved for 72 h and then restimulated with 10% FBS for 4 h while light-labeled HeLa cells were only starved for 72 h. We applied the PS-ChAP workflow to two biological replicates as well. In the first replicate, 260 unique proteins were identified, and most of the proteins had quantifiable ratios. In the second replicate, 223 unique proteins were identified, and still, most of proteins were quantified (Supplemental Table S5). In total, 124 unique proteins were identified in both datasets.
Figure 7 shows the quantitative MS data of the PS-ChAP thiophosphoproteome analysis of serum starvation treatment. We plotted the histogram of the normalized H/L SILAC ratios that were transformed using the binary logarithm (log2) as shown in Fig. 7A. Log2-fold changes ranged from –1.5 to 1.5, and the standard deviation was reduced to 0.25. In Fig. 7B, the same ratios are plotted as a function of peptide intensity. We further analyzed the data to find some significantly abundance-changing protein candidates associated with chromatin that are impacted by the starvation treatment. As shown in Fig. 7C and Fig. 7D, two SILAC peptide pairs are highlighted. One peptide was RGESLDNLDSRP from the protein LMO7 in which the first serine is phosphorylated, while the other peptide is GRPSYVQR from C19orf21 in which the first serine is phosphorylated. Both of these peptides were down-regulated after serum refeeding. We also detected up-regulated phosphorylated proteins such as NUSAP1 isoform 1 of nucleolar and spindle-associated protein 1, SRSF2 serine/arginine-rich splicing factor 2, RBM14-RBM4 isoform 1 of RNA-binding protein 14, etc. Many of these proteins are known to play roles in transcription and protein synthesis, consistent with phosphorylation being linked to restimulus and increased gene transcription. Overall, this strategy based on selective thiophosphoproteome enrichment works well combined with the SILAC approaches to identify changes in phosphorylation status of chromatin-associated proteins involved in gene regulation. To our knowledge, this is the first report of a large-scale technique to identify site-specific phosphorylation changes on chromatin-associated nucleosomes under gene modulating stimuli.
Fig. 7.

PS-ChAP SILAC-based quantitative proteomics analysis during serum deprivation followed by serum replenishing. Light-labeled HeLa cells were serum starved for 72 h and heavy-labeled HeLa cells were starved 72 h then refed with 10% FBS serum for 4 h. (A) Histogram of log2-transformed normalized protein ratios for all quantified proteins identified in the experiment. (B) Plot of the normalized ratios of all quantified proteins plotted against their summed heavy and light peptide intensities. (C) MS-based quantification analysis of the LMO7 peptide (RGESLDNLDpSPR). SILAC peptide pair shows a heavy to light ratio (H/L) of 0.428 corresponding to a more than twofold reduction of the protein level. (D) MS-based quantification analysis of the C19orf21 peptide (GRPpSLYVQR). SILAC peptide pair shows a heavy to light ratio (H/L) of 0.685 corresponding to slightly less than twofold reduction of the protein level.
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-Based Quantitative Proteomic Approaches for Temporal Analysis of EGF Signaling Network
To benchmark our PS-ChAP method on a good model system to track cellular signaling down to the gene level (both characterization of chromatin-associated phosphoproteins and their respective gene targets), we decided to PS-ChAP isolate phosphorylated proteins on nucleosomes following EGF stimulation. Much is already known about the EGF-signaling pathway, thus providing a system that we can compare our PS-ChAP results against, but at the same time, a global view of EGF stimulated phosphoproteome directly on chromatin has also not been achieved. The cellular response to EGF is initiated by rapid kinetics of receptor activation, followed by phosphorylation-dependent activation of signaling cascades. As signaling cascades are highly dynamic, it is very important to follow the space and time ordered sequence of events that occurs as a result of growth factor stimulation. Different time points were selected for temporal analysis of the EGF signaling network. As shown in Supplemental Fig. S3A, HeLa cells were serum starved for 24 h and then stimulated by EGF for the indicated time intervals and then harvested. Then the expression of c-FOS in cell nuclear extracts from HeLa cells was analyzed by Western blotting. According to prior studies, the expression of the c-FOS gene occurs rapidly and transiently (41–43). From the Western blotting result (Supplemental Fig. S3B), we can see that the expression of c-FOS increased after 10 min of EGF stimulation, which indicated that EGF initiates signal transduction pathways.
Combining PS-ChAP with SILAC approaches, we can accurately identify and quantify phosphorylation level changes of the specifically enriched proteins at different time points under EGF stimulation. In order to generate a well-characterized set of EGF-stimulated and control samples, three independent biological replicate experiments were performed where HeLa cells were serum starved for 24 h and then treated with or without EGF stimulation. First, EGF-stimulated cells were grown in the standard “light” medium and nonstimulated cells were grown in the medium containing isotopically “heavy” l-arginine and l-lysine. We then mixed the cells at a 1 to 1 ratio and lysed the cells to get the nuclear pellet. After Mnase digestion, we applied our PS-ChAP for the thiophospho nucleosome enrichment. The affinity-purified sample was digested by trypsin and then analyzed by nanoLC-MS/MS. The intensity of MS signals between light and heavy peptides gives relative protein abundance before and after EGF stimulation.
Two time-course experiments were combined using the common time point (0 min and 10 min of EGF stimulation). Quantitation of phosphorylation sites was performed by pQuant. We identified more than 300 peptides from the PS-ChAP-enriched samples based on sequence and confidently localized phosphosites. Figure 8A shows the ratio distribution of the pull down peptides. We can see that most of the peptides are around the middle area, which indicated that the ratio of most peptides did not change abundance after EGF stimulation. The left portion of Fig. 8A shows the peptides that are highly enriched after 10 min EGF stimulation. As shown in Fig. 8B, ∼89 proteins were found in three biological replicates experimental based on the median quantifiable ratio across all peptides assigned and quantified for a specific protein. Some of the peptides are highly enriched after 10 min of EGF stimulation. Some of the highly enriched phosphoproteins following 10 min EGF stimulation are listed in Fig. 8C. The first line is the gene name, second line is the protein name, the third line is the phosphopeptides ratio, and the last line is the protein ratio according to the ratio coming from detection of many nonphosphopeptides of the protein found in the flow through. According to the results, we can see that the phosphorylation level of the proteins increased after 10 min EGF stimulation while the expression level of the protein remained stable.
Fig. 8.

SILAC-based quantitative proteomics for temporal analysis of the EGF signaling network. 10 min EGF-stimulated cells were grown in the standard “light” medium and nonstimulated cells were grown in the medium containing isotopically heavy l-arginine and l-lysine. (A) Histogram of log2-transformed normalized peptides ratios (heavy/light) for all quantified peptides identified in the experiment. (B) Number of proteins identified in three biological replicates. (C) Table listing some of the highly enriched phosphoproteins after 10 min EGF stimulation. The first line is the gene name, second line is the protein name, third line is the phosphopeptide ratio, and the last line is the protein ratio according to the ratio of many nonphosphopeptides of the protein found in the flow-through.
We also identified around 400 peptides after 120 min EGF stimulation (Fig. 9A), and 109 proteins were found (>60%) between three biological replicates (Fig. 9B). Figure 9A shows the ratio distribution of the pull down peptides, and still, most of the peptides showed similar abundance in two samples. We also listed some of these highly enriched phosphoproteins (120 min EGF) in Fig. 9C. Many proteins showed to be highly enriched both in the 10 min and 120 min EGF stimulations, such as HMGB3, RPL12, HMGA1 et al. Also, there are some new proteins that only showed up-regulation in the 120 min EGF stimulation such as AHNAK, SUB1, and HNRNPA1, demonstrating that those proteins are highly phosphorylated during the later phase of EGF stimulation.
Fig. 9.

SILAC-based quantitative proteomics for temporal analysis of EGF signaling network. 120 min EGF-stimulated cells were grown in the standard “light” medium and non-stimulated cells were grown in the medium containing isotopically heavy l-arginine and l-lysine. (A) Histogram of log2-transformed normalized peptide ratios (heavy/light) for all quantified peptides identified in the experiment. (B) Number of proteins identified in three biological replicates. (C) Table listing some of the highly enriched phosphoproteins after 120 min EGF stimulation. The first line is the gene name, second line is the protein name, third line is the phosphopeptide ratio, and the last line is the protein ratio according to the ratio of many nonphosphopeptides of the protein found in the flow-through.
We also compared our data to the PHOSIDA database (http://www.phosida.com) that was created by Mann lab (44, 45). It is comprised of three main components: the database environment, the prediction platform, and the toolkit section, which provides a wide range of analysis tools. A prior EGF-stimulated phosphoproteomic dataset was entered into the phosphorylation site database (PHOSIDA) (6). Many of the phosphorylated sites were identified in our results, including nucleolar and coiled-body phosphoprotein 1, vang-like protein 2, 40S ribosomal protein S7, chromobox protein homolog 5, high mobility group protein, et al. We also found some new phosphosites that are not included in PHOSIDA that are also highly enriched during EGF stimulation. Also according to the database, some of these enriched proteins show very important function such as transcription factor activity, chromatin binding or RNA binding, such as activated RNA polymerase II transcriptional coactivator p15 and chromobox protein homolog 5 (HP1). By using the toolkit section in this database, we can search for sequence motif matches or identify de novo consensus sequences from large-scale datasets.
Analysis of Kinase-Specific Phosphorylation Sites
Analyses of phosphorylation sites provide valuable information about kinase/substrate relationships and the relative activities of kinases. Most of the kinase groups have conserved amino acids surrounding the phosphorylation sites, and this sequence motif is necessary for the protein kinase to recognize the substrate. To further focus on the affected phosphosites on chromatin and the host kinases potentially responsible for these phosphorylation events during EGF stimulation, we identified consensus kinase phosphorylation motifs among phosphosites that were up-regulated. Several Ser motifs were identified such as the acidic motif [pSxxE], which is selectively recognized by casein kinase (CK) in mammals. Other motifs belonged to CDK2 (in which the consensus phosphorylation site is S/T-P-X-K/R, where X is any amino acid), as we can see that it has specificity for a basic residue in the p + 1 position since the interaction of the kinase and substrate are based on charge, hydrogen bonding, or hydrophobic interactions. Besides these kinase families (CK and CDK), we noted other kinases such as PDK, GSK, CAMK, and CHK were also repeatedly identified within three biological replicates. In summary, the kinase-oriented bioinformatics analysis of the phosphoproteomic data suggests that these kinases may also play a role in directly regulating phosphorylation directly on chromatin during EGF stimulation.
Phosphorylation-Specific Chromatin Affinity Purification (PS-ChAP) for Monitoring Gene Expression Linked to Cellular Signaling
As we now feel the PS-ChAP approach can capture nucleosomes containing phosphorylated proteins linked to transcription, we next applied PS-ChAP to isolate and identify the genes those phosphorylated proteins are found on using PS-ChAP-qPCR. By using this method, we can link the phosphorylation signaling to their intended gene targets initiated by EGF stimulation. As shown in Fig. 10A, EGF-stimulated cells are crosslinked and then dounced to get the nuclear pellet. And then we treated the nuclear extracts with ATP-γ-S. After Mnase digestion, we performed PS-ChAP for the enrichment of the DNA containing nucleosomes. Lastly, crosslinks were reversed and PS-ChAP captured DNA was purified after RNaseA and proteinase K treatment. The purified DNA was then amplified by qPCR using primers directed toward some known EGF-responsive gene related promoters.
Fig. 10.
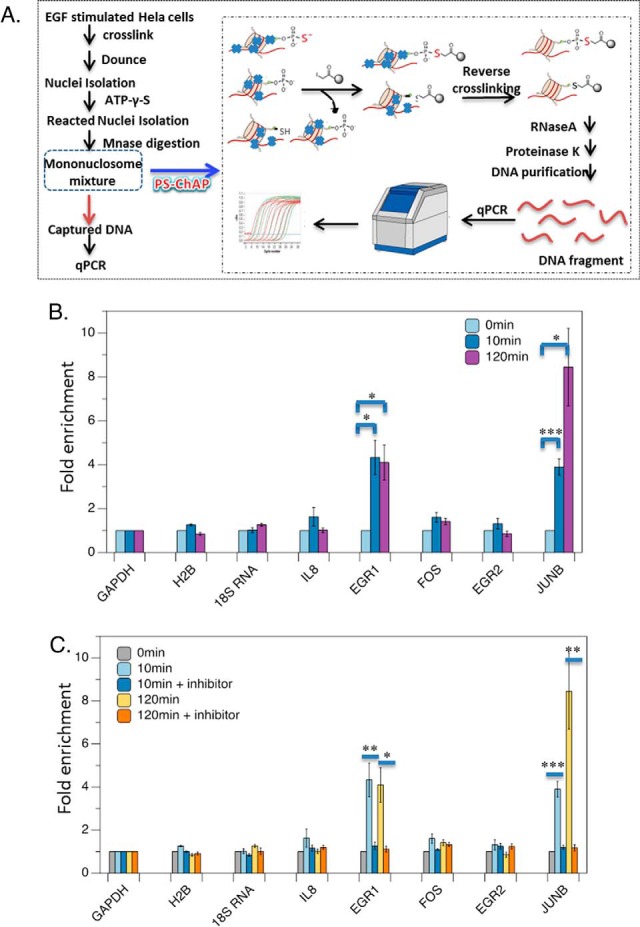
PS-ChAP for monitoring gene expression linked to cellular signaling. (A) EGF stimulated cells are crosslinked and then dounced to get the nuclear pellet followed by treating with ATP-γ-S. Illustration of the steps in the PS-ChAP strategy performed on a mononucleosome mixture after MNase digestion. By using iodoacetyl-beads, thiophosphorylated mononucleosomes can be isolated. Afterward, crosslinks were reversed and PS-ChAP captured DNA was purified and amplified by qPCR. (B) HeLa cells were serum starved for 24 h in media without serum and then treated with 100 ng/ml EGF for 10 min or 120min. Cells were harvested, and DNA was purified and analyzed by qPCR as shown in Fig. 10A. Statistically significant changes are indicated as ***p < .005, **p < .02 and *p < .05. (C) HeLa cells were serum starved for 24 h in media without serum, pretreated with 10 μm PD98059 for 1 h and then treated with 100 ng/ml EGF for 10min or 120min. Cells were harvested, and DNA was purified and analyzed by qPCR as shown in Fig. 10A. Values are relative to MEK-inhibitor-treated cells. Statistically significant changes are indicated as ***p < .005, **p < .02 and *p < .05. p values were calculated using the two-tailed unpaired Student's t test with equal variances. All error bars represent s.d. Data in parts B and C are the result of triplicate independent biological experiments.
HeLa cells were cultured with 100 ng/ml EGF for 10 min and 120 min and then crosslinked before being harvested. Thiophosphorylated proteins binding genes were isolated by the iodoacetyl beads and then amplified by qPCR. We selected some known EGF-responsive genes and designed all the primers at the promoter region. The qPCR results are shown in Fig. 10B. The first line is 0 min, the second line is 10 min EGF stimulation, and the third line is 2 h EGF stimulation. We used three negative controls in which the genes should not be EGF responsive (GAPDH, H2B and 18s RNA) and thus where we would not expect to detect enrichment of those gene via PS-ChAP. As expected, no significant enrichment was observed at a negative control region of DNA from those genes. However, we observed significant enrichment at some selected known EGF responsive genes. Cells harvested after 10 min EGF stimulation showed a total enrichment at most of the known EGF-responsive regions we chose compared with the cells without EGF stimulation, especially at the Epidermal growth factor (EGF) and JUNB gene promoter regions. We found a 4.3-fold increase at the EGR1 promoter gene and a 3.9-fold increase at the JUNB promoter gene after 10 min EGF stimulation. Cells that were treated with 120 min EGF also showed enrichment at three of the chosen DNA regions. It shows fourfold increase at EGR1 promoter region and an even higher increase at JUNB gene promoter region (almost eightfold) after 2 h EGF stimulation. IL8, FOS, and EGR2 also showed a smaller but reproduced enrichment by PS-ChAP. This result indicated that those phosphorylated proteins we found in Fig. 8c and Fig. 9c may potentially bind to EGR1 and JUNB gene promoter region. Some of the proteins are functional feedback regulators of signaling, while others are known to be involved in the transcription regulation of those genes such as activated RNA polymerase II transcriptional coactivator p15 and chromobox protein homolog 5 (HP1).
Then we added an MEK1 inhibitor, PD98059 to cells to block the EGF-induced transcriptional response. HeLa cells were serum starved for 24 h and then treated with PD98059 for 1 h. Afterward, EGF was added and the cells are harvested at different time points. Results are shown in Fig. 10, where the first line is 0 min, the second line is 10 min EGF stimulation, the third line is using MEK inhibitor following EGF for 10min, the fourth line is 2 h EGF stimulation, and the last line is MEK inhibitor for 1h and following EGF for 2 h. We performed the experiment with three biological replicates and calculated p values using the two-tailed unpaired Student's t test with equal variances. As shown in Fig. 10C, statistically significant changes are indicated as ***p < .005, **p < .02, and *p < .05. For 10 min EGF stimulation, there is no significant enrichment at EGR1 and JUNB gene promoter regions in the sample after MEK kinase inhibitor was added as compared with samples before inhibitor was added. The p value is .002 for EGR1 and .018 for JUNB, which indicated there were significant changes of the fold enrichment at EGR1 and JUNB gene promoter regions before and after MEK1 inhibitor treatment. For 120 min EGF stimulation, the enrichment at EGR1 and JUNB gene promoters dropped significantly after we added MEK1 inhibitor. The p value is .019 for EGR1 and .021 and JUNB, which also showed that the changes are significant. From this result, we can determine after we add the MEK kinase inhibitor, there is no significant change of enrichment of the EGR1 and JUNB genes at both 10 min and 120 min time points of EGF treatment. The results indicated that the amount of proteins that is phosphorylated during the EGF stimulation that is captured on these genes through our PS-ChAP enrichment dropped to the level before EGF was added. We expected to see this result as EGF-induced transcription response was blocked by MEK1 inhibitor Also as expected, no significant enrichment was observed at a negative control region of DNA (Fig. 10C). Based on these preliminary results, it is promising that this method can be used for monitoring gene expression linked to cellular signaling.
DISCUSSION
Our goal in this study was to develop a method based on thiophosphorylation enrichment (PS-ChAP) that could be used to isolate phosphorylated proteins on nucleosomes (i.e. capture proteins and DNA) to essentially monitor phosphorylation signaling into chromatin. The overall idea is to map by proteomics the phosphorylated proteins on nucleosomes and then identify the genes those phosphorylated proteins are found to reside on by thiophosphate capture mediated PS-ChAP-qPCR or in the future PS-ChAP-Seq. To accomplish this goal, we first needed to determine the most optimal method for thiophosphorylation enrichment, as this type of work is not readily used in the proteomics community. In our present study, we compared different methods for enrichment of thiophosphorylated peptides and proteins. Results indicated that the selective chemistry-based purification method (pH 8) using iodoacetyl beads was the best choice among all of the methods (TiO2 beads, iodoacetyl beads, and a thiophosphate ester antibody). The iodoacetyl beads can selectively pull down thiophosphopeptides with high efficiency. By reacting thiophosphate peptides attached to the iodoacetyl beads with oxone, we can elute thiophosphopeptides specifically, and the liberated peptides can be detected as phosphopeptides due to replacement of the sulfur atom with oxygen. Titanium dioxide (TiO2) is a highly efficient strategy widely used for phosphopeptide enrichment but fails to efficiently enrich for thiophosphopeptides in our hands. We believe that the reason for this lower efficiency is due to the electron density changes after the replacement of oxygen by sulfur. The thiophosphate-ester-specific antibody purification revealed many thiophosphorylated proteins by Western blotting (Fig. 1C) but suffered very low efficiency in PNBM-modified thiophosphopeptide enrichment (Fig. 2). The antibody may only be effective in Western blotting because it is not designed for peptide or protein isolations or that the ionization efficiency of the thiophosphopeptides could be severely limited after derivatization with PNBM.
For thiophosphoprotein enrichment, the selective chemistry-based iodoacetyl beads purification method again showed the highest efficiency. The iodoacetyl beads at pH 8 are able to release specific binding peptides that can then be detected by mass spectrometry while the thiophosphate-ester-specific antibody revealed very few targeted peptides. By using thiophosphate-ester-specific antibody, we detected all the digested nontargeted peptides of the pulled down proteins together with all the targeted peptides, which greatly decreased the percentage of the targeted peptides. In summary, we find that the best method to specifically purify thiophosphopeptides or thiophosphoproteins is the selective chemistry-based purification method (at pH 8).
While thiophosphorylation affinity purification strategies have been used previously, to our knowledge, this is the first comprehensive study to compare these approaches. By analyzing the results in those experiments using nuclei, we found that nuclear thiophosphorylated proteins have functions in DNA binding, chromatin binding, transcription factor binding function, and transcription cofactor activity modules, which are tightly connected with the regulation of gene transcription.
Although signaling transduction pathways have been well known to elicit defined and precise gene expression changes, still little is known about how these signals are relayed to chromatin to change the transcriptional or epigenetic landscape on a global scale. Several recent studies have indicated kinase activity on chromatin, even discovering chromatin-associated kinases that once were not even believed to be found in the nucleus (46, 47). To study the phosphorylation signaling that directly occurs on chromatin, we applied this chemistry-based purification method to mononucleosomes (PS-ChAP). The results showed that the efficiency of the enrichment of thiophosphoprotein-containing nucleosomes was quite high, which illustrates that this method can be successfully applied to isolate this chromatin. We detected more than 100 proteins from these MS results, identifying among the proteins identified, well-known chromatin-associated proteins such as histone variants, DNA binding proteins, and transcription factors.
The success of this method for mononucleosome enrichment gave us confidence to proceed with experiments to determine how phosphorylated signals on chromatin change under different stimuli by combining the affinity enrichments with quantitative proteomic approaches. Toward investigating phosphorylation directly related to transcriptional events, we used α-amanitin treatment and cell starvation to inhibit global transcription in cells. By analyzing the SILAC-based thiophosphorylation data, we found some interesting phosphorylation sites that directly communicate with chromatin that may be linked to transcriptional activation, which we are now pursuing in more long-term functional experiments. An interesting mark we found linked to transcription was on histone H3 that was phosphorylated at threonine 45 that may play an important role in DNA-histone interactions within the core particle. This region where Thr45 is located makes critical contacts with DNA when assembled into the nucleosome core particle. Thr45 is located precisely at the points of entry and exit of DNA on the nucleosome. It is highly likely that phosphorylation of H3T45 may play an important role in DNA-histone interactions within the core particle. Other research has indicated that six important histone residues (H2A-G107, H2A-I112, H2A-L117, H3-T45, H3-R49, and H3-R52) (Fig. 6D), located at the nucleosome DNA entry exit site, form a surface on the structured nucleosome core and regulate H3-K36me3 deposition. Most of these residues are critical for the chromatin association of RNA polymerase II and Set2 (40), and H3T45 is actually subject to phosphorylation during DNA replication. Together, these data demonstrate the structural importance of H3Thr45 and the relationship between RNA polymerase II and H3Thr45. After α-amanitin treatment, there was a twofold reduction of H3Thr45, which indicated that the phosphorylation site in histone H3 at threonine 45 may be a special epigenetic marker related to general RNA polymerase II mediated gene activation.
Next, we applied this method for studying the EGF-signaling pathway to get a global view of EGF-modulated phosphoproteome directly on chromatin. To our knowledge, this is the first time that the effect of EGF stimulation on the phosphoproteome has been assessed specifically at the chromatin level. Nevertheless, the EGF stimulation model gave us a well-studied system to benchmark our ability to purify out EGF-stimulated phosphorylated proteins and more importantly EGF-activated genes. By combining with SILAC-based quantitative proteomic approaches, we can accurately identify and quantify phosphorylation-level changes of the specifically enriched chromatin proteins at different time points under EGF stimulation. The result reveals that a large proportion of cellular proteins are phosphorylated on chromatin and that only a small subset of total phosphorylation sites are highly enriched in response to stimulus. Those highly enriched phosphopeptides may function as “signal initiators” since they are involved in the early up-regulation of the EGF pathway. Among all the enriched proteins, some of them have been published as being linked to EGF stimulation, such as some high-mobility group proteins. Members of the high-mobility group family of high-mobility-group chromatin proteins appear to play an important role in regulating gene expression. They act as architectural transcription factors and alter the conformation of DNA by modulating nuclear protein-DNA complexes. Our results showed that Ser29 of HMG-I/Y and T13 of high-mobility group protein B3 are highly phosphorylated after EGF stimulation, which may have important implications that those sites are involved in the regulation of gene activity during EGF stimulation. Based on motif analysis, our studies also indicated some specific kinase activity on chromatin, such as from the kinases CK, CDK, PDK, GSK, CAMK, and CHK. Thus, our datasets may greatly accelerate cell signaling research by helping understand the intersection of cellular signaling pathways and chromatin/epigenetic networks to regulate gene expression.
Our mass spectrometry analysis provided a list of proteins that are phosphorylated on genes during EGF stimulation, and next we took the associated nucleosomal DNA and attempted to characterize the genes those phosphorylated proteins were found on by thiophosphate-assisted ChAP-qPCR. Using a slightly modified approach for capturing phosphorylated nucleosomes, we found significant enrichment of EGF-induced kinase substrates at the promoter regions of the EGF-responsive genes, especially the EGR1 and JUNB genes during EGF stimulation. Furthermore, we found that when EGF-induced transcriptional response was blocked, there was no longer significant enrichment found in this region. These results are very promising as proof of principle studies demonstrating that our PS-ChAP methodology can be used for monitoring gene expression linked to cellular phosphorylation mediated signaling. We believe that PS-ChAP would be even more powerful combined with next generation DNA sequencing technology such as in ChIP-seq to identify some specific kinase substrate target genes in an unbiased and more high-throughput manner. Additionally, we also feel that using the analog sensitive kinase approaches developed by the Shokat group (48), specific ATP-γ-S tagging of the protein substrates of one particular kinase on chromatin and its targeted genes can be achieved after being combined with PS-ChAP enrichment. This will solve one of the major challenges in understanding signaling networks by knowing how signaling specificity is achieved when many of the same core signaling pathways are activated by different receptors that then elicit different cellular responses. For instance, if we choose AS PI-3 kinase, we can detect different substrates and targeted genes by using insulin and also growth factor activation queues followed by PS-ChAP enrichment, which would help toward understanding why activation of the PI-3 kinase pathway by the insulin receptor PTK leads to metabolic responses but activation of PI-3 kinase by growth factor receptor PTKs in the same cell type does not.
This quantitative PS-ChAP method we present has the potential to become a powerful tool to characterize thiophosphoproteome-wide chromatin-associated changes related to gene activation or any other chromatin templated processes. Furthermore, this strategy is easily adaptable to fit other phosphoproteomics study designs. For example, it is applicable for use in other cell lines or could be applied to many other stimuli. Our method could be also used with samples that are not amenable to SILAC labeling (such as sample from patient tissue); however, for quantitative analysis, one could also modify this strategy by combining the PS-ChAP enrichment with H2O18 labeling or Tandem Mass Tag (TMT) labeling (49). This strategy demonstrates the possibility of capturing low abundance phosphorylation signaling changes on chromatin-associated nucleosomes, which can significantly impact protein phosphorylation and epigenetic gene expression studies.
Supplementary Material
Acknowledgments
We thank members of the Garcia lab for critical reading of the manuscript.
Footnotes
Author contributions: Y.H. and B.A.G. designed research; Y.H. and R.C.M. performed the experiments; Y.H. and Z.Y. analyzed data; and Y.H. and B.A.G. wrote the paper.
* We also gratefully acknowledge funding support from the National Institutes of Health (R01GM110174 and R01AI118891), the National Science Foundation (NSF) Early Faculty CAREER award, andthe Department of Defense (BC123187P1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material Supplemental Tables S1-S5 and Supplemental Figs. S1-S3.
This article contains supplemental material Supplemental Tables S1-S5 and Supplemental Figs. S1-S3.
1 The abbreviations used are:
- ChAP
- phosphorylation-Specific Chromatin Affinity Purification
- SILAC
- Stable isotope labeling by amino acids in cell culture
- IAP
- immunoaffinity purification
- CAD
- Collision activated dissociation
- HCD
- higher-energy collisional dissociation
- EGF
- Epidermal growth factor
- TMT
- Tandem Mass Tag
- CDK
- CaMK kinase
- GSK
- Glycogen Synthase Kinase
- CAMK
- calmodulin-dependent protein kinase
- CHK
- cyclin-dependent protein kinase.
REFERENCES
- 1. Cohen P. (2002) The origins of protein phosphorylation. Nat. Cell Biol. 4, E127–E130 [DOI] [PubMed] [Google Scholar]
- 2. Hunter T. (1995) Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 [DOI] [PubMed] [Google Scholar]
- 3. Hunter T. (2000) Signaling—2000 and beyond. Cell 100, 113–127 [DOI] [PubMed] [Google Scholar]
- 4. Cohen P. (2001) The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 268, 5001–5010 [DOI] [PubMed] [Google Scholar]
- 5. Hubbard M. J., and Cohen P. (1993) On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci. 18, 172–177 [DOI] [PubMed] [Google Scholar]
- 6. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 7. Futcher B., Latter G. I., Monardo P., McLaughlin C. S., and Garrels J. I. (1999) A sampling of the yeast proteome. Mol. Cell. Biol. 19, 7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marcus K., Immler D., Sternberger J., and Meyer H. E. (2000) Identification of platelet proteins separated by two-dimensional gel electrophoresis and analyzed by matrix assisted laser desorption/ionization-time of flight-mass spectrometry and detection of tyrosine-phosphorylated proteins. Electrophoresis 21, 2622–2636 [DOI] [PubMed] [Google Scholar]
- 9. Mann M., Ong S.-E., Grønborg M., Steen H., Jensen O. N., and Pandey A. (2002) Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 20, 261–268 [DOI] [PubMed] [Google Scholar]
- 10. Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., and White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 11. Aebersold R., and Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 12. Gruhler A., Olsen J. V., Mohammed S., Mortensen P., Færgeman N. J., Mann M., and Jensen O. N. (2005) Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 [DOI] [PubMed] [Google Scholar]
- 13. Nita-Lazar A., Saito-Benz H., and White F. M. (2008) Quantitative phosphoproteomics by mass spectrometry: Past, present, and future. Proteomics 8, 4433–4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oda Y., Nagasu T., and Chait B. T. (2001) Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nat. Biotechnol. 19, 379–382 [DOI] [PubMed] [Google Scholar]
- 15. Feng S., Ye M., Zhou H., Jiang X., Jiang X., Zou H., and Gong B. (2007) Immobilized zirconium ion affinity chromatography for specific enrichment of phosphopeptides in phosphoproteome analysis. Mol. Cell. Proteomics 6, 1656–1665 [DOI] [PubMed] [Google Scholar]
- 16. Han G., Ye M., Zhou H., Jiang X., Feng S., Jiang X., Tian R., Wan D., Zou H., and Gu J. (2008) Large-scale phosphoproteome analysis of human liver tissue by enrichment and fractionation of phosphopeptides with strong anion exchange chromatography. Proteomics 8, 1346–1361 [DOI] [PubMed] [Google Scholar]
- 17. Thingholm T. E., Jørgensen T. J., Jensen O. N., and Larsen M. R. (2006) Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 1, 1929–1935 [DOI] [PubMed] [Google Scholar]
- 18. Rozdzial M. M., and Haimo L. T. (1986) Bidirectional pigment granule movements of melanophores are regulated by protein phosphorylation and dephosphorylation. Cell 47, 1061–1070 [DOI] [PubMed] [Google Scholar]
- 19. Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., and Shokat K. M. (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 [DOI] [PubMed] [Google Scholar]
- 20. Ubersax J. A., Woodbury E. L., Quang P. N., Paraz M., Blethrow J. D., Shah K., Shokat K. M., and Morgan D. O. (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864 [DOI] [PubMed] [Google Scholar]
- 21. Kwon S. W., Kim S. C., Jaunbergs J., Falck J. R., and Zhao Y. (2003) Selective enrichment of thiophosphorylated polypeptides as a tool for the analysis of protein phosphorylation. Mol. Cell. Proteomics 2, 242–247 [DOI] [PubMed] [Google Scholar]
- 22. Badeaux A. I., and Shi Y. (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat. Rev. Mol. Cell Biol. 14, 211–224 [PubMed] [Google Scholar]
- 23. Schreiber S. L., and Bernstein B. E. (2002) Signaling network model of chromatin. Cell 111, 771–778 [DOI] [PubMed] [Google Scholar]
- 24. Suganuma T., and Workman J. L. (2013) Chromatin and signaling. Curr. Opin. Cell Biol. 25, 322–326 [DOI] [PubMed] [Google Scholar]
- 25. Baek S. H. (2011) When signaling kinases meet histones and histone modifiers in the nucleus. Mol. Cell 42, 274–284 [DOI] [PubMed] [Google Scholar]
- 26. Lin S., and Garcia B. A. (2012) Examining histone posttranslational modification patterns by high resolution mass spectrometry. Methods Enzymol. 512, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li D., Fu Y., Sun R., Ling C. X., Wei Y., Zhou H., Zeng R., Yang Q., He S., and Gao W. (2005) pFind: A novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 21, 3049–3050 [DOI] [PubMed] [Google Scholar]
- 28. Wang L. H., Li D. Q., Fu Y., Wang H. P., Zhang J. F., Yuan Z. F., Sun R. X., Zeng R., He S. M., and Gao W. (2007) pFind 2.0: A software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun. Mass Spectrom. 21, 2985–2991 [DOI] [PubMed] [Google Scholar]
- 29. Liu C., Song C.-Q., Yuan Z.-F., Fu Y., Chi H., Wang L.-H., Fan S.-B., Zhang K., Zeng W.-F., He S.-M., Dong M. Q., and Sun R. X. (2014) pQuant improves quantitation by keeping out interfering signals and evaluating the accuracy of calculated ratios. Anal. Chem. 86, 5286–5294 [DOI] [PubMed] [Google Scholar]
- 30. Eden E., Navon R., Steinfeld I., Lipson D., and Yakhini Z. (2009) GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chou M. F., and Schwartz D. (2011) Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinform. 35, 13.15.1–13.15.24 [DOI] [PubMed] [Google Scholar]
- 32. Blethrow J. D., Glavy J. S., Morgan D. O., and Shokat K. M. (2008) Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 105, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Allen J. J., Li M., Brinkworth C. S., Paulson J. L., Wang D., Hübner A., Chou W.-H., Davis R. J., Burlingame A. L., Messing R. O., Katayama C. D., Hedrick S. M., and Shokat K. M. (2007) A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hunter T., and Karin M. (1992) The regulation of transcription by phosphorylation. Cell 70, 375–387 [DOI] [PubMed] [Google Scholar]
- 35. Larochelle S., Amat R., Glover-Cutter K., Sansó M., Zhang C., Allen J. J., Shokat K. M., Bentley D. L., and Fisher R. P. (2012) Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 19, 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao T., Hall H., Kizer K. O., Shibata Y., Hall M. C., Borchers C. H., and Strahl B. D. (2003) Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 17, 654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker S. P., Phillips J., Anderson S., Qiu Q., Shabanowitz J., Smith M. M., Yates J. R. 3rd, Hunt D. F., and Grant P. A. (2010) Histone H3 Thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nat. Cell Biol. 12, 294–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hurd P. J., Bannister A. J., Halls K., Dawson M. A., Vermeulen M., Olsen J. V., Ismail H., Somers J., Mann M., Owen-Hughes T., Gout I., and Kouzarides T. (2009) Phosphorylation of histone H3 Thr-45 is linked to apoptosis. J. Biol. Chem. 284, 16575–16583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ismail H., Hurd P. J., Khalil M. I., Kouzarides T., Bannister A., and Gout I. (2014) S6 kinase 2 is bound to chromatin-nuclear matrix cellular fractions and is able to phosphorylate histone H3 at threonine 45 in vitro and in vivo. J. Cell. Biochem. 115, 1048–1062 [DOI] [PubMed] [Google Scholar]
- 40. Endo H., Nakabayashi Y., Kawashima S., Enomoto T., Seki M., and Horikoshi M. (2012) Nucleosome surface containing nucleosomal DNA entry/exit site regulates H3-K36me3 via association with RNA polymerase II and Set2. Genes Cells 17, 65–81 [DOI] [PubMed] [Google Scholar]
- 41. Kruijer W., Cooper J. A., Hunter T., and Verma I. M. (1984) Platelet-derived growth factor induces rapid but transient expression of the c-fos gene and protein. Nature 312, 711–716 [DOI] [PubMed] [Google Scholar]
- 42. Curran T., and Franza B. R. Jr. (1988) Fos and Jun: The AP-1 connection. Cell 55, 395–397 [DOI] [PubMed] [Google Scholar]
- 43. Sagar S. M., Sharp F. R., and Curran T. (1988) Expression of c-fos protein in brain: Metabolic mapping at the cellular level. Science 240, 1328–1331 [DOI] [PubMed] [Google Scholar]
- 44. Gnad F., Ren S., Cox J., Olsen J. V., Macek B., Oroshi M., and Mann M. (2007) PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gnad F., Gunawardena J., and Mann M. (2011) PHOSIDA 2011: The posttranslational modification database. Nucleic Acids Res. 39, D253–D260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Göke J., Chan Y.-S., Yan J., Vingron M., and Ng H.-H. (2013) Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell 50, 844–855 [DOI] [PubMed] [Google Scholar]
- 47. Klein A. M., Zaganjor E., and Cobb M. H. (2013) Chromatin-tethered MAPKs. Curr. Opin. Cell Biol. 25, 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bishop A., Buzko O., Heyeck-Dumas S., Jung I., Kraybill B., Liu Y., Shah K., Ulrich S., Witucki L., Yang F., Zhang C., and Shokat K. M. (2000) Unnatural ligands for engineered proteins: new tools for chemical genetics. Annu. Rev. Biophys. Biomol. Struct. 29, 577–606 [DOI] [PubMed] [Google Scholar]
- 49. Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 50. Davey C. A., Sargent D. F., Luger K., Maeder A. W., and Richmond T. J. (2002) Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9 Å Resolution†. J. Mol. Biol. 319, 10971–11113 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.