

Abstract
In recent years, cross-linking mass spectrometry has proven to be a robust and effective method of interrogating macromolecular protein complex topologies at peptide resolution. Traditionally, cross-linking mass spectrometry workflows have utilized homogenous complexes obtained through time-limiting reconstitution, tandem affinity purification, and conventional chromatography workflows. Here, we present cross-linking immunoprecipitation-MS (xIP-MS), a simple, rapid, and efficient method for structurally probing chromatin-associated protein complexes using small volumes of mammalian whole cell lysates, single affinity purification, and on-bead cross-linking followed by LC-MS/MS analysis. We first benchmarked xIP-MS using the structurally well-characterized phosphoribosyl pyrophosphate synthetase complex. We then applied xIP-MS to the chromatin-associated cohesin (SMC1A/3), XRCC5/6 (Ku70/86), and MCM complexes, and we provide novel structural and biological insights into their architectures and molecular function. Of note, we use xIP-MS to perform topological studies under cell cycle perturbations, showing that the xIP-MS protocol is sufficiently straightforward and efficient to allow comparative cross-linking experiments. This work, therefore, demonstrates that xIP-MS is a robust, flexible, and widely applicable methodology for interrogating chromatin-associated protein complex architectures.
The structural basis of specific protein–protein interactions and higher-order protein complex multimerization crucially informs our understanding of many molecular and cellular processes. AP-MS-based strategies have been a major player in the elucidation of specific protein–protein interactions and core protein complexes (1–3). However, although quantitative AP-MS/MS experiments identify specific interactors for any given bait, they cannot differentiate between direct and indirect interactors or provide structural or topological information on protein complex assemblies. Furthermore, traditional structural methodologies often fail with large or dynamic protein complexes including chromatin remodeling or chromatin associated protein complexes. Cross-linking mass spectrometry (XL-MS),1 therefore, has emerged as a powerful technique for analyzing protein complex architectures through direct observation of subcomplex interfaces at the peptide level (4–6). Landmark studies used XL-MS to reveal the architectures of the chromatin remodeling complexes Ino80 and SWR1 (7, 8), and other chromatin-associated complexes have been interrogated using traditional XL-MS workflows (9–14).
In general, cross-linked peptides represent a small fraction of the total pool of peptides measured in an MS/MS analysis. Therefore, traditional XL-MS workflows have utilized homogenous complexes obtained through time-limiting reconstitution, tandem affinity purification, and conventional chromatography workflows to facilitate the detection of low abundance cross-linked peptides from the target protein complex. For example, reconstitution of complexes to near homogeneity using tandem tagging systems has been previously reported, using a variety of tagging combinations (13, 15, 16). Similarly, baculovirus over-expression and recombinant yeast systems have also been used effectively to maximize protein yield (7–9, 14). Further strategies often used to enrich cross-link identifications include strong cation exchange chromatography (SCX), size exclusion chromatography (SEC), the use of multiple proteases, or alternate cross-linking strategies including acid-acid cross-linkers, CID-cleavable cross-linkers, or tagged, enrichable cross-linkers (16–20). These steps have been necessary to enrich to an appreciable degree the number of measurable cross-linked peptides from the target complex. However, implementing and optimizing these workflows represents a considerable barrier to entry to the XL-MS field for many molecular biologists.
Single-AP-MS strategies are commonly applied in chromatin biology, where specific interactors can be identified in the presence of a vast amount of background proteins (2, 3, 21–23). Our group and others have previously established workflows for identifying specific protein interactors and interaction stoichiometries using a GFP-tagging BAC transgene systems expressing tagged baits at a near-endogenous level (22, 24–26). However, thus far simple, single-IP methodologies have not been widely used in XL-MS experiments. One main challenge is sufficiently purifying protein complexes to facilitate the consistent identification of low-abundance cross-linked peptides over background. A very recent study used engineered high-affinity, lysine-free GFP-nanobodies to efficiently extract endogenous level tagged baits for on-bead cross-linking studies (27). Here, we present cross-linking immunoprecipitation mass spectrometry, or xIP-MS, utilizing a simple, efficient GFP-AP workflow followed by a high-stringency washing procedure to obtain highly pure protein complex from small volumes of human whole cell lysates. xIP-MS couples single-AP GFP purification with on-bead crosslinking and sample prep for MS/MS analysis. Dynamic exclusion settings, exclusion of lower charge-state peptides, and the high speed of modern mass spectrometers facilitate the fragmentation and identification of low abundance cross-linked peptides. Furthermore, computational analysis of cross-linked peptides is a crucial and widely discussed aspect of XL-MS workflows (28–30). In xIP-MS, we use straightforward data-filtering steps based on peptide length (selectively removing those matches where one long, high scoring peptide compensates for a short, ambiguous peptide match) and reproducibility in multiple experiments (to remove spurious matches) to increase confidence in cross-link identifications.
We benchmark xIP-MS against the structurally well-characterized PRPP complex and provide evidence for a structural ensemble of PRPP complexes in vivo (31). We then apply xIP-MS to the chromatin-associated cohesin, XRCC5/6, and MCM complexes. We use xIP-MS to analyze the cohesion complex and observe numerous cross-links within the well-ordered head region, complementing previous cross-linking experiments and directly supporting a model for the human cohesin head interaction (11, 12). Next, we used xIP-MS to analyze conformational changes in the x-ray repair cross-complementing protein 5/6 complex (XRCC5/6, or Ku70/86) (32). We observe conformational flexibility in the Ku and SAP domains consistent with their involvement in DNA binding during double-stranded break repair (33). Finally, we use xIP-MS to characterize soluble mini-chromosome maintenance (MCM) subcomplexes (34–37). We present an architecture for interactions between MCM subunits and show that soluble MCM subcomplexes are generally cell-cycle invariant.
The xIP-MS workflow requires approximately the time and resources of a standard coIP experiment, yet can provide additional structural information about subunit interfaces and complex topologies. We further demonstrate the biological usefulness of xIP-MS in studying complex structural ensembles, conformational flexibility, or subcomplex architectures under perturbation. Thus, we present here xIP-MS as a simple, efficient, and broadly applicable technique for probing protein complex architectures.
MATERIALS AND METHODS
Cell Culture and Lysate Collection
Cell lysates were collected from stably transgenic HeLa Kyoto cells expressing near-endogenous levels of GFP-tagged bait protein from a recombined BAC transgene system (24, 25). Cell lines were cultured in DMEM plus 10% FBS and 100U/ml penicillin and streptomycin. For 1 week, cells were kept under selection with 400 μg/μl geneticin to ensure stable integration of the tagged bait, then expanded in normal media. Cell lysates were collected by resuspending cells in five cell pellet volumes of lysis buffer (150 mm NaCl, 50 mm Tris pH 8.0, 1 mm EDTA, 20% glycerol) supplemented with 1% Nonidet P-40, 1 mm DTT, and Roche EDTA-free complete protease inhibitors (CPIs, Roche, Basel, Switzerland). Cell lysates were rotated at 4C for two hours. The lysate was then centrifuged for 30 min at 4000 rcf and 4°C, and the supernatant was collected and snap-frozen. This lysis method produced high-quality lysates of ∼10 mg/ml protein concentration.
GFP Affinity Purification for Label-free Quantification (LFQ)
Twenty microliters GFP bead slurry was used per affinity purification (Chromotek, Planegg, Germany). Beads were prewashed three times with Buffer C (300 mm NaCl, 20 mm HEPES pH 7.9, 20% glycerol, 2 mm MgCl2, 0.2 mm EDTA) supplemented with 1% Nonidet P-40, 0.5 mm DTT, and EDTA-free complete protease inhibitors (CPIs, Roche). Four hundred micrograms whole cell lysate was added to the prewashed beads and adjusted to 400 μl with whole cell lysis buffer plus 1% Nonidet P-40, 1 m DTT, and CPIs. Ethidium bromide was added to 50 μg/ml. Reactions were incubated on a rotating wheel for one hour at 4°C. After IP, beads were washed twice with Buffer C increased to 1 m NaCl and supplemented with 1% Nonidet P-40, 0.5 mm DTT, and CPIs. Beads were then washed twice with PBS supplemented with 1% Nonidet P-40, and finally washed twice with PBS. All supernatant was removed carefully with a 30 G syringe before sample prep for AP-MS/MS analysis. Control samples were prepared using binding control agarose beads with the same protocol (Chromotek). All LFQ experiments were performed in triplicate. GFP-AP and control samples from the same experiment were prepared on the same day and analyzed by AP-MS/MS sequentially.
GFP Affinity Purification for xIP-MS
GFP affinity purifications for xIP-MS were performed essentially as described for LFQ GFP affinity purifications. Thirty microliters of GFP bead slurry was used per affinity purification, and 1 ml of total whole cell lysate was used per pulldown (∼10 mg/ml). All xIP-MS experiments were performed in duplicate. Replicates were performed independently and measured by AP-MS/MS separately.
On-bead Chemical Cross-linking for xIP-MS
On-bead cross-linking was performed by immediately resuspending beads in 50 mm borate-buffered saline containing 1 mm BS3 (Thermo Scientific, Waltham, MA) following GFP affinity purification. Cross-linking reactions were performed for 1 h at room temperature with shaking at 1000 rpms. Reactions were quenched by adding 100 mm ammonium bicarbonate and incubated at room temperature for 10 mins with shaking at 1000 rpms. All supernatant was again carefully removed with a 30 G syringe.
Sample Preparation for AP-MS/MS Analysis
Sample preparation was performed in the same manner for LFQ and xIP-MS samples. Purified or cross-linked proteins were denatured and reduced in elution buffer (2 m urea, 100 mm ammonium bicarbonate, 10 mm DTT) for 20 min at room temperature with shaking at 1000 rpms. Iodoacetimide was added to 50 mm, and samples were incubated in the dark for 10 mins at room temperature with shaking at 1000 rpms. To this 0.25 μg of trypsin was added, and samples were digested overnight at room temperature to ensure complete release of tryptic peptides from the beads. Digested peptides were acidified with 10% TFA and stored on C18 StageTips for mass spectrometry analysis.
AP-MS/MS Analysis
All chromatography was performed on an Easy-nLC 1000 (Thermo Scientific). Buffer A was 0.1% formic acid and Buffer B was 80% acetonitrile and 0.1% formic acid. LFQ samples collected on the LTQ-Orbitrap QExactive were measured by developing a gradient from 9 to 32% Buffer B for 94 min before washes at 50% then 95% Buffer B, for 120 min of total data collection time. LFQ samples collected on the LTQ-Orbitrap Fusion Tribrid were measured by developing a gradient from 9 to 32% Buffer B for 114 min before washes at 50% then 95% Buffer B, for 140 min of total data collection time. xIP-MS samples were measured by developing a gradient of 5–32% Buffer B for 214 min before washing with 60% then 95% Buffer B, for 240 min of total data collection time. The flow rate was 250 nl/min for all gradients.
SMC1A-GFP and XRCC6-GFP LFQ samples were measured on an LTQ-Orbitrap QExactive. Full MS scans were collected from 300 to 1650 m/z with a resolution of 70,000 and an AGC target of 3e6. MS/MS scans were collected in the orbitrap with a resolution of 17,500, an AGC target of 1e5, an NCE of 25, and an intensity threshold of 8.3e2. TopN was set to 10, unassigned and 1+ charged ions were excluded, and dynamic exclusion was set at 20 s.
PRPS1-GFP and MCM6-GFP LFQ samples were measured on an LTQ-Orbitrap Fusion Tribrid. Full MS scans were collected from 400 to 1500 m/z with a resolution of 120,000 and an AGC target of 4e5. MS/MS scans were collected in the linear ion trap using CID activation with a resolution of 30,000, an AGC target of 1e4, collision energy of 35, and an intensity threshold of 5e3. Scans were collected in data-dependent top speed mode, ions of charge state 2–7+ were considered, and dynamic exclusion was set at 60 s.
All xIP-MS samples were measured on an LTQ-Orbitrap-QExactive. Full MS scans were collected from 300 to 1650 m/z with a resolution of 70,000 and an AGC target of 3e6. MS/MS scans were collected in the orbitrap with a resolution of 17,500, an AGC target of 1e5, an NCE of 25, and an intensity threshold of 4e2. TopN was set to 10, unassigned, 1+, and 2+ charged ions were excluded, and dynamic exclusion was set at 20 s.
LFQ Peptide Identification and Analysis
Thermo RAW files from LFQ AP-MS/MS measurements were analyzed with MaxQuant version 1.5.1.0 using default settings and searching against the Uniprot curated human proteome (release 03/09/2014) (38, 39). Cysteine carbamidomethyl was used as a fixed modification, and N-terminal acetylation and methionine oxidation were used as variable modifications. Additional options Match between runs, LFQ, and iBAQ were selected. Stoichiometry calculations and volcano plots were produced essentially as described before using a one-way ANOVA test (22). Statistical cutoffs were chosen such that no proteins were present as outliers on the control, non-GFP side of the volcano plot.
xIP-MS Cross-linked Peptide Identification and Analysis
For cross-link identification, Thermo RAW files were converted to mgf format using MSConvert with the peak picking option for levels “1-” selected and the “Prefer Vendor” option checked (40). mgf files were analyzed in pLink version 1.21 with default settings to identify cross-linked peptides with an FDR threshold of 0.05 using a precursor mass tolerance of 10 ppm and a fragment ion mass tolerance of 20 ppm searching against specific interactors as identified by LFQ analysis (30). BS3 was used as the crosslinker, trypsin was used as the enzyme with a maximum of two missed cleavages allowed, cysteine carbamidomethylation was included as a fixed modification, and methionine oxidation was included as a variable modification. pLink identifications were further filtered to include only matches with > = 5 and < = 40 residues per peptide. Only cross-linked sites identified in two out of two independent replicates were considered for further structural analysis (supplemental Fig. S1) (29). Cross-link maps were produced with xiNet, and distance constraints were analyzed with Xlink Analyzer in UCSF Chimera (41–43). All molecular visualizations were performed with UCSF Chimera (41).
Homology Modeling
For PRPS1, only short internal gaps in the crystal structure were modeled using MODELLER in UCSF chimera (PDB: 2h06) (31, 44). All PSPS1 structural alignments were performed using MatchMaker in UCSF Chimera (45).
All cohesin homology models were produced using SWISS-MODEL using alignments calculated with EMBOSS Needle (46–48). For SMC1A (PDB: 1w1w) and SMC3 (PDB: 4ux3) N- and C-terminal head regions were aligned with their respective yeast PDB structure, and then the SMC1A model was further aligned with a second SMC3 head domain present as a dimer in the 4ux3 crystal structure for analysis of distance constraints. For comparative analysis using prokaryote (PDB: 3zgx) and archea (PDB: 4i99) SMC homologs, a similar analysis was performed. The cohesin hinge domain was similarly modeled from the mouse homolog (PDB: 2wd5).
Analysis of Normal Modes
Analysis of normal modes using anisotropic network analysis was conducted for XRCC5/6 (PDB: 1jeq) in Python with the prody package using default settings (49). The twenty slowest frequency modes were kept for visual analysis in VMD (50). Figures were generated in prody, and videos were made using VMD.
Data Access
The mass spectrometry RAW data, maxQuant identifications for LFQ analysis, and pLink identifications for cross-linking analysis have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD002987 (51).
RESULTS
High Stringency Washes Effectively Purify Chromatin-associated Complexes and Permit Chemical Cross-linking Analysis
We first wanted to establish a protocol for effectively purifying stable protein complexes using a simple, single affinity purification workflow. Our group previously optimized workflows for high efficiency purification of GFP-tagged baits using a BAC transgene system (24–26). Using this method as a starting point, we tested whether more stringent washing conditions following bead purification would be sufficient to enrich stable complexes with substantially increased purity. After bead incubation in cell lysate buffer with 1% Nonidet P-40 and physiological salt, we washed our beads twice with cell lysis buffer plus 1% Nonidet P-40 adjusted to 1 m NaCl (Fig. 1A). This straightforward protocol effectively enriched a number of chromatin associated protein complexes over background, on the order of hundreds of nanograms of purified material (Fig. 2A, supplemental Fig. S1A–S1C). By gel and iBAQ analysis, we estimate this strategy isolates stable, specific interactors with >95% purity. We were also able to efficiently cross-link purified proteins on-beads directly following washing steps using the well-characterized BS3 cross-linker in a borate-buffered saline buffer (Fig. 2A).
Fig. 1.
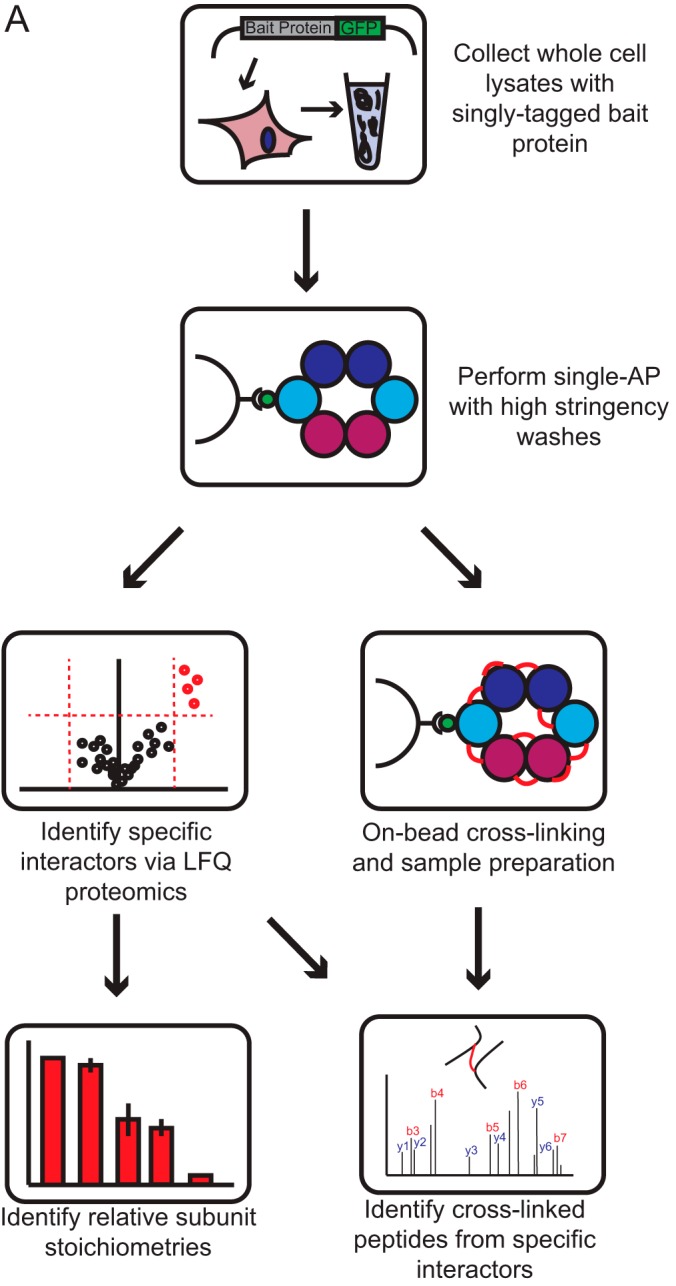
xIP-MS: a workflow for the analysis of protein complex topologies.
Fig. 2.

High stringency washes effectively purify protein complexes for on-bead cross-linking. A, BS3- lanes represent one half of an xIP-MS experiment using 1 ml of cell lysate. The second half was resuspended in cross-linking buffer for on-bead cross-linking as described in text, and is shown in the BS3+ lanes. All lanes were resolved on the same gel at the same exposure; black lines indicated where lanes were cropped together for visibility.
Analysis of cross-linked peptides requires a database search that expands exponentially with the number of peptides included. Such analysis becomes more computationally expensive and less sensitive as the number of proteins included increases. Therefore, we used a standard targeted method of defining a specific database of high-probability complex members for cross-link searching. In our case, this involved identification of stable, specific interactors by LFQ analysis (22, 39). These high-confidence interactors were then included in our cross-link database for targeted searching (Fig. 1A). This approach is justified on two bases; first, cross-linked peptides are already low-abundance in the pool of total peptides, therefore cross-linked peptides from highly enriched, specific interactors are substantially more likely to be measured than those from low-abundance, nonspecific proteins. Second, we used a filtering strategy that effectively removed spurious identifications to background peptides and specifically identified cross-linked samples. We filtered out those cross-links where either the identified peptide was shorter than five residues or longer than 40 residues. This selectively removed matches where one long, high scoring peptide compensated for a short, ambiguous partner in the scoring function. We then additionally filtered out those cross-link identifications that were not observed in both duplicate experiments, thus selectively removing remaining spurious matches (supplemental Fig. S2A, Fig. S3A). To test the specificity of this filtering procedure, we analyzed duplicate LFQ AP-MS/MS runs (without cross-linking) against a database of specific LFQ interactors for each of five baits used in this study. These samples were highly enriched for peptides of specific interactors, yet were not cross-linked and were thus negative controls. We identified only two spurious cross-link matches across all five analyses using this filtering, neither of which were identified as positive identifications when analyzing cross-linked samples, strongly indicating this filtering effectively precludes false positives. We therefore concluded that our high-stringency purification conditions obtained protein complexes with sufficient purity to allow the confident identification of low abundance cross-linked peptides.
Structural Benchmarking Against the PRPP Complex
We used PRPS1-GFP to structurally benchmark xIP-MS against the PRPP complex, a multimeric enzyme associated with nucleotide synthesis, which has a known crystal structure (PDB: 2h06) (31). For the PRPP complex, we identified the bait protein, PRPS1, as well as two associated proteins, PRPSAP1 and PRPSAP2, as significant interactors by LFQ analysis (Fig. 3A). We then used iBAQ values from our LFQ experiment to calculate relative stoichiometries for each subunit to the bait (Fig. 3B). Using xIP-MS, we were able to identify 54 reproducible cross-linked sites between these proteins (Fig. 3C). Importantly, we were able to map all of our unambiguous PRPS1 cross-linked sites onto the hexameric PRPS1 crystal structure within a distance constraint of 34 Å (Fig. 3D showing PRPS1 cross-links and supplemental Fig. S4A–S4B showing all PRPS1 and PRPSAP1/2 cross-links). The distribution of identified cross-links distances for PRPS1 was statistically distinct from the distribution of total distances and was thus structurally informative (Fig. 3E). This provides an important structural verification that xIP-MS provides valid cross-link identifications.
Fig. 3.

xIP-MS analysis of PRPP reveals high confidence distance constraints. A, LFQ analysis of PRPS1 was performed with triplicate pulldowns. Outliers are indicated in red, and background binders are indicated in black. Outlier cutoffs were drawn such that no proteins were present in the non-GFP quadrant on the volcano plot. B, Stoichiometry analysis was performed as described. All ratios are calculated by setting the bait (PRPS1) equal to 1. Error bars indicate standard deviations from triplicate samples. C, Identified cross-links for PRPP. The cross-link map was drawn in xiNet. Purple lines indicate self-links, and green lines indicate interprotein cross-links. Ambiguous cross-links (the same peptide in multiple proteins) are indicated by dashed lines. Homotypic cross-links (the same peptide cross-linked to itself, indicating multimerization) are drawn in red. Uniprot annotated domains are colored variously along the protein sequence bar. D, PRPS1 cross-links mapped onto the PDB: 2h06 crystal structure. Cross-links under 34 Å are colored in blue. PRPS1 monomers are colored in light gray. The silhouette of the PRPS1 holo-hexamer surface is shown in transparent gray.
A Structural Ensemble of PRPP Complexes
PRPS1 has a canonical catalytically active hexameric form composed of three PRPS1 dimers. The crystal structures of PRPSAP1 (PDB: 2c4k, RMSD: 0.901 Å with PRPS1) and PRPSAP2 (PDB: 2JI4, RMSD: 0.889 Å with PRPS1) are also known and indicate high structural homology with PRPS1. PRPSAP1 and PRPSAP2 were observed at stoichiometries of 7–8% relative to PRPS1, indicating either auxiliary subunit is present at slightly lower than one copy per two PRPS1 hexamers (Fig. 3B). We therefore considered whether our cross-linking data could support the presence of an in vivo structural ensemble of PRPP complexes, where PRPSAP1 and PRPSAP2 switch positions with canonical PRPS1 subunits within a single PRPP hexamer. This possibility is particularly intriguing considering the reported role of PRPSAP1 in negative regulation of PRPP catalytic function (52). Their high structural homology enabled high-confidence structural alignment of PRPSAP1 or PRPSAP2 with PRPS1 subunits, and we were able to map all 51 of our unambiguous cross-linked sites onto either PRPS1, PRPS1-PRPSAP1 (6 interlinks), PRPS1-PRPSAP2 (2 interlinks), or PRPSAP1-PRPSAP2 (2 interlinks) subcomplexes within a distance constraint of 34 Å (supplemental Fig. S4A–S4B). High structural homology and our cross-linking data thus suggest an ensemble model of PRPP hexamers, potentially offer a functional explanation for PRPSAP1's negative regulatory function of PRPP, and, again, indicate the structural validity of high confidence, reproducible xIP-MS cross-link identifications.
A Model for Cohesin Head Interactions
We next applied xIP-MS to the topological analysis of chromatin-associated protein complexes. Cohesin is a dynamic and structurally intriguing protein complex involved in maintaining higher order chromosome structure, most canonically during sister chromatid separation at mitosis. We were able to effectively purify the core pentameric cohesin complex using SMC1A-GFP as a bait (Fig. 2A, Fig. 4A). We purified SMC1A and SMC3 at essentially a 1:1 stoichiometric ratio, consistent with their known dimeric interaction implicated in flexibly encircling chromosomes (Fig. 4B). However, we identified cohesin accessory proteins (RAD21, STAG1/2, PDS5B, and WAPAL) at significantly lower stoichiometric ratios to the core cohesin dimer (Fig. 4B). This could indicate lower stoichiometric ratios in vivo, higher dynamism in bait interactions or lower bait affinity, cell cycle effects, or sensitivity to washing conditions. We observed numerous cross-links within the SMC1A and SMC3 head domains (Fig. 4C). This data is therefore complementary to previous studies, which both observed cross-linking predominantly between the coiled-coils separating SMC1A and SMC3 head and hinge domains, potentially because of different protein extraction methods or different cross-linking buffer conditions (11, 12). We conjectured that our cross-linking data would provide direct experimental support for a model of the human SMC1A/SMC3 head interaction derived from available crystal structures. Therefore, we modeled the human SMC1A and SMC3 head regions using crystal structures from their yeast homologs (PDB: 1w1w and PDB: 4ux3, respectively) (53, 54). We then aligned our SMC1A model with a dimeric SMC3 head from the 4ux3 crystal structure (Fig. 4D, supplemental Fig. S5A). Similarly, we modeled the SMC1A and SMC3 hinge regions from their respective chains in the mouse homolog (PDB: 2wd5) (supplemental Fig. S5B) (55). In sum, we found that 24/26 mappable cross-links were within a distance constraint of 34 Å using these models, a number of which sat directly at the SMC1A/SMC3 head interface, indicating that this straightforward modeling approach captures many architectural features of the human cohesin head interaction (Fig. 4D, supplemental Fig. S5C). Using a similar homology modeling approach with prokaryote and archaea SMC homologs resulted in an increased number of violations, therefore indicating eukaryotic cohesin structures represent the best available model for the human complex (supplemental Fig. S6A) (56). Therefore, xIP-MS provides cross-linking data that complements previous XL-MS studies and directly supports a model of the human cohesin head domain.
Fig. 4.

xIP-MS suggests a model for human cohesin head interactions. A, LFQ analysis identifies canonical cohesin tetrameric subunits as interactors of SMC1A. Data is plotted as described previously. B, SMC1A and SMC3 interact at a near 1:1 stoichiometry as indicated by iBAQ values. C, Cross-links between SMC1A and SMC3 preferentially localize toward the head domains, indicated in green on the protein sequence bars. D, Homology modeling allows mapping of SMC1A and SMC3 cross-links onto models for the human proteins. SMC1A is colored light gray, and SMC3 is colored in dark gray. Cross-links below 34 Å are shown in blue, and cross-links above 34 Å are shown in red. The silhouette represents the surface of the SMC3 dimer from the PDB: 4ux3 crystal structure used for aligning the SMC1A and SMC3 models.
Conformational Flexibility in XRCC5/6
Because xIP-MS is efficient enough to facilitate the use of replicates to identify very high-confidence cross-linked sites, we sought to broaden the applications of xIP-MS by using it to study potential conformational changes in flexible proteins. Such studies are typically difficult to interpret because cross-links identified in flexible regions might exceed static spatial constraints and therefore be disregarded as false. However, identification of such cross-links in multiple independent experiments substantially increases confidence in their structural validity and points toward conformational flexibility. Toward this end, we performed xIP-MS on the XRCC5/6 DNA damage repair complex (32). XRCC5/6 has a flexible SAP domain thought to be involved in conformational shifts involved in DNA recognition and binding at double-strand breaks (DSBs) (32, 33). Indeed, the SAP domain is resolved in the XRCC5/6 heterodimer without DNA bound, though a presumably flexible linker region, residues 539–558, is not resolved (PDB: 1jeq). Moreover, the SAP domain is not resolved in the XRCC5/6 crystal structure with DNA bound, further indicating the functional flexibility of this region in DNA contact (PDB: 1jey). We were able to effectively purify the XRCC5/6 heterodimer at nearly 1:1 stoichiometry using xIP-MS (Fig. 2A, Fig. 5A–5B). We observed numerous cross-links within the XRCC5/6 heterodimer (Fig. 5C). Although ∼84% of our identified cross-linked seemed spatially valid, we observed a few reproducible cross-linked sites that exceeded a spatial constraint of even 40 Å (supplemental Fig. S7A–S7B). These cross-links we deemed excessive violations, potentially indicative of conformational flexibility. Upon closer inspection, we realized these cross-links generally correlate with regions of high B-factor in the crystal structure and could be explained by two major conformation shifts (Fig. 5D, supplemental Fig. S7A). First, the DNA-binding Ku domain loop appears to possess substantial flexibility in the vertical axis (supplemental Fig. S7A). This could be a consequence of cross-linking in solution in the absence of DNA, and may represent native flexibility of the unbound dimer. We also observed cross-links connecting the SAP domain with the Ku domain, indicating a conformational shift where the flexible SAP domain approaches the Ku domain DNA binding ring. Analysis of normal modes further supported substantial flexibility in the Ku and SAP domains (supplemental Fig. S7C, supplemental Video S1). Intriguingly, such conformational shifts could be potentially indicative of allowed functional motion involved in stabilizing and capping DNA DSB ends between the Ku and SAP domains (Fig. 5D). Therefore, xIP-MS experiments provide potential functional insight into conformational changes involved in XRCC5/6 DNA-binding action. This highlights the ability of xIP-MS to provide high-confidence cross-link identifications relevant in the study of protein flexibility and conformational states. Thus, importantly, xIP-MS complements traditional crystallographic structural studies, which reveal only static conformational states.
Fig. 5.

xIP-MS reveals conformational changes in XRCC5/6. A, LFQ identifies XRCC5/6 as robust interactors, in agreement with our gel analysis. B, XRCC5/6 stoichiometry is nearly 1:1 as indicated by iBAQ values. C, Cross-linking within the XRCC5/6 heterodimer. The cross-link map is colored as described previously. D, XRCC5/6 are colored by B-factor as indicated on the key; the protein backbone radii are scaled similarly by size. Cross-links within 40 Å are shown in blue, and cross-links longer than this threshold are shown in red. Major conformational shifts explaining observed cross-linking patterns are displayed as yellow arrows. The structure of PDB: 1jeq is used to display cross-links as the SAP domain is resolved only in this structure; however, the surface of the DNA structure bound to XRCC5/6 from PDB: 1jey is displayed in transparent yellow, after alignment with 1jeq using MatchMaker in UCSF Chimera.
Topology of Cell-cycle Independent MCM Subcomplex Interactions
Because xIP-MS is a straightforward protocol requiring relatively small volumes of cell lysates, we sought to show that xIP-MS could methodologically compare protein topologies under states of perturbation. Traditionally, performing comparative cross-linking experiments has been difficult because of the typical requirement of tens or hundreds of micrograms of protein at high purity. However, we were easily able to perform xIP-MS experiments in duplicate qualitatively comparing previously observed soluble MCM complexes in asynchronous and S-phase blocked cells (37). MCM is activated during DNA replication and loaded onto processive replication forks; therefore, we expected MCM subcomplex topologies might change dramatically in association with MCM activation during S-phase. However, we noted similar banding patterns by gel analysis, similar interactors via LFQ proteomics, and similar relative stoichiometries for interactors in both cell cycle states after MCM6-GFP purification (Fig. 2A, Fig. 6A–6F, supplemental Fig. S8A). Furthermore, we observed cross-linking between MCM2/4/6/7 subcomplexes (Fig. 6B, 6D). Our gel analysis indicated three predominant cross-linked species present in both states (Fig. 2A). Our xIP-MS analysis showed a high degree of overlap between cross-linked sites observed in both cell cycle states, indicating soluble MCM complexes are topologically cell-cycle invariant (Fig. 6F). Therefore, our data suggests a sequential model of MCM subcomplex assembly, where ubiquitous soluble MCM subcomplexes are assembled during loading onto processive replication forks during S-phase (34–36). Our data also suggests these soluble MCM subcomplexes may be in vast excess of active replication forks, as they do not seem to be depleted from the soluble fraction during S-phase (Fig. 2A). Our xIP-MS data additionally provides structural insights into MCM subunit interfaces within these subcomplexes. Finally, these experiments indicate the ease with which xIP-MS can be applied to studying protein complex topologies under a variety of possible cellular or molecular perturbations.
Fig. 6.

xIP-MS reveals S-phase independent MCM subcomplex topologies. A, MCM2/4/6/7, MCM10, and MCMBP are observed as interactors with MCM6 in asynchronous cells. B, Cross-linking analysis reveals interprotein contacts within soluble MCM complexes in asynchronous cells. C, Similarly, MCM2/4/6/7, MCM10, and MCMBP are observed as interactors with MCM6 in S-phase blocked cells. D, Cross-linking analysis reveals a similar topology in soluble S-phase MCM complexes. E, Relative stoichiometries to the bait as indicated by iBAQ values show MCM subunit stoichiometries do not change substantially when cells are blocked in S-phase. F, Venn diagram indicates substantial overlap of cross-linked sites between MCM subunits after S-phase block.
DISCUSSION
Chemical cross-linking coupled with mass spectrometry is becoming an increasingly popular technique for studying large or difficult protein complex architectures and topologies at low resolution. XL-MS is a particularly important tool for interrogating chromatin-associated protein complexes, which are often highly dynamic, and subject to large conformational shifts when enacting their functions. However, although affinity purification based strategies are commonly coupled with mass spectrometry to identify specific protein interactions, typical XL-MS workflows have traditionally demanded large (tens or hundreds of micrograms) amounts of pure, homogenous complex coupled with time-consuming enrichment steps to facilitate detection of cross-linked peptides over background. The difficulty of implementing and optimizing complex cross-linking workflows has thus limited their widespread adoption. In general, XL-MS workflows are scaling up toward proteome-wide studies often using cleavable cross-linkers, or scaling down toward computational prediction of protein structures with high resolution using broader-specificity or photo-activatable cross-linkers (16, 57). We envision xIP-MS instead as a workflow that could bridge the gap between proteome-wide and protein-specific XL-MS workflows, offering peptide-resolution architectural and topological information with the ease of coIP experiments particularly in the context of comparative cross-linking workflows (58–61). This study establishes xIP-MS as an additional tool for molecular and structural biologists, offering a flexible and robust methodological platform adaptable to a variety of diverse applications.
It is worth noting that the xIP-MS protocol as presented is designed to enrich stable core complexes with high efficiency and purity from relatively small lysate volumes. The high stringency washing steps necessitated by this workflow can preclude highly dynamic or transient interactions between even stoichiometric interactors. Similarly, salt sensitive interactions may be impeded by xIP-MS. As a general rule, specific purification conditions may be required for different complexes; this should be taken into consideration for the complex in question. However, the workflow we present in this study represents a generally effective starting point, as we have shown by applying xIP-MS successfully to a number of different protein complexes with different downstream applications, including: heterogeneous assemblies, homology modeling, structural dynamics, and comparative cross-linking. xIP-MS therefore represents a simplified and flexible alternatative to existing on-bead cross-linking methodologies.
Although on-bead cross-linking workflows offer many experimental benefits, one notable drawback to on-bead cross-linking workflows is the potential to cross-link purified proteins to the bead itself, potentially facilitating misidentifications. Here, we use a small GFP nanobody (12 kDa) with stringent data filtering to decrease the likelihood of spurious identifications as much as possible. However, alternate strategies include Ni-NTA purification for His-tagged proteins as Ni-NTA is a non-amine containing substrate, using a membrane permeable cross-linker prior to cell lysis and purification, or using engineered high-affinity, lysine-free nanobodies (15, 16, 27). In general, careful experimental design and data analysis should minimize the risk of false positive identifications from on-bead cross-linking workflows.
xIP-MS also offers much promise as a functional follow up to more traditional structural workflows. Because xIP-MS is sufficiently straightforward to allow multiple replicates, high confidence, reproducible cross-linking with xIP-MS can facilitate structural dynamics analysis as we have shown for XRCC5/6. Importantly, perturbation experiments now become substantially streamlined, indicated by our analysis of cell cycle invariance in soluble MCM subcomplexes. Quantitative cross-linking with isotopic linkers offers great potential for comparing conformational states under perturbations, and it is possible xIP-MS could contribute toward further developments in quantitative cross-linking workflows (58–61).
It is likely developments in protein purification reagents, novel cross-linking chemistries and softwares, and faster, more sensitive instruments will improve xIP-MS workflows in the future. For example, xIP-MS would benefit directly from novel ultrahigh-affinity GFP nanobody dimers, as reported recently (62). Also, developments in enrichable or cleavable cross-linkers could facilitate higher cross-link identification rates (16, 20, 63). Therefore, xIP-MS can also be considered as an experimental platform for rapid optimization of novel cross-linking strategies, thereby extending the toolbox of structural proteomics technologies.
Supplementary Material
Footnotes
Author contributions: M.M.M. and M.V. designed research; M.M.M., E.W., P.W.J., and M.V. performed research; M.M.M. analyzed data; M.M.M. and M.V. wrote the paper.
* This work is supported by M.M. and M.V. received funding for this project from the Marie Curie FP7 DevCom ITN.
 This article contains supplemental Figs. S1 to S8, Tables S1 to S3 anf Video S1.
This article contains supplemental Figs. S1 to S8, Tables S1 to S3 anf Video S1.
1 The abbreviations used are:
- XL-MS
- Cross-linking Mass Spectrometry
- xIP-MS
- Cross-linking Immunoprecipitation Mass Spectrometry
- coIP
- Co-immunoprecipitation
- PRPP
- phosphoribosyl pyrophosphate synthetase complex
- SMC
- structural maintenance of chromosomes
- XRCC
- X-ray repair cross-complementing protein
- MCM
- mini-chromosome maintenance
- LFQ
- label free quantification
- DSB
- double-strand breaks
- PTM
- post-translational modification
- GFP
- green fluorescent protein
- SCX
- strong cation exchange
- SEC
- size exclusion chromatography.
REFERENCES
- 1. Mann M., Hendrickson R. C., and Pandey A. (2001) Analysis of proteins and proteomes by mass spectrometry. Ann. Rev. Biochem. 70, 437–473 [DOI] [PubMed] [Google Scholar]
- 2. Gingras A. C., Gstaiger M., Raught B., and Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 [DOI] [PubMed] [Google Scholar]
- 3. Dunham W. H., Mullin M., and Gingras A. C. (2012) Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics 12, 1576–1590 [DOI] [PubMed] [Google Scholar]
- 4. Rappsilber J. (2011) The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Structural Biol. 173, 530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sinz A. (2006) Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrometry Rev. 25, 663–682 [DOI] [PubMed] [Google Scholar]
- 6. Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., and Aebersold R. (2010) Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics 9, 1634–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen V. Q., Ranjan A., Stengel F., Wei D., Aebersold R., Wu C., and Leschziner A. E. (2013) Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. Cell 154, 1220–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tosi A., Haas C., Herzog F., Gilmozzi A., Berninghausen O., Ungewickell C., Gerhold C. B., Lakomek K., Aebersold R., Beckmann R., and Hopfner K. P. (2013) Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell 154, 1207–1219 [DOI] [PubMed] [Google Scholar]
- 9. Ciferri C., Lander G. C., Maiolica A., Herzog F., Aebersold R., and Nogales E. (2012) Molecular architecture of human polycomb repressive complex 2. Elife 1, e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han Y., Luo J., Ranish J., and Hahn S. (2014) Architecture of the Saccharomyces cerevisiae SAGA transcription coactivator complex. EMBO J. 33, 2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huis in 't Veld P. J., Herzog F., Ladurner R., Davidson I. F., Piric S., Kreidl E., Bhaskara V., Aebersold R., and Peters J. M. (2014) Characterization of a DNA exit gate in the human cohesin ring. Science 346, 968–972 [DOI] [PubMed] [Google Scholar]
- 12. Barysz H., Kim J. H., Chen Z. A., Hudson D. F., Rappsilber J., Gerloff D. L., and Earnshaw W. C. (2015) Three-dimensional topology of the SMC2/SMC4 subcomplex from chicken condensin I revealed by cross-linking and molecular modelling. Open Biol. 5, 150005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kloet S. L., Baymaz H. I., Makowski M., Groenewold V., Jansen P. W., Berendsen M., Niazi H., Kops G. J., and Vermeulen M. (2015) Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 282, 1774–1785 [DOI] [PubMed] [Google Scholar]
- 14. Nguyen-Huynh N. T., Sharov G., Potel C., Fichter P., Trowitzsch S., Berger I., Lamour V., Schultz P., Potier N., and Leize-Wagner E. (2015) Chemical cross-linking and mass spectrometry to determine the subunit interaction network in a recombinant human SAGA HAT subcomplex. Protein Sci. 24, 1232–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F. U., Ban N., Malmstrom L., and Aebersold R. (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry, Science 337, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 16. Kaake R. M., Wang X., Burke A., Yu C., Kandur W., Yang Y., Novtisky E. J., Second T., Duan J., Kao A., Guan S., Vellucci D., Rychnovsky S. D., and Huang L. (2014) A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol. Cell. Proteomics 13, 3533–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fritzsche R., Ihling C. H., Gotze M., and Sinz A. (2012) Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun. Mass Spectrom. 26, 653–658 [DOI] [PubMed] [Google Scholar]
- 18. Leitner A., Reischl R., Walzthoeni T., Herzog F., Bohn S., Forster F., and Aebersold R. (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteomics 11, M111 014126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leitner A., Joachimiak L. A., Unverdorben P., Walzthoeni T., Frydman J., Forster F., and Aebersold R. (2014) Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc Natl Acad Sci U.S.A. 111, 9455–9460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nature Methods 12, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 21. Eberl H. C., Mann M., and Vermeulen M. (2011) Quantitative proteomics for epigenetics. Chembiochem 12, 224–234 [DOI] [PubMed] [Google Scholar]
- 22. Smits A. H., Jansen P. W., Poser I., Hyman A. A., and Vermeulen M. (2012) Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. gks941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keilhauer E. C., Hein M. Y., and Mann M. (2015) Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol. Cell. Proteomics 14, 120–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poser I., Sarov M., Hutchins J. R., Hériché J.-K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A. W. Pelletier L., Kittler R., Hua S., Naumann R., Augsburg M., Sykora M. M., Hofemeister H., Zhang Y., Nasmyth K., White K. P., Dietzel S., Mechtler K., Durbin R., Stewart A. F., Peters J. M., Buchholz F., and Hyman A. A. (2008) BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nature Methods 5, 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hubner N. C., Bird A. W., Cox J., Splettstoesser B., Bandilla P., Poser I., Hyman A., and Mann M. (2010) Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 189, 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baymaz H. I., Spruijt C. G., and Vermeulen M. (2014) Identifying Nuclear Protein–Protein Interactions Using GFP Affinity Purification and SILAC-Based Quantitative Mass Spectrometry in Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) pp. 207–226, Springer. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y., Pellarin R., Fridy P. C., Fernandez-Martinez J., Thompson M. K., Li Y., Wang Q. J., Sali A., Rout M. P., and Chait B. T. (2015) A strategy for dissecting the architectures of native macromolecular assemblies. Nature Methods 12, 1135–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Forster F., Beck M., and Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nature Methods 9, 901–903 [DOI] [PubMed] [Google Scholar]
- 29. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang B., Wu Y.-J., Zhu M., Fan S.-B., Lin J., Zhang K., Li S., Chi H., Li Y.-X., and Chen H.-F. (2012) Identification of cross-linked peptides from complex samples, Nature Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 31. Li S., Lu Y., Peng B., and Ding J. (2007) Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 401, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walker J. R., Corpina R. A., and Goldberg J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412, 607–614 [DOI] [PubMed] [Google Scholar]
- 33. Rivera-Calzada A., Spagnolo L., Pearl L. H., and Llorca O. (2007) Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 8, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coué M., Amariglio F., Maiorano D., Bocquet S., and Méchali M. (1998) Evidence for Different MCM Subcomplexes with Differential Binding to Chromatin inXenopus. Exp. Cell Res. 245, 282–289 [DOI] [PubMed] [Google Scholar]
- 35. Maiorano D., Lemaitre J.-M., and Méchali M. (2000) Stepwise regulated chromatin assembly of MCM2–7 proteins. J. Biol. Chem. 275, 8426–8431 [DOI] [PubMed] [Google Scholar]
- 36. Prokhorova T. A., and Blow J. J. (2000) Sequential MCM/P1 subcomplex assembly is required to form a heterohexamer with replication licensing activity. J. Biol. Chem. 275, 2491–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moyer S. E., Lewis P. W., and Botchan M. R. (2006) Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. U.S.A. 103, 10236–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 39. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J. Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Computational Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 42. Combe C. W., Fischer L., and Rappsilber J. (2015) xiNET: cross-link network maps with residue resolution. Mol. Cell. Proteomics 14, 1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kosinski J., von Appen A., Ori A., Karius K., Müller C. W., and Beck M. (2015) Xlink Analyzer: Software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J. Structural Biol. 189, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Z., Lasker K., Schneidman-Duhovny D., Webb B., Huang C. C., Pettersen E. F., Goddard T. D., Meng E. C., Sali A., and Ferrin T. E. (2012) UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Structural Biol. 179, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meng E. C., Pettersen E. F., Couch G. S., Huang C. C., and Ferrin T. E. (2006) Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 7, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rice P., Longden I., and Bleasby A. (2000) EMBOSS: the European molecular biology open software suite. Trends Genetics 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 47. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., and Bordoli L. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res., 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bakan A., Meireles L. M., and Bahar I. (2011) ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Humphrey W., Dalke A., and Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33- 8:27–8, 1996 [DOI] [PubMed] [Google Scholar]
- 51. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kita K., Ishizuka T., Ishijima S., Sonoda T., and Tatibana M. (1994) A novel 39-kDa phosphoribosylpyrophosphate synthetase-associated protein of rat liver. Cloning, high sequence similarity to the catalytic subunits, and a negative regulatory role. J. Biol. Chem. 269, 8334–8340 [PubMed] [Google Scholar]
- 53. Haering C. H., Schoffnegger D., Nishino T., Helmhart W., Nasmyth K., and Löwe J. (2004) Structure and stability of cohesin's Smc1-kleisin interaction. Mol. Cell. 15, 951–964 [DOI] [PubMed] [Google Scholar]
- 54. Gligoris T. G., Scheinost J. C., Bürmann F., Petela N., Chan K.-L., Uluocak P., Beckouët F., Gruber S., Nasmyth K., and Löwe J. (2014) Closing the cohesin ring: Structure and function of its Smc3-kleisin interface. Science 346, 963–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kurze A., Michie K. A., Dixon S. E., Mishra A., Itoh T., Khalid S., Strmecki L., Shirahige K., Haering C. H., Löwe J., and Nasmyth K. (2011) A positively charged channel within the Smc1/Smc3 hinge required for sister chromatid cohesion. EMBO J. 30, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burmann F., Shin H. C., Basquin J., Soh Y. M., Gimenez-Oya V., Kim Y. G., Oh B. H., and Gruber S. (2013) An asymmetric SMC-kleisin bridge in prokaryotic condensing. Nat. Struct. Mol. Biol. 20, 371–379 [DOI] [PubMed] [Google Scholar]
- 57. Belsom A., Schneider M., Fischer L., Brock O., and Rappsilber J. (2015) Serum albumin domain structures in human blood serum by mass spectrometry and computational biology. Mol. Cell. Proteomics ePub article 10.1074/mcp.M115.048504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dimova K., Kalkhof S., Pottratz I., Ihling C., Rodriguez-Castaneda F., Liepold T., Griesinger C., Brose N., Sinz A., and Jahn O. (2009) Structural insights into the calmodulin-Munc13 interaction obtained by cross-linking and mass spectrometry. Biochemistry 48, 5908–5921 [DOI] [PubMed] [Google Scholar]
- 59. Schmidt C., and Robinson C. V. (2014) A comparative cross-linking strategy to probe conformational changes in protein complexes. Nature Protocols 9, 2224–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmidt C., Zhou M., Marriott H., Morgner N., Politis A., and Robinson C. V. (2013) Comparative cross-linking and mass spectrometry of an intact F-type ATPase suggest a role for phosphorylation. Nat. Commun. 4, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fischer L., Chen Z. A., and Rappsilber J. (2013) Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteomics 88, 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fridy P. C., Li Y., Keegan S., Thompson M. K., Nudelman I., Scheid J. F., Oeffinger M., Nussenzweig M. C., Fenyö D., and Chait B. T. (2014) A robust pipeline for rapid production of versatile nanobody repertoires. Nature Methods 11, 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Petrotchenko E. V., Serpa J. J., and Borchers C. H. (2011) An isotopically coded CID-cleavable biotinylated cross-linker for structural proteomics. Mol. Cell. Proteomics 10, M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.