

Abstract
The centromere is the locus on the chromosome that acts as the essential connection point between the chromosome and the mitotic spindle. A histone H3 variant, CENP-A, defines the location of the centromere, but centromeric chromatin consists of a mixture of both CENP-A-containing and H3-containing nucleosomes. We report a surprisingly uniform pattern of primarily monomethylation on lysine 20 of histone H4 present in short polynucleosomes mixtures of CENP-A and H3 nucleosomes isolated from functional centromeres. Canonical H3 is not a component of CENP-A-containing nucleosomes at centromeres, so the H3 we copurify from these preparations comes exclusively from adjacent nucleosomes. We find that CENP-A-proximal H3 nucleosomes are not uniformly modified but contain a complex set of PTMs. Dually modified K9me2-K27me2 H3 nucleosomes are observed at the centromere. Side-chain acetylation of both histone H3 and histone H4 is low at the centromere. Prior to assembly at centromeres, newly expressed CENP-A is sequestered for a large portion of the cell cycle (late S-phase, G2, and most of mitosis) in a complex that contains its partner, H4, and its chaperone, HJURP. In contrast to chromatin associated centromeric histone H4, we show that prenucleosomal CENP-A-associated histone H4 lacks K20 methylation and contains side-chain and α-amino acetylation. We show HJURP displays a complex set of serine phosphorylation that may potentially regulate the deposition of CENP-A. Taken together, our findings provide key information regarding some of the key components of functional centromeric chromatin.
Centromeric location is defined not by a particular DNA sequence but by the presence of a centromere specific nucleosome that contains centromere protein-A (CENP-A)1 in place of histone H3 (1). Human centromeres span megabases of DNA and are typically embedded within repetitive α-satellite DNA elements (2). The location of the CENP-A nucleosome is sufficient to determine the site of centromere formation and kinetochore assembly during mitosis (3). Centromere location is epigenetically maintained by a pathway for nucleosome assembly where deposition of nascent CENP-A (and its partner histone, H4) is mediated by a specific histone chaperone, HJURP (5, 41). Existing CENP-A nucleosomes directly recruit CENP-C, which in turn recruits the Mis18 complex during mitotic exit (6, 7). Mis18 binding recruits HJURP and directs nascent CENP-A nucleosome assembly at an adjacent site (8). Despite the essential presence of the CENP-A nucleosome for centromere formation and maintenance, the centromere-specific nucleosomes are present within a context of chromatin containing post-translational modifications (PTMs) of histones that are thought to be important for centromere function, as outlined below.
The relatively small centromeres of fission yeast and flies are organized with a central CENP-A-containing, kinetochore-forming region that is functionally distinct from and surrounded on both sides by pericentric heterochromatin (9, 10). Pericentric heterochromatin is characterized by a particular set of histone H3 modifications, including H3K9me3, which mediate the formation of repressive chromatin (11). In S. pombe, H3K9me2/3 nucleosomes occupy the dg and dh repeats found outside of the central core of the centromere. The arrangement of H3 PTMs relative to the centromeric nucleosome has been more difficult to assess in human centromeres where the repetitiveness of the DNA hampers the utility of ChIP to define the precise sites of CENP-A and modified H3. This arrangement of methylated H3K9 appears to be preserved based on results from chromatin stretching experiments on centromeres containing typical repetitive DNA (12–14). It should be noted, however, that nonrepetitive human neocentromeres (where ChIP approaches are very useful) are not generally highly enriched for methylated H3K9 (15) suggesting a nonessential role for this mark in chromosome inheritance.
In three-dimensions, many copies of CENP-A-containing nucleosomes cluster in what becomes the surface of centromeric chromatin that forms the massive proteinaceous complex, the kinetochore, which physically attaches to spindle microtubules in mitosis. There are ∼200 CENP-A nucleosomes per typical human centromere (16). CENP-A nucleosomes are homotypic in nature (17–20). The CENP-A and histone H4 subunits of the nucleosome, where we have focused our proteomic efforts, are extremely stable subunits, with H2A and H2B exchanging more rapidly (21, 22). Chromatin stretching experiments show that along a linear DNA derived from interphase nuclei, patches of CENP-A are found interspersed with patches of H3 nucleosomes (10, 14). Indeed, at human centromeres most α-satellite repeats are occupied by H3 nucleosomes, with CENP-A nucleosomes only assembled on a small fraction of them (23). All of this suggests that multiple CENP-A domains are organized together to assemble the mitotic centromere, along with adjacent H3 nucleosomes that are proposed to contribute to centromere function (24). Adjacent H3 nucleosomes also provide the sites for replacement of nascent CENP-A nucleosomes in current models for how centromere location is propagated through HJURP-mediated chromatin assembly in the G1 phase of the cell cycle (25).
Analysis of the PTMs of histone H3 in stretched chromatin fibers shows considerable juxtaposition of CENP-A with the euchromatic marks H3K4me2 and H3K36me2; however, acetylation of histones H3 and H4, which is generally associated with actively transcribed euchromatin, is reported to be in low abundance at centromeres (13, 26, 27). H4K20me1 occurs on CENP-A nucleosomes (28). The PTMs observed thus far on histones at human centromeres indicate that these domains are neither purely heterochromatic nor purely euchromatin. This unique blend of repressive and permissive histone marks has been dubbed “centrochromatin” to reflect this specialized form of chromatin found at centromeres (14).
Nucleosomes modified by H3K36 and H3K4 methylation accumulate in centromere sequences of active human artificial chromosomes (13). Altering the modification state of chromatin within the context of human artificial chromosomes (HACs) by altering transcription or targeting of the LSD1 lysine demethylase has significant effects on centromere specification and the fidelity of segregation. This phenomenon suggests that the modification state of the H3 nucleosomes within the centromere is important for centromere function (13, 29, 30).
Staining stretched chromatin fibers with histone PTM-specific antibodies has allowed the localization of various known epigenetic PTM marks on H3 and H4 relative to CENP-A. To increase resolution using advanced chromatin-optimized proteomics, we undertook an unbiased approach to identify the histone PTMs that are closely associated with CENP-A, likely within the CENP-A nucleosome or present in neighboring nucleosomes. Previously, we used affinity purification and mass spectrometry to show that CENP-A molecules are phosphorylated at serines 16 and 18, and amino-terminally trimethylated (31). Here, using this biochemical system to purify specific populations of CENP-A, we identify and measure the levels of the PTMs on the proximal binding partner proteins, histone H3, H4, and HJURP. We examined two specific sample types: H3 and H4 copurifying with CENP-A-containing small polynuclosomes (i.e. predominantly mono-, di-, and tri-nucleosomes) from asynchronously dividing cells (Asynchronous Nucleosomal sample) and H4 and HJURP copurifying with CENP-A from soluble extracts from mitotically-arrested cells (Mitotic Prenucleosomal sample).
EXPERIMENTAL PROCEDURES
Stable Cell Culture, Nocodazole Treatment, and Affinity Purification of CENP-A-LAP
Stable HeLa S3 cell lines expressing CENP-A tagged with a localization and affinity purification (LAP) tag were established, as described previously (18, 31). HeLa S3 cells expressing CENP-A-LAP were arrested in mitosis using 400 ng/ml nocodazole treatment for 18 h preceding harvest. Stable-expressing CENP-A-LAP HeLa cells were dounce-homogenized on ice in a cell lysis buffer containing 3.75 mm Tris (pH = 7.5), 20 mm KCl, 0.5 mm EDTA, 0.5 mm DTT, 0.05 mm spermidine, 0.125 mm spermine, 0.1% digitonin, 10 mg/ml protease inhibitor mixture (leupeptin, pepstatin, and chymostatin), 0.2 mm Na3VO4, 20 mm β-glycerophosphate, 10 mm NaC4H7O2. Nucleosomes were produced by MNase-digestion as described previously (18, 31). Nucleosomal and Prenucleosomal CENP-A-LAP were recovered from the respective fractions by incubating in wash buffer, 300 mm KCl, and 0.1% Tween-20, with anti-GFP-agarose for 2 h at 4 °C. Mock IP was conducted using the rabbit IgG. Nucleosomal CENP-A-LAP protein was eluted from beads by 1 h incubation with 14 μg of PreScission® protease in wash buffer, 300 mm KCl, and 0.1% Tween-20. Prenucleosomal CENP-A-LAP was eluted using similar conditions although 1 m NaCl was introduced prior to the elution with PreScission protease.
Sample Preparation, Liquid Chromatography, and Mass Spectrometry
Purified CENP-A and associated proteins from a single purification described above or from acid extracted histones (32) (n = 1) were precipitated using ice cold trichloroacetic acid (33% final volume) and were washed using ice cold 100% acetone. Precipitated protein pellets were dried by vacuum centrifugation and reconstituted with 100 μl of 0.1% RapiGest SF surfactant in 100 mm ammonium acetate, pH = 8.0. Resuspended proteins were reduced using 4 mm dithiothreitol, carbamidomethylated with 10 mm iodoacetamide, and digested using endoproteinases trypsin, LysC, GluC, or AspN (1:20, enzyme/protein) for 16 h at 20 °C. Trifluoroacetic acid was added to 0.1% total volume to quench enzymatic proteolysis and hydrolyze the RapiGest SF (Waters). Samples were centrifuged at 20,000 × g for 10 min to precipitate the hydrophobic RapiGest SF cleavage product. The supernatant was stored at −40 °C until analysis.
Digested protein samples were separated by nano-flow HPLC using in-house fabricated microcapillary fused silica columns (Polymicro Technologies, Phoenix AZ). Precolumns measured 75 μm in internal diameter and were packed with 4–5 cm length of 3 μm C4 Vydac beads (Grace, Columbia MD) or 6–8 cm of 15 μm beads (YMC). Analytical columns were 50 μm in internal diameter with packed 6–8 cm in length and were packed with 3 μm C4 Vydac beads. Analytical columns were laser-pulled (Sutter Instruments) to produce an electrospray ionization emitter 2–5 μm in width. Digested peptides sample (5–20 pmol) was mixed with angiotensin I peptide (DRVYIHPFHL, 100 fmol) and vasoactive intestinal peptide fragment (HSDAVFTDNYTR, 100 fmol) and pressure bomb-loaded onto a precolumn. After loading, pre- and analytical-columns were assembled and connected to an Agilent 1100-series HPLC in-line with an Orbitrap-based mass spectrometer (LTQ-FT, LTQ-Orbitrap, or Velos-Orbitrap; Thermo Fisher Scientific, Waltham MA). A gradient of 0–60% B in 120 min was used to elute peptides (Mobile phase A, 0.1% acetic acid in water; Mobile phase B, 0.1% acetic acid and 70% acetonitrile in water).
Electrospray ionization was performed using 2 kV applied through a liquid junction. The inlet heated capillary of each mass spectrometer was set at 200 °C. Data dependent analysis was performed as described previously (31). Data were acquired using an MS1 scan (m/z 300–2000) in the orbitrap (60 k resolution) or in the ion cyclotron resonance cell (100 k resolution). Ions were selected for isolation in the ion trap (3 m/z isolation width) and 6 ETD scans (30 ms reaction time using azulene radical anion) or 10 CID scans (35% normalized collision energy). To optimize data-dependent selection we used a dynamic exclusion of 30 s and a repeat count of 3.
Mass Spectrometry Data Analysis
MS2 peak lists were acquired using Thermo Fisher Scientific Xcalibur software (version 2.1) and searched against human entries (taxonomy id 9606) in the NCBI RefSeq database (June, 2012) using OMSSA (version 2.1.8) (33). Three missed cleavages were allowed for all searches, and each search was performed with the enzyme appropriate to the proteases used for sample digestion (trypsin, LysC, GluC, or AspN). For all searches, the precursor mass tolerance was set to ±0.05 Da, and the fragment mass tolerance was set to ±0.50 Da. Cysteine carbamidomethylation (Δ 57.021464 Da) was considered as a static modification. Variable modifications considered were acetylation (Δ 42.010565 Da) on K, N-terminal; mono-, di- and tri-methylations (Δ 14.015650 Da) as appropriate on K, R, N-terminal; oxidation (Δ 15.994915 Da) on M; phosphorylation (Δ 79.966331 Da) on S, T, Y. Identified peptides with the lowest E-Values (E-Value threshold set to 1.0) were considered for validation. Spectra were further inspected manually using Thermo Fisher Scientific Qual Browser software (version 2.1) for combinations of PTMs on histone H3, histone H4, and HJURP resulting from the specific enzymatic digests above. Peptides abundances were quantified by generating an extracted ion chromatogram (XIC) plot for each charge state observed and summing total peak areas. For peptides longer than 20 residues in length, Isotope Pattern Calculator software (Pacific Northwest National Laboratories, Richland WA, http://omics.pnl.gov/) was used to calculate the expected dominant isotopic peak (based on natural abundance of carbon as 1.070% 13C and nitrogen as 0.368% 15N) for plotting XICs. MS1 spectra of peptides from histones H3 and H4 were analyzed manually using a mass tolerance of 0–5 ppm to interpret and assign combinations of acetylation and methylation. For comparison of H3.1 A1-E50 N-terminal tail we measured the most abundant charge state (z = +11) of the isotope peak corresponding to the 13C x 3 (A+3) species. For H4 S1-R23 N-terminal tail we measured the most abundant charge state (z = +7 or +6) of the isotope peak corresponding to the 13C x 1 (A+1) species.
RESULTS
Nucleosomal CENP-A-bound H3.1 in Asynchronously Cycling Cells
Affinity purification of LAP-tagged CENP-A from chromatin that had been digested with MNase to produce primarily mono-, di- and tri-nucleosome stretches of chromatin was used identify the PTMs present on histone H3 nucleosomes that are interspersed among CENP-A nucleosomes at the centromere (Fig. 1A, 1B, supplemental Fig. S1). Using the affinity purification of CENP-A-containing nucleosomes described previously we observed that H3 protein is in nearly 1:1 stoichiometry with CENP-A in asynchronous nucleosomal samples (31) (Fig. 1C). The presence of histone H3 and H4 in CENP-A affinity purified fractions was verified by Western blot (Fig. 1D).
Fig. 1.
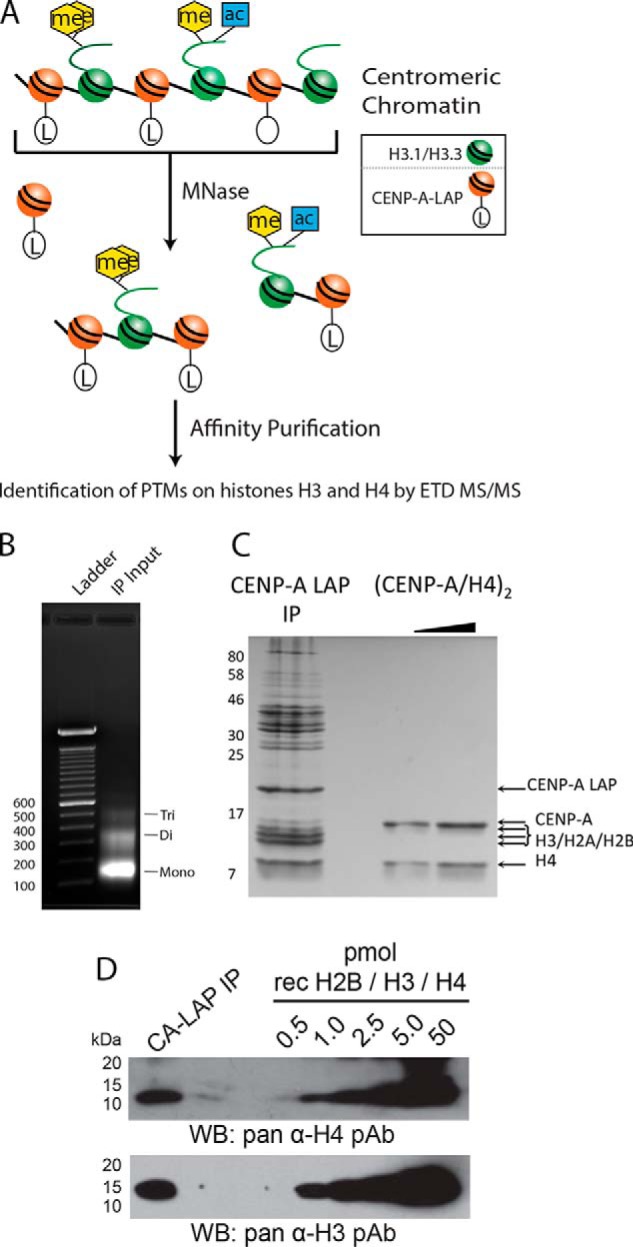
Copurification of histones H3 and H4 with affinity-tagged CENP-A in nucleosomal and prenucleosomal fractions. A, Affinity purification of CENP-A was used to identify the post-translational modifications present on centromeric histone H3 present in neighboring nucleosomes and H4 present in either neighboring nucleosomes or the CENP-A nucleosome itself by digestion of chromatin with MNase followed by affinity purification. B, Chromatin digested with Micrococcal Nuclease (MNase) to mostly Mono-, Di- and Tri- nucleosomes was used as the input for the anti-GFP affinity purificiation (IP). Ethidium bromide stained gel of purified DNA is shown. C, Coomassie stained gel of the CENP-A LAP IP shows that the CENP-A LAP nucleosomes are isolated as complexes, which include the core histones. Recombinant (CENP-A/H4)2 is shown for comparison. D, Western blots show quantitative amounts of histones H3 and H4 are present in CENP-A-LAP immunoprecipitated samples when compared with a mixture of recombinant histones.
The H3 tail contains several lysine and arginine residues. Lysine and arginine residues are typically no longer substrates for the trypsin or LysC proteases when they are post-translationally modified. Use of either of these endoproteinases allows recovery of variable-length H3 peptides that provide important information about combinations of PTMs present on the histone H3 tail. Unmodified arginines and lysines on the H3 N-terminal tail allow generation of trypsin- and LysC-derived peptides, which are too hydrophilic to be retained on reverse phase columns or are too small to be detected with the operating conditions of our mass spectrometer. The abundances of these modified H3 tail peptides reflect the relative abundances of specific PTM combinations present within the H3-tail, although the overall distribution of these modifications cannot be determined from these experiments. In the LysC and trypsin digests of the asynchronous nucleosomal CENP-A-LAP sample, many peptides from canonical H3 (H3.1) were observed (Fig. 2A). Data from both of these digests combined allowed us to cover 124 of the 134 residues, or 93% of the H3.1 sequence (Fig. 2A, 2B, supplemental Fig. S2). We identified varying degrees of methylation on lysine residues K4, K9, K27, K36, of the amino-terminal tail and at K79 within the histone fold. Acetylation was found on lysines K14, K18, and K23 (Fig. 2A, supplemental Fig. S2).
Fig. 2.

Post-translational modification of CENP-A-associated histone H3. A, Histone H3 amino-terminal tail peptides and post-translational modifications identified by LC/MS (ETD) following LysC digestion of CENP-A purified chromatin. The absolute amounts of recovered peptides were estimated by comparing peptides to internal standard peptides. Signature fragment ions in MS2 spectra were used to measure the degree of methylation at specific sites on combinatorially modified peptides containing multiple methylation sites. B, Histone H3 peptides and posttranslational modifications identified by LC/MS (ETD) following trypsin digestion of CENP-A purified chromatin. Peptide identification and quantitation was as descried in A. C, Full MS spectrum of H3 A1-E50 intact tail peptide PTM forms. Expected and observed m/z ratios for +11 ions used to determine the PTM state of each peptide are shown in supplemental Fig. S3A. A highly abundant form (asterisk, box) corresponding to four methylations was selected for MS2 analysis by data-dependent acquisition and is shown in panel D. D, H3 A1-E50 tail containing four methylations was fragmented using ETD (25 ms) and observed in an averaged 13 MS2 scans. E, Fragment ion data conclusively show dimethylation of K9 and dimethylation of K27.
Analysis of the asynchronous nucleosomal CENP-A-LAP sample using the protease GluC allowed observation of the intact H3 amino tail (IAT) (amino acids 1–50). We exclusively detected the canonical H3.1 IAT, and not that of replication independent histone variant H3.3. We made this determination based on peptides that contain H3 amino acid at position 31, which differs between H3.1 and H3.3 (H3.1 alanine, H3.3 serine), whereas other differences are located in the core of H3.
We identified a broad distribution of masses potentially corresponding to the combinatorially modified H3.1 IAT (Fig. 2C). These peptides showed a series of mass defects of ∼14 Da. This mass defect is equivalent to the addition of 1 methyl group; a nominal mass defect of 42 Da can be the result of three methylations or one acetylation. Accordingly, each of the forms observed were given a “methyl equivalent” (mass defect/14 Da) (34). To determine the nature of the combinations of added methylations versus acetylations, the observed accurate masses of the PTM-forms of the GluC-generated H3 tail were compared with calculated masses. For this manual analysis, we used a 0–5 ppm mass accuracy tolerance, which was based on mass accuracies of ∼1–5 ppm measured for our two standard peptides. We used the calculated mass of the most abundant isotopic peak (3 × 13C) of the observed H3.1 tail PTM-forms to determine presence and measure abundance. We found that the centromere-associated H3.1 IAT is dominantly modified with methylations on lower mass forms; on higher mass forms exist increasing additions of acetylation and less methylation. Similar marks (H3K4me1/2, H3K9me3, H3K27me1/2/3, and H3K36me2/3) were observed previously by antibody-based tools (13, 14, 35).
We generated XIC plots to determine the absolute amount of each modified form of the Histone H3 IAT. The most abundant form of the H3 IAT (m/z = 491.6573) reflects the presence of 4 methyl groups, and no acetylations (2.24 ppm) (Fig. 3A, supplemental Fig. S3). Because the maximum number of methylations per lysine is 3, the presence of 4 methyl groups means that this peptide must be combinatorially modified at more than one site within the IAT. In fact 73% of the peptides contained more than three methyl groups, suggesting that the vast majority of H3 within the centromere is combinatorially modified. These data indicate that centromeric chromatin contains a complex mixture of H3.1 modifications states, rather than a uniform set of post-translational modification.
Fig. 3.

Centromeric chromatin is enriched for hypoacetylated H3 and limited methylation. A, Relative abundances of methylated and acetylated PTM-forms of GluC-generated H3.1 IAT found in the Asynchronous Nucleosomal sample. B, The enrichment of H3 tail PTM forms in centromeric H3 compared with bulk chromatin was compared as the log fold difference between the relative abundance of the modified peptides. Combinations of methylation and acetylation that are enriched in centromeric chromatin are indicated in red. N.D. indicates that the modified form was not detected in the analysis of general chromatin. Absent from centromere indicates that the modified form was observed in the general chromatin but not in the CENP-A associated fraction.
We used data-dependent selection of the four methyl-containing H3 tail peptide to identify the nature and sites of PTM marks (Fig. 2D). Manual inspection of ETD MS2 spectra of this peptide revealed that the peptide featured dimethylation specifically and purely on K9 and dimethylation on K27. We did not find evidence to support the presence of mixed isobaric species, which would indicate alternate PTM occupancy sites. This result suggests that a significant amount of dimethylated H3 peptides K9-K14 (trypsin digest) and Q5-K14 (LysC digest) evaded detection (Fig. 2A–2B). To determine the enrichment of histone H3 modification states found in centromeric chromatin relative to H3 nucleosomes in general chromatin we analyzed acid extracted histone H3 from CENP-A-LAP expressing HeLa S3 cells (Fig. 3B, S4). The majority of H3 we identified contained 0, 1 or 2 acetylations, and 4–5 methylations. We observed that histone H3 nucleosomes containing no acetylation and either zero or 1 methylations were slightly enriched in centromeric chromatin relative to general chromatin.
Nucleosomal CENP-A-associated H4 in Asynchronously Cycling Cells
Similar to histone H3, LysC digestion of the Asynchronous Nucleosomal CENP-A-LAP sample derived from MNase digested chromatin produced many combinatorially modified peptides from H4 (Fig. 4A). In total, LysC-generated peptides including 92 of 102 residues (90%) of the total H4 sequence (Fig. 4A, supplemental Fig. S2). We detected several combinations of PTMs on histone H4 including acetylation at K8, K12, and K16, and methylation at K20. The most abundant peptides recovered were those containing both K16ac and K20me2, or the singly-modified K12ac or K20me2 peptides. Peptides not methylated at K20 were not recovered at significant amounts, suggesting that K20 is heavily methylated in centromeric chromatin and is likely present on H4 within both CENP-A- and H3-containing nucleosomes.
Fig. 4.

Post-translational modification of CENP-A-associated histone H4. A, Histone H4 peptides and posttranslational modifications identified by LC/MS (ETD) following LysC digestion of CENP-A purified chromatin from asynchronously dividing cells. The absolute amounts of recovered peptides were estimated by comparing peptides to internal standard peptides. B, Full MS spectrum showing the parent ions for histone H4 tails marked by 1–2 acetylations and 0–3 methylations. C, D, H4 S1-R23 tail generated by AspN digestion containing one acetylation and two methylations was fragmented using ETD (25 ms) and observed in an averaged 15 MS2 scans. Acetylation was localized to the N terminus and both methylations were localized to K20.
AspN digestion cleaves H4 to produce an N-terminal tail peptide of 23 amino acids (1–23), which encompasses all of the PTMs that were observed in the LysC digestion. MS1 spectra of the centromeric nucleosomal H4 tail showed a broad distribution of PTM-forms (Fig. 4B). We determined that these PTM-forms reflect combinations of 1–3 acetylations and 1–3 methylations (Fig. 5A, supplemental Fig. S5). The most abundant form of the AspN-generated H4 tail contained one acetylation and two methylations. Data-dependent selection and ETD fragmentation of this form produced MS2 spectra that allowed us to localize these PTM sites (Fig. 4C, 4D). The most abundant form of histone H4 is α-N acetylated at S1 and dimethylated at K20. The second most abundant form of the histone H4 tail was modified by one acetylation and a single methylation, suggesting that it may be S1ac-K20me1 on H4. In contrast to the H3 tail, which showed a distribution of modified forms with similar abundances, these mono- and dimethylated forms accounted for the vast majority of H4 at the centromere.
Fig. 5.

Comparison of H4 PTMs between centromeric and general chromatin. A, Relative abundances of methylated and acetylated PTM-forms of AspN-generated H4 S1-R23 amino-terminal tail peptide found in the Asynchronous Nucleosomal sample. B, Comparison of H4 tail PTM forms from bulk chromatin versus centromeric chromatin. Histone H4 tail PTM forms were compared using the log of the fold difference between the relative abundance of centromeric H4 modified forms versus those observed in general chromatin. Centromeric chromatin enriched methylation and acetylation are indicated in red. N.D. indicates that the modified form was not detected in the analysis of general chromatin. Absent from centromere indicates that the modified form was observed in the general chromatin but not in the CENP-A associated fraction.
By comparing the modifications identified on CENP-A associated histone H4 with the modification states of bulk chromatin (supplemental Fig. S6), we identified the combinations of PTMs that are unique to centromeric H4 (Fig. 5B). We found that compared with bulk chromatin, histone H4 containing 1 acetylation and 1 methylation is enriched in centromeres. This may represent the S1ac-K20me1 form described above. Histone H4 containing 2 methylations and either 1 or 2 acetylations were the most abundant species observed in general chromatin. Although the 1 acetyl state was equally represented in centromeric and general chromatin the protein containing two acetylations was slighted underrepresented in centromeric chromatin. Unacetylated histone H4 was not observed in centromeric associated histones, whereas it was readily detected in general chromatin; albeit at low levels.
Prenucleosomal CENP-A-bound H4 is Acetylated
To understand more about the events surrounding deposition of CENP-A and the timing of CENP-A-associated H4 modification we sought to identify the PTMs on H4 that is part of the prenucleosomal CENP-A complex. To access this information, we analyzed H4 protein which was co-isolated by affinity-purification of our Mitotic Prenucleosomal CENP-A-LAP sample (31). In contrast to H4 derived from CENP-A containing chromatin, the H4 from the prenucleosomal prep was exclusively associated with CENP-A and not histone H3 (Fig. 6A).
Fig. 6.

Histone H4 present in the CENP-A prenucleosomal complex is acetylated and hypomethylated. A, Schematic showing the purification approach to identify posttranslational modification on components of the CENP-A prenucleosomal complex from mitotically arrested cells. B, Full MS spectrum of AspN digested samples showing the parent ions for H4 tails marked by 3 acetylations and 0–3 methylations. C, H4 S1-R23 tail containing three acetylations was fragmented using ETD (25 ms) and observed in an averaged 6 MS2 scans. Acetylation was localized to the N terminus, K5, and K12.
We used AspN to generate the H4 N-terminal S1-R23 tail peptides, which were shown in our Asynchronous Nucleosomal CENP-A-LAP sample to contain numerous PTM sites. In contrast to our observation in the Asynchronous Nucleosomal CENP-A-LAP sample, we found that Mitotic Prenucleosomal H4 had a narrow distribution of PTMs comprised of almost entirely 2–3 acetylations and 0–3 methylation marks (Fig. 7A). The large majority of these H4 molecules contained three combinatorial acetylations: α-N acetylation on S1 and lysine acetylation specifically on K5 and K12 (Fig. 6B). We found minor contributions of methylated forms of the H4 tails. Similar to the Asynchronous Nucleosomal sample H4 methylation in prenucleosomal H4 was localized to K20. In the case of the Mitotic Prenucleosomal sample, the abundance of H4K20 methylation was inversely proportional to the number of methyl additions at this site (supplemental Fig. S7). Finally, we quantitatively compared the combinatorial PTM forms of the H4 tails identified in the Mitotic Prenucleosomal CENP-A-LAP sample with the H4 tails in the Asynchronous Nucleosomal sample (Fig. 7B). This comparison indicates that canonical prenucleosomal acetylations on H4 are removed after deposition at centromeres.
Fig. 7.

Prenucleosomal CENP-A-Bound H4 is N-terminally acetylated and lacks H4K20 methylation. A, Relative abundances of methylated and acetylated PTM-forms of AspN-generated H4 S1-R23 amino-terminal tail peptide found in the Mitotic Prenucleosomal sample. B, Comparison of H4 tail PTM forms from Mitotic Prenucleosomal CENP-A-LAP versus Asynchronous Nucleosomal H4 forms observed in this study shows H4 molecules bound to prenucleosomal CENP-A are enriched in forms that lack H4K20 methylation and contain α-N-terminal acetylation as well as acetylations at K5 and K12.
Mitotic Phosphorylations of the CENP-A Assembly Factor HJURP
Holliday Junction Recognition Protein (HJURP) is the dedicated chaperone required for targeting and deposition of new CENP-A at centromeres (3, 5, 41). Current proposals suggest that the N-terminal 80 amino acids of HJURP (the Scm3 homology domain) recognizes CENP-A specific residues within the CENP-A targeting domain (CATD) with S68 outside of the CATD providing a stabilizing contact point that can be lost by phosphorylation of that side chain (36–40). HJURP-mediated CENP-A deposition is restricted to the early G1 phase of the cell cycle (5, 41, 42). Deposition of CENP-A is inhibited during G2 and mitosis by Cdk1 dependent phosphorylation of Mis18BP1 (43) and then promoted by the phosphorylation of the Mis18 complex at other sites by PLK1 (44). We focused on HJURP phosphorylation because it is physically bound to CENP-A, and is a prime candidate for kinase-mediated cell cycle regulation.
We detected peptides corresponding to CENP-A chaperone HJURP in LysC, GluC, AspN, and trypsin digests of the Mitotic Prenucleosomal CENP-A-LAP sample. Our initial OMSSA searches identified several phosphorylations throughout the sequence of HJURP (supplemental Fig. S8). Based on initial identification of phosphorylated HJURP peptides by OMSSA we selected peptides and determined the exact sites of phosphorylation and quantified the prevalence of phosphorylation at each of these sites. From our initial list of potential phospho-sites generated by the OMSSA search results, we validated and localized nine distinct serine phosphorylation sites (Fig. 8). Of the nine total phosphorylation sites we identified on prenucleosomal CENP-A-bound HJURP, six sites were previously identified (S123, S140, S412, S486, S557, S559) (supplemental Fig. S8) (45–49). We identified three sites of serine phosphorylation that have not been previously identified; S382, S595, and S686. Quantitative measurement of the HJURP phosphorylated sites shows varying degrees of phosphorylation on each site (Fig. 8). Relatively low levels of phosphorylation were observed on S382, S486, and S686. In contrast, phosphorylation was present at levels of >60% on S123, S140, S412. Among these highly phosphorylated sites, we found that S140 and S412 are located within CDK consensus sites ([S/T]XP[K/R]). In the cases of phosphorylation of S123/S140 and S557/S559 we observed combinatorial phosphorylation at neighboring sites indicating that many of these phosphorylations are often present simultaneously on the same molecule.
Fig. 8.

Phosphorylation of the CENP-A chaperone HJURP. A, Schematic showing the location of previously known and novel sites of phosphorylation detected in this study. The percentage phosphorylation for each site is shown below. Two peptides that contained phosphorylation simultaneously at two sites were identified. B, The phosphorylated peptides that were identified and the integrated peak area used to determine the relative degree of phosphorylation.
DISCUSSION
Taking advantage of the co-purification of CENP-A-associated proteins we have identified PTMs on H3.1, H4 and HJURP, which closely associate with or are adjacent to CENP-A containing nucleosomes, or are components of the centromere chromatin assembly complex. Our analysis suggests that CENP-A chromatin is enriched in a form of the canonical H3.1 that is hypoacetylated and contains limited methylations. Centromeric H3.1 has previously been reported to be hypoacetylated (35). In our experiments, the low recovery of peptides acetylated at K14, K18, and K23 is consistent with the idea that H3.1 is hypoacetylated at centromeres of cycling cells.
Our work is particularly important because it provides the first evidence to indicate the combinatorial nature of core histone PTMs on centromeric chromatin. Several groups relying on antibody-based detections have identified K4 dimethylation and K36 di- and tri-methylation as highly abundant at centromeres and functionally-important for accurate and efficient targeting of CENP-A to centromeres (13, 35). Our results also confirm the presence of methylation at K4 and K36. Using trypsin- and LysC-digestions we found mono- and dimethylation on K4 as well as mono-, di-, and trimethylation on K36. However, our data indicate, that K4 and K36 methylation usually exist in combination with other acetylations and methylations on the same molecules. In fact, although the K4 and K36 are reported to be important PTM sites for the centromeric H3, the predominant sites of centromeric H3.1 methylation appear to be at K9 (mono-, di-, and trimethylation) and K27 (mono-, di-, and trimethylation) (Fig. 2A–2E) (13, 14). When analyzing the most abundant form of the intact H3 tail, a form containing 4 total methylations, we determined the natures of these modifications are, within measurements, only dimethylations of K9 and K27. These data demonstrate that the most abundant forms of centromeric H3 observed in cycling HeLa cells contain methylation of K9 and K27 rather than K4 or K36. Our data do not rule out that H3K4me2 and H3K36me2 are not important for centromere function or that at specific times these marks are predominating.
We found that histone H4 present in the prenucleosomal complex with CENP-A is predominantly modified with three acetylations: α-N acetylation of S1 and side chain acetylation of K5 and K12. We are not surprised to find N-terminal α-N acetylation of prenucleosomal H4 as this PTM is added constitutively during translation (50). Acetylation of Histone H4 at K5 and K12 was previously shown in prenucleosomal H3/H4 and is added by in the cytosol by Histone Acetyltransferase B (51). Upon deposition, these marks are removed and H4 can be subjected to other marks in a locus-specific manner. From our work, it appears the H4 within the CENP-A prenucleosomal complex undergoes the same modifications, as does prenucleosomal H3/H4.
We found that in cycling cells the most abundant form of centromeric H4 contains dimethylation of H4K20 at the interface of the histone H4 tail and globular domain, near the surface of nucleosomes (52). However, H4K20me2 is also a common modification within general chromatin, accounting for ∼80% of H4K20 modification states in flies and humans (53). So, although H4K20me2 is a prominent form at centromeres, it was not uniquely enriched there relative to bulk chromatin; therefore, it is unlikely to play a specific role in centromere identity.
Our study provides quantitative comparisons of H4K20 methylations that are pertinent to current investigation of the functional consequence of histone PTMs at the centromere. In contrast to H4K20me2, we found that monomethylated H4K20 (H4K20me1) was specifically enriched in the Asynchronous Nucleosomal CENP-A-LAP sample relative to in general chromatin. Recently, H4K20me1 was detected in the vicinity of centromeric CENP-A nucleosomes and that targeting H4K20 demethylase activity to centromeres resulted in impaired kinetochore assembly (28). Our study provides quantitative proteomic data to highlight that H4K20me1 is indeed directly generated on CENP-A nucleosomes and adjacent H3.1 nucleosomes. Moreover, our data demonstrates that centromeric associated H4 is combinatorial modified with both K20me1 and acetylation of the amino terminus.
Consistent with other marks of centrochromatin, H4K20me1 has been directly correlated with seemingly conflicting roles of both activation and repression of transcription. Monomethylation of H4K20 is catalyzed solely by PR-Set7. Monomethylation in this case could also potentially exist as a result of demethylase activity on di- or trimethylated H4K20; however, whereas a demethylase exists for transitioning H4K20me1 to an unmethylated form, no such enzyme has yet been identified to demethylate H4K20me2 or H4K20me3.
Our quantification of HJURP PTMs is pertinent to the current investigation of the processes that closely restrict the cell cycle timing of the delivery (and subsequent deposition) of nascent CENP-A to centromeres. Our data corroborates the phosphorylation of several sites in the CENP-A chaperone HJURP that have been previously reported, and identified three previously unidentified sites. We demonstrated that in mitotic extracts when HJURP is not centromere associated, S123, S412, and S557 are the most highly phosphorylated sites, suggesting that these sites may be involved in negatively regulating HJURP centromere association. Previously, mutation of serines 412, 448, and 472 to alanine resulted in precocious loading of CENP-A during G2 (48). Serine 412 was highly phosphorylated in mitosis in our CENP-A prenucleosomal complex (84%), whereas phosphorylation of serines 448 and 472 was not detected, suggesting that S412 is the major site of phosphorylation that negatively regulates HJURP recruitment outside of G1 phase. We observed low or nondetectable levels of phosphorylation on several other sites. It is possible that these sites are modulated during the cell cycle and mediate other aspects of HJURP function, perhaps acting as positive regulators of HJURP activity.
Although the direct or indirect targeting of CENP-A leads to the generation of functional centromeres, it is important to consider the individual nucleosomal context and the higher-order chromatin context that ultimately forms the chromatin at the foundation of the mitotic kinetochore. Our findings here of the modification state of key chromatin components provide the framework for understanding the precise makeup of this essential epigenetically defined chromosomal locus.
Supplementary Material
Acknowledgments
We are grateful for access to Isotope Pattern Calculator software, developed by Pacific Northwest National Laboratories.
Footnotes
Author contributions: A.O.B., T.P., J.S., D.F.H., B.E.B., and D.R.F. designed research; A.O.B., T.P., and S.M.L. performed research; A.O.B., J.S., S.M.L., D.L.B., D.F.H., B.E.B., and D.R.F. analyzed data; A.O.B., T.P., B.E.B., and D.R.F. wrote the paper.
* This work was supported by National Institutes of Health (NIH) Grants R01 GM037537 (to D.F.H.), R01 GM082989 (to B.E.B.), R01 GM111907 (to D.R.F.), and the American Cancer Society (D.R.F.). A.O.B. and T.P. were supported by the following training grants: T32 GM08715 T32 (to A.O.B.) GM08275 (to T.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental materials.
This article contains supplemental materials.
1 The abbreviations used are:
- CENP-A
- centromere protein-A
- PTM
- post-translational modification
- XIC
- extracted ion chromatogram.
REFERENCES
- 1. Stellfox M. E., Bailey A. O., and Foltz D. R. (2013) Putting CENP-A in its place. Cell Mol. Life Sci. 70, 387–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wevrick R., and Willard H. F. (1989) Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability. Proc. Natl. Acad. Sci. U.S.A. 86, 9394–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnhart M. C., Kuich P. H., Stellfox M. E., Ward J. A., Bassett E. A., Black B. E., and Foltz D. R. (2011) HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol. 194, 229–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunleavy E. M., Almouzni G., and Karpen G. H.. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foltz D. R., Jansen L. E., Bailey A. O., Yates J. R. 3rd, Bassett E. A., Wood S., Black B. E., and Cleveland D. W. (2009) Centromere-specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell 137, 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moree B., Meyer C. B., Fuller C. J., and Straight A. F.. CENP-C recruits M18BP1 to centromeres to promote CENP-A chromatin assembly. J. Cell Biol. 194, 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dambacher S., Deng W., Hahn M., Sadic D., Frohlich J., Nuber A., Hoischen C., Diekmann S., Leonhardt H., and Schotta G. (2012) CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin. Nucleus 3, 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Liu X., Dou Z., Chen L., Jiang H., Fu C., Fu G., Liu D., Zhang J., Zhu T., Fang J., Zang J., Cheng J., Teng M., Ding X., and Yao X. (2014) Mitotic Regulator Mis18beta Interacts with and Specifies the Centromeric Assembly of Molecular Chaperone Holliday Junction Recognition Protein (HJURP). J. Biol. Chem. 289, 8326–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Partridge J. F., Borgstrom B., and Allshire R. C. (2000) Distinct protein interaction domains and protein spreading in a complex centromere. Genes Dev. 14, 783–791 [PMC free article] [PubMed] [Google Scholar]
- 10. Blower M. D., and Karpen G. H. (2001) The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. Nat. Cell Biol. 3, 730–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schalch T., Job G., Noffsinger V. J., Shanker S., Kuscu C., Joshua-Tor L., and Partridge J. F. (2009) High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell. 34, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blower M. D., Sullivan B. A., and Karpen G. H. (2002) Conserved organization of centromeric chromatin in flies and humans. Dev. Cell 2, 319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bergmann J. H., Rodriguez M. G., Martins N. M., Kimura H., Kelly D. A., Masumoto H., Larionov V., Jansen L. E., and Earnshaw W. C. (2011) Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 30, 328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sullivan B. A., and Karpen G. H. (2004) Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 11, 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alonso A., Hasson D., Cheung F., and Warburton P. E. (2010) A paucity of heterochromatin at functional human neocentromeres. Epigenetics Chromatin 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bodor D. L., Mata J. F., Sergeev M., David A. F., Salimian K. J., Panchenko T., Cleveland D. W., Black B. E., Shah J. V., and Jansen L. E. (2014) The quantitative architecture of centromeric chromatin. Elife 3, e02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shelby R. D., Vafa O., and Sullivan K. F. (1997) Assembly of CENP-A into centromeric chromatin requires a cooperative array of nucleosomal DNA contact sites. J. Cell Biol. 136, 501–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Foltz D. R., Jansen L. E., Black B. E., Bailey A. O., Yates J. R. 3rd, and Cleveland D. W. (2006) The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 8, 458–469 [DOI] [PubMed] [Google Scholar]
- 19. Padeganeh A., Ryan J., Boisvert J., Ladouceur A. M., Dorn J. F., and Maddox P. S. (2013) Octameric CENP-A nucleosomes are present at human centromeres throughout the cell cycle. Curr. Biol. 23, 764–769 [DOI] [PubMed] [Google Scholar]
- 20. Lacoste N., Woolfe A., Tachiwana H., Garea A. V., Barth T., Cantaloube S., Kurumizaka H., Imhof A., and Almouzni G. (2014) Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell 53, 631–644 [DOI] [PubMed] [Google Scholar]
- 21. Bodor D. L., Valente L. P., Mata J. F., Black B. E., and Jansen L. E. (2013) Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol. Biol. Cell 24, 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Falk S. J., Guo L. Y., Sekulic N., Smoak E. M., Mani T., Logsdon G. A., Gupta K., Jansen L. E., Van Duyne G. D., Vinogradov S. A., Lampson M. A., and Black B. E. (2015) Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science 348, 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasson D., Panchenko T., Salimian K. J., Salman M. U., Sekulic N., Alonso A., Warburton P. E., and Black B. E. (2013) The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Structural Mol. Biol. 20, 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hori T., Amano M., Suzuki A., Backer C. B., Welburn J. P., Dong Y., McEwen B. F., Shang W. H., Suzuki E., Okawa K., Cheeseman I. M., and Fukagawa T. (2008) CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell 135, 1039–1052 [DOI] [PubMed] [Google Scholar]
- 25. Black B. E., and Cleveland D. W. (2011) Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144, 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ribeiro S. A., Vagnarelli P., Dong Y., Hori T., McEwen B. F., Fukagawa T., Flors C., and Earnshaw W. C.. A super-resolution map of the vertebrate kinetochore. Proc. Natl. Acad. Sci. U.S.A. 107, 10484–10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lam A. L., Boivin C. D., Bonney C. F., Rudd M. K., and Sullivan B. A. (2006) Human centromeric chromatin is a dynamic chromosomal domain that can spread over noncentromeric DNA. Proc. Natl. Acad. Sci. U.S.A. 103, 4186–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hori T., Shang W. H., Toyoda A., Misu S., Monma N., Ikeo K., Molina O., Vargiu G., Fujiyama A., Kimura H., Earnshaw W. C., and Fukagawa T. (2014) Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Developmental cell 29, 740–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cardinale S., Bergmann J. H., Kelly D., Nakano M., Valdivia M. M., Kimura H., Masumoto H., Larionov V., and Earnshaw W. C. (2009) Hierarchical inactivation of a synthetic human kinetochore by a chromatin modifier. Mol. Biol. Cell 20, 4194–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakano M., Cardinale S., Noskov V. N., Gassmann R., Vagnarelli P., Kandels-Lewis S., Larionov V., Earnshaw W. C., and Masumoto H. (2008) Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell 14, 507–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bailey A. O., Panchenko T., Sathyan K. M., Petkowski J. J., Pai P. J., Bai D. L., Russell D. H., Macara I. G., Shabanowitz J., Hunt D. F., Black B. E., and Foltz D. R. (2013) Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proc. Natl. Acad. Sci. U.S.A. 110, 11827–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shechter D., Dormann H. L., Allis C. D., and Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 33. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., and Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 34. Young N. L., DiMaggio P. A., Plazas-Mayorca M. D., Baliban R. C., Floudas C. A., and Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8, 2266–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergmann J. H., Martins N. M., Larionov V., Masumoto H., and Earnshaw W. C. (2012) HACking the centromere chromatin code: insights from human artificial chromosomes. Chromosome Res. 20, 505–519 [DOI] [PubMed] [Google Scholar]
- 36. Shuaib M., Ouararhni K., Dimitrov S., and Hamiche A. (2010) HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc. Natl. Acad. Sci. U.S.A. 107, 1349–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu H., Liu Y., Wang M., Fang J., Huang H., Yang N., Li Y., Wang J., Yao X., Shi Y., Li G., and Xu R. M. (2011) Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev. 25, 901–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bassett E. A., DeNizio J., Barnhart-Dailey M. C., Panchenko T., Sekulic N., Rogers D. J., Foltz D. R., and Black B. E.. HJURP uses distinct CENP-A surfaces to recognize and to stabilize CENP-A/histone H4 for centromere assembly. Dev. Cell 22, 749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu Z., Zhou X., Wang W., Deng W., Fang J., Hu H., Wang Z., Li S., Cui L., Shen J., Zhai L., Peng S., Wong J., Dong S., Yuan Z., Ou G., Zhang X., Xu P., Lou J., Yang N., Chen P., Xu R. M., and Li G. (2015) Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Dev. Cell 32, 68–81 [DOI] [PubMed] [Google Scholar]
- 40. Logsdon G. A., Barrey E. J., Bassett E. A., DeNizio J. E., Guo L. Y., Panchenko T., Dawicki-McKenna J. M., Heun P., and Black B. E. (2015) Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J. Cell Biol. 208, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dunleavy E. M., Roche D., Tagami H., Lacoste N., Ray-Gallet D., Nakamura Y., Daigo Y., Nakatani Y., and Almouzni-Pettinotti G. (2009) HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485–497 [DOI] [PubMed] [Google Scholar]
- 42. Jansen L. E., Black B. E., Foltz D. R., and Cleveland D. W. (2007) Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol. 176, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silva M. C., Bodor D. L., Stellfox M. E., Martins N. M., Hochegger H., Foltz D. R., and Jansen L. E. (2012) Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev. Cell 22, 52–63 [DOI] [PubMed] [Google Scholar]
- 44. McKinley K. L., and Cheeseman I. M. (2014) Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell 158, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dephoure N., Zhou C., Villen J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayya V., Lundgren D. H., Hwang S. I., Rezaul K., Wu L., Eng J. K., Rodionov V., and Han D. K. (2009) Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal 2, ra46. [DOI] [PubMed] [Google Scholar]
- 47. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3, ra3. [DOI] [PubMed] [Google Scholar]
- 48. Muller S., Montes de Oca R., Lacoste N., Dingli F., Loew D., and Almouzni G. (2014) Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3(CENP-A) loading. Cell Reports 8, 190–203 [DOI] [PubMed] [Google Scholar]
- 49. Luhn P., Wang H., Marcus A. I., and Fu H. (2007) Identification of FAKTS as a novel 14-3-3-associated nuclear protein. Proteins 67, 479–489 [DOI] [PubMed] [Google Scholar]
- 50. Hole K., Van Damme P., Dalva M., Aksnes H., Glomnes N., Varhaug J. E., Lillehaug J. R., Gevaert K., and Arnesen T. (2011) The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS ONE 6, e24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang L., Loranger S. S., Mizzen C., Ernst S. G., Allis C. D., and Annunziato A. T. (1997) Histones in transit: cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry 36, 469–480 [DOI] [PubMed] [Google Scholar]
- 52. Lu X., Simon M. D., Chodaparambil J. V., Hansen J. C., Shokat K. M., and Luger K. (2008) The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Structural Mol. Biol. 15, 1122–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang H., and Mizzen C. A. (2009) The multiple facets of histone H4-lysine 20 methylation. Biochem. Cell Biol. 87, 151–161 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.