

Abstract
Histones represent a class of proteins ideally suited to analyses by top-down mass spectrometry due to their relatively small size, the high electron transfer dissociation-compatible charge states they exhibit, and the potential to gain valuable information concerning combinatorial post-translational modifications and variants. We recently described new methods in mass spectrometry for the acquisition of high-quality MS/MS spectra of intact proteins (Anderson, L. C., English, A. M., Wang, W., Bai, D. L., Shabanowitz, J., and Hunt, D. F. (2015) Int. J. Mass Spectrom. 377, 617–624). Here, we report an extension of these techniques. Sequential ion/ion reactions carried out in a modified Orbitrap Velos Pro/EliteTM capable of multiple fragment ion fills of the C-trap, in combination with data-dependent and targeted HPLC-MS experiments, were used to obtain high resolution MS/MS spectra of histones from butyrate-treated HeLa cells. These spectra were used to identify several unique intact histone proteoforms with up to 81% sequence coverage. We also demonstrate that parallel ion parking during ion/ion proton transfer reactions can be used to separate species of overlapping m/z that are not separated chromatographically, revealing previously indiscernible signals. Finally, we characterized several truncated forms of H2A and H2B found within the histone fractions analyzed, achieving up to 93% sequence coverage by electron transfer dissociation MS/MS. Results of follow-up in vitro experiments suggest that some of the truncated histone H2A proteoforms we observed can be generated by cathepsin L, an enzyme known to also catalyze clipping of histone H3.
Chromatin is the structural framework that packages DNA into chromosomes within the nucleus of a cell (2). Histones comprise the principal protein component of chromatin and are involved in the regulation of gene expression (3, 4). This epigenetic regulation is achieved through complex patterns of post-translational modifications (PTMs),1 the incorporation of histone variants, and through controlled histone proteolysis (5–10). Comprehensive characterization of histones by mass spectrometry (MS) has proven technically difficult for a number of reasons. Traditional methods (bottom-up MS) of sequence determination and PTM site localization are not practical. Histone N-terminal regions are rich in lysine and arginine residues, and thus proteolysis using trypsin generates peptides that are too small or that are poorly retained on reverse-phase HPLC C18 resins for subsequent MS detection (11). With the advent of electron transfer dissociation (ETD) and more efficient electron capture dissociation fragmentation methods, which are better suited for larger, more highly charged peptides (12, 13), several studies utilizing other endoproteases to generate longer peptides have emerged (14–16). Although these methodologies do well to preserve the combinatorial PTM profiles of histone tails, in some cases it is still impossible to identify the proteoforms from which these peptides originate. This is why analyzing histones intact, as they exist in the cells from which they are derived, is the best method for identifying unique histone proteoforms.
The results of several recent studies involving top-down analyses of histones highlight the complexity of the histone proteome as well as important biological implications that are not easily captured unless the proteoforms are analyzed intact (17–20). Although these results are very promising, this approach is not routinely utilized because of the many challenges with which it is associated. In a 2014 Proteomics article authored by members of the Consortium for Top-Down Proteomics, Dang et al. concluded that analyses of intact proteins could be greatly improved by the development of higher resolution isolation capabilities and by the ability to acquire rich sequence informative MS/MS spectra on a time scale more compatible with chromatography (18).
We have previously described modifications to a Thermo Fisher Scientific Orbitrap Velos ProTM mass spectrometer that make it possible to perform multiple fills of the C-trap with product ions from sequential ion/ion reactions of intact proteins prior to high resolution mass analysis (1, 21). The result is a dramatic enhancement of the observed fragment ion current and significant improvement of signal-to-noise ratios (SNRs) in MS/MS spectra. This eliminates the need for the acquisition of several time-consuming transients and allows for the acquisition of high quality spectra on a time scale compatible with chromatography. The front-end ETD (FETD) source can also be used to ionize ion/ion proton transfer (IIPT) reagents. IIPT reactions are used to disperse fragment ions over the available m/z range in a controlled manner and to simplify ETD fragment ion spectra (1, 22, 23). Additionally, we have enabled parallel ion parking, first reported by McLuckey and co-workers (24, 25), which involves harmonic excitation of selected ions within the ion trap to reduce their reactivity in gas-phase ion/ion reactions. This allows us to perform IIPT without reacting the precursor outside of the targeted product m/z range. Here, we demonstrate the power of these new techniques through their coupling with on-line, nano-LC chromatography and application to the analysis of intact histones derived from butyrate-treated HeLa cells.
EXPERIMENTAL PROCEDURES
Unless otherwise stated, all MS analyses were carried out on an in-house modified (21) hybrid Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA). In the time that the experiments described below were performed, the instrument was upgraded to include a high-field Orbitrap mass analyzer (26). Experiments involving recombinant H4 and parallel ion parking were performed following the upgrade and so they make use of enhanced FT (27). Otherwise, spectra are acquired in magnitude mode. All spectra were acquired in reduced profile mode, using instrument default noise reduction.
Direct Infusion of Recombinant H4
Recombinant H4 was purchased from New England Biolabs (Ipswich, MA), desalted using an Amicon® Ultra-0.5 3K centrifugal filter (Millipore, Billerica, MA), and reconstituted in a solution containing 40% acetonitrile and 60% of 0.1% acetic acid in water (all percentages expressed as v/v). The sample was directly infused at a concentration of 5 pmol/μl and ionized using microelectrospray ionization as described previously (28) into the front end of an in-house modified ETD/IIPT-enabled Orbitrap EliteTM. MS/MS spectra were acquired using the following instrument parameters: resolution (r) = 60,000 at 400 m/z; 190–2000 m/z scan range; 1E5 ion counts precursor automatic gain control (AGC) target; 3E5 reagent AGC target; 1–20 fragment ion fills of the C-trap; 1 μscan/spectrum; 2 m/z precursor isolation window centered at 708.4 m/z ([M + 16H]+16 charge state of H4); 6–8-ms ETD; 0–20-ms IIPT.
Histone Extraction from HeLa S3 Cells
Cultured HeLa S3 cells were treated with 10 mm sodium butyrate overnight and harvested, and the nuclei were isolated as described previously (29). The nuclei pellet was resuspended in 0.4 m NaCl containing 1 mm dithiothreitol (DTT), 0.3 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 10 mm sodium butyrate, and 5 μm Halt protease inhibitor mixture at a ratio of 10:1 (v/v), salt buffer/pellet. This was incubated with constant rotation for 2 h at 4 °C and then spun at 3000 × g for 5 min. Supernatant was discarded, and the pellet was resuspended in a 2.5 m NaCl solution containing DTT, 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, sodium butyrate, and protease inhibitors as above in a ratio of 5:1 (v/v), salt buffer/pellet. The histones were then acid-extracted and TCA-precipitated as described previously (29).
Off-line Histone Fractionation
Histones purified from HeLa S3 cells (∼100 μg) were dissolved in water and chromatographically separated on a Vydac C18 column (4.6-mm inner diameter, 250-mm length, 5-μm particle size) at a flow rate of 0.8 ml/min using a gradient of 30–60% solvent B in 100 min, 60–100% B in 20 min, and 100–30% B in 10 min (solvent A = 5% (v/v) acetonitrile, 0.2% (v/v) trifluoroacetic acid in HPLC-grade water; solvent B = 95% (v/v) acetonitrile, 0.19% (v/v) trifluoroacetic acid in HPLC-grade water). Fractions were collected using an automatic sample collector in 1-min intervals between 15 and 80 min from the start of the gradient. The UV detector was set to monitor absorbance at 214 nm, and fractions containing histones H1, H2A, H2B, and H4 were identified and appropriately combined based on their known chromatographic retention times (29). These were transferred to microcentrifuge tubes, taken to dryness using a SpeedVac concentrator, and stored at −35 °C.
On-line RP-HPLC for Mass Spectrometry
Dried samples were reconstituted in water at concentrations of 5–10 pmol/μl. Aliquots of 1–2.5 pmol of protein along with 100 fmol of peptide standards were pressure-loaded onto a 360-μm outer diameter × 75-μm inner diameter fused-silica micro-capillary analytical column (packed 11-cm of POROSHELL 300SB-C18 resin (Agilent Technologies, Palo Alto, CA) (30) equipped with a laser-pulled electrospray emitter tip (31). The column was washed with ∼20 column volumes of 0.3% (v/v) formic acid in water. Samples were eluted at 60 nl/min using a gradient of 0–33% B in 10 min, 33–48% B in 60 min, 48–100% B in 10 min and 100–0% B in 10 min. The elution gradient utilized solvent A, 0.3% formic acid in water, and solvent B, 0.3% formic acid, 72% acetonitrile, 18% isopropyl alcohol, and 9.7% water (all v/v). Following reverse-phase HPLC separation, samples were directly ionized using microelectrospray ionization as described (28). For some samples, initial screens revealed that better chromatography might be achieved by enacting simple changes in the elution gradient. These changes included doubling the 0–33% B portion of the gradient to a time of 20 min for samples containing truncated histones and altering the gradient used for H1 to 0–20% B in 10 min, 20–40% B in 60 min, and then 40–100% B in 10 min. All other aspects of the RP-HPLC separation and ionization for mass spectrometry remained the same across all analyses.
On-line MS Analyses
Samples were ionized by microelectrospray into the front-end of an in-house modified ETD/IIPT-enabled Orbitrap Velos ProTM. Except for the occasional targeted analyses, the instrument was operated in data-dependent mode in which the MS1 scan was taken from m/z 300–2000 in the Orbitrap mass analyzer (r = 60,000 at m/z 400) followed by a second MS1 scan acquired in the linear ion trap over a limited m/z range, upon which data-dependent selection was based. In this way, same species/different charge state reselection was reduced. A single ETD/IIPT MS/MS scan was also taken in the Orbitrap. MS/MS scan parameters were set as follows: r = 30k, 60k or 100k at m/z 400; 200–2000 m/z scan range; 1E5 ion counts AGC target; 3E5 ion counts reagent AGC target; 5–30 fragment ion fills of the C-trap; 1–3 μscans; 2–3 m/z precursor isolation window; 4–25-ms ETD (fluoranthene reagent); and 20–40-ms IIPT (sulfur hexafluoride reagent). The instrument was operated using charge state exclusion (+1, +2, and +3) and dynamic exclusion with a repeat count of 1–2, repeat duration of 60 s, and an exclusion duration of 90 s. Analytes of different sizes and charge densities require different ETD/IIPT reaction times, C-trap fill counts, and resolving power; these were varied depending on the fraction being analyzed. For example, when analyzing H1, a 21.9-kDa protein, shorter ETD and IIPT reaction times, more multiple fills, and higher resolution settings were used than those used for the analysis of H4, an 11.4-kDa protein. Duty cycle times also varied accordingly, but in general they were within 6–15 s/cycle.
Some fractions contained larger proteins that required MS/MS scan ranges over 2000 m/z for sequence determination and PTM site mapping. However, the instrument control software did not allow the acquisition of spectra with scan ranges exceeding 2000 m/z for ETD/IIPT MS/MS scan types in data-dependent mode. This prompted us to perform “on-the-fly” targeted analyses based upon full MS data acquired in the initial sample screenings. Instrument method parameters were varied at our discretion based on our evaluation of the MS/MS spectra generated as data were acquired.
High resolution full MS and MS/MS data were manually inspected using Qual Browser software (Thermo Fisher Scientific). Interpretation of all ETD/IIPT MS/MS data was performed manually on the unprocessed raw spectra. Theoretical fragment ion masses were calculated using an in-house-developed fragment mass calculator. Theoretical isotopic distributions were modeled using Isotope Pattern Calculator software (Pacific Northwest National Laboratories, omics.pnl.gov/). Percent sequence coverage was calculated by dividing the number of observed N–Cα bond cleavages by the total number of predicted N–Cα bond cleavages (note that cleavage of the N–Cα bond that is N-terminal to proline does not produce an observable fragment). Masses for all intact proteoforms identified agreed to within 5 ppm and were confirmed by MS/MS. Relative abundance information for each truncated histone species was determined by taking the area under the curve of the extracted ion chromatogram for the most abundant isotope for every charge state present utilizing a 10-ppm mass window (32).
Parallel Ion Parking during IIPT
For parallel ion parking experiments, the MS1 scan was taken from m/z 300 to 2000 in the high-field Orbitrap mass analyzer (1E6 ion counts AGC target, r = 120,000 at 400 m/z). A 40 m/z window centered at m/z 810 (790–830 m/z) was selected and subject to 140 ms of IIPT. The instrument control software was modified to apply a custom RF waveform to the x-rods of the linear ion trap during IIPT. The waveform included frequencies mapping to m/z 2300–3900, corresponding to the desired final charge state of the precursor. For the sample analyzed (fraction H2B), the [M + 5H]+5 charge state was the desired product (∼m/z 2770). The amplitude of the waveform was adjusted such that maximum activation of the target product ions was achieved without dissociation or ejection. Product ions were analyzed in the Orbitrap (1E5 ion counts AGC target, r = 120,000 at 400 m/z) after five fills of the C-trap.
In Vitro H2A Proteolysis and nano-HPLC-MS/MS Analysis
The in vitro proteolysis of recombinant histone H2A type 2 (Uniprot ID, Q6FI13) was carried out in 50 mm MES, pH 6.0, and 5 mm DTT. Recombinant cathepsin L (R&D Systems 1515-CY-010) was used in a 5000:1 (histone/enzyme) ratio during a 1-h time course reaction. 20 μg of the sample reaction was quenched with 10% acetic acid after 15, 30, and 60 min. Samples were desalted using in-house packed Stage tips as described previously (29). After Stage tip desalting, samples were resuspended in 0.1% formic acid prior to nano-LC-MS/MS analysis. 1 μg of sample was loaded onto a 75-μm inner diameter × 17-cm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany) column using an EASY-nLC nano-HPLC (Thermo Fisher Scientific). The HPLC gradient was 0–100% solvent B (A = 0.1% formic acid; B = 95% acetonitrile, 0.1% formic acid) over 60 min at a flow rate of 250 nl/min. nano-LC was coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). MS1 spectra (m/z 300–1200) were acquired in the Orbitrap with a resolution of 120,000 (at 200 m/z) and an AGC target of 5E5. Data-dependent selection was performed using TopSpeed with a cycle time of 3 seconds. MS/MS for charge states +2–5 was performed using higher-energy collisional dissociation (HCD), with a normalized collision energy of 27%, a maximum injection time of 150 ms, and an AGC target of 5E4. Higher charge states were fragmented using ETD, using a reaction time of 20 ms, a maximum injection time of 400 ms, 3 μscans, and an AGC target of 5E5. Both types of MS/MS fragmentation were acquired in the Orbitrap with a resolution of 30,000 (at 200 m/z). Data were analyzed using Proteome Discoverer (version 1.4, Thermo Scientific, Bremen, Germany) with Mascot (Matrix Science, London, UK) as the database search engine (database: human histones, Uniprot, March, 2014, 58 entries). Search parameters were as follows: HCD spectra were searched using a precursor ion mass tolerance of 10 ppm and a fragment ion mass tolerance of 0.05 Da; ETD spectra were deconvoluted using Xtract (Thermo Fisher Scientific) and searched using a precursor ion mass tolerance of 10 ppm and a fragment ion mass tolerance of 0.02 Da. Unspecific cleavage (no enzyme) and no modifications were specified. Only identifications with a false discovery rate of <1% (calculated using fixed value PSM validator) were accepted. Chromatograms of the relevant peptides were manually extracted using Xcalibur (Thermo Fisher Scientific) after deconvolution for peptide quantification.
RESULTS
Multiple Fills and IIPT
A Thermo Fisher Scientific Orbitrap Velos ProTM was modified to introduce reagent anions from the front by adding a glow discharge ionization source between the stacked ring ion guide and multipole 00, as described previously (21). The discharge is used to ionize ETD and IIPT reagents (fluoranthene and sulfur hexafluoride, respectively) for ion/ion reactions. By placing the reagent ion source in the front of the instrument, the C-trap is made available to store fragment ions from multiple iterations of ion/ion reactions. Once sufficient amounts of fragment ions are collected, they are sent to the Orbitrap for high resolution mass analysis.
Fig. 1, A and B, shows 8-ms ETD/20-ms IIPT MS/MS spectra of intact recombinant H4 using 1 and 20 fragment ion fills of the C-trap, respectively. SNRs for fragments shared between the two spectra are indicated in parentheses. By increasing the number of fragment ion volumes analyzed from 1 to 20, total fragment ion current is increased by an order of magnitude. This is also demonstrated in supplemental Fig. 1, which shows a trace of total ion current as the number of multiple fills is varied. The spectrum shown in Fig. 1C is an average of 20 spectra like that shown in Fig 1A and represents a cumulative acquisition time of 11.2 s (0.56 s/spectrum). Elapsed scan time for the spectrum shown in Fig. 1B is 6.24 s. Average SNR gains for the fragments indicated are 10.4 in Fig. 1B versus 3.5 in Fig. 1C. Hand-annotated spectra indicating the most abundant fragments observed are shown in supplemental Fig. 2.
Fig. 1.
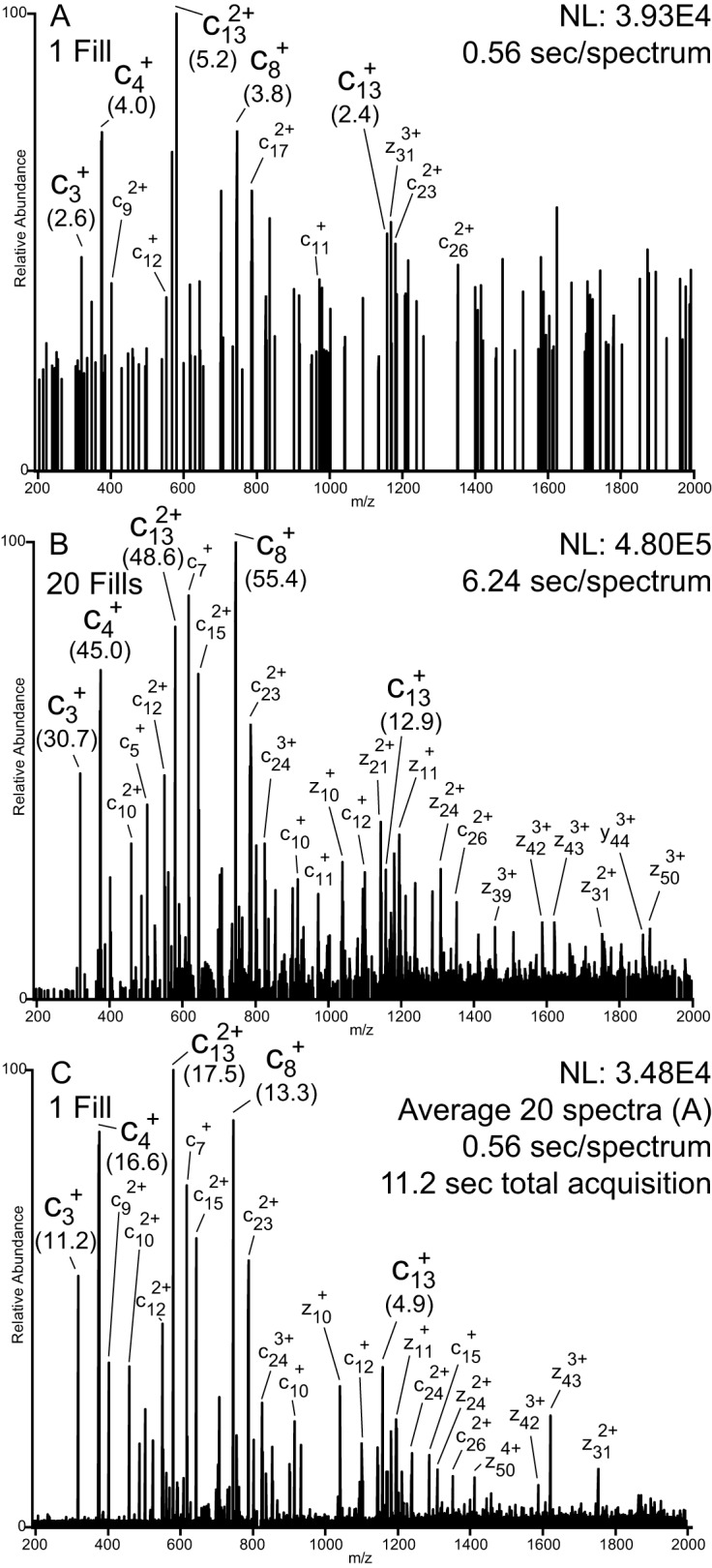
Multiple fills versus spectral averaging. MS/MS spectra of intact recombinant H4 (precursor charge state +16, m/z 703.40) following ETD (8 ms) and IIPT (20 ms) reactions with 1 and 20 fragment ion fills of the C-trap. SNRs for fragments shared among the spectra are indicated in parentheses. A, spectrum produced from a single iteration of ion/ion reactions (1 fill, 1 μscan, r = 60,000 at 400 m/z, 0.56-s elapsed scan time). B, spectrum produced from 20 iterations of ion/ion reactions (20 fills, 1 μscan, r = 60,000 at 400 m/z, 6.24-s elapsed scan time). C, spectrum produced by averaging 20 (A) spectra (1 fill, 1 μscan, r = 60,000 at 400 m/z, 0.56-s elapsed scan time/spectrum, 11.2-s total acquisition time).
The use and benefits of IIPT to charge reduce ETD-produced fragment ions have been described previously (1, 23). Briefly, following fragmentation by ETD, fragment ions are allowed to react with a proton transfer reagent. These reagents act as a base and abstract protons from highly charged fragments. The amount of charge reduction is tuned by varying the amount of time the reaction is allowed to take place. By extending IIPT reaction time such that fragment ions are significantly charge-reduced, the fragment ion signals are increasingly distributed over the full scan range (example shown in supplemental Fig. 3).
Characterization of Intact Histones Derived from Butyrate-treated HeLa Cells
HeLa cells were treated with sodium butyrate prior to histone isolation and separation. Butyrate treatment of cells results in histone hyperacetylation; butanoate acts as an inhibitor of histone deacetylase activity (33). The histones were acid-extracted from the nuclei and purified by off-line reverse-phase HPLC fractionation. The histones were identified based on their well known chromatographic retention times (34). Intact protein was directly loaded onto a reverse-phase C18 Poros-shell column and gradient-eluted into the mass spectrometer.
Fig. 2 summarizes the data that can be obtained from a single LC-MS experiment. Fig. 2A shows the base-peak chromatogram of the H2A2 fraction revealing three distinct regions of intact protein elution. Peaks 1 and 2 correspond to H4 in various modified forms containing oxidized and unoxidized methionines, respectively, whereas peak 3 corresponds to various proteoforms of intact H2A. Displayed in Fig. 2, B and C, are the averaged high resolution ESI spectra recorded on peaks 2 and 3 in the base peak chromatogram, respectively. Signals in the observed charge state distribution carry 7–18 and 8–23 positive charges for H4 and H2A, respectively. An ETD/IIPT MS/MS spectra recorded on an [M + 19H]19+ ion of an intact form of H2A is shown in Fig. 2D. This spectrum was obtained using a 4-ms ETD and a 25-ms IIPT and is an average of six spectra, each taking 11.7 s to acquire utilizing 3 μscans and 15 multiple fills with r = 30,000 at 400 m/z. Note that equivalent spectra taken as the sum of 45 μscans (transients) of single fragment ion fills would take ∼19.8 s/scan. Residues identified by ions of types c and z observed in Fig. 2D are indicated in Fig. 2E (hand annotated version shown in supplemental Fig. 4). These define 80% sequence coverage and identify the intact H2A as H2A.1 (Q93077).
Fig. 2.

Example data obtained from a single LC-MS experiment. A, base peak chromatogram obtained from LC-MS analysis of histone fraction H2A2. Peaks corresponding to 100 fmol of co-loaded peptide standards are labeled, and regions of intact protein elution are numbered. B and C, averaged ESI full MS spectra depicting the charge state distributions of intact H4 and H2A, respectively (peaks 2 and 3 from A, respectively). D, ETD/IIPT spectrum recorded on [M + 19H]19+ ions from H2A type 1 (P0C0S8) using 4-ms ETD and 25-ms IIPT. Precursor m/z 738.0, 3 μscans, 15 multiple-fills, r = 30,000, average of 6 spectra, 11.7-s elapsed scan time/spectrum (∼19.8 s for single fill/transient equivalent spectrum). E, sequence coverage obtained from MS/MS spectrum shown in D.
Depicted in supplemental Fig. 5 is a full MS spectrum indicating signals for intact H2A proteoforms that were identified and confirmed by ETD/IIPT MS/MS. In addition to H2A.1, also detected were H2A.1-C and H2A.1-H (Uni Parc Q93077 and Q96KK5, respectively). H2A.1-C differs from H2A.1 by the substitution of Thr-16 with a serine residue and by substitution of Lys-99 with an arginine residue. These substitutions give rise to a δ mass of +13.9904 Da from H2A.1, which is very close to the δ mass that would be exhibited in the event of a monomethylation (+14.0157 Da). The calculated and observed mass differences for intact H2A.1-C and the monomethylated form of H2A.1 are 3.5 and 5.3 ppm, respectively, making H2A.1-C the better fit by mass accuracy. However, based on this information alone, neither could be conclusively excluded. The MS/MS spectra contained c ions that exhibited a δ mass of −14 Da following Thr-16 and z ions that exhibited a mass shift of +28 Da following Lys-99. The +28-Da mass shift is also indicative of a dimethylated lysine, but this possibility was ruled out based on the mass accuracy of the fragment ions observed (z313⨥, 13C m/z 1101.9823: 0.2 ppm for Lys → Arg, 7.8 ppm for Lys → di-methylation Lys). Thus, we were able to conclude that this is H2A.1-C. This demonstrates the importance of obtaining high quality MS/MS spectra for the confirmation of proteoform identity. H2A.1-H differs from H2A.1 by the deletion of the last two C-terminal residues, emphasizing the importance of analyzing the intact forms of these proteins, as digestions would produce identical peptides with the exception of those containing the C termini. This difference was easily discerned upon examination of the z fragment ion series in the MS/MS spectra. H2A.1-C and H2A.1-H are present at levels roughly 20–25 and 15–20%, respectively, relative to H2A.1. Eluting later in the gradient and also confirmed by MS/MS are the respective mono-Lys-acetylated forms of these proteins. The acetylation is localized to Lys-5 in all H2A types.
Fig. 3 displays a spectrum recorded on [M + 16H]16+ (m/z 718.0) from intact H4 using a 4-ms ETD reaction time and 40-ms IIPT. This is an average of eight spectra, each taking ∼8.75 s to acquire utilizing 3 μscans at r = 30,000 (at 400 m/z) with 10 multiple fills of the C-trap per μscan (∼13.9-s acquisition for single fill/transient equivalent spectrum). Fig. 3B offers an expanded view of the 1250–1500 m/z region of the spectrum. With the exception of the extreme C terminus, which is devoid of charge, a near contiguous series of both c′ and z•′ can be observed for ∼60 residues from either end of the protein, yielding 81% sequence coverage and site localization of five acetylations as well as Arg-20 dimethylation (fully annotated spectrum shown in supplemental Fig. 6).
Fig. 3.

ETD/IIPT MS/MS spectrum of intact H4 proteoform. A, ETD/IIPT spectrum recorded on [M + 16H]16+ ions from H4 (P62805) using 4-ms ETD and 40 ms IIPT. Precursor m/z 718.0, 3 μscans, 10 multiple-fills, r = 30,000, average of 8 spectra, 8.75 sec elapsed scan time/spectrum. B, expanded view of the 1250–1500 m/z region of A (indicated in red). Ions of type c′ and z•′ are indicated. C, sequence coverage obtained from MS/MS spectrum shown in A. The locations of PTMs are indicated.
Compared with other histones, H1 presents a more difficult situation for on-line LC-MS analysis of the intact form. The most common human isoforms of H1 range from 194 to 226 amino acids in length and, due to their larger size, require higher resolution, shorter gas-phase ion/ion reaction times, more multiple fills of the C-trap, and an extended mass range. Initial sample runs revealed the presence of at least two forms of H1, H1.4 and H1.2. MS/MS spectra acquired in data-dependent mode using 2-ms ETD/25-ms IIPT are shown in supplemental Fig. 7 along with the observed sequence coverage (H1.4, 72%, and H1.2, 67%, P16403 and P10412, respectively). Additional targeted analyses elucidated several additional unique H1 proteoforms. One of these was a form of H1.2 containing a previously uncharacterized single amino acid substitution, A141T. The spectrum and coverage are shown in Fig. 4 (hand-annotated version shown in supplemental Fig. 8).
Fig. 4.

ETD/IIPT MS/MS spectrum of intact H1 proteoform. A, ETD/IIPT spectrum recorded on [M + 23H]23+ ions from H1.2 (P16403) with an undocumented single amino acid substitution (A141T) using 4-ms ETD and 20-ms IIPT. Precursor m/z 927.5, 1 μscan, 25 multiple-fills, r = 100,000 at 400 m/z, average of 11 spectra, 9.38-s elapsed scan time/spectrum (27.6 s for single fill/transient equivalent spectrum). B, sequence coverage obtained from MS/MS spectrum shown in A. The location of the Ala → Thr amino acid substitution is indicated with an asterisk.
Parallel Ion Parking during IIPT
Histone fraction H2B presented the most difficult challenge due to the number of unique proteoforms it contained. Fig. 5, A and B, displays the averaged (five spectra, r = 120,000 at 400 m/z) high resolution ESI spectra recorded on intact H2B proteoforms eluting over ∼45 s. Isolation of the region containing the [M + 17H]+17 (Fig. 5C, m/z 790–830) reveals several distinct isotopic clusters, each spaced less than 1 m/z apart. Using a 2 m/z isolation window, all MS/MS spectra acquired on precursors possessing ETD-compatible charge states contained fragment ions unique to at least two H2B proteoforms. Upon close inspection of Fig. 5C, two +8 precursors can be seen between m/z 825–830 (labeled D and E, also see supplemental Fig. 9, D and E). This same 40 m/z window was isolated and subjected to 140-ms IIPT. During the IIPT reaction, a waveform, including frequencies mapping to m/z 2300–3900, was applied. As the precursor ions are charge-reduced, their m/z values increase, and their frequencies of oscillation come into resonance with the applied waveform. This occurs when an ion's m/z reaches the 2300–3900 m/z range. At this point, these ions are activated, increasing their velocity relative to the reagent ions and slowing the rate of further proton transfer.
Fig. 5.
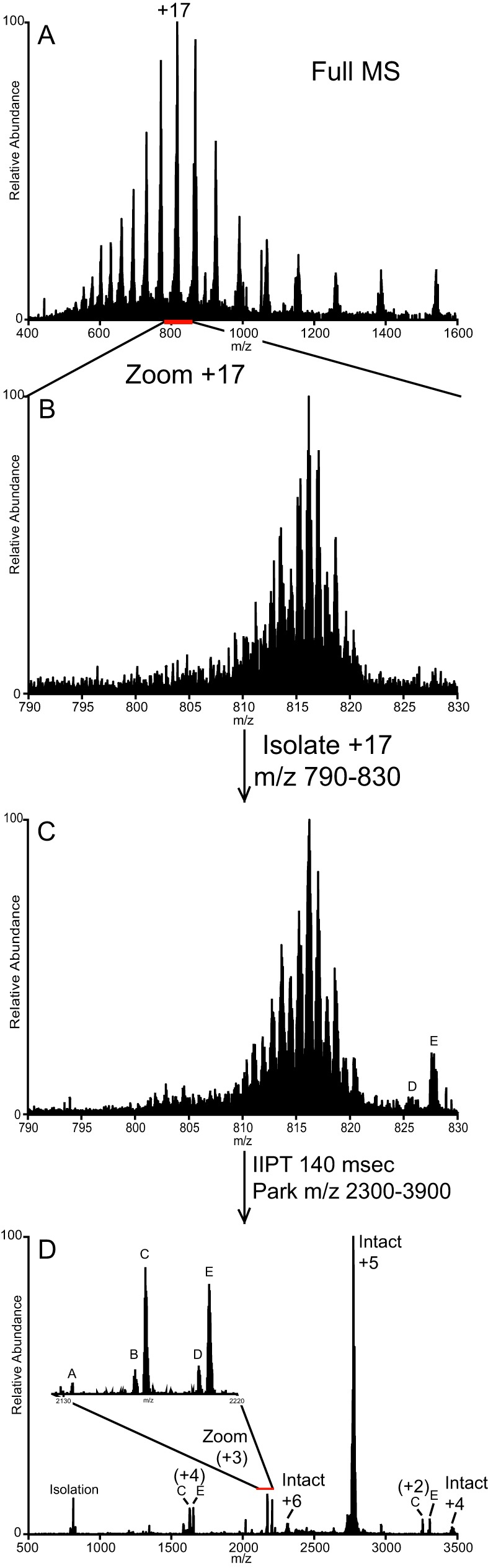
Parallel ion parking during IIPT reveals previously unobserved signals. A, averaged ESI full MS spectrum depicting the charge state distribution of intact H2B acquired over 45 s (five spectra total). The +17 change state is indicated. B, expanded view of the region indicated in red (m/z 790–830) from A. C, spectrum produced upon isolation of ions possessing m/z 790–830. Truncated proteoforms of +8 charge are labeled D and E. D, MS/MS spectrum produced upon isolation of ions possessing m/z 790–830 and 140-ms IIPT with charge-reduced precursor “parked” in the 2300–3900 m/z range. The inset shows an expanded view of newly observed truncated histone H2B proteoforms (labeled A–C).
Fig. 5D displays the product ion spectrum following 140-ms IIPT/parking. The vast majority of the ion current lies within 2730–2800 m/z range where the “parked” intact H2B [M + 5H]+5 ions lie. An enlarged inset of this region is shown in supplemental Fig. 10. Signals corresponding to precursor ions that were not fully reacted to the +5 charge state or that were not fully parked and underwent further proton transfer to the +4 are indicated. Close inspection of the spectrum shown in Fig. 5D reveals several signals of lesser charged species (labeled A–E). Their observed charge state and m/z as well as their calculated m/z and charge state prior to IIPT are given in Table I. Comparisons of the observed masses to calculated theoretical values suggest that these signals are derived from truncated forms of H2B that co-eluted with intact H2B during the time these spectra were acquired (identity and mass accuracy of the most abundant isotopologue in each envelope are shown in Table I). Peptides containing residues 66–125, 67–125, and 68–125 derived from two isoforms of H2B match within 5 ppm mass error. Expected signals for these species A–C are not observed in Fig. 5C (also see supplemental Fig. 9), presumably due to the density of unique species within the window. Note that the assigned identities for these truncated H2Bs are not confirmed by MS/MS and are based on mass accuracy and on MS/MS-confirmed identities of the intact H2Bs observed in this fraction. These truncated H2Bs could be derived from several other proteoforms of intact H2B due to their sequence similarity.
Table I. Truncated H2B proteoforms observed in IIPT/parallel ion parking MS/MS spectrum.
Observed m/z and theoretical precursor m/z values are shown. The masses reported are those of the most abundant isotopologue in the charge state envelope. UniProt sequences from isoforms of H2B with residues matching those contained in the truncated H2B are given. Note these are not confirmed by MS/MS, and identification is based on accurate mass and MS/MS spectra of other truncated H2Bs seen in LC-MS analyses performed without parallel ion parking. Observed mass accuracies are reported in ppm and were calculated from the most abundant isotopologue.
| Peak | Observed m/z | Precursor m/z | Possible H2B isoform | Residues | Observed mass accuracy (ppm) |
|---|---|---|---|---|---|
| A | 2134.4929 (+3) | 801.0661 (+8) | P62807 | 68–125 | −2.16 |
| B | 2167.1735 (+3) | 813.3221 (+8) | O60814 | 67–125 | −3.09 |
| C | 2172.5071 (+3) | 815.3215 (+8) | P62807 | 67–125 | −2.16 |
| D | 2200.1957 (+3) | 825.7057 (+8) | O60814 | 66–125 | −3.32 |
| E | 2205.5313 (+3) | 827.7050 (+8) | P62807 | 66–125 | −1.50 |
Characterization of Truncated H2B and H2A Proteoforms Derived from Butyrate-treated HeLa Cells
Eluting after the intact H2B proteoforms were several fairly abundant forms of truncated H2B. The base peak chromatogram and the sequence of the most abundant intact H2B (H2B-1C, P62807) observed in the fraction are shown in Fig. 6. The solid black lines in Fig. 6 indicate “primary” cleavage sites mapped by MS. Less abundant cleavage sites in Fig. 6 are marked with gray lines. This is not a comprehensive account of all the cleavages that are observed, but rather only those that are most abundant. Complementary N-terminal peptides, 1–25 and 1–51, are present and elute near the front of the gradient. They are less abundant and are more difficult to characterize because their signals are split among multiple acetylated forms (+1–3 lysine-acetyl confirmed by MS/MS). The acetylations are shared among multiple sites, including Lys-11, Lys-12, Lys-15, Lys-16, and Lys-20, which are all sites that were confirmed by MS/MS in the intact form and that have previously been identified by MS in an earlier study (35). Truncations derived from two additional H2B isoforms (possibly H2B.2-E, Q16778, and H2B.1-K, O60814) were also present. Note that the sequences of the intact forms found in the sample differ by only one residue (V39I for H2B.2-E or S124A for H2B.1-K), and so distinction between truncated peptides can only be made if both unique residues are contained within the peptide. If only one of these residues is present, then only one of the two additional isoforms can be excluded. There is also no way to distinguish the modified (acetylated) form of the intact protein from which the C-terminal peptides are derived. Up to 93% sequence coverage is obtained by ETD/IIPT MS/MS of the three most abundant C-terminal containing peptides (90%, 26–125; 93%, 52–125; and 84%, 50–125). The spectrum and sequence coverage of the 26–125 peptide are shown in supplemental Fig. 11.
Fig. 6.

Fraction H2B contains truncated H2B proteoforms. A, base peak chromatogram from LC-MS analysis of fraction H2B. B, sequence of the most abundant and intact H2B contained within the fraction (H2B.1-C, P62807). Solid bold lines indicate “primary” cleavage sites mapped by MS. The gray lines indicate additional less abundant cleavage sites.
Additionally, dozens of truncated H2As were observed in the H2A1 fraction. These ranged in size from 10 to 75 residues in length, and each account for less than 1% of the total ion current relative to intact H2A from the same fraction. The base peak chromatogram is shown in Fig. 7, A–C, and indicates where truncated species elute relative to intact H2A. Fig. 7D shows the sequence of the most abundant intact H2A observed in the fraction (H2A2A, Q6FI13). Again, the solid black lines in Fig. 7 indicate “primary” cleavage sites, and the less abundant cleavage sites are indicated with gray lines. This is not a comprehensive account of all the truncated histone forms observed, only the most abundant. Supplemental Table 1 lists these truncated proteoforms along with their respective abundances. The most abundant of these was the N terminus-containing proteoform cleaved C-terminal to Gly-44. Intact forms of H2A identified on the fraction were H2A2A and H2A2C (Q16777), which differ by their last 5–6 C-terminal residues. Because of this, there is no way to determine whether the N terminus-containing truncations were derived from H2A2A or H2A2C. MS/MS spectra of truncated species are shown in supplemental Fig. 12.
Fig. 7.

Fraction H2A1 contains truncated H2A proteoforms. Inspection of the total ion chromatogram and full MS spectra revealed the presence of several peptidic species in a wide range of sizes. A, base peak chromatogram from LC-MS analysis of fraction H2A1. B and C, ESI full MS spectra averaged over the time periods indicated in red from A. Highly charged peptidic species are observed. D, sequence of the most abundant and intact H2A isoform contained within the fraction (H2A.2-A, Q6FI13). Solid bold lines indicate primary cleavage sites mapped by MS. The gray lines indicate additional less abundant cleavage sites.
Identification of Histone H2A Clipping Enzyme by in Vitro proteolytic Assay
Given the large variety of truncated histone proteoforms identified, we further investigated whether cathepsin L, a protease responsible for histone H3 and H3.3 clipping (9, 37, 38), is capable of cleaving histone H2A. To do so, we performed an in vitro assay incubating recombinant histone H2A with cathepsin L and characterized the cleavage specificity using nano-LC coupled to an Orbitrap Fusion with FETD. Supplemental Fig. 13 and supplemental Table 2 show that the signal of intact histone H2A decreased by ∼3 orders of magnitude after 1 h of incubation, while the clipped forms increased in abundance over time. Specifically, we determined the clipped proteoform Gly-44 (cleaved after glycine 44) as one of the most abundant cleaved proteoforms and confirmed its sequence by ETD MS/MS (supplemental Fig. 13C). We also observed several other C-terminal peptides (supplemental Table 2). Taken together, our data suggest that cathepsin L might have a larger variety of targets than currently described.
DISCUSSION
In the commercial configuration of the Orbitrap Velos Pro/EliteTM, the reagent ion source is located at the rear of the instrument, requiring that reagent anions traverse the C-trap to reach the ion trap, where ion/ion reactions are performed. By putting the reagent ion source in the front of the instrument, the C-trap is made available for the storage of fragments from multiple iterations of ion/ion reactions prior to transient acquisition in the Orbitrap. In this way, we are able to dramatically increase fragment ion current. The multiple fill strategy for improving SNRs has a number of advantages over transient summing or spectral averaging. Chief among these is that the time that it takes to acquire spectra of multiple C-trap fills is less than the time it takes to acquire transients from the same number of single C-trap fills to be averaged or summed. Specifically, if a transient takes 380 ms to acquire, an additional fragment ion fill can be accomplished in less than ½ that time, i.e. ∼130 ms for overhead plus time for ETD/IIPT activation. Additionally, SNRs improve faster using multiple fills compared with summing transients or averaging spectra and so fewer transients are needed to produce a useful spectrum for analysis. This enables the acquisition of high quality spectra on a time scale more compatible with chromatography.
The power of top-down mass spectrometry lies in its ability to provide an unambiguous view of protein expression dynamics by distinguishing closely related proteoforms from one another while also observing any combinations of PTMs. Bottom-up MS strategies are not capable of this, which is why there has been a significant push in the field of proteomics to develop instruments and methodologies that enable high throughput, top-down mass spectrometry of intact proteins. Here, we demonstrated the power of FETD-enabled sequential ion/ion reactions along with multiple fragment ion fills through their coupling with on-line HPLC and application to the analysis of intact histones. Data-dependent and targeted LC-MS experiments were employed to record high resolution MS/MS spectra of histones from butyrate-treated HeLa cells. Using these spectra, several unique intact histone proteoforms were identified with up to 81% sequence coverage. Examples shown here contain multiple modifications, including single amino acid substitutions, methylations, and acetylations. Additionally, many truncated forms of histones H2A and H2B were characterized with up to 93% sequence coverage.
Histone proteolysis has been implicated in several important regulatory processes, yet it remains poorly understood. Only eight histone-specific proteases are known, and their biological significance has not been extensively defined (39). This area of study is clearly poised for significant expansion, as evidenced by the identification of two H2A-specific proteases and several novel histone clipping events in just the past few years (39–42). Discerning whether any histone-cleaving event is the result of continuous histone turnover or sample degradation versus an alternative biological process with epigenetic implications is challenging (36). Our in vitro data suggest that cathepsin L might be a potential candidate to explain some of the truncated histone H2A proteoforms we observed in the butyrate-treated HeLa cell histone samples. We found that cleavage C-terminal to Gly-44 produced the most abundant truncated H2A proteoforms observed in both the in vitro experiments and those derived in vivo from HeLa cells. Differences between the in vitro and the HeLa cell data in the abundance of other truncated proteoforms might be explained by improper folding of the recombinant histone or by the absence of a proper nucleosome structure in the in vitro experiments. In the future, we hope that the tools we describe here will aid in deciphering whether or not these truncated histone proteoforms are biologically regulated and contribute to epigenetic mechanisms.
That we are able to observe these low level truncated histone proteoforms in simple top-down LC-MS experiments further demonstrates the utility of our methodologies as it is very likely that bottom-up- or middle-down-based proteomics approaches would have missed cleavage events like those we describe in our analyses. This could happen for a number of reasons. First, given the low levels at which these truncations were observed (which, for any single truncated form, is less than 1% relative to the intact histone), along with the added complexity that is generated when samples are digested for MS proteomic studies, peptides of this type may have not been detected or may have simply gone unnoticed by investigators. Several of the generated peptides derived from originally truncated species might have been too small to be retained by the HPLC column. Quantitative analysis of the relative abundance of unique truncated forms would be difficult to perform as evidence for some unique truncated species could be lost or could be below the limit of detection. Also, the ability to correlate the endogenous histone proteolysis with any specific histone variant or PTM state would not be possible unless those PTMs or sequence variations and the cleavage sites were contained within the same peptide produced in the sample preparative digestion. Only by determining the primary structures of these species, as they existed in the cell, can one draw these potentially important biological parallels.
Furthermore, the use of parallel ion parking during IIPT is demonstrated in an analysis of H2B proteoforms. Chromatographic separation is made difficult by the fact that several H2B proteoforms differ from one another by just a single amino acid residue. At high ETD-compatible charge states, multiple proteoforms are present within a relatively small m/z range. Upon data-dependent selection, often more than one proteoform is isolated and fragmented, resulting in mixed MS/MS spectra, making identification difficult. Additionally, as demonstrated here, overlapping signals make it difficult to tease out the number of unique species present. By using IIPT to charge-reduce the precursors contained within a wide isolation window and parking the products in a desired m/z range, we are able separate species that, prior to IIPT, possessed roughly the same m/z but a significantly different mass. This reveals signals that can be attributed to less abundant H2B proteoforms that were not observed in the MS1 spectrum nor the isolation-only spectrum due to their proximity to more abundant signals derived from other species. This is the first time ion parking has been demonstrated on a commercially available linear ion trap using ion cloud densities that lend themselves to high resolution MS/MS analyses. We believe that parallel ion parking will become a powerful tool in identifying proteins in a mixture when used in conjunction with MS/MS fragmentation methods and multiple fill technology. Most exciting is the potential for this technology to allow differentiation and identification of closely related proteoforms that are difficult to separate chromatographically, like H2B. As demonstrated in supplemental Fig. 10, following IIPT/parking, we are able to spread apart isotopic clusters such that their centers are ∼3 m/z apart (compared with <1 m/z prior to IIPT) without losing signal due to further proton transfer. This is a stepping stone to enabling higher resolution isolation on the MS3 level. However, doing so will require further instrument modification to increase the precursor ion capacity of the linear ion trap to be able to observe fragments in MS3 spectra following IIPT.
Supplementary Material
Acknowledgments
We thank John E. P. Syka, Christopher Mullen, and Lee Early of Thermo Fisher Scientific for aiding in the modification of the Orbitrap Velos Pro to include FETD and for helpful discussion. We also thank Dina L. Bai for critical reading of early manuscript revisions and Greg T. Blakney for helpful discussion.
Footnotes
Author contributions: L.C.A., J.S., B.A.G., and D.F.H. designed the research; L.C.A., K.R.K., S.A.U., M.C., and S.S. performed the research; A.M.E. contributed new reagents or analytic tools; L.C.A., S.A.U., M.C., S.S., and J.S. analyzed data; and L.C.A., K.R.K., S.A.U., M.C., S.S., and B.A.G. wrote the paper.
* This work was supported in part by National Institutes of Health Grants GM037537 and AI033993 (to D.F.H.). This work was also supported by NIH Grants GM110174 and AI118891 and a National Science Foundation Faculty Early Career Award (to B.A.G). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental materials.
This article contains supplemental materials.
The authors declare that they have no conflicts of interest with the contents of this article.
1 The abbreviations used are:
- PTM
- post-translational modification
- HCD
- higher-energy collisional dissociation
- ETD
- electron transfer dissociation
- ECD
- electron capture dissociation
- MS/MS
- tandem mass spectrometry
- SNR
- signal-to-noise ratio
- FETD
- front-end electron transfer dissociation
- IIPT
- ion/ion proton transfer
- m/z
- mass to charge ratio
- v/v
- volume/volume percent
- AGC
- automatic gain control
- RP-HPLC
- reverse-phase high-performance liquid chromatography
- ppm
- parts per million.
REFERENCES
- 1. Anderson L. C., English A. M., Wang W., Bai D. L., Shabanowitz J., and Hunt D. F. (2015) Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry. Int. J. Mass Spectrometry 377, 617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kornberg R. D. (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871 [DOI] [PubMed] [Google Scholar]
- 3. Kornberg R. D., and Thomas J. O. (1974) Chromatin structure; oligomers of the histones. Science 184, 865–868 [DOI] [PubMed] [Google Scholar]
- 4. Allis C. D., Jenuwein T., and Reinberg D. eds; Caparros M.-L. assoc. ed. (2007) Epigenetics 1st Ed., 28–29, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 5. Cheung P., Allis C. D., and Sassone-Corsi P. (2000) Signaling to chromatin through histone modifications. Cell 103, 263–271 [DOI] [PubMed] [Google Scholar]
- 6. Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 7. Franklin S. G., and Zweidler A. (1977) Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature 266, 273–275 [DOI] [PubMed] [Google Scholar]
- 8. Bernstein E., and Hake S. B. (2006) The nucleosome: A little variation goes a long way. Biochem. Cell Biol. 84, 505–517 [DOI] [PubMed] [Google Scholar]
- 9. Duncan E. M., Muratore-Schroeder T. L., Cook R. G., Garcia B. A., Shabanowitz J., Hunt D. F., and Allis C. D. (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin R., Cook R. G., and Allis C. D. (1991) Proteolytic removal of core histone amino termini and dephosphorylation of histone H1 correlate with the formation of condensed chromatin and transcriptional silencing during Tetrahymena macronuclear development. Genes Dev. 5, 1601–1610 [DOI] [PubMed] [Google Scholar]
- 11. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., and Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., and Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zubarev R. A., Kelleher N. L., and McLafferty F. W. (1998) Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120, 3265–3266 [DOI] [PubMed] [Google Scholar]
- 14. Bonenfant D., Coulot M., Towbin H., Schindler P., and van Oostrum J. (2006) Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 5, 541–552 [DOI] [PubMed] [Google Scholar]
- 15. Arnaudo A. M., Molden R. C., and Garcia B. A. (2011) Revealing histone variant induced changes via quantitative proteomics. Crit. Rev. Biochem. Mol. Biol. 46, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sidoli S., Schwämmle V., Ruminowicz C., Hansen T. A., Wu X., Helin K., and Jensen O. N. (2014) Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 14, 2200–2211 [DOI] [PubMed] [Google Scholar]
- 17. Tian Z., Tolić N., Zhao R., Moore R. J., Hengel S. M., Robinson E. W., Stenoien D. L., Wu S., Smith R. D., and Paša-Tolić L. (2012) Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 13, R86–2012-13–10-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dang X., Scotcher J., Wu S., Chu R. K., Tolić N., Ntai I., Thomas P. M., Fellers R. T., Early B. P., Zheng Y., Durbin K. R., Leduc R. D., Wolff J. J., Thompson C. J., Pan J., Han J., Shaw J. B., Salisbury J. P., Easterling M., Borchers C. H., Brodbelt J. S., Agar J. N., Paša-Tolić L., Kelleher N. L., and Young N. L. (2014) The first pilot project of the consortium for top-down proteomics: a status report. Proteomics 14, 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y., Hoover M. E., Dang X., Shomo A. A., Guan X., Marshall A. G., Freitas M. A., and Young N. L. (2015) Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence. Mol. Cell. Proteomics, 3, 818–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tvardovskiy A., Wrzesinski K., Sidoli S., Fey S. J., Rogowska-Wrzesinska A., and Jensen O. N. (2015) Top-down and middle-down protein analysis reveals that intact and clipped human histones differ in post-translational modification patterns. Mol. Cell. Proteomics 14, 3142–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Earley L., Anderson L. C., Bai D. L., Mullen C., Syka J. E., English A. M., Dunyach J. J., Stafford G. C. Jr, Shabanowitz J., Hunt D. F., and Compton P. D. (2013) Front-end electron transfer dissociation: a new ionization source. Anal. Chem. 85, 8385–8390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stephenson J. L. Jr., and McLuckey S. A. (1998) Simplification of product ion spectra derived from multiply charged parent ions via ion/ion chemistry. Anal. Chem. 70, 3533–3544 [DOI] [PubMed] [Google Scholar]
- 23. Chi A., Bai D. L., Geer L. Y., Shabanowitz J., and Hunt D. F. (2007) Analysis of intact proteins on a chromatographic time scale by electron transfer dissociation tandem mass spectrometry. Int. J. Mass. Spectrom. 259, 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLuckey S. A., Reid G. E., and Wells J. M. (2002) Ion parking during ion/ion reactions in electrodynamic ion traps. Anal. Chem. 74, 336–346 [DOI] [PubMed] [Google Scholar]
- 25. Chrisman P. A., Pitteri S. J., and McLuckey S. A. (2006) Parallel ion parking of protein mixtures. Anal. Chem. 78, 310–316 [DOI] [PubMed] [Google Scholar]
- 26. Makarov A., Denisov E., and Lange O. (2009) Performance evaluation of a high-field Orbitrap mass analyzer. J. Am. Soc. Mass Spectrom. 20, 1391–1396 [DOI] [PubMed] [Google Scholar]
- 27. Lange O., Damoc E., Wieghaus A., and Makarov A. (2014) Enhanced Fourier transform for orbitrap mass spectrometry. Int. J. Mass Spectrom. 369, 16–22 [Google Scholar]
- 28. Udeshi N. D., Compton P. D., Shabanowitz J., Hunt D. F., and Rose K. L. (2008) Methods for analyzing peptides and proteins on a chromatographic timescale by electron-transfer dissociation mass spectrometry. Nat. Protoc. 3, 1709–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin S., and Garcia B. A. (2012) Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roth M. J., Plymire D. A., Chang A. N., Kim J., Maresh E. M., Larson S. E., and Patrie S. M. (2011) Sensitive and reproducible intact mass analysis of complex protein mixtures with superficially porous capillary reversed-phase liquid chromatography mass spectrometry. Anal. Chem. 83, 9586–9592 [DOI] [PubMed] [Google Scholar]
- 31. Martin S. E., Shabanowitz J., Hunt D. F., and Marto J. A. (2000) Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 72, 4266–4274 [DOI] [PubMed] [Google Scholar]
- 32. Ong S. E., and Mann M. (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 33. Candido E. P., Reeves R., and Davie J. R. (1978) Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14, 105–113 [DOI] [PubMed] [Google Scholar]
- 34. Shechter D., Dormann H. L., Allis C. D., and Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 35. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., and Jensen O. N. (2006) Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics 5, 1314–1325 [DOI] [PubMed] [Google Scholar]
- 36. Dhaenens M., Glibert P., Meert P., Vossaert L., and Deforce D. (2015) Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’. Bioessays 37, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams-Cioaba M. A., Krupa J. C., Xu C., Mort J. S., and Min J. (2011) Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat. Commun. 2, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duarte L. F., Young A. R., Wang Z., Wu H. A., Panda T., Kou Y., Kapoor A., Hasson D., Mills N. R., Ma'ayan A., Narita M., and Bernstein E. (2014) Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 5, 5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Azad G. K., and Tomar R. S. (2014) Proteolytic clipping of histone tails: the emerging role of histone proteases in regulation of various biological processes. Mol. Biol. Rep. 41, 2717–2730 [DOI] [PubMed] [Google Scholar]
- 40. Panda P., Chaturvedi M. M., Panda A. K., Suar M., and Purohit J. S. (2013) Purification and characterization of a novel histone H2A specific protease (H2Asp) from chicken liver nuclear extract. Gene 512, 47–54 [DOI] [PubMed] [Google Scholar]
- 41. Dhaenens M., Glibert P., Lambrecht S., Vossaert L., Van Steendam K., Elewaut D., and Deforce D. (2014) Neutrophil elastase in the capacity of the “H2A-specific protease”. Int. J. Biochem. Cell Biol. 51, 39–44 [DOI] [PubMed] [Google Scholar]
- 42. Vossaert L., Meert P., Scheerlinck E., Glibert P., Van Roy N., Heindryckx B., De Sutter P., Dhaenens M., and Deforce D. (2014) Identification of histone H3 clipping activity in human embryonic stem cells. Stem Cell Res. 13, 123–134 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.