

Abstract
We document a unique DNA recombination between polyomavirus JC (JC virus [JCV]) and Epstein-Barr virus (EBV) at sequences of JCV found infecting the brain. Archetype JCV is present in bone marrow and uroepithelial cells of most adults. During immunosuppression, JCV can infect the brain, causing a demyelinating disease, progressive multifocal leukoencephalopathy. Rearrangements in the archetype noncoding control region are necessary for neurovirulence. Two NCCR deletions and a duplication occur at sequences of homology with EBV, present latently in B cells, which may be coinfected with both viruses. Recombination between JCV and EBV occurs in B lymphoblasts at a sequence essential for JCV neurovirulence and in cerebrospinal fluid of immunosuppressed patients with multiple sclerosis, those susceptible to progressive multifocal leukoencephalopathy. Interviral recombination is a model for conferring advantages on JCV in the brain. It can alter a critical noncoding control region sequence and potentially facilitate use of EBV DNA abilities to transfer among different cell types.
Keywords: progressive multifocal leukoencephalopathy, PML, multiple sclerosis, AIDS, DNA replication, interviral recombination
JC virus (JCV) and Epstein-Barr virus (EBV) are DNA viruses of the polyomavirus and herpesvirus families, respectively. EBV (170–180 kb, linear with latent circular episomes) is nearly ubiquitous in adults, where it is normally latent in B lymphocytes. In addition to infectious mononucleosis, EBV is causally linked to nasopharyngeal carcinoma, and lymphomas, often under conditions of immunosuppression (IS) [1]. A recent report documents that nearly 75% of adults have been infected by JCV although there are age-related variations in this number [2]. JCV (5.1 kb, circular) normally inhabits bone marrow and uroepithelial cells as an archetypal form existing asymptomatically. In AIDS and in persons treated with certain IS agents, however, the virus infects oligodendroglial cells in the brain, causing progressive multifocal leukoencephalopathy (PML), a frequently fatal demyelinating disease [3–5].
The sequence of JCV infecting glial brain cells almost always differs from that of the archetype in the noncoding control region (NCCR), the region containing the origin of DNA replication and separating the early and late gene transcription units. There are 3 NCCR DNA rearrangements that characterize PML: a deletion of approximately 23 base pairs (bp), another deletion of 66 bp and a duplication of 98 bp that remain after the 2 archetype deletions [6, 7]. Although archetype JCV can be detected in some cases of PML, all 3 rearrangements are associated with the worst prognoses [8]. These rearrangements in PML can all be derived from the archetype sequence, and they may do so within a single individual in a sequential order, possibly in different cell types in transit from distal tissues to the brain [9]. In cerebellar granular cell degeneration, distinct from PML, patients frequently have mutations in the JCV VP1 coding region. In several JCV clones from this disease the virus had both 23- and 66-bp NCCR deletions seen in PML but not the duplication [10–12], essentially in agreement with our concept of sequentiality. Because PML does not develop in all persons with these 3 rearrangements, they are considered necessary but not sufficient.
Coinfection of individual B cells by both JCV and EBV has been visualized in a central nervous system lymphoma [13]. JCV reportedly replicates in B cells, including those infected with EBV [14]. B cells reportedly can carry JCV to the brain [15] even in the absence of persistent replication. JCV can induce DNA double-strand breaks [16], in cells including B cells [17]. These breaks can initiate recombination [18]. JCV is present in bone marrow [19, 20] as well as in CD19+ and CD34+ B cells in patients with multiple sclerosis (MS) [21]. A regulatory interaction between JCV and EBV in B cells has previously been implied [22]. Here we present evidence that JCV and EBV can undergo a unique interviral recombination that may help facilitate the ability of JCV to adapt to neurovirulence in glial cells. This recombination reveals a mechanism of opportunistic creation of virus variants that represents a new consideration in disease development.
METHODS
Cell Lines and Primary Cells
Cell lines used were Raji, a B lymphoma cell type (American Type Culture Collection [ATCC] CCL-86), and SVG cells, astroglia constitutively expressing SV40 large T antigen (ATCC CRL-8621). CD19+ primary B cells were ordered from HemaCare Bioresearch Products. Raji and B cells were cultured in suspension [23]. SVG cells were cultured as described in Supplementary Methods.
DNA From Cultured B Lymphoblasts
DNA recovered from Raji cells was amplified using polymerase chain reaction (PCR), as described in the Supplementary Methods.
DNA From Cerebrospinal Fluid of Patients With MS
Patients' cerebrospinal fluid (CSF) samples were obtained from PrecisionMed. All of the patients were properly consented volunteer donors, and CSF samples were collected using institutional review board–approved protocols. All samples are from the United States and were obtained through several participating physicians. Four available samples were from patients with MS having received treatment with natalizumab alone as the only immunomodulatory or IS agent. Samples from patients and MS stages of patients studied are given in Supplementary Methods, as are purification and amplification methods for CSF DNA.
Cell Nucleofection
Raji cells and B cells were subjected to nucleofection, as described in the Supplementary Methods.
Preparation for Indirect Immunofluorescence Staining
Raji cells, primary B cells and SVG cells were fixed and stained as described in the Supplementary Methods [24]. Throughout the study, all data points were repeated at least 3 times. With regard to patient samples, which are difficult to obtain, data from individual patients were repeated.
RESULTS
We used a temperature range of PCR amplifications to reveal the most prominent secondary structure sites of DNA polymerase stalling [9] (Supplementary Figure 1). These sites in the NCCR sequence are noted by red arrows in Figure 1. NCCR rearrangements, shown for the Mad-1 strain of neurovirulent JCV, illustrated with an archetype sequence in Figure 1, include 23-bp and 66-bp deletions. A 98-bp duplication of the sequence remaining after the deletions begins at the first black-lettered c after the origin. An analysis of our own sequencing data, those of several groups, including Reid et al [4], and of nearly 3000 JCV entries in GenBank has allowed us to draw certain conclusions as to the frequencies of the different NCCR deletions before PML. There are multiple NCCR entries containing one of the PML-related deletions but not the other. Of these, 93% contain the larger, approximately 66-bp deletion, and 7% contain the smaller, approximately 23-bp deletion. Thus the 66-bp deletion is more frequent and probably occurs before the 23-bp deletion.
Figure 1.
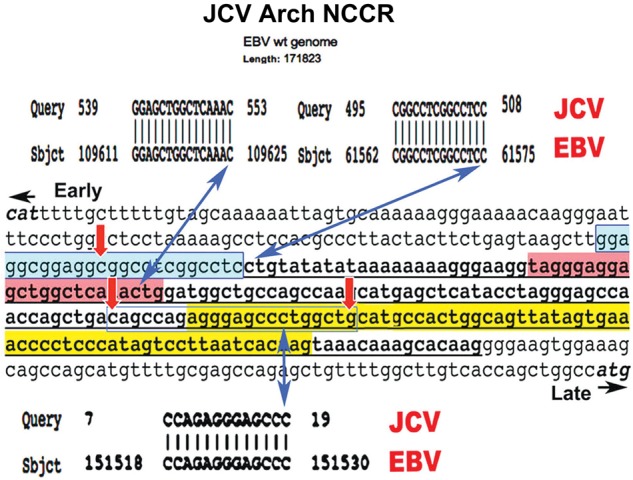
Sequence identities at 3 strategic sites in both Epstein-Barr virus (EBV) and the JC virus (JCV) noncoding control region (NCCR). The National Center for Biotechnology Information BLAST algorithm was used to compare the JCV NCCR with EBV. In each case the query was the JCV archetype NCCR (in this case, GenBank JX273163.1) and the subject was the entire EBV wild-type (wt) genome (GenBank 507799.2). The entire NCCR is presented bracketed by start codons for T antigen (CAT) and agnoprotein (ATG). The T-antigen binding region is shaded in blue, the 23–base pair (bp) deletion in red, and the 66-bp deletion in yellow. The 98-bp duplication is underlined. Three sequences identical between the NCCR and EBV were found, and no others. Three double-headed blue arrows indicate the identities' 3′ end points. All 3 identities correspond to sites in the NCCR critical for recombination leading to JCV neurovirulence. The 15-bp identity corresponds to the center of the 23-bp JCV deletion. Based on the frequency of sequence occurrence in DNA, the odds of the presence of these 15 bp once in both EBV and JCV genomes is P < 10−8. The 14-bp identity contains 2 canonical JCV T-antigen–binding elements and ends at the first nucleotide of the JCV 98-bp duplication. The 13-bp identity is central to the palindrome beginning the JCV 66-bp deletion.
Elements at Key Recombination Sites in Neurovirulent JCV Identical to Sequences in EBV
Because we intended to determine whether JCV DNA recombination can occur in B cells, which may harbor EBV, we used the National Center for Biotechnology Information BLAST algorithm to compare the entire JCV archetype NCCR sequence with the entire wild-type EBV genome (GenBank 507799.2), using a window of 10 nucleotides to look for identities. Only 3 identities longer than that were found (Figure 1), containing 15, 14, and 13 consecutive nucleotide identities. All 3 identities are at ends of the 3 major JCV rearrangements (blue arrows in Figure 1; JCV sequences from GenBank, JO2226.1 [Mad-1] and JX273163.1 [10]). The 15-bp identity is a complex sequence expected to occur once per 109 nucleotides. The probability of this occurring in the EBV genome is P < 104. The probability that it would occur in both viral genomes is infinitesimally small. The 14-bp identity is at an end of the large T-antigen–binding palindrome and at the precise early region end of the 98-bp duplication. It is a degenerate and repetitive sequence and can be found at several sites in human and herpes virus genomes. The 13-bp identity is at the end of a palindrome marking the early region end of the 66-bp deletion. Again, the probability that this identity would occur in both EBV and JCV is very small. The proximity to palindromes and to elongation stop points (Figure 1) renders all identities good candidates for recombination between JCV and EBV initiated by abortive JCV DNA replication. The 15-bp identity is within the 23-bp JCV deletion and is the first sequence information linking this deletion to a potentially specific recombination event.
Sequences Identical to Those in the JCV NCCR in Coding Sequences of EBV Proteins Expressed in Lytic Infection
The 15-bp identity between JCV and EBV is in the EBV gene encoding the protein, BGLF5 (Figure 2A). No alkaline exonuclease of any other herpesvirus possesses the VFEPAP motif (Figure 2A) encoded partially by the 15-bp homology. The 14-bp identity with JCV is in the EBV gene encoding BOLF1, a putative tegument protein (Figure 2B). The 13-bp identity is in the EBV gene encoding LF1, a multifunctional dUTPase (Figure 2C). It is unlikely that recombination events at these sites would be detected in vivo in EBV. There is no foreseeable advantage to EBV for recombination at any of these sites, in which case recombinants would not prevail. In focusing on the 15-bp identity, we can address an important question: can recombination between these 2 viral genomes be detected in a cell harboring both?
Figure 2.
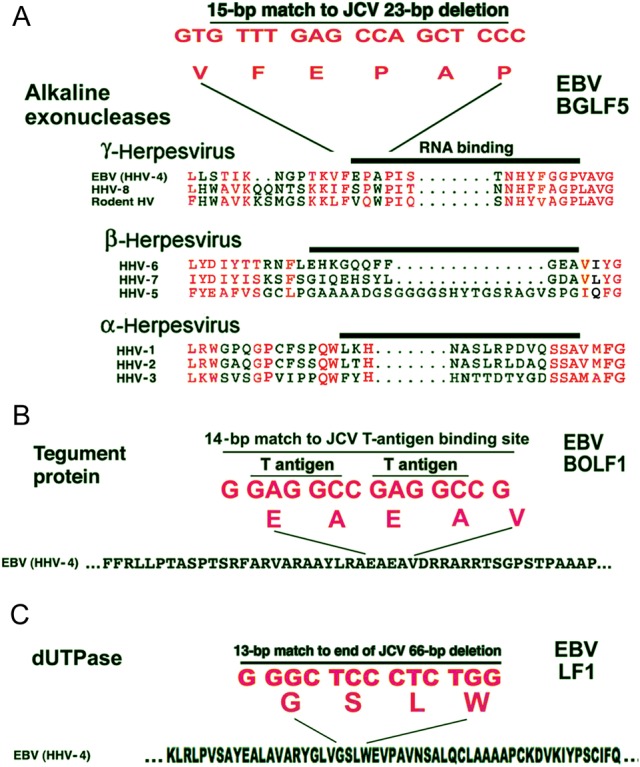
All 3 JC virus (JCV) noncoding control region (NCCR) homologies to Epstein-Barr virus (EBV) are located in EBV proteins essential for lytic infection. A, Alignments were with CLUSTAL W. Red lettering in amino acids denotes identity or strong conservation within a given class of herpesvirus. No other herpesvirus has the 15–base pair (bp) nucleotide sequence identical between JCV and EBV and in the EBV BGLF5 alkaline exonuclease coding sequence. HHV, human herpesvirus; HSV, herpes simplex virus. B, A 14-bp identity between the EBV BOLF1 tegument protein coding sequence and the JCV origin of DNA replication. C. A 13-bp identity to the EBV LF1 dUTPase coding sequence at the early end of a 66-bp sequence deleted in neurovirulent JCV.
Production of JCV Large T Antigen in EBV-Containing B Lymphoblasts and in CD19+ B Cells
To examine recombination at the 15-bp identity, we used Raji cells, a Burkitt lymphoma line harboring EBV. In Raji cells, EBV is latent and nonproductive owing to viral genomic deletions. Raji cells contain 50–80 circular EBV episomes that replicate once per cell cycle using EBV latent oriP and cellular minichromosome maintenance helicase machinery [25, 26]. It has been reported that EBV-containing B cells can support JCV replication [14]. Supplementary Figure 2 shows staining for JCV T antigen in Raji cells, primary CD19+ B cells and positive control SVG glial cells. All have been nucleofected with complete archetype JCV circular DNA. Proteins influencing both JCV and EBV DNA replication are colocalized near the nuclear periphery during times of viral replication [27]. In Supplementary Figure 2F, F’ shows T antigen at the nuclear periphery of B cells. T-antigen binding sites are present throughout human chromosomal DNA [28], but their distance from the 15-bp homology is most likely much greater than it is in EBV.
Recombination Between JCV and EBV at a Site Critical for JCV Neurovirulence
To directly assay for the occurrence of recombination at the 15-bp identity, we initially used nucleofected Raji cells. For Figure 3, a plasmid bearing the complete archetype JCV genome was transfected into Raji cells. At 72 hours, DNA was extracted, and PCR was performed (Figure 3A, Nucl. Arch, nucleofected archetype lane). Several bands were obtained, including a dense one at 177 bp. DNA was similarly analyzed from nonnucleofected cells to which the archetype plasmid was added after lysis. No bands were seen in this mock-archetype lane. The plasmid-only and the no-DNA-template lanes were similarly free of bands. Thus, PCR products containing sequences from both viruses were only obtained when Raji cells were nucleofected with the JCV genome. The band at 177 bp was excised and sequenced using a primer from either JCV or EBV (Figure 3B and 3C). Completely complementary overlapping sequences were obtained using these primers (electropherograms of sequencing in Figure 3). The 15-bp homology was nearly central in the PCR segment.
Figure 3.

Demonstration of homologous recombination in Raji cells between the JC virus (JCV) noncoding control region (NCCR) and Epstein-Barr virus (EBV) at a sequence essential for development of JCV neurovirulence. A, An 8% polyacrylamide gel showing polymerase chain reaction (PCR) products indicative of recombination between JCV and EBV. These products are seen only in one lane, that from which JCV and EBV were both within the living Raji cells together. DNA extraction and PCR are detailed in Supplementary Materials in “Methods” section. Lanes are as follows: Nucl archetype, DNA from Raji cells nucleofected with pUC19-JCV archetype and subjected to hemi-nested PCR; mock archetype, a control in which the JCV-containing plasmid was added after cell lysis and treated exactly as for Nucl archetype; Nucl plasmid, a control in which nucleofection was with pUC19 alone; no template, a control PCR with primers only. The Nucl archetype band (arrow) was excised for sequencing as in B. The higher bands could conceivably represent larger recombinants, which would fit with the model we present in the Supplemental Data. They could also represent recombinants for which one PCR primer has extended beyond its intended stop point, making it seem larger than it is. These bands were not sequenced. B, Sequencing, performed in 2 directions using the primers shown at each end, yielded a segment of 177 base pairs (bp), corresponding to the indicated band in A. The sequences of JCV and EBV overlap at the 15-bp homology, indicated in red. The same recombination transition point is detected if nested PCR primers from the reverse strands of both viruses are used. C, Electropherograms of part of the sequence shown in B, highlighting a single-nucleotide polymorphism within the 15-bp JCV-EBV homology. Abbreviations: M, markers; Nucl. Arch, nucleofected archetype.
The possibility that this result is a PCR artifact is negligible. First, controls in which both viral genomes are present do not yield any recombinant bands. Second, the transition point for recombination occurs exactly at the 15-bp identity. Finally, JCV bears a single-nucleotide polymorphism in the sequence. The strain of JCV used in Figure 3 contains the sequence GTAAAAC, whereas EBV contains the sequence GTCAAAC. Sequencing shows that some products begin with GTA, and others with GTC (boxes in Figure 3). This polymorphism indicates that the products sequenced came from one virus or the other and not because of PCR infidelity. Approximately 0.1% of archetype sequences in GenBank begin the 15-bp homology with GTC, and the others begin with GTA. This single-nucleotide polymorphism helps verify recombination.
Another concern was that our recombination product could be an artifact of overlap-extension synthesis occurring during PCR. We controlled extensively for that. Our mock-archetype lane in Figure 3A uses supercoiled plasmid containing JCV DNA. As another control, we have added single-stranded JCV DNA replication intermediates, purified from a standard NCCR-containing PCR reaction, to an EBV-containing B-cell lysate before PCR, using the same primers as for our mock lane. In this case, we do not see any bands containing EBV-JCV recombined sequences. These negative control results are not shown. They strengthen the genomic sequencing data of Figure 3, suggesting JCV-EBV recombination.
JCV-EBV Recombination Detected in CSF of Patients With MS Treated With IS
MS is an ideal model to search for disease-relevant recombination because MS is strongly serologically linked to EBV [29, 30], and IS treatment is a known cofactor in causing PML [4, 31]. Although natalizumab is frequently described as an IS agent, its action can be categorized as immunomodulatory. Here we have used IS to denote natalizumab treatment. CSF was obtained from several persons with or without MS and with or without IS treatment. Lanes 1 and 2 of Figure 4A reveal no significant EBV bands in persons without MS using PCR with primers for EBV alone. We detected higher EBV levels in patients with MS not treated with IS than in controls without MS. Owing to the small number of subjects, the significance of this difference should be further evaluated. Lanes 3–6 in Figure 4, from patients with MS treated with IS, show EBV bands of varying intensity. Lanes 7 and 8, from patients with MS not treated with IS, show faint bands. Because MS stages differ between patients, however, we cannot at this time assume any causal relationship among natalizumab treatment, CSF EBV levels, and JCV recombinant formation. None of the patients tested has had PML diagnosed. The band for Raji cells in lane 10 is presented as a control.
Figure 4.

Differential presence of Epstein-Barr virus (EBV) DNA and a JC virus (JCV)–EBV recombination product in patients with multiple sclerosis (MS) either receiving or not receiving immunosuppression (IS) with natalizumab. In this experiment IS was with a single agent, natalizumab (Biogen). A, Polymerase chain reaction (PCR) was performed on cerebrospinal fluid (CSF) samples from patients using primers from EBV alone chosen to detect sequence surrounding the 15–base pair (bp) homology to the JCV noncoding control region (NCCR). PCR followed by a nested PCR used the primers described in “Methods” section. Numbers above gel lanes refer to samples from individual patients. B, PCR was performed using one primer from JCV and one from EBV on DNA isolated from the CSF of patients using a method that purifies and concentrates DNA; see “Methods” section for details and primers used. PCR was not nested, so the expected recombination intermediate is larger (421 bp, arrow) than the product containing the same intermediate in Figure 3 (177 bp). No recombination is detected in a patient with no MS. The samples MS + IS3 and MS no IS7 refer to samples from patients 3 and 7 in the gel shown in A; the recombination band is much more intense in the former sample than in the latter. Data from real-time Cts are presented in C. C, Relative DNA amounts of samples with bands from A and B, based on real-time PCR. Cts from samples obtained from patients without MS were similar and high, and they were assigned a value of 1 for calculating relative amounts of DNA for all other samples based on their Cts. Levels of amplification in C cannot be directly compared with the bands in A and B, because the latter were subjected to more PCR cycles to visualize them. D, Sequencing of the 421-bp band (arrow in B), derived from a patient with MS treated with IS (MS + IS3). Sequencing was in either direction using primers EBV7F and JCV7R (blue shading). The EBV-JCV homology corresponding to the smaller progressive multifocal leukoencephalopathy (PML)–related JCV NCCR deletion is shown in red. Note that this sequence cannot be derived as an artifact of nucleofection because it is from a living patient. The sequence demonstrates an EBV-JCV recombination product, occurring in patients with MS, switching sequences specifically at the 15-bp NCCR-EBV homology. (Additional control data are provided in the Supplementary Notes to Figure 4D.). Abbreviations: Cts, cycle threshold crossings; Std, standard size markers.
Figure 4B shows PCR reactions performed on CSF with a forward primer from EBV and a reverse primer from JCV, that, the opposite orientation to that of Figure 3. The band expected if recombination occurs at the 15-bp homology would be 421 bp. PCR of CSF from a patient without MS shows no potential recombination band. Both patients with MS (corresponding to lanes 3 and 7 in Figure 4A) showed bands of the anticipated size. Figure 4C shows real-time PCR for the bands of Figure 4A and 4B. It can be seen that the relative amount of DNA for the MS + IS3 CSF sample is much higher than those for the other samples. This result is not reflected in Figure 4A because by the time bands in 4A are highly visible, the other samples have had several cycles to catch up. Patients without MS have essentially no JCV-EBV recombined DNA in their CSF, whereas the patients with the highest CSF levels of EBV and recombined DNA were those treated with IS, that is, those susceptible to PML (Figure 4C). Again, we cannot presently conclude a causal relationship between natalizumab treatment and JCV-EBV recombinant formation; larger sample sizes would be required to establish any possible causal relationship. Although neither EBV nor recombined DNA is present at high levels in CSF, based on ease of detection by PCR, the levels of unrecombined EBV are much higher than those of the recombined form. The EBV level in the highest MS + IS sample is 860 pg/mL CSF. This is in comparison with a control standard of EBV plasmid DNA (BGLF5-pCDNA3.1). All of the patients' CSF samples were tested for JCV DNA using several sets of PCR primers. We do not detect intact, independent JCV DNA in CSF from any of our patient samples. This indicates that any JCV DNA detected in the CSF is in the form of JCV-EBV recombinants. It is also conceivable that recombination between JCV and EBV occurs in a cell outside the CSF before gaining access to that compartment.
The sequence DNA of the MS + IS3 lane (Figure 4D) is larger than the 421 bp between the primers because each primer was used to sequence separately in reverse order. Sequencing reveals the same JCV-EBV recombination transition as seen in Figure 4. Although the number of patient samples here is small, the proof of principle is that JCV-EBV recombination can be detected in patients with MS susceptible to PML. More samples will be analyzed to affirm and extend these CSF studies.
We verified recombination between JCV and EBV with a second method using EBV PCR primers only to generate a hybridization template. No JCV primers have been used, and no JCV sequences have been added to the patients' nonprocessed CSF samples. Therefore, if JCV sequences are recombined with EBV, they have not been artifactually derived by our methods. Our EBV primers all bracket the 15-bp homology with JCV in the BGLF5 gene. Agarose gel bands were subjected to Southern blotting using either a phosphorus 32–labeled JCV archetype genome or a labeled EBV BGLF5 fragment. The EBV band from the BGLF5 coding region containing the 15-bp homology, using one of the primer pairs specified in “Methods” section, would be 378 bp. There is no band of this size hybridizing to JCV. Figure 5 shows results of hybridization of blots to either JCV or EBV probes. Results show that patients 3–5 (with MS and no IS) have no JCV sequences in bands higher than the expected EBV band, although the patient of lane 5 does harbor EBV. No JCV is associated with any of the EBV bands at 378 bp. In contrast, there are strong JCV bands at the positions of EBV bands of approximately 5 kb and higher. These bands contain both EBV DNA (white arrows) and potentially incorporated JCV DNA (black arrows).
Figure 5.

Recombination between JV virus (JCV) and Epstein-Barr virus (EBV) demonstrated by Southern blotting. Here we used a second method to demonstrate JCV-EBV recombination, one that does not rely on polymerase chain reaction (PCR) amplification using one primer from each virus. This method uses no JCV primers at all in any step. Therefore, if JCV sequences are detected with EBV, they are there before amplification and are not PCR artifacts. Cerebrospinal fluid (CSF) samples from patients with multiple sclerosis (MS) with no natalizumab treatment (MS no IS) and from those with treatment (MS + IS) were subjected to PCR with primers both from the EBV BGLF5 gene on either side of the 15–base pair (bp) identity with the JCV noncoding control region (NCCR). The size of the EBV band thus generated with no insert would be 378 bp, as shown at the bottom of the panel (EBV primers, EBV probe). Probing with JCV sequences is shown in red. The JCV probe is a random primed JCV archetype. Probing with EBV sequences. is shown in green. The black and white arrows indicate positions of JCV and EBV higher than anticipated from amplification with the EBV primers. The sizes of these bands, but not their existence, vary in different experiments. There is consistently a band slightly below the position of the full-length JCV archetype. The black and white arrows in this figure indicate the black bands (JCV) that coincide with the white bands (EBV). Arrows denote positions of bands in this experiment rather than positions of bands consistent in all experiments. The positions of these arrows shows that JCV and EBV bands coincide. JCV complete archetype standard, 5.1 kb, is hybridized to JCV (left) and EBV (right). Abbreviations: BL, blank lane; CC, column control; no viral sequences are derived from the PCR cleanup column.
DISCUSSION
To our knowledge this is the first demonstration that 2 entirely different classes of DNA virus can undergo recombination in a cell harboring both. Although this was demonstrated in a B-lymphoblast line, it may not necessarily occur in B cells in persons, and the possibility that it occurs in bone marrow remains speculative. Estimates of the fraction of B cells that harbor latent EBV in approximately 90% of adults vary from 1 to 50 per 106 cells. EBV infects a variety of epithelial cells [32–34], and JCV infects renal tubular epithelial cells [3, 35]. Epithelial cells, lytically infected with EBVl including nasopharyngeal cells, can fuse [36] The proximity of these cells to tonsillar cells harboring JCV [31, 37] provides a potential mechanism for transfer of recombinant JCV among cell types. Nanbo and colleagues [34] have described cell-to-cell contact-mediated EBV transmission.
BGLF5 (mutated in Figures 3 and 4) is an alkaline nuclease, expressed only during active infection, that shuts off host cell messenger RNA function and would also shut off JCV protein expression. The strategic RNA-binding “bridge” in BGLF5 of gamma herpesviruses is a flexible motif that spans the active sites of alkaline nuclease activity [38]. Mutations in it are especially prone to disrupting host cell shutoff. Because latent EBV has 50–80 episomes, it is unlikely that mutation in this region in one or a few episomes would have any effect on EBV. Its advantage to JCV could be in allowing JCV to gain a foothold in a new cell type after cell-cell transfer [34].
MS represents an intriguing intersection of EBV, JCV and IS, although neither virus is firmly linked to MS etiology. Natalizumab, an antibody to integrin α4, is associated with many current non-AIDS cases of PML, and PML has developed in a small percentage of patients receiving this treatment [4, 31, 39]. Prior infection with EBV is strongly serologically linked to MS, although the virus is not consistently found in MS lesions [29]. IS activates proliferation of B cells infected with EBV [1], presumably predisposing any JCV-coinfected cells to opportunistic recombination. All of the NCCR rearrangements described can be derived from interviral recombination among JCV molecules alone [9], so that recombination with EBV is not necessarily the exclusive route for rearrangement of JCV. What advantage, then, is accrued by JCV through recombination with EBV? Several intriguing possibilities present themselves. Insertion of JCV sequences into EBV at the 15-bp homology can create a duplication of the homology at each end of the insert. That creates a potential fork-stalling palindrome. There is no such fork-stalling site at the 23-bp deleted sequence in JCV alone. Thus recombination with EBV generates a favorable feature promoting JCV recombination. No known single model of recombination can account for the types of recombinants seen in Figures 3 and 4. Supplementary Figure 3 presents a hypothetical model involving cooperation of several known recombination modes [43, 44].
Figure 4 is of key importance to this study because it seems to imply a relationship between natalizumab treatment, CSF EBV levels, and levels of JCV-EBV recombinants. The nuances and caveats of this interpretation are discussed in the Supplementary Notes to Figure 4.
Under the right circumstances, EBV can mediate cell fusion and syncytium formation [36, 40, 41]. Glial cells transfected with an EBV-based CD4 expression vector have been observed to undergo fusion [42]. Furthermore, induction of lytic EBV infection of Burkitt lymphoma cells reportedly stimulates cell-to-cell contact-mediated EBV transmission [34]. By capitalizing on such EBV capabilities, JCV DNA may be transported “piggy-back” into cell types not normally infected by JCV. It may then recombine with itself, or as a JCV-EBV hybrid, to advance progression of JCV to neurovirulence.
JCV-EBV recombination is potentially critical because, even if it occurs on a small scale, cells bearing JCV-EBV recombinants can gain advantages regarding transit of JCV into new cell types and enhanced ability of JCV to undergo further recombination capable of generating neurovirulent rearrangements. Interviral recombination involving DNA viruses may be applicable to several viruses in addition to JCV and EBV. This possibility may have implications for the etiology or treatment of several diseases in which interaction of viruses can confer an adaptive advantage on certain of these viruses.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Dr Lindsey Hutt-Fletcher, PhD, of Louisiana State University Health Sciences Center, Shreveport, for review, comments and discussion regarding EBV and Dr H. J. Delecluse, PhD, University of Heidelberg, for generously providing EBV BGLF5 complementary DNA clone, plasmid BGLF5-pCDNA3.1. Malcolm Hall, BA, reviewed the manuscript for scientific clarity.
Financial support. This work was supported by the PML Consortium (E. M. J.) and the National Institutes of Health (D. C. D.).
Conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
REFERENCES
- 1.Longnecker RM, Kieff E, Cohen JI. Epstein Barr virus. In: Knipe D, Howley PM, eds. Fields virology. Philadelphia, PA: Williams & Wilkins, 2013; pp. 1898–959. [Google Scholar]
- 2.Berger JR, Houff SA, Gurwell J, Vega N, Miller CS, Danaher RJ. JC virus antibody status underestimates infection rates. Ann Neurol 2013; 74:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger JR, Major EO. Progressive multifocal leukoencephalopathy. Semin Neurol 1999; 19:193–200. [DOI] [PubMed] [Google Scholar]
- 4.Reid CE, Li H, Sur G et al. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J Infect Dis 2011; 204:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tavazzi E, White MK, Khalili K. Progressive multifocal leukoencephalopathy: clinical and molecular aspects. Rev Med Virol 2012; 22:18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol 1984; 51:458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yogo Y, Kitamura T, Sugimoto C et al. Sequence rearrangement in JC virus DNAs molecularly cloned from immunosuppressed renal transplant patients. J Virol 1991; 65:2422–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfister LA, Letvin NL, Koralnik IJ. JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol 2001; 75:5672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson EM, Wortman MJ, Dagdanova AV, Lundberg PS, Daniel DC. Polyomavirus JC in the context of immunosuppression: a series of adaptive, DNA replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin Dev Immunol 2013; doi:10.1155/2013/197807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koralnik IJ, Wuthrich C, Dang X et al. JC virus granule cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol 2005; 57:576–80. [DOI] [PubMed] [Google Scholar]
- 11.Agnihotri SP, Dang X, Carter JL et al. JCV GCN in a natalizumab-treated MS patient is associated with mutations of the VP1 capsid gene. Neurology 2014; 83:727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Pasquier RA, Corey S, Margolin DH et al. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology 2003; 61:775–82. [DOI] [PubMed] [Google Scholar]
- 13.Del Valle L, Enam S, Lara C, Miklossy J, Khalili K, Gordon J. Primary central nervous system lymphoma expressing the human neurotropic polyomavirus, JC virus, genome. J Virol 2004; 78:3462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atwood WJ, Amemiya K, Traub R, Harms J, Major EO. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology 1992; 190:716–23. [DOI] [PubMed] [Google Scholar]
- 15.Chapagain ML, Nerurkar VR. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J Infect Dis 2010; 202:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darbinyan A, White MK, Akan S et al. Alterations of DNA damage repair pathways resulting from JCV infection. Virology 2007; 364:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neel JV, Major EO, Awa AA et al. Hypothesis: “rogue cell”-type chromosomal damage in lymphocytes is associated with infection with the JC human polyoma virus and has implications for oncopenesis. Proc Natl Acad Sci U S A 1996; 93:2690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costantino L, Sotiriou SK, Rantala JK et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014; 343:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houff SA, Major EO, Katz DA et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med 1988; 318:301–5. [DOI] [PubMed] [Google Scholar]
- 20.Rieckmann P, Michel U, Kehrl JH. Regulation of JC virus expression in B lymphocytes. J Virol 1994; 68:217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frohman EM, Monaco MC, Remington G et al. JC virus in CD34+ and CD19+ cells in patients with multiple sclerosis treated with natalizumab. JAMA Neurol 2014; 71:596–602. [DOI] [PubMed] [Google Scholar]
- 22.Houff SA, Berger J. The curious incident of the dog in the nighttime: does the absence of virus replication in Epstein-Barr virus-transformed B cells point to an important feature of JC virus biology? J Infect Dis 2010; 202:181–3. [DOI] [PubMed] [Google Scholar]
- 23.Johnson EM, Karn J, Allfrey VG. Early nuclear events in the induction of lymphocyte proliferation by mitogens. Effects of concanavalin A on the phosphorylation and distribution of non-histone chromatin proteins. J Biol Chem 1974; 249:4990–9. [PubMed] [Google Scholar]
- 24.Johnson EM, Kinoshita Y, Weinreb DB et al. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res 2006; 83:929–43. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc Natl Acad Sci U S A 2001; 98:10085–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frappier L. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr Opin Virol 2012; 2:733–9. [DOI] [PubMed] [Google Scholar]
- 27.Daniel DC, Kinoshita Y, Khan MA et al. Internalization of exogenous human immunodeficiency virus-1 protein, Tat, by KG-1 oligodendroglioma cells followed by stimulation of DNA replication initiated at the JC virus origin. DNA Cell Biol 2004; 23:858–67. [DOI] [PubMed] [Google Scholar]
- 28.Johnson EM, Jelinek WR. Replication of a plasmid bearing a human Alu-family repeat in monkey COS-7 cells. Proc Natl Acad Sci U S A 1986; 83:4660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. I. The role of infection. Ann Neurol 2007; 61:288–99. [DOI] [PubMed] [Google Scholar]
- 30.Goodin DS. The causal cascade to multiple sclerosis: a model for MS pathogenesis. PLoS One 2009; 4:e4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger JR, Houff S. Progressive multifocal leukoencephalopathy: lessons from AIDS and natalizumab. Neurol Res 2006; 28:299–305. [DOI] [PubMed] [Google Scholar]
- 32.Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 2002; 8:594–9. [DOI] [PubMed] [Google Scholar]
- 33.Hutt-Fletcher LM. Epstein-Barr virus entry. J Virol 2007; 81:7825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanbo A, Terada H, Kachi K, Takada K, Matsuda T. Roles of cell signaling pathways in cell-to-cell contact-mediated Epstein-Barr virus transmission. J Virol 2012; 86:9285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 1992; 5:49–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato H, Takimoto T, Tanaka S, Ogura H, Shiraishi K, Tanaka J. Cytopathic effects induced by Epstein-Barr virus replication in epithelial nasopharyngeal carcinoma hybrid cells. J Virol 1989; 63:3555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato A, Kitamura T, Takasaka T et al. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J Neurovirol 2004; 10:244–9. [DOI] [PubMed] [Google Scholar]
- 38.Horst D, Burmeister WP, Boer IG et al. The “bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J Virol 2012; 86:9175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall LJ, Ferenczy MW, Daley EL, Jensen PN, Ryschkewitsch CF, Major EO. Lymphocyte gene expression and JC virus noncoding control region sequences are linked with the risk of progressive multifocal leukoencephalopathy. J Virol 2014; 88:5177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc Natl Acad Sci U S A 2009; 106:20464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinkovics JG. Hodgkin's disease revisited: Reed-Sternberg cells as natural hybridomas. Crit Rev Immunol 1991; 11:33–63. [PubMed] [Google Scholar]
- 42.Volsky B, Sakai K, Reddy MM, Volsky DJ. A system for the high efficiency replication of HIV-1 in neural cells and its application to anti-viral evaluation. Virology 1992; 186:303–8. [DOI] [PubMed] [Google Scholar]
- 43.Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, Haber JE. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 2010; 24:1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenab S, Johnson EM. A dual-circular plasmid structure dependent on DNA replication generated in monkey COS7 cells and cell extracts. Biochem Biophys Res Commun 1989; 160:53–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.